

Radical SAM, a novel protein superfamily linking unresolved steps in familiar biosynthetic pathways with radical mechanisms: functional characterization using new analysis and information visualization methods
- PMID: 11222759
- PMCID: PMC29726
- DOI: 10.1093/nar/29.5.1097
Radical SAM, a novel protein superfamily linking unresolved steps in familiar biosynthetic pathways with radical mechanisms: functional characterization using new analysis and information visualization methods
Abstract
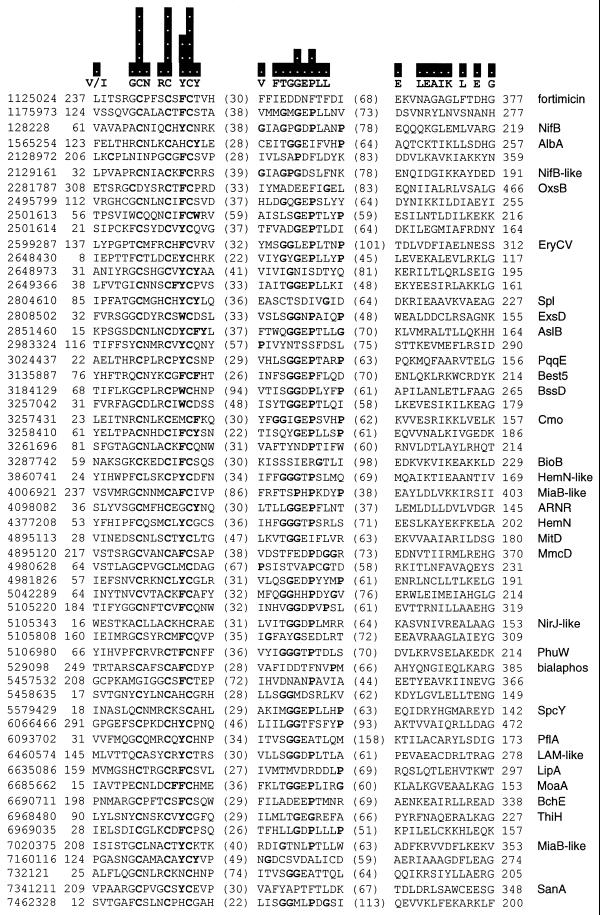
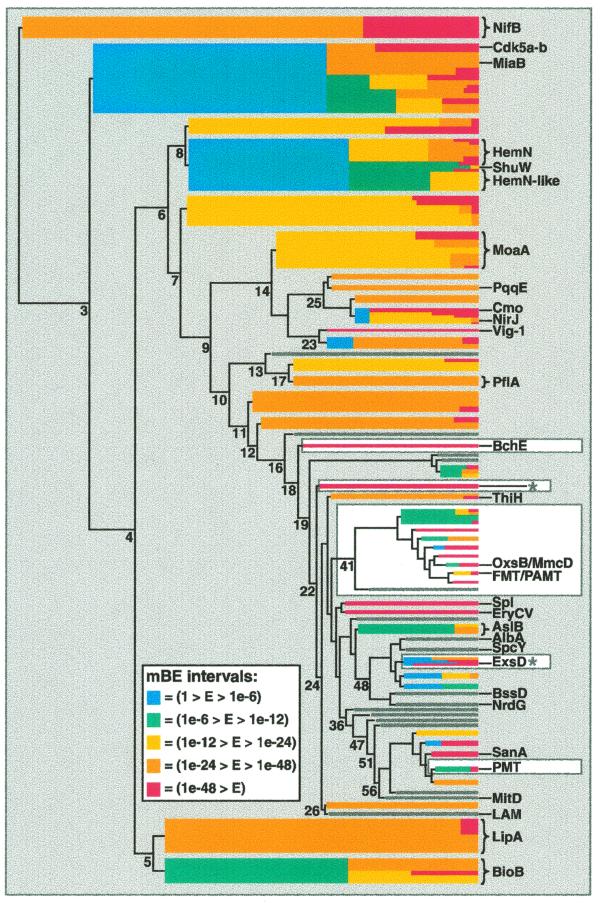
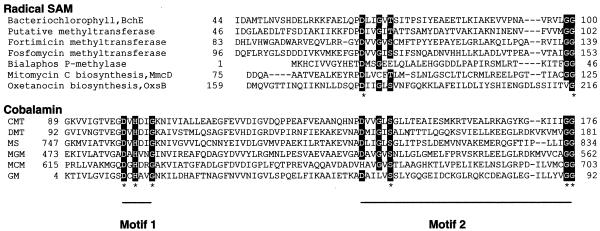
A novel protein superfamily with over 600 members was discovered by iterative profile searches and analyzed with powerful bioinformatics and information visualization methods. Evidence exists that these proteins generate a radical species by reductive cleavage of S:-adenosylmethionine (SAM) through an unusual Fe-S center. The superfamily (named here Radical SAM) provides evidence that radical-based catalysis is important in a number of previously well- studied but unresolved biochemical pathways and reflects an ancient conserved mechanistic approach to difficult chemistries. Radical SAM proteins catalyze diverse reactions, including unusual methylations, isomerization, sulfur insertion, ring formation, anaerobic oxidation and protein radical formation. They function in DNA precursor, vitamin, cofactor, antibiotic and herbicide biosynthesis and in biodegradation pathways. One eukaryotic member is interferon-inducible and is considered a candidate drug target for osteoporosis; another is observed to bind the neuronal Cdk5 activator protein. Five defining members not previously recognized as homologs are lysine 2,3-aminomutase, biotin synthase, lipoic acid synthase and the activating enzymes for pyruvate formate-lyase and anaerobic ribonucleotide reductase. Two functional predictions for unknown proteins are made based on integrating other data types such as motif, domain, operon and biochemical pathway into an organized view of similarity relationships.
