

Heterologous dimerization domains functionally substitute for the double-stranded RNA binding domains of the kinase PKR
- PMID: 11447114
- PMCID: PMC125533
- DOI: 10.1093/emboj/20.14.3728
Heterologous dimerization domains functionally substitute for the double-stranded RNA binding domains of the kinase PKR
Abstract
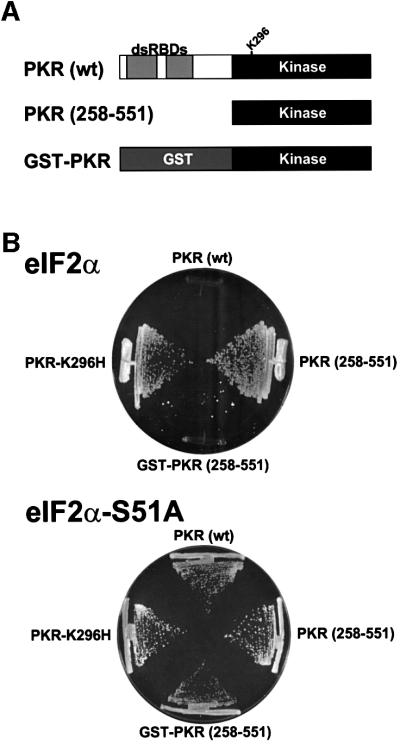
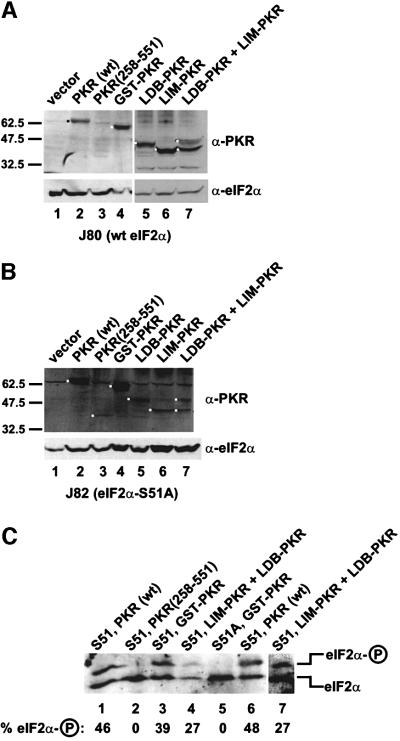
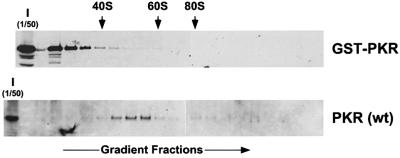
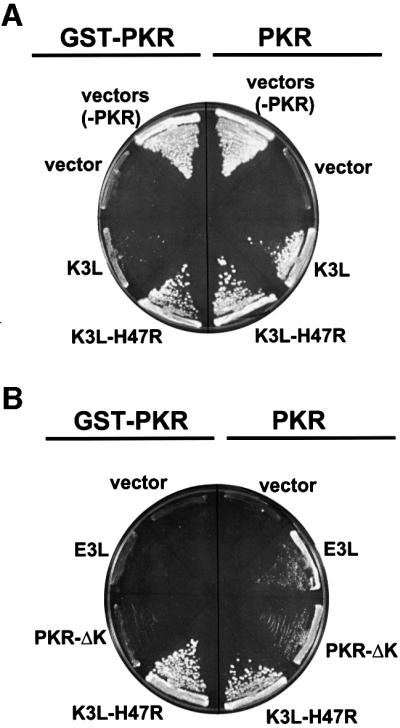
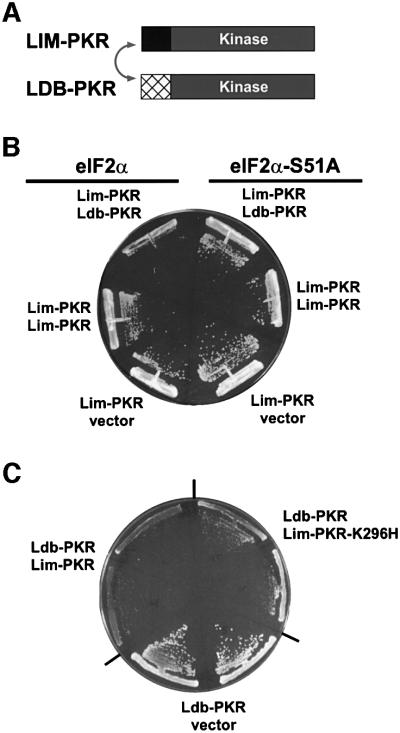
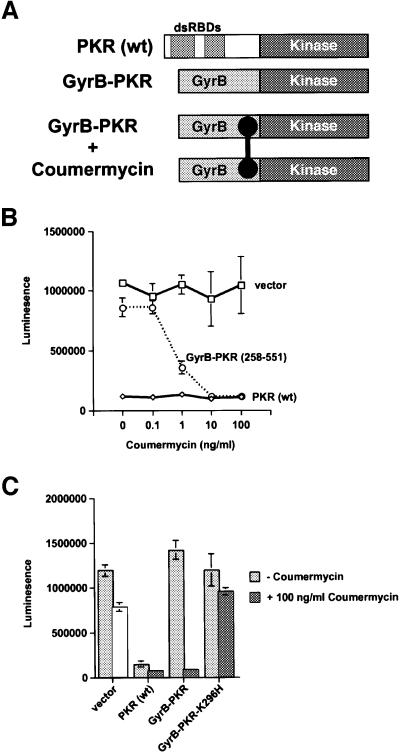
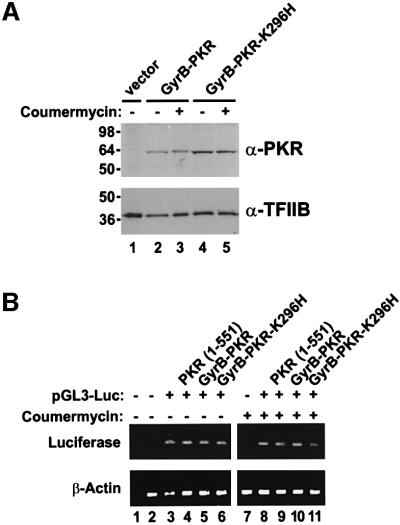
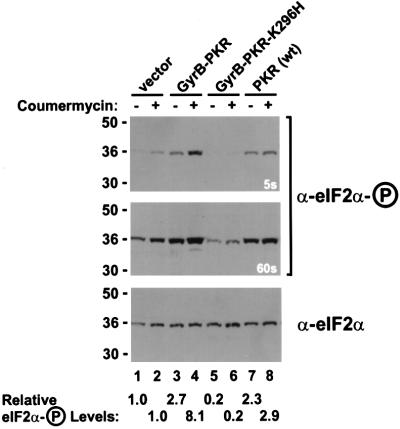
The protein kinase PKR (dsRNA-dependent protein kinase) phosphorylates the eukaryotic translation initiation factor eIF2alpha to downregulate protein synthesis in virus-infected cells. Two double-stranded RNA binding domains (dsRBDs) in the N-terminal half of PKR are thought to bind the activator double-stranded RNA, mediate dimerization of the protein and target PKR to the ribosome. To investigate further the importance of dimerization for PKR activity, fusion proteins were generated linking the PKR kinase domain to heterologous dimerization domains. Whereas the isolated PKR kinase domain (KD) was non-functional in vivo, expression of a glutathione S-transferase-KD fusion, or co-expression of KD fusions containing the heterodimerization domains of the Xlim-1 and Ldb1 proteins, restored PKR activity in yeast cells. Finally, coumermycin-mediated dimerization of a GyrB-KD fusion protein increased eIF2alpha phosphorylation and inhibited reporter gene translation in mammalian cells. These results demonstrate the critical importance of dimerization for PKR activity in vivo, and suggest that a primary function of double-stranded RNA binding to the dsRBDs of native PKR is to promote dimerization and activation of the kinase domain.
