

Activation of p38 mitogen-activated protein kinase and c-Jun NH(2)-terminal kinase by double-stranded RNA and encephalomyocarditis virus: involvement of RNase L, protein kinase R, and alternative pathways
- PMID: 10611240
- PMCID: PMC85147
- DOI: 10.1128/MCB.20.2.617-627.2000
Activation of p38 mitogen-activated protein kinase and c-Jun NH(2)-terminal kinase by double-stranded RNA and encephalomyocarditis virus: involvement of RNase L, protein kinase R, and alternative pathways
Abstract
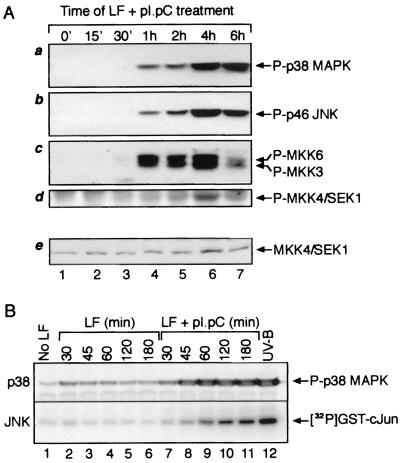
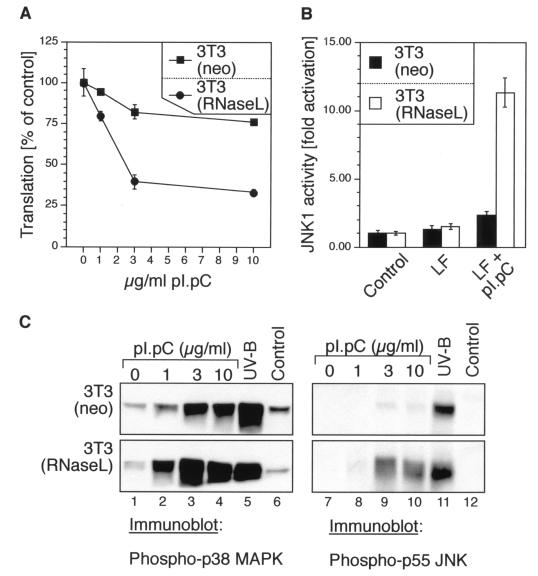
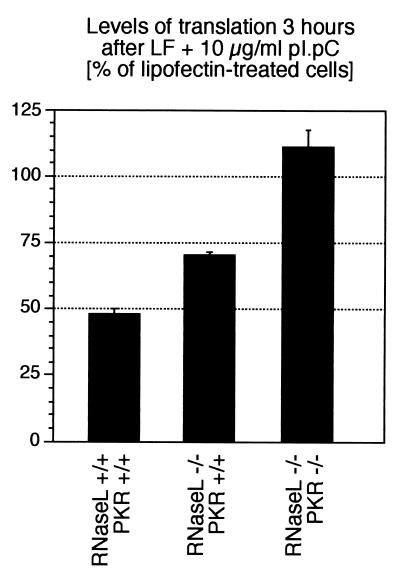
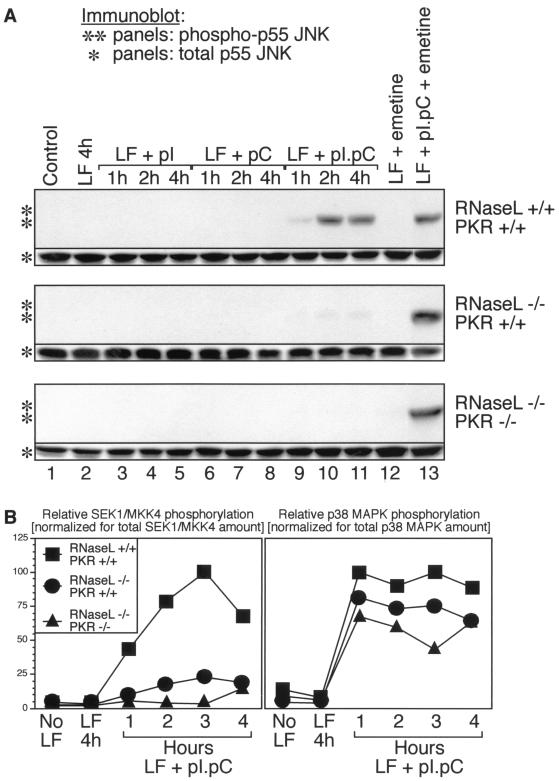
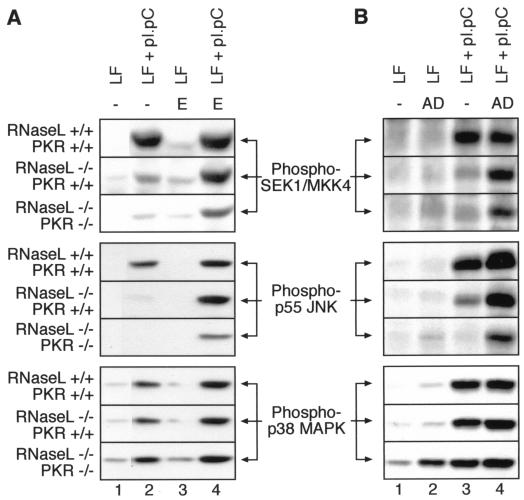
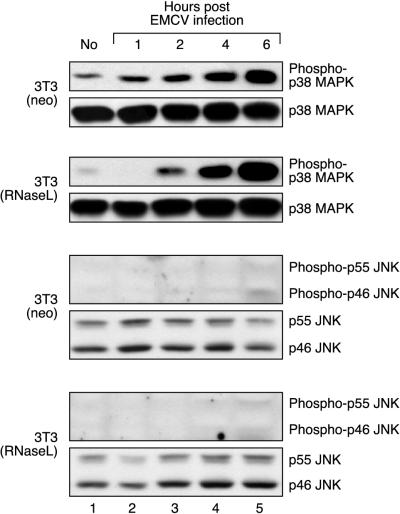
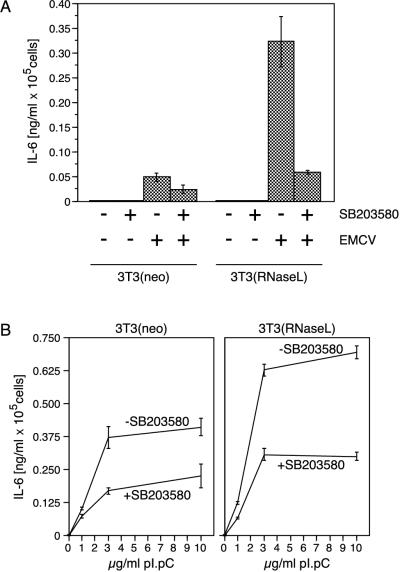
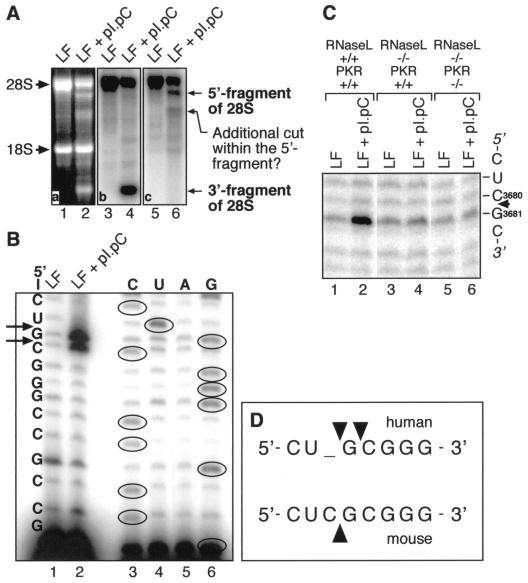
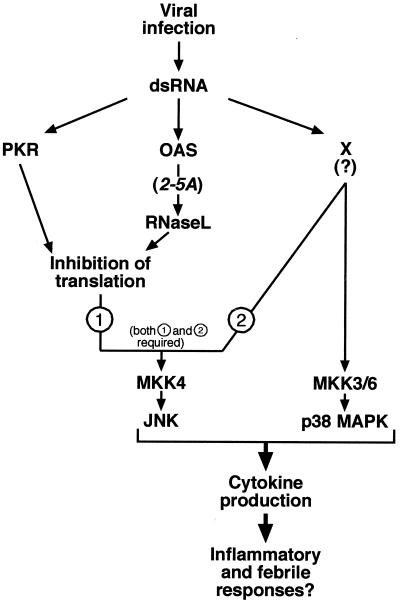
Double-stranded RNA (dsRNA) accumulates in virus-infected mammalian cells and signals the activation of host defense pathways of the interferon system. We describe here a novel form of dsRNA-triggered signaling that leads to the stimulation of the p38 mitogen-activated protein kinase (p38 MAPK) and the c-Jun NH(2)-terminal kinase (JNK) and of their respective activators MKK3/6 and SEK1/MKK4. The dsRNA-dependent signaling to p38 MAPK was largely intact in cells lacking both RNase L and the dsRNA-activated protein kinase (PKR), i. e., the two best-characterized mediators of dsRNA-triggered antiviral responses. In contrast, activation of both MKK4 and JNK by