

Functional analysis of human herpesvirus 8-encoded viral interferon regulatory factor 1 and its association with cellular interferon regulatory factors and p300
- PMID: 10438822
- PMCID: PMC104259
- DOI: 10.1128/JVI.73.9.7334-7342.1999
Functional analysis of human herpesvirus 8-encoded viral interferon regulatory factor 1 and its association with cellular interferon regulatory factors and p300
Abstract
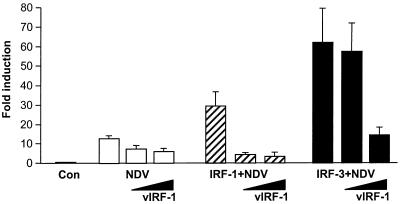
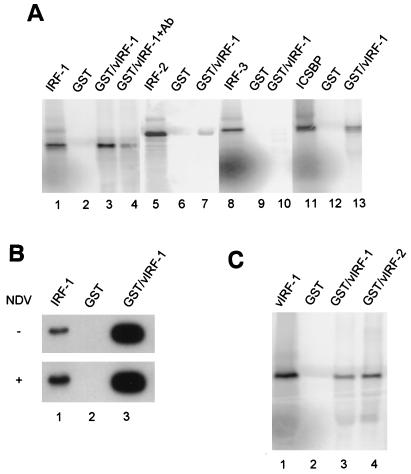
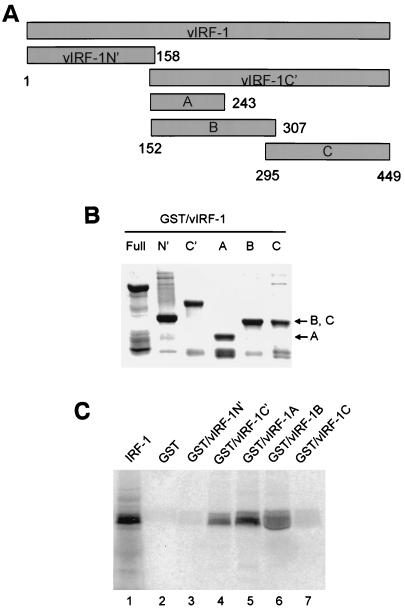
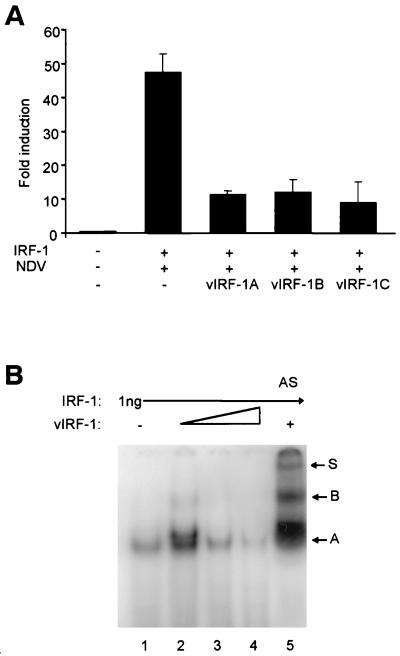
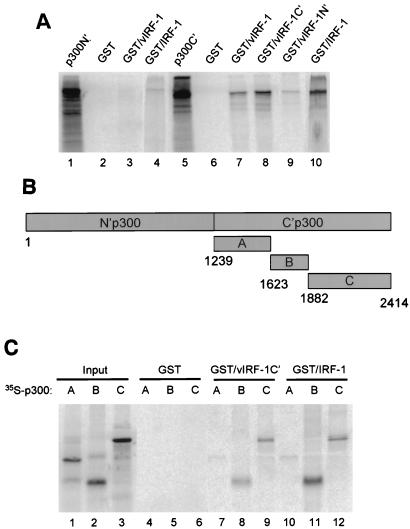
Human herpesvirus 8/Kaposi sarcoma-associated virus (HHV-8/KSHV) contains, in addition to genes required for viral replication, a unique set of nonstructural genes which may be part of viral mimicry and contribute to viral replication and pathogenesis in vivo. Among these, HHV-8 encodes four open reading frames (ORFs) that showed homology to the transcription factors of the interferon regulatory factor (IRF) family. The ORF K9, viral IRF 1 (vIRF-1), has been cloned, and it was shown that, when overexpressed, it down modulates the interferon-mediated transcriptional activation of the interferon-stimulated gene 15 (ISG 15) promoter, and the role of vIRF-1 in viral mimicry was implied. However, the molecular mechanism of this effect has not been clarified. Here, we extend this observation and show that vIRF-1 also downregulates the transcriptional activity of IFNA gene promoter in infected cells by interfering with the transactivating activity of cellular IRFs, including IRF-1 and IRF-3. We further show that ectopic expression of vIRF-1 in NIH 3T3 cells confers resistance to tumor necrosis factor alpha-induced apoptosis. While vIRF-1 is unable to bind DNA with the same specificity as cellular IRFs, we demonstrate by in vitro binding assay that it can associate with the family of cellular IRFs, such as IRF-1 and the interferon consensus sequence binding protein. vIRF-1 interaction domain was localized between amino acids (aa) 152 and 243. While no binding between the full-size IRF-3 and vIRF-1 could be detected by the same assay, we show that vIRF-1 also targets the carboxy-terminal region (aa 1623 to 2414) of the transcriptional coactivator p300 which could also bind IRF-3 and IRF-1. These results demonstrate that vIRF-1 can modulate the transcription of the IFNA genes by direct heterodimerization with members of the IRF family, as well as by competitive binding with cellular transcription factors to the carboxy-terminal region of p300.
