

Demonstration of the in vivo interaction of key cell death regulators by structure-based design of second-site suppressors
- PMID: 11027303
- PMCID: PMC17269
- DOI: 10.1073/pnas.210391597
Demonstration of the in vivo interaction of key cell death regulators by structure-based design of second-site suppressors
Abstract
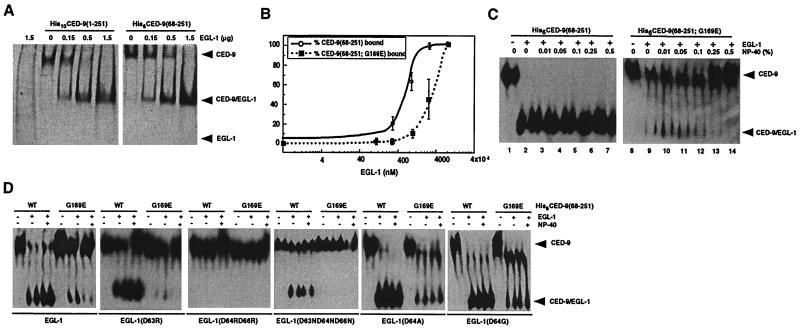
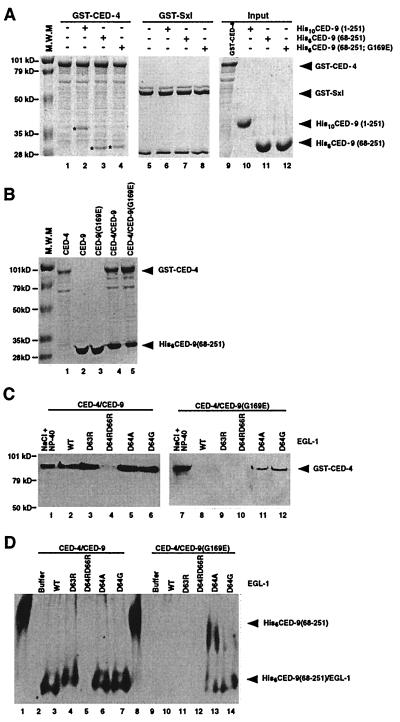
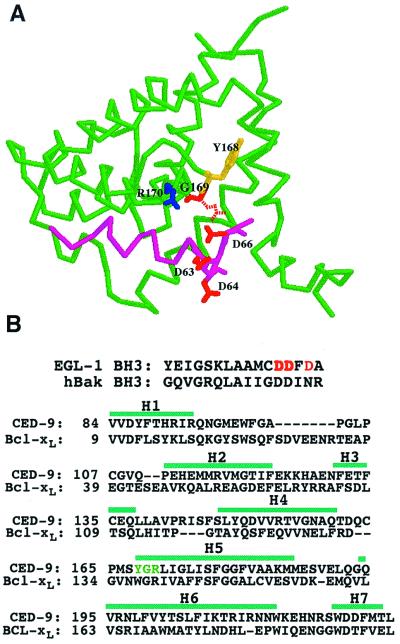
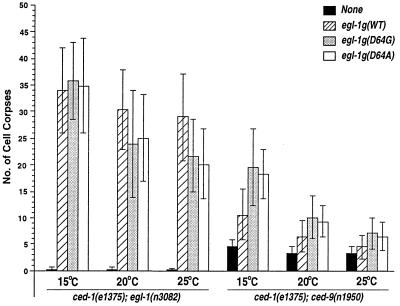
Demonstrating in vivo interaction of two important biomolecules and the relevance of the interaction to a biological process have been difficult issues in biomedical research. Here, we report the use of a homology modeling approach to establish the significance of protein interactions in governing the activation of programmed cell death in Caenorhabditis elegans. A protein interaction cascade has been postulated to mediate activation of cell death in nematodes, in which the BH3-domain-containing (Bcl-2 homology region 3) protein EGL-1 binds the cell-death inhibitor CED-9 and induces release of the death-activating protein CED-4 from inhibitory CED-4/CED-9 complexes. We show here that an unusual gain-of-function mutation in ced-9 (substitution of glycine 169 to glutamate) that results in potent inhibition of most nematode cell deaths impairs the binding of EGL-1 to CED-9 and EGL-1-induced release of CED-4 from CED-4/CED-9 complexes. Based on a modeled EGL-1/CED-9 complex structure, we generated second-site compensatory mutations in EGL-1 that partially restore the binding of EGL-1 to CED-9(G169E) and EGL-1-induced release of CED-4 from CED-4/CED-9(G169E) complexes. Importantly, these mutations also significantly suppress the death-protective activity of CED-9(G169E) in vivo. These results establish that direct physical interaction between EGL-1 and CED-9 is essential for the release of CED-4 and the activation of cell death. The structure-based design of second-site suppressors via homology modeling should be widely applicable for probing important molecular interactions that are implicated in fundamental biological processes.
