

Abstract
For over two decades, the development of B-cell lymphoma-2 (Bcl-2) family therapeutics has primarily focused on anti-apoptotic proteins, resulting in the first-in-class drugs called BH3 mimetics, especially for Bcl-2 inhibitor Venetoclax. The pro-apoptotic protein Bcl-2-associated X protein (BAX) plays a crucial role as the executioner protein of the mitochondrial regulated cell death, contributing to organismal development, tissue homeostasis, and immunity. The dysregulation of BAX is closely associated with the onset and progression of diseases characterized by pathologic cell survival or death, such as cancer, neurodegeneration, and heart failure. In addition to conducting thorough investigations into the physiological modulation of BAX, research on the regulatory mechanisms of small molecules identified through biochemical screening approaches has prompted the identification of functional and potentially druggable binding sites on BAX, as well as diverse all-molecule BAX modulators. This review presents recent advancements in elucidating the physiological and pharmacological modulation of BAX and in identifying potentially druggable binding sites on BAX. Furthermore, it highlights the structural and mechanistic insights into small-molecule modulators targeting diverse binding surfaces or conformations of BAX, offering a promising avenue for developing next-generation apoptosis modulators to treat a wide range of diseases associated with dysregulated cell death by directly targeting BAX.
Key words: Apoptosis, Pro-apoptotic protein BAX, Dynamic conformational activation, Small-molecule apoptosis modulators
Graphical abstract
The elucidation of the physiologic and pharmacologic regulatory mechanisms of BAX and characterization of potential druggable binding sites offer an innovative avenue for developing next-generation apoptosis modulators to treat a wide range of diseases in the context of dysregulated cell death by directly targeting BAX.

1. Introduction
The precise control of apoptosis by the Bcl-2 family proteins plays a critical role in organismal development, tissue homeostasis, and immunity1. Bcl-2 family proteins are characterized by the presence of Bcl-2 homology domains 1–4 (BH1–BH4). These proteins are classified into three subsets based on their structural and functional characteristics: multi-domain pro-apoptotic proteins, pro-apoptotic BH3-only proteins, and multi-domain anti-apoptotic proteins. Both multi-domain pro-apoptotic and anti-apoptotic proteins adopt similar globular structures featuring a hydrophobic surface groove. The pro-apoptotic effector proteins BAX2 and BAK (Bcl-2 antagonist/killer)3 contain three Bcl-2 homology domains (BH1–BH3), while BOK (Bcl-2-related ovarian killer protein)4 contains all four Bcl-2 homology domains. The multi-domain anti-apoptotic proteins Bcl-25, Bcl-2-like protein 1 (Bcl-XL)6, Bcl-2-related gene expressed in fetal liver (Bfl-1)7 consist of all four Bcl-2 homology domains, whereas myeloid cell leukemia sequence 1 (Mcl-1)8 and Bcl-2-like protein 2 (Bcl2L2, also known as Bcl-W)9 consist of three Bcl-2 homology domains (BH1–BH3 and BH1/BH2/BH4, respectively). Bcl-2-like protein 10 (Bcl2L10, also known as Bcl-B)10, 11, 12 consists of two Bcl-2 homology domains (BH1–BH2). BH3-only proteins share only the BH3 domain and form an amphipathic helix, enabling them to interact with multi-domain pro-apoptotic and anti-apoptotic proteins. BH3-only proteins are further divided into two functional groups13: activators, such as BH3-interacting domain death agonist (BID)14, Bcl-2-interacting mediator of cell death (BIM)15, p53 upregulated modulator of apoptosis (PUMA)16, and sensitizers, such as phorbol-12-myristate-13-acetate-induced protein 1 (PMAIP1, also known as NOXA)17, Bcl-2 antagonist of cell death (BAD)18, and activator of apoptosis harakiri (HRK)19.
Bcl-2 family proteins play a pivotal role in determining the cell's survival or death through intricate interactions between anti-apoptotic and pro-apoptotic proteins. The subtle differences in the BH3 domains of pro-apoptotic proteins and the BH3-binding grooves of the anti-apoptotic proteins are in charge of the selectivity in interactions among Bcl-2 family proteins. BAX appears to be restrained by all of the anti-apoptotic proteins, whereas BAK is primarily restrained by the anti-apoptotic Bcl-XL, Mcl-1, and Bfl-120, 21, 22. Certain BH3-only proteins, such as BIM, BID, and PUMA likely sequester all of the anti-apoptotic proteins, while others exhibit selectivity for specific anti-apoptotic proteins within the Bcl-2 family, such as BAD for Bcl-2, Bcl-XL, and Bcl-W, as well as NOXA for Mcl-1 and Bfl-123, 24, 25.