dsRNA was greatly reduced in cells lacking RNase L (or lacking both RNase L and PKR) but was restored in these cells when introduction of dsRNA was followed by inhibition of ongoing protein synthesis or transcription. These results are consistent with the notion that the role of RNase L and PKR in the activation of MKK4 and JNK is the elimination, via inhibition of protein synthesis, of a labile negative regulator(s) of the signaling to JNK acting upstream of SEK1/MKK4. In the course of these studies, we identified a long-sought site of RNase L-mediated cleavage in the 28S rRNA, which could cause inhibition of translation, thus allowing the activation of JNK by dsRNA. We propose that p38 MAPK is a general participant in dsRNA-triggered cellular responses, whereas the activation of JNK might be restricted to cells with reduced rates of protein synthesis. Our studies demonstrate the existence of alternative (RNase L- and PKR-independent) dsRNA-triggered signaling pathways that lead to the stimulation of stress-activated MAPKs. Activation of p38 MAPK (but not of JNK) was demonstrated in mouse fibroblasts in response to infection with encephalomyocarditis virus (ECMV), a picornavirus that replicates through a dsRNA intermediate. Fibroblasts infected with EMCV (or treated with dsRNA) produced interleukin-6, an inflammatory and pyrogenic cytokine, in a p38 MAPK-dependent fashion. These findings suggest that stress-activated MAPKs participate in mediating inflammatory and febrile responses to viral infections.
