

Abstract
Murine thioglycolate-induced peritoneal macrophages (MPMs) and the murine RAW264.7 macrophage-like cell line (RAW cells) constitutively produce vascular endothelial growth factor (VEGF). VEGF production is increased under hypoxic conditions or after cell activation with interferon-γ (IFNγ) and endotoxin (lipopolysaccharide, LPS). In contrast, tumor necrosis factor-α is produced only by IFNγ/LPS-activated cells. Lactate (25 mmol/L) does not increase VEGF production by these cells. However, hypoxia, lactate, and IFNγ/LPS-activated MPMs express angiogenic activity, whereas normoxic, nonactivated MPMs do not. Lack of angiogenic activity is not due to an antiangiogenic factor(s) in the medium of these cells. Angiogenic activity produced by hypoxia and lactate-treated MPMs is neutralized by anti-VEGF antibody, which also neutralizes most of the angiogenic activity produced by IFNγ/LPS-activated MPMs. The inducible nitric oxide synthase inhibitors Ng-nitro-l-arginine-methyl ester (1.5 mmol/L) and aminoguanidine (1 mmol/L) block production of angiogenic activity by MPMs and RAW cells. In RAW cells, Ng-nitro-l-arginine-methyl ester and AG block IFNγ/LPS-activated, but not constitutive, VEGF production, whereas in MPMs, neither constitutive nor IFNγ/LPS-activated VEGF synthesis is affected. Synthesis of tumor necrosis factor-α is also unaffected. In contrast to normoxic, nonactivated MPMs, inducible nitric oxide synthase-inhibited, IFNγ/LPS-activated MPMs produce an antiangiogenic factor(s). We conclude that VEGF is a major contributor to macrophage-derived angiogenic activity, and that activation by hypoxia, lactate, or IFNγ/LPS switches macrophage-derived VEGF from a nonangiogenic to an angiogenic state. This switch may involve a posttranslational modification of VEGF, possibly by the process of ADP-ribosylation. ADP-ribosylation by MPM cytosolic extracts or by cholera toxin switches rVEGF165 from an angiogenic to a nonangiogenic state. In IFNγ/LPS-activated MPMs, the inducible nitric oxide synthase-dependent pathway also regulates the expression of an antiangiogenic factor(s) that antagonizes the bioactivity of VEGF and provides an additional regulatory pathway controlling the angiogenic phenotype of macrophages.
Macrophages play a key role in the induction of angiogenesis in fibroproliferative states, including wound repair, rheumatoid arthritis, and solid tumor development. 1-5 Production of angiogenic activity by macrophages depends on the balance of production of positive angiogenic regulators and inhibitors of angiogenesis. 6-8 Positive angiogenic regulators previously shown to be produced by monocytes and macrophages include the cytokines tumor necrosis factor-α (TNFα) and interleukin (IL-8) 9-11 ; negative regulators include thrombospondin-1, interferon-γ (IFNγ)-inducible protein-10, and other as yet uncharacterized protein inhibitors. 12-14 The mechanisms controlling the balance of positive and negative angiogenesis regulators are not well understood. Nonactivated monocytes and macrophages exhibit a nonangiogenic phenotype. 1,4 After activation with agents such as IFNγ and/or endotoxin (lipopolysaccharide, LPS), macrophages express angiogenic activity, characterized by the expression of angiogenic cytokines, as well as of inhibitors of angiogenesis. 15-18 Activated cells also produce and release oxygen radicals, nitric oxide, and their derivatives. 17,19 These radicals have been shown to play an important role in regulating the angiogenic phenotype of activated macrophages. 20,21 Agents such as IFNγ and LPS, as well as reduced oxygen tension (hypoxia) and elevated lactate levels, induce macrophages to express angiogenic activity. 1-3,9,22 Recently, macrophages in vivo have been shown to express vascular endothelial growth factor (VEGF), an endothelial cell-specific mitogen that is potently angiogenic. 18,23-30 In this study, the expression of the angiogenic growth factor VEGF by murine thioglycolate-induced peritoneal macrophages (MPMs) and RAW cells was examined and compared with that of TNFα. The effects of hypoxia, lactate, and the l-arginine-dependent inducible nitric oxide synthase (iNOS) pathway on the production of VEGF and TNFα by these cells was also examined. We find that VEGF production is regulated both transcriptionally and translationally by hypoxia and the iNOS pathway, and that posttranslational modification may play an important role in regulating the bioactivity of VEGF as an angiogenic factor. In addition, the iNOS pathway in IFNγ/LPS-activated macrophages regulates the expression of an antiangiogenic factor that antagonizes the angiogenic effects of VEGF, providing an additional regulatory pathway to control the angiogenic phenotype of macrophages.
Materials and Methods
MPMs and RAW264.7 Cells
Balb c mice (male, 6 to 8 weeks old, Taconic, Germantown, NY) were injected intraperitoneally with 2.5 ml sterile Brewer’s thioglycollate broth (3% w/v) (DIFCO Laboratories, Detroit, MI). Five days later, the mice were sacrificed and MPMs were harvested using phosphate-buffered saline containing 100 U/ml of heparin. Cells were centrifuged at 300 × g for 5 minutes at 4°C, washed twice with serum-free (DMEM), and resuspended in DMEM containing 10% fetal calf serum (FCS) and 50 μg/ml gentamycin (DMEM-10% FCS). Cells were seeded into 60-mm tissue culture dishes (Costar, Cambridge, MA) (4 × 106 cells/dish) and incubated at 37°C in a humidified incubator in 95% air/5% CO2 for 4 hours to allow the cells to adhere. In some experiments, cells were seeded in Contur Permanox gas-permeable dishes (Miles, Naperville, IL) rather than regular tissue culture dishes, to increase the availability of ambient gases to the cells on the base of the dishes. Nonadherent cells were removed by washing with serum-free DMEM, and the cells were refed with DMEM-1% FCS. MPMs were activated using 100 U/ml murine IFNγ (Sigma Chemical Co., St. Louis, MO) and 100 ng/ml of LPS (Escherichia coli serotype 055:B5, Sigma) either in the presence or absence of either of the iNOS inhibitors, Ng-nitro-l-arginine-methyl ester (l-NAME) (1.5 mmol/L) or aminoguanidine (AG) (1 mmol/L). To test the effects of lactate on MPMs, sodium lactate (25 mmol/L) was added to the cultures at the start of the incubation period. To test the effects of hypoxia, MPMs were incubated in Permanox dishes, either under normoxic conditions (95% air/5% CO2) or under hypoxic conditions (95% N2/5% CO2). Media and cells were harvested at the indicated time points after addition of IFN-γ/LPS and/or lactate. Aliquots of media were sampled immediately after incubation and analyzed in a Blood Gas Analyzer (Instrumentation Laboratories, Lexington, MA). The remaining media were centrifuged at 4°C for 5 minutes at 15,000 × g to remove cellular debris and stored at −80°C before analysis.
RAW264.7 cells were obtained from American Type Culture Collection (Manassas, VA) and routinely maintained in DMEM-10% FCS. Cells were passaged by scraping and plated in either regular or Permanox dishes, with or without IFNγ/LPS, with or without sodium lactate, and under hypoxic conditions, as described above. The effects of l-NAME and AG on the production of VEGF and TNFα by these cells were also tested. Media and cells were harvested and treated as described above.
Isolation of Total Cellular RNA
Total cellular RNA was isolated from macrophage cell cultures using TRI reagent (Molecular Research Center, Inc., Cincinnati, OH). Medium was removed from the cells, TRI reagent was added directly to the culture dishes, and the cell lysate was passed several times through a 21-gauge syringe needle. Samples were stored at room temperature for 5 minutes, 0.2 ml chloroform was then added per milliliter lysis reagent, and the mixture was vortexed for 15 seconds and then incubated at room temperature for 10 minutes. The resultant mixture was centrifuged at 12,000 × g for 15 minutes at 4°C. The aqueous (upper) phase was transferred to a fresh microfuge tube, and RNA was precipitated by adding 0.5 ml isopropanol per 1 ml TRI reagent used for the original extraction. Samples were incubated at room temperature for 5 minutes and then centrifuged at 12,000 × g for 10 minutes at 4°C. The RNA pellets were washed with 75% ethanol, air dried for 5 minutes, and dissolved in RNase-free water.