Figures
Similar articles
-
trans-Autophosphorylation by the isolated kinase domain is not sufficient for dimerization or activation of the dsRNA-activated protein kinase PKR.Biochemistry. 2004 Aug 31;43(34):11027-34. doi: 10.1021/bi0360105. Biochemistry. 2004. PMID: 15323561
-
Identification and requirement of three ribosome binding domains in dsRNA-dependent protein kinase (PKR).Biochemistry. 1998 Sep 29;37(39):13816-26. doi: 10.1021/bi981472h. Biochemistry. 1998. PMID: 9753471
-
Uncoupling of RNA binding and PKR kinase activation by viral inhibitor RNAs.J Mol Biol. 2006 May 19;358(5):1270-85. doi: 10.1016/j.jmb.2006.03.003. Epub 2006 Mar 20. J Mol Biol. 2006. PMID: 16580685
-
PKR and eIF2alpha: integration of kinase dimerization, activation, and substrate docking.Cell. 2005 Sep 23;122(6):823-5. doi: 10.1016/j.cell.2005.09.007. Cell. 2005. PMID: 16179248 Review.
-
PKR; a sentinel kinase for cellular stress.Oncogene. 1999 Nov 1;18(45):6112-20. doi: 10.1038/sj.onc.1203127. Oncogene. 1999. PMID: 10557102 Review.
Cited by
-
Membrane association of the Arabidopsis ARF exchange factor GNOM involves interaction of conserved domains.Plant Cell. 2008 Jan;20(1):142-51. doi: 10.1105/tpc.107.056515. Epub 2008 Jan 18. Plant Cell. 2008. PMID: 18203920 Free PMC article.
-
Analysis of PKR structure by small-angle scattering.J Mol Biol. 2009 Apr 10;387(4):910-20. doi: 10.1016/j.jmb.2009.02.019. Epub 2009 Feb 14. J Mol Biol. 2009. PMID: 19232355 Free PMC article.
-
Design and Applications of Bifunctional Small Molecules: Why Two Heads Are Better Than One.ACS Chem Biol. 2008 Nov 21;3(11):677-692. doi: 10.1021/cb8001792. ACS Chem Biol. 2008. PMID: 19112665 Free PMC article. Review.
-
Analysis of PKR activation using analytical ultracentrifugation.Macromol Biosci. 2010 Jul 7;10(7):703-13. doi: 10.1002/mabi.201000069. Macromol Biosci. 2010. PMID: 20533534 Free PMC article.
-
Controlling activation of the RNA-dependent protein kinase by siRNAs using site-specific chemical modification.Nucleic Acids Res. 2006;34(17):4900-11. doi: 10.1093/nar/gkl464. Epub 2006 Sep 18. Nucleic Acids Res. 2006. PMID: 16982647 Free PMC article.
References
-
- Bertolotti A., Zhang,Y., Hendershot,L., Harding,H. and Ron,D. (2000) Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nature Cell Biol., 2, 326–332. - PubMed
-
- Breen J.J., Agulnick,A.D., Westphal,H. and Dawid,I.B. (1998) Interactions between LIM domains and the LIM domain-binding protein Ldb1. J. Biol. Chem., 273, 4712–4717. - PubMed
-
- Carpick B.W., Graziano,V., Schneider,D., Maitra,R.K., Lee,X. and Williams,B.R.G. (1997) Characterization of the solution complex between the interferon-induced, double-stranded RNA-activated protein kinase and HIV-1 trans-activating region RNA. J. Biol. Chem., 272, 9510–9516. - PubMed
-
- Cesareni G. and Murray,J.A.H. (1987) Plasmid vectors carrying the replication origin of filamentous single-stranded phages. In Setlow,J.K. and Hollaender,A. (eds), Genetic Engineering: Principles and Methods. Vol. 9. Plenum Press, New York, NY, pp. 135–154.
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