Figures

Similar articles
-
Unique properties of a second human herpesvirus 8-encoded interferon regulatory factor (vIRF-2).J Hum Virol. 1999 Jan-Feb;2(1):19-32. J Hum Virol. 1999. PMID: 10200596
-
Kaposi's sarcoma-associated herpesvirus-encoded vIRF-3 stimulates the transcriptional activity of cellular IRF-3 and IRF-7.J Biol Chem. 2004 Feb 27;279(9):7643-54. doi: 10.1074/jbc.M309485200. Epub 2003 Dec 10. J Biol Chem. 2004. PMID: 14668346
-
Characterization of a novel human herpesvirus 8-encoded protein, vIRF-3, that shows homology to viral and cellular interferon regulatory factors.J Virol. 2000 Sep;74(17):8194-201. doi: 10.1128/jvi.74.17.8194-8201.2000. J Virol. 2000. PMID: 10933732 Free PMC article.
-
On the role of IRF in host defense.J Interferon Cytokine Res. 2002 Jan;22(1):59-71. doi: 10.1089/107999002753452665. J Interferon Cytokine Res. 2002. PMID: 11846976 Review.
-
Interferon regulatory factors: the next generation.Gene. 1999 Sep 3;237(1):1-14. doi: 10.1016/s0378-1119(99)00262-0. Gene. 1999. PMID: 10524230 Review.
Cited by
-
USP7-Dependent Regulation of TRAF Activation and Signaling by a Viral Interferon Regulatory Factor Homologue.J Virol. 2020 Jan 6;94(2):e01553-19. doi: 10.1128/JVI.01553-19. Print 2020 Jan 6. J Virol. 2020. PMID: 31666375 Free PMC article.
-
Molecular biology of Kaposi's sarcoma-associated herpesvirus and related oncogenesis.Adv Virus Res. 2010;78:87-142. doi: 10.1016/B978-0-12-385032-4.00003-3. Adv Virus Res. 2010. PMID: 21040832 Free PMC article. Review.
-
Herpes simplex virus 1 has multiple mechanisms for blocking virus-induced interferon production.J Virol. 2004 Aug;78(16):8411-20. doi: 10.1128/JVI.78.16.8411-8420.2004. J Virol. 2004. PMID: 15280450 Free PMC article.
-
Innate antiviral response: role in HIV-1 infection.Viruses. 2011 Jul;3(7):1179-203. doi: 10.3390/v3071179. Epub 2011 Jul 14. Viruses. 2011. PMID: 21994776 Free PMC article. Review.
-
Histone acetyltransferases as regulators of nonhistone proteins: the role of interferon regulatory factor acetylation on gene transcription.J Biomed Biotechnol. 2011;2011:640610. doi: 10.1155/2011/640610. Epub 2010 Dec 29. J Biomed Biotechnol. 2011. PMID: 21234331 Free PMC article. Review.
References
-
- Au W-C, Moore P A, LaFleur D W, Tomball B, Pitha P M. Characterization of intereferon regulatory factor-7, and its potential role in the transcriptional activation of interferon A genes. J Biol Chem. 1998;273:29210–29217. - PubMed
-
- Baer R, Bankier A T, Biggin M D, Deininger P L, Farrell P J, Gibson T J, Hatfull G, Hudson G S, Satchwell S C, Seguin C, et al. DNA sequence and expression of the B95-8 Epstein-Barr virus genome. Nature. 1984;310:207–211. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
