

Transcriptional induction of slit diaphragm genes by Lmx1b is required in podocyte differentiation
- PMID: 11956244
- PMCID: PMC150942
- DOI: 10.1172/JCI13954
Transcriptional induction of slit diaphragm genes by Lmx1b is required in podocyte differentiation
Abstract
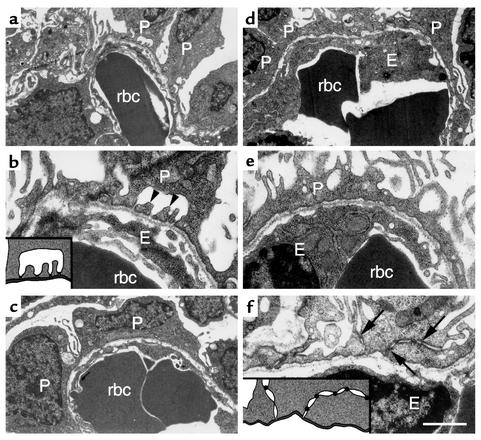
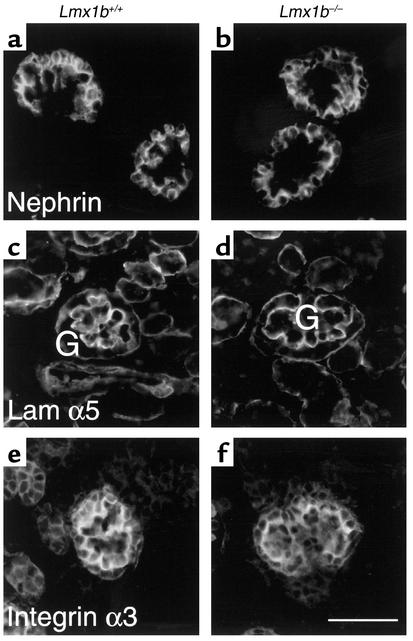
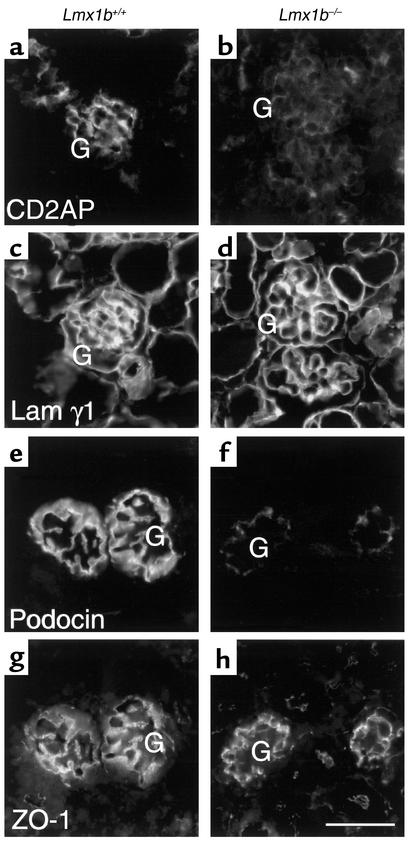
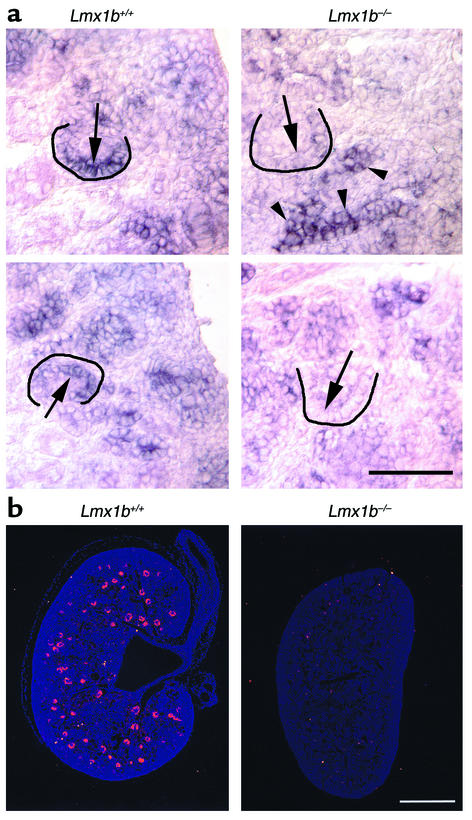
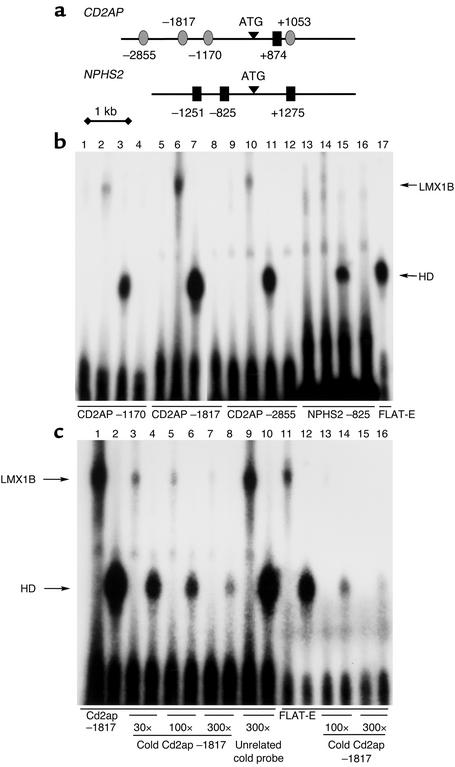
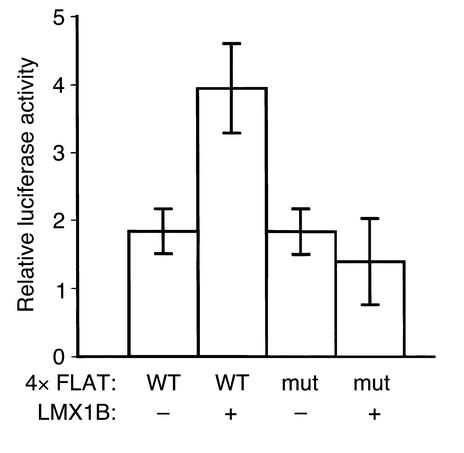
LMX1B encodes a LIM-homeodomain transcription factor. Mutations in LMX1B cause nail-patella syndrome (NPS), an autosomal dominant disease with skeletal abnormalities, nail hypoplasia, and nephropathy. Expression of glomerular basement membrane (GBM) collagens is reduced in Lmx1b(-/-) mice, suggesting one basis for NPS nephropathy. Here, we show that Lmx1b(-/-) podocytes have reduced numbers of foot processes, are dysplastic, and lack typical slit diaphragms, indicating an arrest in development. Using antibodies to podocyte proteins important for podocyte function, we found that Lmx1b(-/-) podocytes express near-normal levels of nephrin, synaptopodin, ZO-1, alpha3 integrin, and GBM laminins. However, mRNA and protein levels for CD2AP and podocin were greatly reduced, suggesting a cooperative role for these molecules in foot process and slit diaphragm formation. We identified several LMX1B binding sites in the putative regulatory regions of both CD2AP and NPHS2 (podocin) and demonstrated that LMX1B binds to these sequences in vitro and can activate transcription through them in cotransfection assays. Thus, LMX1B regulates the expression of multiple podocyte genes critical for podocyte differentiation and function. Our results indicate that reduced levels of proteins associated with foot processes and the glomerular slit diaphragm likely contribute, along with reduced levels of GBM collagens, to the nephropathy associated with NPS.
Figures
Similar articles
-
The LIM-homeodomain transcription factor Lmx1b plays a crucial role in podocytes.J Clin Invest. 2002 Apr;109(8):1073-82. doi: 10.1172/JCI13961. J Clin Invest. 2002. PMID: 11956245 Free PMC article.
-
In vivo expression of putative LMX1B targets in nail-patella syndrome kidneys.Am J Pathol. 2003 Jul;163(1):145-55. doi: 10.1016/S0002-9440(10)63638-3. Am J Pathol. 2003. PMID: 12819019 Free PMC article.
-