Figures
Similar articles
-
Disruption of the CED-9.CED-4 complex by EGL-1 is a critical step for programmed cell death in Caenorhabditis elegans.J Biol Chem. 2000 Sep 1;275(35):27205-11. doi: 10.1074/jbc.M000858200. J Biol Chem. 2000. PMID: 10846174
-
The C. elegans protein EGL-1 is required for programmed cell death and interacts with the Bcl-2-like protein CED-9.Cell. 1998 May 15;93(4):519-29. doi: 10.1016/s0092-8674(00)81182-4. Cell. 1998. PMID: 9604928
-
Caenorhabditis elegans EGL-1 disrupts the interaction of CED-9 with CED-4 and promotes CED-3 activation.J Biol Chem. 1998 Dec 11;273(50):33495-500. doi: 10.1074/jbc.273.50.33495. J Biol Chem. 1998. PMID: 9837929
-
Programmed cell death in Caenorhabditis elegans.Curr Opin Genet Dev. 1994 Aug;4(4):581-6. doi: 10.1016/0959-437x(94)90076-f. Curr Opin Genet Dev. 1994. PMID: 7950327 Review.
-
2:1 Stoichiometry of the CED-4-CED-9 complex and the tetrameric CED-4: insights into the regulation of CED-3 activation.Cell Cycle. 2006 Jan;5(1):31-4. doi: 10.4161/cc.5.1.2263. Epub 2006 Jan 18. Cell Cycle. 2006. PMID: 16294007 Review.
Cited by
-
Control of sex-specific apoptosis in C. elegans by the BarH homeodomain protein CEH-30 and the transcriptional repressor UNC-37/Groucho.Genes Dev. 2007 Dec 1;21(23):3195-207. doi: 10.1101/gad.1607807. Genes Dev. 2007. PMID: 18056429 Free PMC article.
-
Cell death specification in C. elegans.Cell Cycle. 2008 Aug 15;7(16):2479-84. doi: 10.4161/cc.7.16.6479. Epub 2008 Aug 19. Cell Cycle. 2008. PMID: 18719375 Free PMC article. Review.
-
Cell Death in C. elegans Development.Curr Top Dev Biol. 2015;114:1-42. doi: 10.1016/bs.ctdb.2015.07.018. Epub 2015 Sep 9. Curr Top Dev Biol. 2015. PMID: 26431562 Free PMC article. Review.
-
Programmed cell death and clearance of cell corpses in Caenorhabditis elegans.Cell Mol Life Sci. 2016 Jun;73(11-12):2221-36. doi: 10.1007/s00018-016-2196-z. Epub 2016 Apr 5. Cell Mol Life Sci. 2016. PMID: 27048817 Free PMC article. Review.
-
Hepatitis B virus X protein targets Bcl-2 proteins to increase intracellular calcium, required for virus replication and cell death induction.Proc Natl Acad Sci U S A. 2012 Nov 6;109(45):18471-6. doi: 10.1073/pnas.1204668109. Epub 2012 Oct 22. Proc Natl Acad Sci U S A. 2012. PMID: 23091012 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