Figures
Similar articles
-
The Radical SAM Superfamily.Crit Rev Biochem Mol Biol. 2008 Jan-Feb;43(1):63-88. doi: 10.1080/10409230701829169. Crit Rev Biochem Mol Biol. 2008. PMID: 18307109 Review.
-
Atlas of the Radical SAM Superfamily: Divergent Evolution of Function Using a "Plug and Play" Domain.Methods Enzymol. 2018;606:1-71. doi: 10.1016/bs.mie.2018.06.004. Epub 2018 Jul 24. Methods Enzymol. 2018. PMID: 30097089 Free PMC article.
-
Radical mechanisms of enzymatic catalysis.Annu Rev Biochem. 2001;70:121-48. doi: 10.1146/annurev.biochem.70.1.121. Annu Rev Biochem. 2001. PMID: 11395404 Review.
-
S-adenosylmethionine as an oxidant: the radical SAM superfamily.Trends Biochem Sci. 2007 Mar;32(3):101-10. doi: 10.1016/j.tibs.2007.01.002. Epub 2007 Feb 8. Trends Biochem Sci. 2007. PMID: 17291766 Review.
-
Radical-mediated enzymatic methylation: a tale of two SAMS.Acc Chem Res. 2012 Apr 17;45(4):555-64. doi: 10.1021/ar200202c. Epub 2011 Nov 18. Acc Chem Res. 2012. PMID: 22097883 Free PMC article. Review.
Cited by
-
Resistance to linezolid caused by modifications at its binding site on the ribosome.Antimicrob Agents Chemother. 2012 Feb;56(2):603-12. doi: 10.1128/AAC.05702-11. Epub 2011 Dec 5. Antimicrob Agents Chemother. 2012. PMID: 22143525 Free PMC article. Review.
-
Radical SAM enzymes in methylation and methylthiolation.Metallomics. 2012 Nov;4(11):1149-54. doi: 10.1039/c2mt20136d. Epub 2012 Sep 19. Metallomics. 2012. PMID: 22992596 Free PMC article. Review.
-
Mechanistic Insights from the Crystal Structure and Computational Analysis of the Radical SAM Deaminase DesII.Adv Sci (Weinh). 2024 Sep;11(33):e2403494. doi: 10.1002/advs.202403494. Epub 2024 Jun 28. Adv Sci (Weinh). 2024. PMID: 38943270 Free PMC article.
-
Radical SAM-Mediated Methylation of Ribosomal RNA.Methods Enzymol. 2015;560:355-76. doi: 10.1016/bs.mie.2015.03.002. Epub 2015 Apr 25. Methods Enzymol. 2015. PMID: 26253978 Free PMC article.
-
Computational Approaches: An Underutilized Tool in the Quest to Elucidate Radical SAM Dynamics.Molecules. 2021 Apr 29;26(9):2590. doi: 10.3390/molecules26092590. Molecules. 2021. PMID: 33946806 Free PMC article. Review.
References
-
- Eddy S.R. (1998) Profile hidden Markov models. Bioinformatics, 14, 755–763. - PubMed
-
- Aravind L. and Koonin,E.V. (1999) Gleaning non-trivial structural, functional, and evolutionary information about proteins by iterative database searches. J. Mol. Biol., 287, 1023–1040. - PubMed
-
- Wu W., Booker,S., Lieder,K.W., Bandarian,V., Reed,G.H. and Frey,P.A. (2000) Lysine 2,3-aminomutase and trans-4,5-dehydrolysine: characterization of an allylic analogue of a substrate-based radical in the catalytic mechanism. Biochemistry, 39, 9561–9570. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