The crucial step in the process of apoptosis is the mitochondrial outer membrane permeabilization (MOMP), which leads to the release of mitochondrial proteins (e.g., cytochrome c) and consequently activates the caspase cascade to trigger apoptosis26, 27. MOMP is stimulated by multi-domain pro-apoptotic or BH3-only proteins and is inhibited by anti-apoptotic Bcl-2 family proteins1,28, 29, 30. Upon exposure to cellular stress or cytostatic agents, activator proteins such as BIM utilize their BH3 domain to bind the hydrophobic groove of anti-apoptotic Bcl-2 proteins, thereby neutralizing their anti-apoptotic function, and engage the trigger site of cytosolic BAX monomers to initiate their activation and mitochondrial translocation31, 32 (Fig. 1A). In contrast, the interaction between the BH3 domain of activators and the canonical site of membrane protein BAK triggers their activation and oligomerization33, 34, 35, 36. Whereas sensitizers neutralize the function of anti-apoptotic Bcl-2 proteins and displace activator or activated effector proteins from anti-apoptotic grooves, indirectly activating dormant effector protein BAX13,20,37, 38 (Fig. 1A). Ultimately, the activated effector proteins BAX/BAK execute MOMP by forming pores in the mitochondrial membrane. Unlike BAX and BAK, the functional and regulatory mechanisms of BOK are still under investigation. Multi-domain anti-apoptotic proteins such as Bcl-2 utilize the hydrophobic groove (BH3-binding groove) to trap the BH3 domain of unleashed BH3-only and activated effector proteins, thereby restraining their pro-apoptotic activity2,39, 40. In summary, the activation of effector proteins BAX/BAK is a critical requirement for MOMP41.
Figure 1.

Regulation of apoptosis during health and disease. (A) Diverse cellular stress and cytostatic agents activate pro-apoptotic BH3-only proteins, which sequestrate anti-apoptotic proteins or trigger BAX activation, thereby permeabilizing the mitochondrial outer membrane to activate apoptosis. (B) Tumor cell populations avoid apoptosis through the overexpression of anti-apoptotic proteins during their evolution to the malignant state. (C) The overexpressed anti-apoptotic proteins are neutralized by BH3 mimetics and subsequently reactivate apoptosis in cancer. (D) Diverse BAX agonists directly bind to and conformationally activate BAX to restore apoptosis.
The disruption of apoptosis mediated by Bcl-2 family proteins is closely associated with the development and advancement of various diseases, which can be categorized into two groups: diseases characterized by resisting cell death such as cancer and autoimmune diseases, and diseases marked by excessive cell death such as neurodegeneration and heart failure42.
1.1. The pathologic cell survival with BAX
Tumor cell populations evolve various apoptosis-avoiding mechanisms during their evolution to a malignant state, particularly through the overexpression of Bcl-2 anti-apoptotic proteins, such as Bcl-2, Bcl-XL, and Mcl-143. These overexpressed proteins can inhibit MOMP by trapping BH3-only proteins to prevent BAX activation and by neutralizing the activated pro-apoptotic effector proteins (Fig. 1B). Consequently, therapeutical suppressing Bcl-2 anti-apoptotic proteins in cancer has been extensively investigated for cancer treatment, resulting in the diverse approved therapeutic apoptosis indcucers44, 45. These inducers can be segmented into those that indirectly target the apoptosis pathway, such as the proteasome, histone deacetylase (HDAC)46, and epidermal growth factor receptor (EGFR) inhibitors47, and those that directly target the apoptosis pathway, such as BH3 mimetics48, 49, 50.
The loss-of-function strategy of directly inhibiting Bcl-2 anti-apoptotic proteins has been successfully translated to innovative apoptosis inducers known as ‘BH3 mimetics’51, 52, 53, including the first-in-class Bcl-2 inhibitor Venetoclax50, the Bcl-2/Bcl-XL dual inhibitor Navitoclax49,54, 55 and Mcl-1 selective inhibitors AZD599156, AMG-17657, and S6341558 (Fig. 1C). However, the limitations of BH3 mimetics are gradually exposed, mainly including 1) limited monotherapy activity, 2) resistance mechanisms related to aberrant expression of Bcl-2 pro-survival members59, 60 and multiple acquired mutations in target proteins61, 62, 63, 64, 3) on-target platelet toxicity of Bcl-XL inhibitors65 and cardiac toxicity of Mcl-1 inhibitors66, 67, 68, 69.