Quantitative Reverse Transcription-Polymerase Chain Reaction (RT-PCR) Analysis of VEGF mRNA Levels
VEGF mRNA levels were determined by RT-PCR using an internal minigene RNA standard that is present through both the RT and the PCR reaction stages. The 293-bp VEGF minigene RNA standard, containing a 69-bp gene deletion, was prepared as follows. Total RNA from MPMs was subjected to RT and PCR through 35 cycles, using the following primers: sense minigene primer (18-mer) in exon 1 (positions 41 to 58), 5′-GGACCCTGGCTTTACTGC-3′, and antisense minigene primer (39-mer), starting in exon 5, spanning an intron, and continuing into exon 4 to position 387, deleting 69 bp of the gene to position 318, and continuing to position 300. The primer thus spans an intron, and contains a 69 bp deletion: 5′-TTGGTCTGCATTCACATCGGC-GTGATGTTGCTCTCTGAC-3′. The PCR band was purified from primers by ethanol precipitation and blunt end ligated into the pCR-Script AmpSK(+) vector (Stratagene, La Jolla, CA). The orientation of the minigene fragment in the vector was determined by dideoxy sequence analysis. A clone containing the minigene insert in an antisense orientation was used for subsequent in vitro transcription for the preparation of the RNA minigene. The vector was linearized with NotI, treated with proteinase-K (4 μg/ml) for 1 hour at 37°C, and purified by phenol extraction and ethanol precipitation. The linearized plasmid was then transcribed in vitro using a VTRAN-7 transcription kit (Sigma), with T7 RNA polymerase, yielding sense RNA. The reaction product was treated with RNase-free DNase-1 (10 U/mg DNA in the transcription reaction) (Promega, Madison WI) for 2 hours/at 37°C. The reaction mixture was then heated to 90°C for 5 minutes and cooled, and 10 × transcription stop solution (5 mmol/L ammonium acetate and 0.1 mmol/L ethylenediaminetetraacetic acid) was added, followed by phenol extraction and isopropanol precipitation. The RNA concentration was determined spectrophotometrically. VEGF RNA minigene (2.5 pg per reaction) was then incorporated into the RT-PCR reactions. Total RNA from macrophages treated under various conditions was added to the RT-PCR reactions in amounts ranging from 1 to 200 ng/reaction. The oligonucleotide primers used for the competitive RT-PCR reaction were 18-mers nested into the initial primers used to prepare the minigene: sense primer in exon 1, 5′-ACCCTGGCTTTACTGCTG-3′, and antisense primer (intron spanning), 5′-GGTCTGCATTCACATCGG-3′. The antisense primer was used for the initial RT reaction; the reverse transcriptase was inactivated at 99°C for 5 minutes, and added to a PCR mix containing an equivalent amount of sense primer. PCR was then carried out for 25 cycles. The reactions were analyzed by electrophoresis on 1.5% agarose gels in Tris-acetate-ethylenediamine tetraacetic acid (TAE) buffer, stained with ethidium bromide, and scanned using the Molecular Dynamics FluorImage Analyzer. The concentrations of input RNA that gave bands of equal intensity to that of the internal VEGF RNA minigene were then determined. Although intron-spanning primers were used throughout, controls for genomic DNA contamination of total RNA preparations were routinely carried out. These controls involved the performance of parallel reactions in the absence of reverse transcriptase.
As a control for a housekeeping gene that is not markedly modulated by the various culture conditions used, an RT-PCR procedure for the enzyme glyceraldehyde-3-phosphate dehydrogenase (G3PDH) was also developed (details not shown). Parallel reactions for G3PDH mRNA levels were performed on the various macrophage RNA samples, and the VEGF mRNA levels determined by RT-PCR were normalized to the G3PDH levels.
RT-PCR Analysis of VEGF mRNA Isoforms
For RT, 1.0 μg of total RNA was reverse transcribed using 100 ng of the reverse VEGF-specific primer indicated below, using 50 U murine leukemia virus reverse transcriptase with an RNA PCR Kit (Perkin Elmer, Foster City, CA), following the manufacturer’s protocol. After the initial RT reaction step, the 20-μl reaction volumes were boiled for 5 minutes to inactivate the reverse transcriptase. One hundred ng forward primer (see below) was added, together with 80 μl of a PCR master mix, to give a final concentration of 1 mmol/L MgCl2, 1× PCR buffer II, and 2.5 U Taq polymerase (Perkin Elmer) per reaction. PCR primers were selected to enable the amplification of the three differentially spliced murine isoforms of VEGF mRNA formed from the VEGF gene. These VEGF mRNA isoforms are derived from a gene containing eight exons. 31 The largest, VEGF-1, is formed using all eight exons. VEGF-2 lacks exon 7, and VEGF-3 lacks exons 6 and 7. By using PCR primers in exons 3 and 8, the three different isoforms of VEGF generate PCR amplification products of different sizes, and because they amplify from the same primers, the ratio of intensities of the three bands gives an estimate of the relative abundance of the three differentially spliced mRNA isoforms. The primers selected for the PCR amplifications were: forward primer, located in exon 3, 5′-GATGAAGCCCTGGAGTGC-3′, and reverse primer, located in exon 8, 5′-TCCCAGAAACAACCCTAA-3′.
The following cycling program for PCR was used: Denaturation at 94°C for 1 minute, annealing at 54°C for 1 minute, and extension for 2 minutes at 72°C, for 25 cycles, with a final extension at 72°C for 15 minutes. PCR reactions were then analyzed by electrophoresis on 1.5% agarose gels using TAE buffer, and stained with ethidium bromide. Gels were scanned using a Molecular Dynamics FluorImage analyzer, and the staining intensities of the PCR-amplified VEGF isoform bands were analyzed using the ImageQuant image analysis software package (Molecular Dynamics).
Assay of VEGF Protein Levels by Enzyme-Linked Immunosorbent Assay (ELISA)
VEGF in conditioned media was assayed using a sandwich ELISA kit (Quantikine M, R&D Systems, Minneapolis, MN), following the manufacturer’s protocol. This assay detects murine VEGF with sensitivity in the range of 3 to 500 pg/ml. Samples with VEGF concentrations above this range were diluted with RPMI and reassayed. All samples were assayed in triplicate. Results are presented as means ± standard deviations of the mean (SD).
Assay of TNFα by ELISA
Murine TNFα was assayed using a sandwich ELISA kit (TNF-A Minikit, Endogen, Woburn, MA), following the procedure of the manufacturer. All samples were assayed in triplicate. Results are presented as means ± SD.
Assay of Nitrite
To determine the production of nitric oxide by the cells under the various conditions tested, the media were analyzed for nitrite using the Griess reaction, as described previously. Briefly, 50 μl of culture medium was placed in a 96-well plate, followed by 50 μl of cold 350 mmol/L ammonium chloride, pH 9.6. One hundred μl of a mixture of 1 part 5 mmol/L sulfanilic acid, 1 part 5 mmol/L N-(1-naphthyl)ethylenediamine, and 3 parts glacial acetic acid was added. After 10 minutes of incubation in the dark at room temperature, absorbance at 570 nm was determined using a microplate scanner (BioTek Instruments, Burlington, VT). The system was calibrated using freshly prepared standard nitrite solutions. A linear regression line was determined from the standards, and the experimental nitrite concentrations calculated. Results are means ± SD.
Assay of Angiogenic and Antiangiogenic Activity
Conditioned media from MPM cultures were concentrated 20-fold and diafiltered using Amicon centrifugal spin filters (3-kd cutoff) (Beverly, MA). Five μl of concentrated media was incorporated into equal volumes of slow-release Hydron (12% w/v in 95% ethanol) (Interferon Sciences, New Brunswick, NJ) and allowed to dry. Hydron pellets were implanted aseptically into pockets within rat corneal stromas 2 mm from the limbal vasculature, as described previously. 1,2,4,9 Corneas were examined daily for 7 days using a stereomicroscope and perfused with colloidal carbon at the end of the observation period to provide a permanent record of the angiogenic responses. Corneas were examined histologically for any evidence of nonspecific inflammation. Angiogenic responses were assessed on a graded scale as follows: 0, no response, or slight budding of the limbal vasculature that regresses rapidly; 1, formation of a few capillary buds and sprouts that progress less then 0.2 mm from the limbus and start to regress; 2, persistent growth of a network of capillary buds and sprouts that grow at least 1 mm toward the implant, but do not reach and invade the implant and 3, strong growth of a dense network of capillary buds and sprouts that reaches and surrounds the implant. Four corneal implants were prepared per test sample, and the responses were summed. A maximal response thus has a score of 12, whereas a minimal response has a score of 0. For the assay of antiangiogenic activity, test-conditioned media (20× concentrated) were combined with 20 ng recombinant human VEGF165 (gift of Dr. Napoleone Ferrara, Genentech Inc., South San Francisco, CA). The effects of the test media on the angiogenic activity of the rVEGF were then determined using the corneal bioassay.
Effects of Anti-VEGF Antibodies on Macrophage Angiogenic Activity
To determine the contribution of VEGF to the angiogenic activity of the MPM-conditioned media, an affinity-purified neutralizing polyclonal antibody to VEGF (gift of Dr. Napoleone Ferrara) was used. Concentrated conditioned media prepared as described above were incubated with anti-VEGF antibody at a final concentration of 10 μg/ml at 37°C for 2 hours. Controls were incubated with preimmune immunoglobulin at the same concentration. These treated media were then assayed for angiogenic activity in the rat corneal bioassay.
ADP-Ribosylation of rVEGF
Initial attempts to metabolically label VEGF endogenously synthesized in MPMs, using 32P-labeled nicotinamide adenine dinucleotide ([32P]NAD+) were unsuccessful, as macrophages are impermeable to NAD+, which cannot enter the cells and provide a substrate for the cytoplasmic ADP-ribosyl transferases. 32-34 We therefore used either permeabilized MPMs (data not shown) or macrophage cytoplasmic extracts to determine whether exogenous rVEGF is a substrate for macrophage ADP-ribosyl transferases. Similarly, rVEGF was tested as a substrate for cholera toxin (an arginine-specific ADP-ribosyl transferase) and for pertussis toxin (a cysteine-specific ADP-ribosyl transferase). 35,36
Cytosolic extracts of MPMs were prepared as follows: MPMs were plated in 100-mm culture dishes (10 × 106 cells per dish in 10 ml of medium) in RPMI 1640 containing 10% FCS and incubated at 37°C overnight. The medium was then removed, and the cells were washed two times with cold phosphate-buffered saline. The cells were then harvested by scraping into cold phosphate-buffered saline (1 ml/dish). The cells were spun down at 300 × g and resuspended on ice in 20 mmol/L Tris-HCl pH 7.5, 1 mmol/L ethylenediaminetetraacetic acid, 5 mmol/L MgCl2, 1 mmol/L dithiothreitol DTT, 2 mmol/L mercaptoethanol, 1 mmol/L phenylmethylsulfonyl fluoride, 1 μg/ml leupeptin, 1 μg/ml aprotinin, and 0.25 mol/L sucrose (1 ml/50 × 106 cells) and sonicated briefly. The extract was centrifuged in the cold for 15 minutes at 1100 × g to remove nuclei and insoluble debris. The protein content of the extracts was determined using the Bradford method (BioRad, Richmond, CA), and the extracts were stored at −80°C until use. To determine whether these extracts were able to ADP-ribosylate rVEGF165, labeling reactions were set up containing: 500 ng VEGF165, 10 μg macrophage protein extract, 20 mmol/L Tris-HCl pH 7.8, 20 mmol/L isoniazid, 120 mmol/L MgCl2, 10 mmol/L NaF, 0.02% leupeptin, 0.54 mmol/L NAD phosphate, 0.4 mmol/L isobutyl-methylxanthine, 0.1% lubrol, 2 mmol/L dithiothreitol, 10 mmol/L thymidine, and 7 μCi 32P-labeled NAD+ (800 Ci/mmol) (DuPont-NEN, Wilmington, DE). After 2 hours of incubation at 30°C the reaction mixture was placed on ice and precleared for 30 minutes with 10 μl of protein A/G-agarose (Santa Cruz Biotechnology, Santa Cruz, CA). Ten μg of a murine anti-VEGF monoclonal antibody (gift of Texas Biotechnology, Inc., Dallas, TX) was added to the supernatant, and the mixture was incubated on ice for 2 hours. Protein A/G-agarose beads (10 μl) were then added, and the mixture was further incubated for 2 hours at 4°C with gentle rocking. The beads were harvested by centrifugation and washed three times with cell lysis buffer. The beads were then incubated in an equal volume of 2× electrophoresis sample buffer (final concentration of 100 mmol/L dithiothreitol), and heated at 95°C for 10 minutes to elute bound VEGF from the beads. The samples were then separated using 0.1% sodium dodecyl sulfate (SDS) 15% polyacrylamide gel electrophoresis (PAGE), and the fractionated proteins were transferred to a nitrocellulose membrane by semidry electrophoretic transfer. The filters were then immunostained using anti-VEGF antibody, and the VEGF bands were detected using enhanced fluorecence detection reagents (Amersham Vistra reagents, Amersham, Arlington Heights, IL) and a Fluorimage Analyzer (Molecular Dynamics, Sunnyvale, CA). The nitrocellulose blots were then analyzed using a PhosphorImager analyzer (Molecular Dynamics), to determine the localization of 32P-labeled bands.
rVEGF165 was incubated for up to 2 hours at 30°C with cholera toxin as follows: 500 ng rVEGF and 250 μg cholera toxin (A-subunit, Sigma Chemical Co.), in the reaction buffer described above. The reaction was terminated by the addition of an equal volume of cold 10% trichloroacetic acid. The precipitated protein was washed three times with water-saturated chloroform and finally resuspended in an equal volume of 2× PAGE sample buffer, as above. The samples were separated by SDS-PAGE and transferred to a nitrocellulose membrane as described above.
Five hundred ng rVEGF165 was incubated for up to 2 hours at 30°C with 25 μg pertussis toxin (Sigma Chemical Co., cat. no. P-0317) in the reaction mixture described above. Pertussis toxin was preactivated by incubation for 30 minutes with 10 mmol/L ATP and 20 mmol/L dithiothreitol before addition to the VEGF reaction mixture. The reaction was terminated and analyzed as described above.
Effects of ADP-Ribosylation on the Angiogenic Activity of VEGF
To determine whether ADP-ribosylation of VEGF modulates its bioactivity as an angiogenic factor rVEGF165 was treated as described above with either cholera toxin or macrophage cytosolic extract, but in the presence of unlabeled NAD+. To facilitate the recovery of rVEGF from the reaction mixture, rather than using immunoprecipitation for the recovery of VEGF, which requires the use of harsh, denaturing conditions for the recovery of VEGF from the protein A/G-agarose beads, heparin-Sepharose binding was used to recover the VEGF. After the labeling reaction, 10 μl washed heparin-Sepharose beads were added, and the mixture was incubated at 4°C for 4 hours with gentle agitation. The beads were then washed three times with 100 μl 20 mmol/L Tris-HCl pH 7.8 containing 0.4 mmol/L NaCl. VEGF was eluted from the beads by incubation with 20 μl Tris-HCl containing 1.5 mol/L NaCl. Recovery of VEGF was determined by specific ELISA. Control reactions were carried out in the absence of bacterial toxins and macrophage extract. To ensure that antiangiogenic activity was not present in the macrophage extracts or the cholera toxin preparations, similar labeling reactions were carried out in the absence of VEGF, and the heparin-Sepharose eluates from these reactions were tested in the antiangiogenesis assay.
Results
Production of Nitrite by RAW264.7 Cells and MPMs
Figure 1 ▶ shows the production of nitrite by MPMs. Nitrite was not produced by nonactivated cells, either with or without lactate. After challenge with IFNγ/LPS, nitrite production was strongly induced, with nitrite accumulating over the 48-hour incubation period. l-NAME (1.5 mmol/L) blocked nitrite production by about 70 to 80%; AG (1 mmol/L) blocked nitrite production by >95%. RAW264.7 cells produced nitrite in a similar manner, and l-NAME and AG blocked nitrite synthesis by RAW264.7 cells to a similar extent (data not shown).
Figure 1.

Nitrite production by MPMs. Cells were incubated in DMEM-1% FCS with or without sodium lactate (25 mmol/L), IFNγ (100 U/ml) and LPS (100 ng/ml), l-NAME (1.5 mmol/L), or AG (1 mmol/L), as indicated. Media were harvested 18 and 48 hours after challenge with IFNγ/LPS. Results are means ± SD of triplicate determinations in a typical experiment. Similar results were found in at least three separate experiments.
Production of VEGF by RAW264.7 Cells and MPMs
The production of VEGF by RAW cells is shown in Figure 2A ▶ . Nonstimulated RAW cells produced VEGF in an apparently constitutive manner over the 48-hour incubation period. This spontaneous production of VEGF was similar in regular culture plates and in gas-permeable Permanox plates. Stimulation of cells with IFNγ and LPS increased the production of VEGF by RAW cells over the constitutive level produced by nonstimulated cells by about 3 to 4-fold by 18 hours. By 48 hours, the stimulated VEGF levels were only two-fold increased over the constitutive level. The iNOS inhibitors AG (1.0 mmol/L) and l-NAME (1.5 mmol/L) did not block the constitutive production of VEGF by nonstimulated RAW cells, but reduced the production of VEGF by IFNγ/LPS-activated RAW cells to a level markedly below that of the nonstimulated cells. Sodium lactate (25 mmol/L) did not alter the production of VEGF by these cells, either with or without IFNγ/LPS activation. RAW cells cultured under hypoxic conditions produced increased amounts of VEGF. After 18 hours, VEGF levels in the media of cells cultured under hypoxic conditions were about three-fold greater than those in the media of control, normoxic cells. This differential was less marked by 48 hours. Analyses of the dissolved oxygen levels in the conditioned media directly after harvesting indicated clearly that under normoxic conditions, oxygen levels were consistently high (partial pressure of oxygen > 145). After 24 and 48 hours of incubation under hypoxic conditions (95% N2/5% CO2), the partial pressures of oxygen were 71 mm and 46 mm, respectively.
Figure 2.
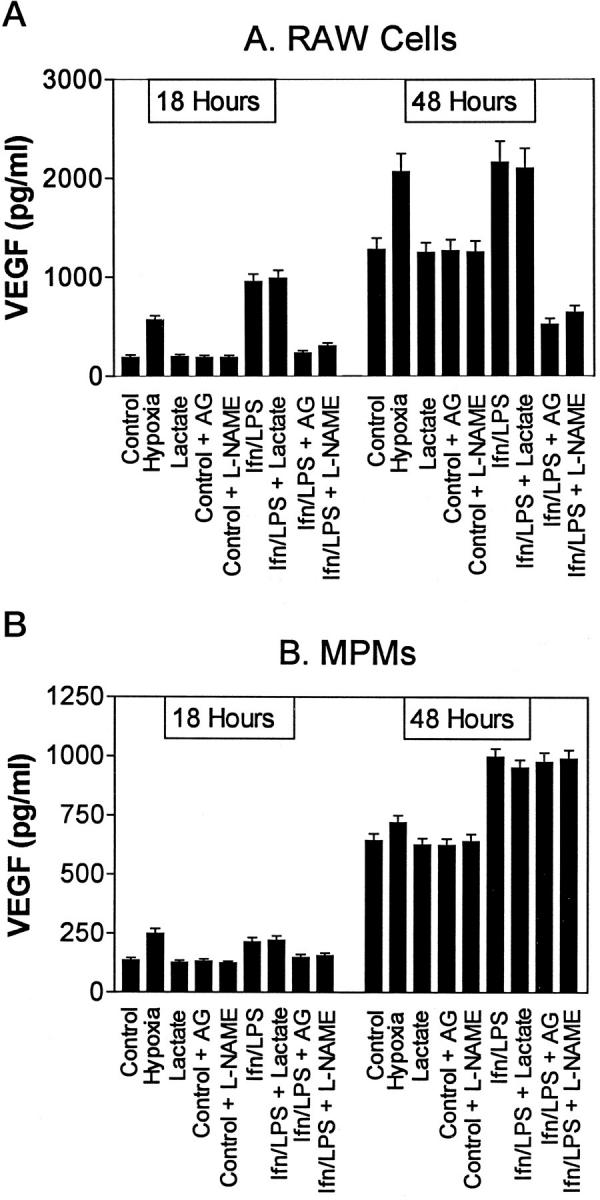
VEGF production by RAW264.7 cells (A) and MPMs (B). Cells were incubated in DMEM-1% FCS with or without sodium lactate (25 mmol/L), IFNγ (100 U/ml) and LPS (100 ng/ml), l-NAME (1.5 mmol/L), or AG (1 mmol/L), as indicated. Media were harvested 18 and 48 hours after challenge with IFNγ/LPS. Results are means ± SD of triplicate determinations in a typical experiment. Similar results were found in at least three separate experiments.
The production of VEGF by MPMs was similar to that of RAW cells, with constitutive production occurring over 48 hours (Figure 2B) ▶ . Increased production was induced by IFNγ/LPS. However, in contrast to RAW cells, iNOS inhibitors did not significantly reduce the production of VEGF by IFNγ/LPS-activated MPMs. As was observed for RAW cells, sodium lactate did not modulate the production of VEGF by these cells. Culture of MPMs under hypoxic conditions resulted in an increase in VEGF production in the first 18 hours; after 48 hours, however, constitutive production of VEGF was only slightly higher than that of hypoxic cells. Oxygen levels determined in the conditioned media of MPMs were similar to those found in RAW cell media.
Quantitative RT-PCR Analysis of VEGF mRNA Levels
A typical example of a quantitative RT-PCR dilution series using the VEGF RNA minigene as internal standard is shown in Figure 3 ▶ . The PCR amplification product of the minigene is 293 bp in size. The native mRNA PCR amplification band is 362 bp in size. The point of equivalence for the amplified minigene and the amplified native mRNA is readily determined from the dilution series. The values determined from these analyses were normalized to the levels of G3PDH mRNA determined in parallel samples, although little variation in the G3PDH mRNA levels were in fact observed between samples. On this basis, the relative amounts of VEGF mRNA in the various macrophage preparations are shown in Table 1 ▶ . Both hypoxia and IFNγ/LPS activation up-regulated VEGF steady-state mRNA levels in MPMs at 4 and 10 hours. By 24 hours, however, the levels of VEGF mRNA were similar in all the groups. In RAW cells, VEGF mRNA levels remained elevated at 24 hours. AG treatment of IFNγ/LPS-treated MPMs did not significantly reduce their steady-state VEGF mRNA levels at any time point; in RAW cells, however, the VEGF mRNA levels were reduced by 70 to 80% at 4, 10, and 24 hours.
Figure 3.

Competitive RT-PCR analysis of VEGF mRNA levels in control (nonstimulated) MPMs 24 hours after plating. Varying amounts of total RNA (1 to 200 ng) isolated from MPMs were reverse transcribed and amplified by PCR through 25 cycles in the presence of a VEGF RNA minigene (2.5 pg) that amplifies using the same primers as the native VEGF mRNA, as described in Materials and Methods. The RNA minigene yields an amplified PCR product of 293 bp, and the native VEGF mRNA yields a 362-bp fragment. The amount of total RNA that yields an amplification band of the same intensity as the minigene is determined from these analyses.
Table 1.
Relative VEGF mRNA Levels* in Macrophages Determined by Competitive RT-PCR
| Time (hours) | |||
|---|---|---|---|
| 4 | 10 | 24 | |
| Control (unstimulated) MPMs | 1 | 1 | 1 |
| Hypoxic MPMs | 2.8 | 5 | 1.4 |
| IFNγ/LPS-activated MPMs | 2.2 | 4.8 | 1 |
| IFNγ/LPS-activated MPMs+ AG (1 mmol/L) | 2 | 4.7 | 1 |
| Control (unstimulated) RAW cells | 1 | 1.3 | 1.2 |
| Hypoxic RAW cells | 3 | 5.8 | 2.5 |
| IFNγ/LPS-activated RAW cells | 2.4 | 5.4 | 2.2 |
| IFNγ/LPS-activated RAW cells+ AG (1 mmol/L) | 0.9 | 1.4 | 1.2 |
*VEGF mRNA levels for each group are compared with the G3PDH mRNA level in the same RNA samples.
RT-PCR Analysis of VEGF mRNA Isoforms
Three isoforms of VEGF were found to be produced by both nonactivated and IFNγ/LPS-activated MPMs. These isoforms corresponded to VEGF-1 (652 bp), VEGF-2 (580 bp), and VEGF-3 (448 bp). 33 The relative proportions of the VEGF isoforms expressed by MPMs at each time point after IFNγ/LPS activation were only slightly modulated by IFNγ/LPS activation and by inhibition of iNOS with AG (Figure 4) ▶ . In RAW cells, VEGF mRNA isoforms were similarly unaffected by IFNγ/LPS activation and by AG treatment.
Figure 4.

RT-PCR analysis of VEGF isoforms produced by IFNγ/LPS-activated MPMs with or without AG treatment. Total RNA isolated from MPMs was reverse transcribed and amplified by PCR, as described in Materials and Methods. PCR primers were located in exons 3 and 8, resulting in the amplification of three PCR products corresponding to 652, 580, and 448 bp.
Production of TNFα by MPMs and RAW264.7 Cells
TNFα was not produced by either nonstimulated MPMs or by RAW264.7 cells over the 48-hour test period. Production of TNFα by MPMs is shown in Figure 5 ▶ . After stimulation with IFNγ/LPS, TNFα expression was strongly induced, with increased TNFα in the conditioned media being apparent by 8 hours after challenge. There was no significant difference in TNFα production in cells treated with or without sodium lactate. Similarly, culture of cells in Permanox dishes, under either normoxic or hypoxic conditions, did not modulate TNFα production. The iNOS inhibitors l-NAME and AG had no significant effect on the production of TNFα by MPMs. Production of TNFα by RAW cells was similar to that observed in MPMs (data not shown).
Figure 5.

TNFα production by MPMs. Cells were incubated in DMEM-1% FCS with or without sodium lactate (25 mmol/L), IFNγ (100 U/ml) and LPS (100 ng/ml), l-NAME (1.5 mmol/L), or AG (1 mmol/L), as indicated. Media were harvested 8, 24, and 48 hours after challenge with IFNγ/LPS. Results are means ± SD of triplicate determinations in a typical experiment. Similar results were found in at least three separate experiments.
ADP-Ribosylation of VEGF by Bacterial Toxins and Macrophage Extracts
Labeling of rVEGF with 32P-labeled NAD was observed using cholera toxin and macrophage cytosolic extracts (Figure 6) ▶ . Labeling with cholera toxin resulted in a single 32P-labeled band corresponding to the size of the rVEGF165 standard (Figure 6C) ▶ . Labeling with macrophage cytosolic extracts resulted in the 32P labeling of a large number of bands, because of the endogenous labeling of macrophage cytosolic proteins (Figure 6A) ▶ . To clearly demonstrate labeling of rVEGF165 in this mixture, immunoprecipitation of the macrophage cytosolic labeling mixture with anti-VEGF antibody was necessary. After immunoprecipitation, a prominent labeled band corresponding to rVEGF165 was clearly visible (Figure 6B) ▶ . This band was not present in control reactions carried out in the absence of rVEGF165. Labeling of VEGF using pertussis toxin was not observed (Figure 6E) ▶ .
Figure 6.

ADP-ribosylation of rVEGF165 by bacterial toxins and by macrophage cytosolic extract. Lane A: rVEGF (500 ng) was incubated with macrophage cytosolic extract (see Materials and Methods) in the presence of [32P]NAD+. The total labeling reaction was analyzed on the 0.1% SDS-15% PAGE gel. Lane B: The rVEGF165-macrophage cytosolic extract labeling mixture was immunoprecipitated with anti-VEGF antibody, and the immunoprecipitated VEGF was analyzed by SDS-PAGE. A dominant 32P-labeled band migrating in the same position as rVEGF165 (determined by Western analysis of the same blot) is indicated. Lane C: rVEGF165 was incubated with cholera toxin subunit A and [32P]NAD+ as described in Materials and Methods. Lane D: Cholera toxin was incubated with [32P]NAD+ in the absence of rVEGF165. Lane E: rVEGF165 was incubated with pertussis toxin and [32P]NAD+, as described in Materials and Methods.
Angiogenic and Antiangiogenic Responses in Rat Corneas
The angiogenic responses induced in rat corneas by the concentrated conditioned media from the MPMs cultured under various conditions are shown in Table 2 ▶ . Medium from nonactivated MPMs cultured under normoxic conditions did not induce angiogenesis. This medium did not contain antiangiogenic activity, as the angiogenic effects of VEGF (25 ng) were unaffected by this medium. Medium from IFNγ/LPS-activated MPMs was potently angiogenic, whereas medium from iNOS-inhibited IFNγ/LPS-activated MPMs showed markedly reduced angiogenic activity. In contrast to medium from normoxic, nonactivated MPMs, this medium was found to contain antiangiogenic activity, as we have reported previously. 37 Medium from normoxic, lactate-treated nonactivated MPMs showed significant angiogenic activity. Similarly, medium from nonactivated MPMs cultured under hypoxic conditions showed significant angiogenic activity. In both of these cases, a polyclonal antibody to VEGF neutralized the angiogenic activity in the conditioned media. Angiogenic responses induced by rVEGF165 were neutralized by anti-VEGF antibody in control experiments, whereas those induced by basic fibroblast growth factor (20 ng/implant) and TNFα (20 ng/implant) were unaffected.
Table 2.
Angiogenic and Antiangiogenic Responses Induced in Rat Corneas by Conditioned Media from MPMs Cultured under Various Conditions in Vitro
| Group | Macrophage culture conditions* | Angiogenic score† |
|---|---|---|
| 1 | Normoxia | 1 |
| 2 | Hypoxia | 9 |
| 3 | Normoxia+ lactate (25 mmol/L) | 8 |
| 4 | IFNγ (100 U/ml)/LPS (100 ng/ml) | 11 |
| 5 | IFNγ/LPS+ AG (1 mmol/L) | 2 |
| 6 | Group 2+ anti-VEGF antibody (10 μg/ml) | 1 |
| 7 | Group 3+ anti-VEGF antibody (10 μg/ml) | 2 |
| 8 | Group 4+ anti-VEGF antibody (10 μg/ml) | 4 |
| 9 | rVEGF165 (20 ng) | 11 |
| 10 | Basic fibroblast growth factor (20 ng) | 12 |
| 11 | TNFα (20 ng) | 10 |
| 12 | Group 9+ anti-VEGF antibody (10 μg/ml) | 2 |
| 13 | Group 10+ anti-VEGF antibody (10 μg/ml) | 11 |
| 14 | Group 11+ anti-VEGF antibody (10 μg/ml) | 11 |
| 15 | Group 1+ rVEGF165 (20 ng) | 11 |
| 16 | Group 5+ rVEGF165 (20 ng) | 2 |
*Macrophages were incubated for 48 hours under the indicated conditions, concentrated (×20), and diafiltered using Centricon 3 (3000 Mr cutoff) filters (Amicon). Samples were then combined with equal volumes of Hydron (Interferon Sciences, Inc.) (12% w/v in 95% ethanol). Ten-microliter droplets were then allowed to dry on the cut ends of 2 mm-diameter Teflon rods. These pellets were then implanted aseptically in the corneas of rats.
†Angiogenic responses were assessed 7 days after implantation. The angiogenic score represents the sum of the graded angiogenic responses from four individual corneas for each test sample. A maximal response would score 12; a minimal response, 0 (see Materials and Methods).
The angiogenic responses induced by rVEGF165 that was ADP-ribosylated using cholera toxin or MPM cytosolic extract are shown in Table 3 ▶ . Whereas control VEGF (taken through a sham labeling procedure in the absence of cholera toxin and MPM cytoplasmic extracts) strongly induced angiogenesis, both cholera toxin-mediated and MPM cytoplasmic extract-mediated ADP-ribosylated VEGF showed greatly reduced angiogenic responses, indicating that the ADP-ribosylation abrogated the angiogenic activity of the VEGF. Because the VEGF was purified from the reaction mixtures using heparin-Sepharose binding and elution, we also tested eluates from control VEGF-free reactions prepared with cholera toxin or macrophage cytosolic extract, to determine, first, whether these extracts contained angiogenic activity in their own right, and second, whether any antiangiogenic activity might be enriched in the eluates through this procedure and interfere with the angiogenic activity of the VEGF. The eluates were therefore tested alone, and then with the postreaction addition of rVEGF165. The sham eluates did not exhibit direct angiogenic activity, nor did they exhibit antiangiogenic activity when combined with VEGF.
Table 3.
Effects of ADP-Ribosylation* on the Angiogenic Activity of rVEGF165
| Test material | Angiogenic score |
|---|---|
| 1. Sham-reacted rVEGF165 (20 ng) | 11 |
| 2. Cholera toxin-treated rVEGF165 (20 ng) (heparin-Sepharose eluate) | 2 |
| 3. Cholera toxin control (heparin- Sepharose eluate) | 1 |
| 4. rVEGF165 (20 ng)+ cholera toxin control | 10 |
| 5. Macrophage cytosolic extract-treated rVEGF165 (20 ng) (heparin-Sepharose eluate) | 3 |
| 6. Macrophage cytosolic extract control | 2 |
| 7. rVEGF165 (20 ng)+ macrophage cytosolic extract control | 11 |
*rVEGF165 was treated in a reaction mixture with either cholera toxin or macrophage cytosolic extracts, as described in Materials and Methods. Controls of VEGF treated in the absence of cholera toxin or macrophage cytosolic extract were performed to determine the effects of the buffers on VEGF. Controls of the cholera toxin and macrophage cytosolic extract incubated without VEGF were also performed, to determine whether extraneous angiogenic or antiangiogenic factors were present in these reagents. All reactions were treated with heparin-Sepharose as described in Materials and Methods, to recover the VEGF from the reaction mixtures.
Discussion
In this study, we show that murine macrophages (MPMs) produce VEGF, a potent, endothelial cell-specific, angiogenic growth factor. 23,24 VEGF production by MPMs does not require activation, with significant VEGF levels being released into the conditioned media over 18 to 48 hours without the addition of external stimulants. This constitutive level of VEGF production was, however, markedly increased by stimulation of the cells with IFNγ/LPS (Figure 2) ▶ . In contrast, the production of TNFα was strictly dependent on macrophage activation with IFNγ and LPS, as has been shown in many previous studies (Figure 4) ▶ . 38,39
VEGF expression has been shown to be regulated by oxygen tension both in vivo and in vitro, 40-44 with low levels of oxygen (hypoxia) resulting in the up-regulation of VEGF expression. This increased expression has been shown to be regulated both at the transcriptional level and at the level of mRNA stability, depending on the cell type. In our studies, oxygen concentrations were measured in the conditioned media of macrophages cultured in both normal and Permanox culture dishes. These measurements indicated that under these conditions, the media on the MPMs and RAW cells were normoxic, suggesting that the constitutive VEGF production observed was not due to induction of VEGF gene expression by low oxygen tension. However, when cells were specifically incubated under hypoxic conditions, significant up-regulation of VEGF, but not of TNFα or nitrite production, was observed in both cell types. This up-regulation of VEGF expression was apparent both at the mRNA and the protein levels. These observations suggest that the expression of the VEGF gene is regulated by oxygen tension in macrophages, as observed in other cell types. It is not yet clear, however, whether this regulation occurs at the level of transcription or at the level of mRNA stability.
Knighton and coworkers have shown previously that the expression of angiogenic activity by rabbit bone marrow-derived macrophages is regulated by hypoxia, and that the high levels of lactate that accumulate in the conditioned media of hypoxic macrophages are important in regulating the expression of macrophage-derived angiogenic activity. 22 In MPMs and RAW cells, culture in the presence of high lactate concentrations (25 mmol/L), under normoxic conditions, did not modulate the level of expression of VEGF mRNA or protein. However, it is important to note that, although nonstimulated MPMs express significant levels of VEGF, the conditioned media from these cells is nonangiogenic. 4,8,22 After lactate or hypoxia treatments, the media exhibit angiogenic activity (Table 1) ▶ . This raises the important question of how the angiogenic activity of VEGF is regulated. First, VEGF may operate in synergy with TNFα to stimulate the microvasculature in the conditioned media from IFNγ/LPS-activated macrophages. However, the fact that medium from lactate-treated or hypoxia-treated nonactivated macrophages, which do not contain TNFα, express potent angiogenic activity suggests that under the appropriate conditions, VEGF can be angiogenic in the absence of TNFα. This is supported by the fact that the angiogenic activity in these media is neutralized by anti-VEGF antibodies (Table 1) ▶ . A second possibility tested was that medium from normoxic, nonactivated MPMs might contain an antiangiogenic factor(s) that blocks the angiogenic effects of VEGF. This hypothesis was tested using the rat corneal bioassay, by combining concentrated conditioned medium from these cells with rVEGF165, to determine whether the angiogenic effects of the VEGF were inhibited. No inhibition of the effects of VEGF were in fact observed in this system, clearly indicating that antiangiogenic factor(s) were not present in this conditioned medium. This is in contrast to the conditioned medium from iNOS-inhibited, IFNγ/LPS-activated macrophage medium, as discussed further below.
We then hypothesized that the VEGF produced by nonstimulated MPMs may differ structurally from the VEGF produced by stimulated MPMs. This structural difference could relate to alternatively spliced isoforms of VEGF with differing angiogenic activities, or to posttranslational modification of VEGF by, for example, ADP ribosylation-dependent mechanisms. 32,33,46 Our results using RT-PCR indicate that the isoforms of VEGF are not markedly changed during macrophage activation, by lactate, or by inhibition of iNOS. VEGF-1, -2, and -3 mRNA isoforms are produced in similar proportions under all conditions tested. It thus seems that the most likely mechanism for regulation of VEGF angiogenic activity might involve posttranslational mechanisms, as has been suggested recently by Hussain et al. 33,47 In support of this hypothesis, rVEGF was shown to be a substrate for ADP-ribosylation, and ADP-ribosylation was shown to abrogate the angiogenic activity of rVEGF. Because macrophages are impermeable to NAD+, metabolic labeling of endogenously synthesized VEGF by macrophages using 32P-labeled NAD+, is not possible. 33,34 However, we demonstrated labeling of rVEGF165 in vitro, using cytosolic extracts of macrophages, as well as by the bacterial arginine-specific ADP-ribosyl transferase, cholera toxin subunit A (Figure 6) ▶ . 35,36 Pertussis toxin, on the other hand, which is a cysteine-specific ADP-ribosyl transferase, did not modify rVEGF165. 48 In addition, we showed that ADP-ribosylation of VEGF abrogates its angiogenic activity. In contrast to unmodified rVEGF165, rVEGF165 derivatized using either cholera toxin or macrophage cytosolic extract was found to be nonangiogenic (Table 3) ▶ .
We have shown previously that the production of angiogenic activity by human monocytes and by murine macrophages is induced by activation of the cells with IFNγ/LPS. 1-3,20,21,49 In addition, the l-arginine-dependent iNOS-dependent pathway plays an important role in regulating the expression of angiogenic activity by IFNγ/LPS-activated macrophages. 21 Inhibitors of iNOS, such as l-NAME, Ng-monomethyl-l-arginine, diphenyleneiodonium, and AG block the production of angiogenic activity by activated macrophages, without inhibiting the production of the angiogenic cytokines TNFα and Il-8. 21,49,50 In this study, we show that the iNOS inhibitors l-NAME and AG markedly inhibit the production of VEGF by IFNγ/LPS-activated RAW cells (>70% inhibition) but have little effect on the constitutive (nonstimulated) production of VEGF by these cells. Interestingly, in IFNγ/LPS-activated RAW cells, l-NAME and AG inhibit VEGF production to a level significantly below that of nonstimulated cells. This suggests that the pathways involved in the regulation of VEGF production in nonactivated and activated RAW cells are different, with only the activated pathway being sensitive to iNOS products. This might relate to the nature of the transcriptional promotors involved in the expression of the VEGF gene under constitutive and activated conditions. In MPMs, on the other hand, the iNOS inhibitors had no significant effect on the production of either the constitutive or IFNγ/LPS-stimulated VEGF. However, it is again important to note that the angiogenic activity of the MPM conditioned media was markedly down-regulated by the iNOS inhibitors. Our results suggest that two mechanisms are involved in the regulation of expression of angiogenic activity by the iNOS-inhibited, IFNγ/LPS-activated MPMs. The first is analogous to that observed in the activation of macrophages by hypoxia and lactate, namely, the regulation of the ADP-ribosylation of VEGF, and hence of its angiogenic activity. IFNγ/LPS activation switches the production of VEGF from the ADP-ribosylated, nonangiogenic form to the unmodified, angiogenic form. Secondly, the iNOS-dependent pathway regulates the expression of an inhibitor of angiogenesis. When the iNOS pathway is active and nitric oxide is produced, the inhibitor is inactive or absent; when the iNOS pathway is blocked with AG or l-NAME, the inhibitor is active. We have previously reported that this antiangiogenic activity is present in the conditioned medium of iNOS-inhibited IFNγ/LPS-activated MPMs. 37 The nature of this inhibitor is not yet clear; however, it is not neutralized by specific antibodies to thrombospondin-1 or IFNγ-inducible protein-10, both of which are potent antiangiogenic agents that may be produced by macrophages. 51,52 Specific antibodies to TNFα and transforming growth factor-β also do not neutralize the antiangiogenic activity. The inhibitor binds weakly to heparin-Sepharose and has an apparent molecular weight >100 kd. 37 Further characterization of this antiangiogenic factor is underway in our laboratory.
Hussain and coworkers 33,47 have suggested that ADP-ribosylation-dependent mechanisms may be involved in the posttranslational modification of angiogenic factors, resulting in nonangiogenic forms. Our results suggest that this may indeed be one of the mechanisms regulating the production of angiogenic activity by macrophages. We suggest that VEGF produced by the constitutive pathway is normally in the ADP-ribosylated, nonangiogenic form, whereas VEGF produced by IFNγ/LPS-activated MPMs is in the unribosylated, angiogenic form. Activation may thus regulate the posttranscriptional modification of VEGF from the ADP-ribosylated nonangiogenic form to the unmodified angiogenic form. In addition, the iNOS pathway in activated MPMs appears to regulate the production (or bioactivity) of an antiangiogenic factor that is apparent only in IFNγ/LPS-activated, iNOS-inhibited MPM medium.
