

Long RNA hairpins that contain inosine are present in Caenorhabditis elegans poly(A)+ RNA
- PMID: 10339539
- PMCID: PMC26833
- DOI: 10.1073/pnas.96.11.6048
Long RNA hairpins that contain inosine are present in Caenorhabditis elegans poly(A)+ RNA
Abstract
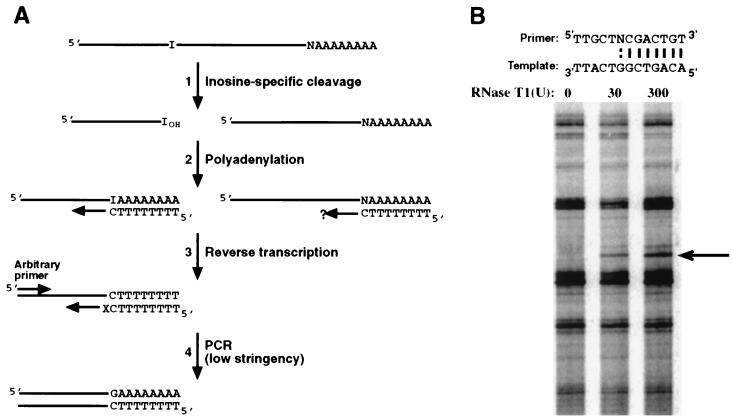
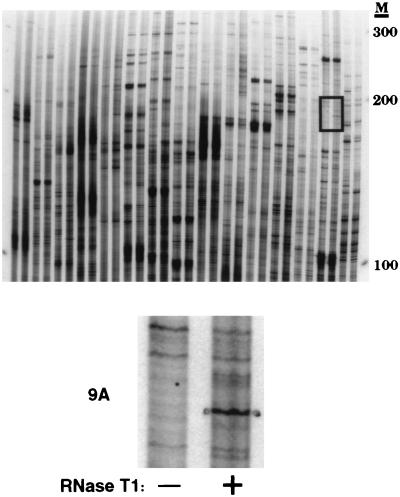
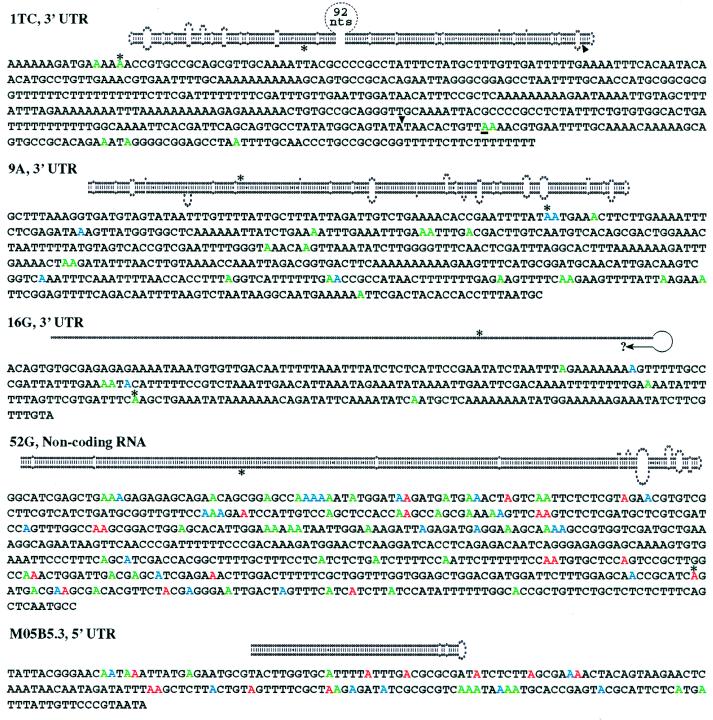
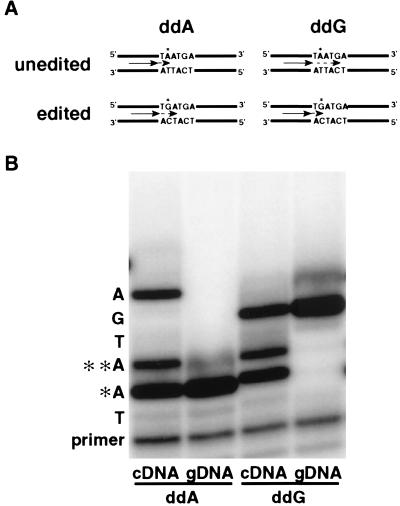
Adenosine deaminases that act on RNA (ADARs) are RNA-editing enzymes that convert adenosine to inosine within double-stranded RNA. In the 12 years since the discovery of ADARs only a few natural substrates have been identified. These substrates were found by chance, when genomically encoded adenosines were identified as guanosines in cDNAs. To advance our understanding of the biological roles of ADARs, we developed a method for systematically identifying ADAR substrates. In our first application of the method, we identified five additional substrates in Caenorhabditis elegans. Four of those substrates are mRNAs edited in untranslated regions, and one is a noncoding RNA edited throughout its length. The edited regions are predicted to form long hairpin structures, and one of the RNAs encodes POP-1, a protein involved in cell fate decisions.