To address these concerns, a diversity of apoptosis inducers targeting Bcl-2 anti-apoptotic proteins have been identified and progressed in clinical studies. These include Bcl-2/Bcl-XL dual inhibitors APG-1252 (prodrug)70 and AZD0466 (nanomedicine)71 for poor therapeutic effect and on-target platelet toxicity, Bcl-XL degrader DT-221672 and antibody–drug conjugate (ADC) ABBV-155 for on-target platelet toxicity73, second-generation Bcl-2 inhibitor S5574674, 75 for multiple gene mutations of BCL-2-mediated Venetoclax resistance, among others. Indeed, current Bcl-2 family therapeutics, including BH3 mimetics, degraders, or ADCs restore tumor cell apoptosis by disarming anti-apoptotic proteins to liberate BH3-only proteins or activated effector proteins76, suggesting their powerful anti-tumor potency is mainly dependent on the indirect activation of pro-apoptotic effector proteins BAX and BAK.
The capacity of various chemotherapeutic drugs to prompt apoptosis is attributed to BH3-only proteins that trigger BAX-mediated apoptosis45. Downregulation of pro-apoptotic BH3-only proteins at the transcriptional, translational, and post-translational levels is an important mechanism by which tumor cells limit or circumvent apoptosis77, leading to resistance to current apoptosis inducers78. However, studies have underscored the limited efficacy and resistance of current apoptosis inducers that rely on additional anti-apoptotic proteins78. Recent evidence stresses the importance of apoptosis inducers that trigger apoptosis through the indirect activation of BAX79.
BAX is extensively expressed in nearly all cell types, including cancer cells. Accumulating evidence has shown that the vast majority of cancer cells contain dormant conformers of BAX or the active form of BAX suppressed by anti-apoptotic proteins, such as lung, colon, and breast cancer. Cancer cells also exhibit elevated levels of cytosolic dimers compared to normal cells, which is a key mechanism by which cancer cells resist apoptotic insults. Dysregulated levels of the active form of BAX (e.g., 6A7 BAX) and the inactive form of BAX (e.g., the phospho-specific S184 BAX, pBAX) are strongly linked to the occurrence and development of cancers, such as the downregulation of BAX in colon cencer80, non-small cell lung cancer81, and melanoma82, 83, and relatively increased levels of pBAX in lung cancer84. The enhanced AKT-mediated phosphorylation of BAX at serine 184 (S184) inactivates its pro-apoptotic activity85, contributing to the acquired resistance to chemotherapy, BH3 mimetics86, and mammalian target of rapamycin (mTOR) inhibitor rapalog84. The wild-type p53-induced phosphatase 1 (Wip1)-mediated BAX dephosphorylations (T172, T174, and T186) also suppress its membrane translocation and inhibit γ-radiation-induced apoptosis in prostate cancer87. Reduced BAX expression is associated with cisplatin resistance in ovarian carcinoma cell systems88. The acquisition of missense BAX mutation (G179E) abrogates BAX membrane translocation, thereby conferring the resistance to Bcl-2 inhibitor Venetoclax and partial cross-resistance to other antineoplastic agents in lymphoma89. Accumulating evidence indicates that low levels of BAX (mRNA and/or protein expression) are implicated in the poor prognosis of chronic lymphocytic leukemia90, 91, 92, 93. Decreased expression of the BAX gene is implicated in both carcinogenesis and gaining resistance to 5-fluorouracil (5-FU) in colorectal cancer94. Increased proteasomal degradation of BAX is a common feature of poor prognosis CLL, while the proteasome inhibitor bortezomib sensitizes CLL cells to tumor necrosis factor-related apoptosis inducing ligand (TRAIL)-induced apoptosis via stabilizing BAX95. Additionally, decreased BAX/Bcl-2 ratio and downstream caspase activity are partially responsible for acquired chemoresistance in glioma96 and breast cancer97. Several studies suggest that reduced expression of BAX is strongly related to the tumorigenesis of breast cancer98, 99, 100. Taken together, deregulation of BAX including phosphorylation, dephosphorylation inactivated mutation, and downregulation, results in decreased BAX/Bcl-2 ratio and confers acquired resistance to multiple anti-tumor agents targeting the apoptotic pathways including chemotherapies (e.g., 5-FU), BH3 mimetics and mTOR inhibitors.
Accumulating evidence suggests that cancer cells are ‘primed’ for cell death because of constitutively elevated stress levels compared to normal cells25. This is due to a delicate balance between anti-apoptotic and pro-apoptotic proteins in cancer cells, resulting in a lower threshold for apoptosis induction. In contrast, normal tissues have higher levels of unbound anti-apoptotic proteins, providing them with resistance to apoptosis induction. The difference in apoptotic threshold between cancer and normal cells indicates a favorable therapeutic window for direct BAX activation.
Therefore, the gain-of-function strategy of directly activating pro-apoptotic protein BAX in tumor cells may offer the opportunity to overcome resistance to existing small-molecule apoptosis inducers in cancer and provide a new paradigm for the pharmacological induction of apoptosis101. Over the last decade, diverse binding sites responsible for BAX activation have been identified and characterized102, 103, leading to the development of BAX modulators that directly bind to the functional binding surface on BAX to activate or sensitize its function for cancer treatment (Fig. 1D).