Our results clearly indicate that VEGF is a substrate for ADP-ribosylation, and that ADP-ribosylation of VEGF abrogates its angiogenic activity. Preliminary results (manuscript in preparation) also indicate that vitamin K3 and novobiocin, both inhibitors of mono-ADP-ribosylation reactions, 35,53 result in the production of angiogenically active VEGF by nonactivated normoxic macrophages, without affecting the level of VEGF production or the production of TNFα, suggesting the involvement of mono-ADP-ribosylation in the regulation of angiogenic activity in macrophages. Ultimate proof of the role of mono-ADP-ribosylation in the regulation of VEGF bioactivity by macrophages however, will require the direct demonstration that VEGF is differentially ADP-ribosylated in macrophages under conditions that modify oxygen tension or IFNγ/LPS-induced macrophage activation and the iNOS-dependent pathway. These studies are underway in our laboratory.
In summary, we suggest on the basis of our observations that VEGF is an important contributor to macrophage-dependent angiogenic activity. VEGF production in macrophages is regulated at several levels. Constitutively expressed VEGF is normally angiogenically inactive. Hypoxia and IFNγ/LPS activation increase the absolute amount of VEGF produced but also result in the expression of angiogenic VEGF. High lactate does not increase the amount of VEGF produced, but it also results in the production of angiogenic VEGF. The change in the angiogenic phenotype of VEGF may be due to posttranslational modification, perhaps by the process of ADP-ribosylation, that modulates VEGF bioactivity. rVEGF165 is a substrate for ADP-ribosylation by cholera toxin and by MPM cytoplasmic extracts, and ADP-ribosylation of rVEGF165 was shown to abrogate its angiogenic activity. In hypoxic and IFNγ/LPS-activated MPMs, activation up-regulated VEGF mRNA expression and also shifted the balance of posttranslational modification of VEGF from the nonangiogenic to the angiogenic form. In RAW264.7 cells, the IFNγ/LPS activation-dependent modulation of VEGF mRNA levels is regulated in part by the iNOS pathway, but the constitutive production of VEGF in nonactivated cells is not. In MPMs, on the other hand, the regulation of VEGF mRNA level by IFNγ/LPS activation is not significantly dependent on the iNOS pathway. VEGF angiogenic activity in these cells appears to be regulated at the level of posttranslational modification. Finally, when the iNOS pathway is inhibited in IFNγ/LPS-activated MPMs, an antiangiogenic factor is expressed that blocks the angiogenic activity of VEGF. Together, regulation of VEGF bioactivity by posttranslational modification and iNOS-dependent regulation of the expression of an antiangiogenic factor provide novel mechanisms for controlling the angiogenic phenotype of macrophages and may play a key role in the regulation of macrophage-dependent angiogenic activity in vivo in wound repair, fibroproliferation, and possibly in solid tumor development.
Acknowledgments
The affininity-purified anti-VEGF polyclonal antibody was a kind gift of Dr. Napoleone Ferrara (Genentech, Inc., South San Francisco, CA). An affinity-purified antibody to murine IFNγ-inducible protein-10 was a kind gift of Dr. Gerald Luster (Harvard University, Boston, MA).
Footnotes
Address reprint requests to Dr. S.J. Leibovich, Ph.D., Department of Anatomy, Cell Biology & Injury Sciences, New Jersey Medical School, UMDNJ, 185 South Orange Avenue, Newark, NJ 07103. E-mail: leibovic@umdnj.edu.
References
- 1.Polverini PJ, Cotran RS, Gimbrone MA, Jr, Unanue ER: Activated macrophages induce vascular proliferation. Nature 1977, 269:804-806 [DOI] [PubMed] [Google Scholar]
- 2.Koch AE, Polverini PJ, Leibovich SJ: Induction of neovascularization by activated human monocytes. J Leukocyte Biol 1985, 37:279-288 [DOI] [PubMed] [Google Scholar]
- 3.Polverini PJ: Macrophage-induced angiogenesis: a review. Cytokines 1989, 1:54-73 [Google Scholar]
- 4.Polverini PJ, Leibovich SJ: Induction of neovascularization and nonlymphoid mesenchymal cell proliferation by macrophage cell lines. Lab Invest 1985, 51:635-642 [DOI] [PubMed] [Google Scholar]
- 5.Sunderkotter C, Steinbrink K, Goebeler M, Bhardwaj R, Sorg C: Macrophages and angiogenesis. J Leukocyte Biol 1994, 55:410-422 [DOI] [PubMed] [Google Scholar]
- 6.DiPietro LA, Polverini PJ: Angiogenic macrophages produce the angiogenic inhibitor thrombospondin-1. Am J Pathol 1993, 143:678-684 [PMC free article] [PubMed] [Google Scholar]
- 7.Polverini PJ: The pathophysiology of angiogenesis. Crit Rev Oral Biol Med 1995, 6:230-247 [DOI] [PubMed] [Google Scholar]
- 8.Folkman J: Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med 1995, 1:27-31 [DOI] [PubMed] [Google Scholar]
- 9.Leibovich SJ, Polverini PJ, Shepard HM, Wiseman DM, Shively V, Nuseir N: Production of angiogenic activity by human monocytes requires an l-arginine/nitric oxide synthase-dependent effector mechanism. Nature 1987, 329:630-6322443857 [Google Scholar]
- 10.Koch A, Polverini PJ, Kunkel SL, Harlow LA, DiPietro LA, Elner VM, Elner SG, Strieter RM: Interleukin-8 as a macrophage-derived mediator of angiogenesis. Science 1992, 258:1798-1801 [DOI] [PubMed] [Google Scholar]
- 11.Frater-Schroder M, Risau W, Hallmann P, Gautschi R, Bohlen P: Tumor necrosis factor type-α, a potent inhibitor of endothelial cell growth in vitro, is angiogenic in vivo. Proc Natl Acad Sci USA 1987, 84:5277-5281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.DiPietro LA, Polverini PJ: Angiogenic macrophages produce the angiogenic inhibitor thrombospondin-1. Am J Pathol 1993, 143:678-684 [PMC free article] [PubMed] [Google Scholar]
- 13.Xiong M, Lanahan M, Elson E, Leibovich SJ: P53 protein levels in murine peritoneal macrophages are modulated by the inducible-nitric oxide-synthase (iNOS) pathway. Mol Biol Cell 1996, 7(suppl):23a [Google Scholar]
- 14.Besner EB, Klagsbrun M: Macrophages secrete a heparin-binding inhibitor of endothelial cell growth. Microvasc Res 1991, 42:187-197 [DOI] [PubMed] [Google Scholar]
- 15.Polverini PJ: Cytokines 1989, 1:54-73 [Google Scholar]
- 16.Sunderkotter C, Goebeler M, Schulze-Osthoff K, Bhardwaj R, Sorg C: Macrophage-derived angiogenesis factors. Pharmacol Ther 1991, 51:195-216 [DOI] [PubMed] [Google Scholar]
- 17.Nathan CF: Secretion of oxygen intermediates: role in effector functions of activated macrophages. Fed Proc 1982, 41:2206-2211 [PubMed] [Google Scholar]
- 18.Nagashima M, Yoshiro S, Ishiwata T, Asano G: Role of vascular endothelial growth factor in angiogenesis of rheumatoid arthritis. J Rheumatol 1995, 22:1624-1630 [PubMed] [Google Scholar]
- 19.Hibbs JB, Jr, Vavrin Z, Taintor RR: l-Arginine is required for the expression of the activated macrophage effector mechanism causing selective metabolic inhibition in target cells. J Immunol 1987, 138:550-565 [PubMed] [Google Scholar]
- 20.Koch AE, Cho M, Burrows JC, Polverini PJ, Leibovich SJ: Inhibition of production of monocyte/macrophage-derived angiogenic activity by oxygen free-radical scavengers. Cell Biol Int Rep 1992, 16:415-411 [DOI] [PubMed] [Google Scholar]
- 21.Leibovich SJ, Polverini PJ, Fong TW, Harlow LA, Koch AE: Production of angiogenic activity by human monocytes requires an l-arginine/nitric oxide-synthase-dependent effector mechanism. Proc Natl Acad Sci USA 1994, 91:4190-4194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Knighton DR, Hunt TK, Scheuenstahl H, Halliday BJ, Werb Z, Banda MJ: Oxygen tension regulates the expression of angiogenesis factor by macrophages. Science 1984, 221:1283-1285 [DOI] [PubMed] [Google Scholar]