Figures
Similar articles
-
RNase L induces autophagy via c-Jun N-terminal kinase and double-stranded RNA-dependent protein kinase signaling pathways.J Biol Chem. 2012 Dec 21;287(52):43651-64. doi: 10.1074/jbc.M112.399964. Epub 2012 Oct 29. J Biol Chem. 2012. PMID: 23109342 Free PMC article.
-
Activation of NF-kappaB by double-stranded RNA (dsRNA) in the absence of protein kinase R and RNase L demonstrates the existence of two separate dsRNA-triggered antiviral programs.Mol Cell Biol. 2001 Jan;21(1):61-72. doi: 10.1128/MCB.21.1.61-72.2001. Mol Cell Biol. 2001. PMID: 11113181 Free PMC article.
-
Role of MAPK in the regulation of double-stranded RNA- and encephalomyocarditis virus-induced cyclooxygenase-2 expression by macrophages.J Immunol. 2006 Sep 1;177(5):3413-20. doi: 10.4049/jimmunol.177.5.3413. J Immunol. 2006. PMID: 16920983
-
PKR; a sentinel kinase for cellular stress.Oncogene. 1999 Nov 1;18(45):6112-20. doi: 10.1038/sj.onc.1203127. Oncogene. 1999. PMID: 10557102 Review.
-
Metastasis and MAPK Pathways.Int J Mol Sci. 2022 Mar 31;23(7):3847. doi: 10.3390/ijms23073847. Int J Mol Sci. 2022. PMID: 35409206 Free PMC article. Review.
Cited by
-
Double-stranded RNA-induced activation of activating protein-1 promoter is differentially regulated by the non-structural protein 1 of avian influenza A viruses.Viral Immunol. 2012 Feb;25(1):79-85. doi: 10.1089/vim.2011.0059. Epub 2012 Jan 12. Viral Immunol. 2012. PMID: 22239235 Free PMC article.
-
The antiviral response to gamma interferon.J Virol. 2002 Sep;76(18):9060-8. doi: 10.1128/jvi.76.18.9060-9068.2002. J Virol. 2002. PMID: 12186889 Free PMC article.
-
Comparative induction of 28S ribosomal RNA cleavage by ricin and the trichothecenes deoxynivalenol and T-2 toxin in the macrophage.Toxicol Sci. 2008 Sep;105(1):67-78. doi: 10.1093/toxsci/kfn111. Epub 2008 Jun 4. Toxicol Sci. 2008. PMID: 18535001 Free PMC article.
-
Selection and cloning of poly(rC)-binding protein 2 and Raf kinase inhibitor protein RNA activators of 2',5'-oligoadenylate synthetase from prostate cancer cells.Nucleic Acids Res. 2006;34(22):6684-95. doi: 10.1093/nar/gkl968. Epub 2006 Dec 1. Nucleic Acids Res. 2006. PMID: 17145707 Free PMC article.
-
A systems-based approach to analyse the host response in murine lung macrophages challenged with respiratory syncytial virus.BMC Genomics. 2013 Mar 18;14:190. doi: 10.1186/1471-2164-14-190. BMC Genomics. 2013. PMID: 23506210 Free PMC article.
References
-
- Baldassare J J, Bi Y, Bellone C J. The role of p38 mitogen-activated protein kinase in IL-1 beta transcription. J Immunol. 1999;162:5367–5373. - PubMed
-
- Brimacombe R. The structure of ribosomal RNA: a three-dimensional jigsaw puzzle. Eur J Biochem. 1995;230:365–383. - PubMed
-
- Chebath J, Benech P, Hovanessian A, Galabru J, Revel M. Four different forms of interferon-induced 2′,5′-oligo(A) synthetase identified by immunoblotting in human cells. J Biol Chem. 1987;262:3852–3857. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous