

Multiple diverse ligands binding at a single protein site: a matter of pre-existing populations
- PMID: 11790828
- PMCID: PMC2373439
- DOI: 10.1110/ps.21302
Multiple diverse ligands binding at a single protein site: a matter of pre-existing populations
Abstract
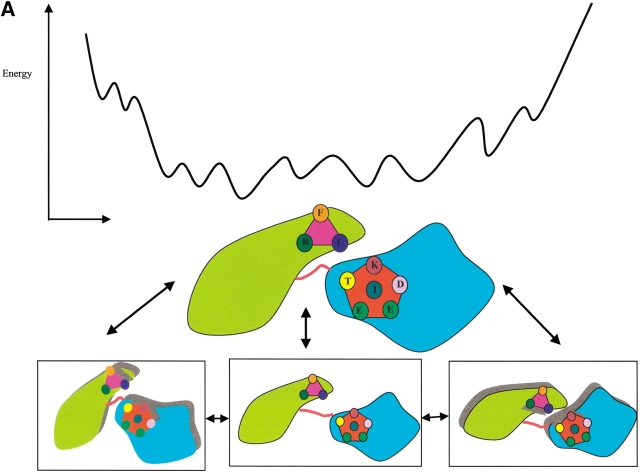
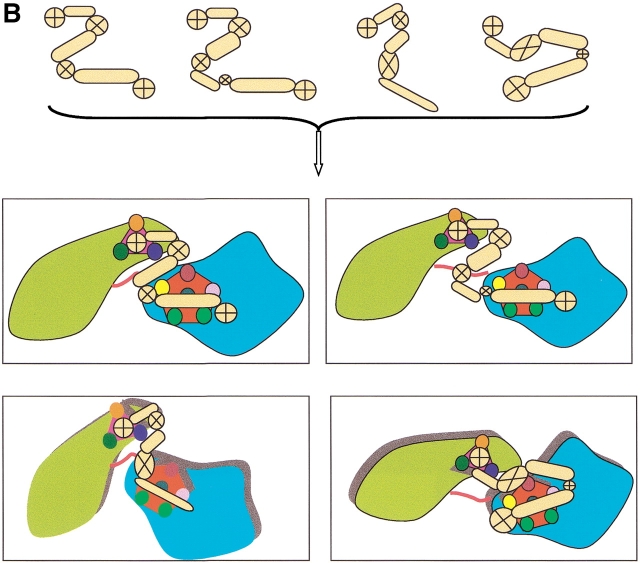
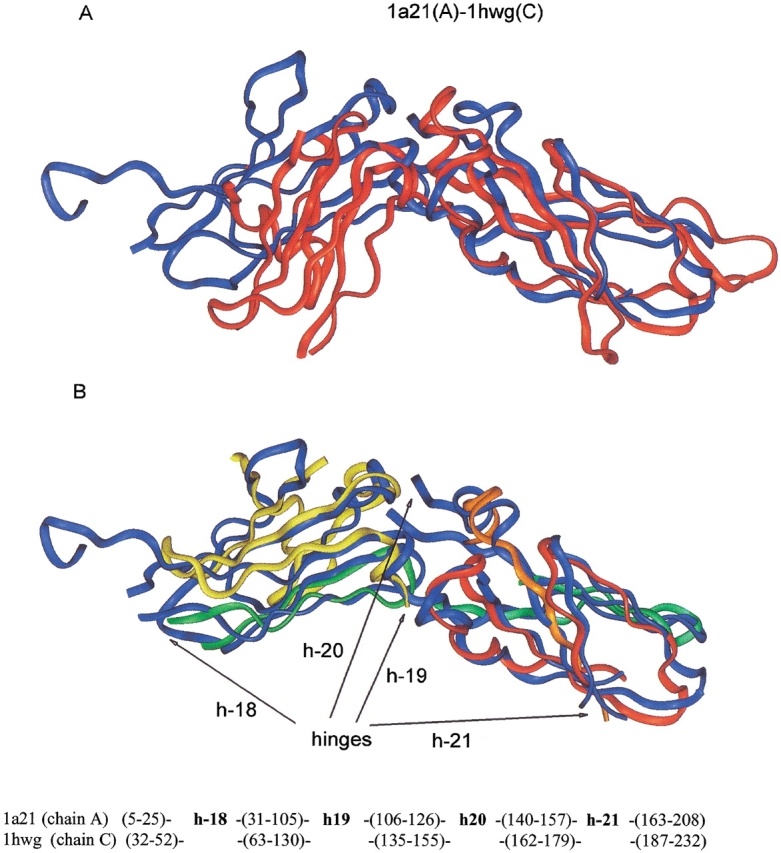
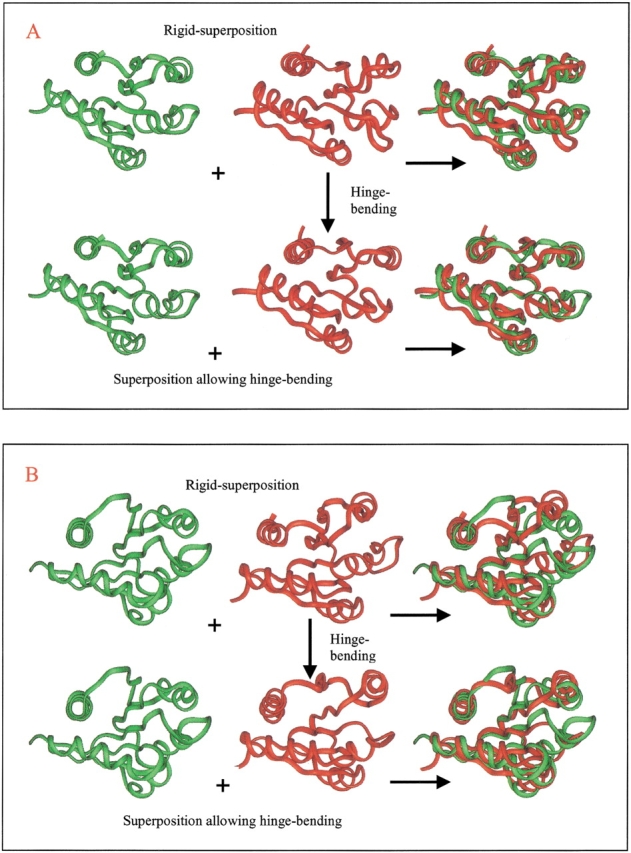
Here, we comment on the steadily increasing body of data showing that proteins with specificity actually bind ligands of diverse shapes, sizes, and composition. Such a phenomenon is not surprising when one considers that binding is a dynamic process with populations in equilibrium and that the shape of the binding site is strongly influenced by the molecular partner. It derives implicitly from the concept of populations. All proteins, specific and nonspecific, exist in ensembles of substates. If the library of ligands in solution is large enough, favorably matching ligands with altered shapes and sizes can be expected to bind, with a redistribution of the protein populations. Point mutations at spatially distant sites may exert large conformational rearrangements and hinge effects, consistent with mutations away from the binding site leading to population shifts and (cross-)drug resistance. A similar effect is observed in protein superfamilies, in which different sequences with similar topologies display similar large-scale dynamic motions. The hinges are frequently at analogous sites, yet with different substrate specificity. Similar topologies yield similar conformational isomers, although with different distributions of population times, owing to the change in the conditions, that is, the change in the sequences. In turn, different distributions relate to binding of different sizes and shapes. Hence, the binding site shape and size are defined by the ligand. They are not independent entities of fixed proportions and cannot be analyzed independently of the binding partner. Such a proposition derives from viewing proteins as dynamic distributions, presenting to the incoming ligands a range of binding site shapes. It illustrates how presumably specific binding molecules can bind multiple ligands. In terms of drug design, the ability of a single receptor to recognize many dissimilar ligands shows the need to consider more diverse molecules. It provides a rationale for higher affinity inhibitors that are not derived from substrates at their transition states and indicates flexible docking schemes.
Figures
Similar articles
-
Ligand binding remodels protein side-chain conformational heterogeneity.Elife. 2022 Mar 21;11:e74114. doi: 10.7554/eLife.74114. Elife. 2022. PMID: 35312477 Free PMC article.