- 23.Leung DW, Cachianes G, Kuang WJ, Goeddel DV, Ferrara N: Vascular endothelial growth factor is a secreted angiogenic mitogen. Science 1989, 244:1306-1309 [DOI] [PubMed] [Google Scholar]
- 24.Connolly DT, Olander JV, Heuvelman D, Nelson R, Monsell R, Siegel N, Haymore BL, Leimgruber R, Feder J: Human vascular permeability factor. J Biol Chem 1989, 264:20017-20024 [PubMed] [Google Scholar]
- 25.Gospodarowicz D, Abraham JA, Schilling J: Isolation and characterization of a vascular endothelial cell mitogen produced by pituitary-derived folliculostellate cells. Proc Natl Acad Sci USA 1989, 86:7311-7315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Berse B, Brown LF, Van de Water L, Dvorak HF, Senger DR: Vascular permeability factor (vascular endothelial growth factor) gene is expressed differentially in normal tissues, macrophages, and tumors. Mol Biol Cell 1992, 3:211-220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sharkey AM, Charnock-Jones DS, Boocock CA, Brown KD, Smith SK: Expression of mRNA for vascular endothelial growth factor in human placenta. J Reprod Fertil 1993, 99:609-615 [DOI] [PubMed] [Google Scholar]
- 28.Fava RA, Olsen NJ, Spencer-Green G, Yeo TK, Berse B, Jackman RW, Senger DR, Dvorak HF, Brown LF: Vascular permeability factor/endothelial growth factor (VPF/VEGF): accumulation and expression in human synovial fluids and rheumatoid synovial tissue. J Exp Med 1994, 180:341-346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Torry RJ, Labarrere CA, Torry DS, Holt VJ, Faulk WP: Vascular endothelial growth factor expression in transplanted human hearts. Transplantation 1995, 60:1451-1457 [DOI] [PubMed] [Google Scholar]
- 30.McLaren J, Prentice A, Charnock-Jones DS, Millican SA, Muller KH, Sharkey AM, Smith SK: Vascular endothelial growth factor is produced by peritoneal fluid macrophages in endometriosis and is regulated by ovarian steroids. J Clin Invest 1996, 98:482-489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shima DT, Kuroki M, Deutsch U, Ng YS, Adamis AP, D’Amore PA: The mouse gene for vascular endothelial growth factor. J Biol Chem 1996, 271:3877-3887 [DOI] [PubMed] [Google Scholar]
- 32.Brune B, Lapetina EG: Activation of a cytosolic ADP-ribosyl transferase by nitric oxide-generating agents. J Biol Chem 1989, 264:8455-8458 [PubMed] [Google Scholar]
- 33.Zabel DD, Feng JJ, Scheuenstahl H, Hunt TK, Hussain MZ: Lactate stimulation of macrophage-derived angiogenic activity is associated with inhibition of poly(ADP-ribose) synthesis. Lab Invest 1996, 74:644-649 [PubMed] [Google Scholar]
- 34.Aktories K, Just I: In vitro ADP-ribosylation of Rho by bacterial ADP-ribosyl transferases. Methods Enzymol 1995, 256:184-195 [DOI] [PubMed] [Google Scholar]
- 35.Okazaki IJ, Moss J: Mono-ADP-ribosylation: a reversible post-translational modification of proteins. Adv Pharmacol 1996, 35:247-280 [DOI] [PubMed] [Google Scholar]
- 36.Moss J, Vaughan M: Mechanism of action of cholaregen: evidence for ADP-ribosyl transferase activity with arginine as an acceptor. J Biol Chem 1997, 252:2455-2457 [PubMed] [Google Scholar]
- 37.Leibovich SJ, Xiong M, Elson E, Sharma S, Seo C, Lanahan M: The role of macrophages in the control of angiogenesis in wound repair: nitric oxide (NO), angiogenesis inhibitors and tumor suppresser genes. Rabie AM Urist MR eds. Bone Formation and Repair. 1997, :pp 101-111 Elsevier, Amsterdam [Google Scholar]
- 38.Beutler B, Cerami A: Cachectin and tumor necrosis factor as two sides of the same biological coin. Nature 1986, 320:584-588 [DOI] [PubMed] [Google Scholar]
- 39.Burchett SK, Weaver WM, Westall JA, Larsen A, Kronheim S, Wilson CB: Regulation of tumor necrosis factor/cachectin and IL-1 secretion in human mononuclear phagocytes. J Immunol 1988, 140:3473-3481 [PubMed] [Google Scholar]
- 40.Mukaida N, Harada A, Yasumoto K, Matsushima K: Properties of pro-inflammatory cell type-specific leukocyte chemotactic cytokines, interleukin-8 (IL-8) and monocyte chemotactic and activating factor (MCAF). Microbiol Immunol 1992, 36:773-789 [DOI] [PubMed] [Google Scholar]
- 41.Shweiki D, Itin A, Soffer D, Keshet E: Patterns of expression of vascular endothelial growth factor (VEGF) and VEGF receptors in mice suggest a role in hormonally regulated angiogenesis. Nature 1992, 359:843-845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ladoux A, Frelin C: Hypoxia is a strong inducer of vascular endothelial growth factor messenger RNA expression in the heart. Biochem Biophys Res Commun 1993, 195:1005-1010 [DOI] [PubMed] [Google Scholar]
- 43.Goldberg MA, Schneider TJ: Similarities between the oxygen-sensing mechanisms regulating the expression of vascular endothelial growth factor and erythropoietin. J Biol Chem 1994, 269:4355-4359 [PubMed] [Google Scholar]
- 44.Shima DT, Adamis AP, Ferrara N, Yeo KT, Allende R, Folkman J, D’Amore P: Hypoxic induction of endothelial cell growth factors in retinal cells: identification and characterisation of vascular endothelial growth factor (VEGF) as the mitogen. Mol Med 1995, 1:182-193 [PMC free article] [PubMed] [Google Scholar]
- 45.Levy AP, Levy NS, Wegner S, Goldberg MA: Transcriptional regulation of the rat vascular endothelial growth factor gene by hypoxia. J Biol Chem 1995, 270:13333-13340 [DOI] [PubMed] [Google Scholar]
- 46.Molina-Vedia L, McDonald B, Reep B, Brune B, DiSilvio M, Billiar TR, Lapetina EG: Nitric oxide induced S-nitrosylation of glyceraldehyde-3-phosphate dehydrogenase inhibits enzymatic activity and increases endogenous ADP-ribosylation. J Biol Chem 1992, 267:24929-24932 [PubMed] [Google Scholar]
- 47.Feng JJ, Hunt TK, Ghani P, Hussain MZ: Macrophage-derived angiogenic activity potential can be reversibly inhibited by ADP-ribosylation. Wound Repair Regen 1997, 5:A111 [Google Scholar]
- 48.West RE, Moss J, Vaughan M, Liu T-Y: Pertussis toxin-catalyzed ADP-ribosylation of transducin. Cysteine 347 is the ADP-ribose acceptor site. J Biol Chem 1985, 260:14428-14430 [PubMed] [Google Scholar]
- 49.Leibovich SJ, Golczewski J: Expression of inducible nitric oxide synthase (iNOS) is required for production of angiogenic activity by murine macrophages and IC-21 cells. Mol Biol Cell (suppl):373a
- 50.Upperman JS, Leibovich SJ, Xiong M, Golczewski J, Deitch EA: Diphenyleneiodonium (DPI), an inhibitor of nucleotide-requiring flavoproteins, inhibits the production of macrophage-derived angiogenic activity. Surg Forum 1996, 47:708-710 [Google Scholar]
- 51.Good DJ, Polverini PJ, Rastinejad F, LeBeau MM, Lemons RS, Frazier WA, Bouck NP: A tumor suppressor-dependent inhibitor of angiogenesis is immunologically indistinguishable from a fragment of thrombospondin. Proc Natl Acad Sci USA 1990, 87:6624-6628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Angiolillo AL, Sgadari C, Taub DD, Liao F, Farber JM, Maheshawari S, Kleinman HK, Reaman GH, Tosato G: Human interferon-inducible protein 10 is a potent inhibitor of angiogenesis in vivo. J Exp Med 1995, 182:155-159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Banasik M, Ueda K: Inhibitors and activators of ADP-ribosylation reactions. Mol Cell Biochem 1994, 138:185-197 [DOI] [PubMed] [Google Scholar]