How are podocytes affected in nail-patella syndrome?Pediatr Nephrol. 2008 Jul;23(7):1017-20. doi: 10.1007/s00467-007-0714-9. Epub 2008 Feb 6. Pediatr Nephrol. 2008. PMID: 18253764 Free PMC article.
-
Insight into podocyte differentiation from the study of human genetic disease: nail-patella syndrome and transcriptional regulation in podocytes.Pediatr Res. 2002 May;51(5):551-8. doi: 10.1203/00006450-200205000-00002. Pediatr Res. 2002. PMID: 11978876 Review.
-
Nail-patella syndrome.Pflugers Arch. 2017 Aug;469(7-8):927-936. doi: 10.1007/s00424-017-2013-z. Epub 2017 Jul 5. Pflugers Arch. 2017. PMID: 28681095 Review.
Cited by
-
Quality assessment and refinement of chromatin accessibility data using a sequence-based predictive model.Proc Natl Acad Sci U S A. 2022 Dec 20;119(51):e2212810119. doi: 10.1073/pnas.2212810119. Epub 2022 Dec 12. Proc Natl Acad Sci U S A. 2022. PMID: 36508674 Free PMC article.
-
Novel missense mutation affecting the LIM-A domain of LMX1B in a family with Nail-Patella syndrome.Intractable Rare Dis Res. 2019 Feb;8(1):14-19. doi: 10.5582/irdr.2018.01131. Intractable Rare Dis Res. 2019. PMID: 30881852 Free PMC article.
-
Spectrum of LMX1B mutations: from nail-patella syndrome to isolated nephropathy.Pediatr Nephrol. 2017 Oct;32(10):1845-1850. doi: 10.1007/s00467-016-3462-x. Epub 2016 Jul 23. Pediatr Nephrol. 2017. PMID: 27450397 Review.
-
Genetics of focal segmental glomerulosclerosis.Pediatr Nephrol. 2007 May;22(5):638-44. doi: 10.1007/s00467-007-0445-y. Epub 2007 Mar 9. Pediatr Nephrol. 2007. PMID: 17347836 Free PMC article.
-
Neph1 and nephrin interaction in the slit diaphragm is an important determinant of glomerular permeability.J Clin Invest. 2003 Jul;112(2):209-21. doi: 10.1172/JCI18242. J Clin Invest. 2003. PMID: 12865409 Free PMC article.
References
-
- Smoyer WE, Mundel P. Regulation of podocyte structure during the development of nephrotic syndrome. J Mol Med. 1998;76:172–183. - PubMed
-
- Somlo S, Mundel P. Getting a foothold in nephrotic syndrome. Nat Genet. 2000;24:333–335. - PubMed
-
- Tryggvason K, Ruotsalainen V, Wartiovaara J. Discovery of the congenital nephrotic syndrome gene discloses the structure of the mysterious molecular sieve of the kidney. Int J Dev Biol. 1999;43:445–451. - PubMed
-
- Tryggvason K. Unraveling the mechanisms of glomerular ultrafiltration: nephrin, a key component of the slit diaphragm. J Am Soc Nephrol. 1999;10:2440–2445. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