Figures
Similar articles
-
RNA hairpins in noncoding regions of human brain and Caenorhabditis elegans mRNA are edited by adenosine deaminases that act on RNA.Proc Natl Acad Sci U S A. 2002 Jun 11;99(12):7906-11. doi: 10.1073/pnas.112704299. Epub 2002 Jun 4. Proc Natl Acad Sci U S A. 2002. PMID: 12048240 Free PMC article.
-
Noncoding regions of C. elegans mRNA undergo selective adenosine to inosine deamination and contain a small number of editing sites per transcript.RNA Biol. 2015;12(2):162-74. doi: 10.1080/15476286.2015.1017220. RNA Biol. 2015. PMID: 25826568 Free PMC article.
-
C. elegans and H. sapiens mRNAs with edited 3' UTRs are present on polysomes.RNA. 2008 Oct;14(10):2050-60. doi: 10.1261/rna.1165008. Epub 2008 Aug 21. RNA. 2008. PMID: 18719245 Free PMC article.
-
A-to-I editing of coding and non-coding RNAs by ADARs.Nat Rev Mol Cell Biol. 2016 Feb;17(2):83-96. doi: 10.1038/nrm.2015.4. Epub 2015 Dec 9. Nat Rev Mol Cell Biol. 2016. PMID: 26648264 Free PMC article. Review.
-
Proteome diversification by adenosine to inosine RNA editing.RNA Biol. 2010 Mar-Apr;7(2):205-12. doi: 10.4161/rna.7.2.11286. Epub 2010 Mar 25. RNA Biol. 2010. PMID: 20200492 Review.
Cited by
-
The difficult calls in RNA editing. Interviewed by H Craig Mak.Nat Biotechnol. 2012 Dec;30(12):1207-9. doi: 10.1038/nbt.2452. Nat Biotechnol. 2012. PMID: 23222792 No abstract available.
-
Age-related gene-specific changes of A-to-I mRNA editing in the human brain.Mech Ageing Dev. 2010 Jun;131(6):445-7. doi: 10.1016/j.mad.2010.06.001. Epub 2010 Jun 9. Mech Ageing Dev. 2010. PMID: 20538013 Free PMC article.
-
RNA binding-independent dimerization of adenosine deaminases acting on RNA and dominant negative effects of nonfunctional subunits on dimer functions.J Biol Chem. 2007 Jun 1;282(22):16054-61. doi: 10.1074/jbc.M611392200. Epub 2007 Apr 11. J Biol Chem. 2007. PMID: 17428802 Free PMC article.
-
Biochemical analysis and scanning force microscopy reveal productive and nonproductive ADAR2 binding to RNA substrates.RNA. 2003 Jul;9(7):839-46. doi: 10.1261/rna.2167603. RNA. 2003. PMID: 12810917 Free PMC article.
-
RNA interference: advances and questions.Philos Trans R Soc Lond B Biol Sci. 2002 Jan 29;357(1417):65-70. doi: 10.1098/rstb.2001.0952. Philos Trans R Soc Lond B Biol Sci. 2002. PMID: 11839183 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