-
Protein promiscuity: drug resistance and native functions--HIV-1 case.J Biomol Struct Dyn. 2005 Jun;22(6):615-24. doi: 10.1080/07391102.2005.10531228. J Biomol Struct Dyn. 2005. PMID: 15842167
-
Folding funnels and conformational transitions via hinge-bending motions.Cell Biochem Biophys. 1999;31(2):141-64. doi: 10.1007/BF02738169. Cell Biochem Biophys. 1999. PMID: 10593256 Review.
-
Flexible docking allowing induced fit in proteins: insights from an open to closed conformational isomers.Proteins. 1998 Aug 1;32(2):159-74. Proteins. 1998. PMID: 9714156
-
Allostery and population shift in drug discovery.Curr Opin Pharmacol. 2010 Dec;10(6):715-22. doi: 10.1016/j.coph.2010.09.002. Epub 2010 Sep 29. Curr Opin Pharmacol. 2010. PMID: 20884293 Free PMC article. Review.
Cited by
-
Substrate binding and protein conformational dynamics measured by 2D-IR vibrational echo spectroscopy.Proc Natl Acad Sci U S A. 2007 Feb 20;104(8):2637-42. doi: 10.1073/pnas.0610027104. Epub 2007 Feb 12. Proc Natl Acad Sci U S A. 2007. PMID: 17296942 Free PMC article.
-
Seven transmembrane receptors as shapeshifting proteins: the impact of allosteric modulation and functional selectivity on new drug discovery.Pharmacol Rev. 2010 Jun;62(2):265-304. doi: 10.1124/pr.108.000992. Epub 2010 Apr 14. Pharmacol Rev. 2010. PMID: 20392808 Free PMC article. Review.
-
Investigation of Molecular Details of Keap1-Nrf2 Inhibitors Using Molecular Dynamics and Umbrella Sampling Techniques.Molecules. 2019 Nov 12;24(22):4085. doi: 10.3390/molecules24224085. Molecules. 2019. PMID: 31726716 Free PMC article.
-
Antigen binding allosterically promotes Fc receptor recognition.MAbs. 2019 Jan;11(1):58-74. doi: 10.1080/19420862.2018.1522178. Epub 2018 Oct 5. MAbs. 2019. PMID: 30212263 Free PMC article.
-
Trapping of peptide-based surrogates in an artificially created channel of cytochrome c peroxidase.Protein Sci. 2003 Feb;12(2):278-87. doi: 10.1110/ps.0228403. Protein Sci. 2003. PMID: 12538891 Free PMC article.
References
-
- Abad-Zapatero, C., Rydel, T.G., and Erickson, J. 1990. Revised 2.3 Åstructure of porcine pepsin: Evidence of a flexible subdomain. Proteins 8 62–81. - PubMed
-
- Baldwin, E.T., Bhat, T.N., Gulnik, S., Hosur, M.V., Sowder, R.C., Cachau, R.E., Collins. J., Silva, A.M., and Erickson, J.W. 1993. Crystal structures of native and inhibited forms of human cathepsin D: Implications for lysosomal targeting and drug design. Proc. Natl. Acad. Sci. 90 6796–6800. - PMC - PubMed
-
- Banner, D.W., D'Arcy, A., Chene, C., Winkler, F.K., Guha, A., Konigsberg, W.H., Nemerson, Y., and Kirchhofer, D. 1996. The crystal structures of the complex of blood coagulation factor VIIa with soluble tissue factor Nature 380 41–46. - PubMed
-
- Barak, R. and Eisenbach, M. 1992. Correlation between phosphorylation of the chemotaxis protein Che Y and its activity at the flaggelar motor. Biochemistry 31 1821–1826. - PubMed
-
- Bernstein, F.C., Koetzle, T.F., Williams, G.J.B., Meyer, E.F., Brice, M.D., Rodgers, J.R., Kennard, O., Shimanouchi, T., and Tasumi, M. 1977. The Protein Data Bank: A computer based archival file for macromolecular structures. J. Mol. Biol. 112 535–542. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