Furthermore, Bcl-2 protein family-mediated apoptosis plays a major role in the maintenance of lymphocytes, particularly in the elimination of autoreactive cells during lymphocyte development104. Dysregulated anti-apoptotic signals provoke excessive activated and/or autoreactive lymphocytes, contributing to the occurrence and progression of human autoimmune diseases, such as systemic lupus erythematosus (SLE) and rheumatoid arthritis (RA)1. The overexpressed anti-apoptotic Bcl-2 proteins105 and loss of BH3-only proteins106 or BAX107, 108 are implicated in the disruption of the balance between pro- and anti-apoptotic signals in the context of autoimmunity. This suggests that BH3 mimetics could be utilized for the treatment of autoimmune diseases. Notably, BH3 mimetic ABT-737 has demonstrated significant efficacy in SLE or RA models109, offering potential strategies for the development of alternative immune modulatory drugs that target the Bcl-2 protein family to reactivate lymphocyte apoptosis.
Additionally, studies have shown that the murine cytomegalovirus (MCMV) encodes a specific BAX inhibitor known as m38.5 protein, which promotes viral dissemination via blocking BAX-mediated apoptosis110, 111, 112, 113, underscoring the critical role of BAX-mediated apoptosis to potentiating pathogen-targeting immune responses.
1.2. The pathologic cell death with BAX
In contrast to diseases characterized by resisting cell death, the development and progression of various conditions associated with unwanted cell death, such as acute or chronic degenerative diseases114, 115 and heart failure, are closely related to the hyperactivation of BAX-mediated apoptosis42. Neurons, in particular, rely solely on BAX-regulated apoptosis in the absence of full-length BAK during brain development 116. Alzheimer's disease (AD), the most prevalent type of dementia117, is marked by the accumulation of nonvascular amyloid β-protein (Aβ) and phosphorylated tau deposition, leading to a decrease in the ratios of pro/anti-apoptotic proteins (Bcl-2/BAX) through pathways including rat sarcoma-extracellular regulated protein kinase (RAS-ERK), glycogen synthase kinase-3β (GSK-3β) and stress-activated protein kinase (JNK), ultimately triggering neuronal apoptosis 118. The pathologic neuronal death in the whole brain is closely related to AD's multiple pathological symptoms, such as cognitive deficiencies and emotional/behavioral abnormalities. Ablation of BAX prevents neuronal cell death and promotes pronounced neuroprotection in mice subjected to various experimental brain injuries amyotrophic lateral sclerosis119 or several neurodegenerative diseases such as neonatal hypoxia-ischemia120. BAX deletion also suppresses neuromuscular disease progression in mice models of congenital muscular dystrophy121. Likewise, BAX deficiency displays increased survival both in mouse models of traumatic brain and spinal cord injuries122, 123. BAX-mediated dynamic Ca2+ signaling contributes to excitotoxic and ischemic neuronal injury, and BAX deficiency protects neurons in mice subjected to transient middle cerebral artery occlusion or oxygen-glucose deprivation124.
Multiple forms of regulated cell death are implicated in heart failure, although the mechanistic connections between BAX and cardiomyocyte death are not fully understood42. Notably, cardiomyocyte-specific deletion of BAX significantly reduces infarct size in BAX−/− mice subjected to experimental myocardial ischemia-reperfusion125. Research has shown that doxorubicin-induced cardiomyopathy is closely linked to BAX-mediated cell apoptosis and necrosis. However, the combination of small-molecule BAX inhibitor BAI1 effectively protects the heart without compromising the anti-tumor potency of doxorubicin in zebrafish and mice126, suggesting that BAX may be an actionable cardioprotection target.
Furthermore, the deletion of BAX has been shown to protect nephrons in mice exposed to experimental renal ischemia/reperfusion127. Simultaneous deletion of BAX and BAK has also been found to prevent tubular apoptosis, subsequently reducing kidney inflammation and fibrosis in mouse models of unilateral ureteral obstruction-induced fibrosis128, 129. These findings suggest that BAX-mediated cell death plays a significant role in kidney diseases.
As a central death regulator, the pharmacological modulation of BAX presents promising prospects for managing diseases characterized by dysregulated cell death whether it involves the context of pathologic cell survival or unwanted cell demise130. In the past decade, various binding sites capable of modulating BAX activity have been identified and functionally characterized, providing a foundation for the development of next-generation apoptosis modulators that directly target BAX. This review summarizes recent advancements in the discovery of next-generation BAX-targeted apoptosis modulators, with a focus on the characteristics of binding sites and the structure–activity relationships of different small-molecule BAX modulators.
2. The physiological modulation of BAX
2.1. Physiologic activation of BAX
The effector protein BAX is composed of Bcl-2 homology domains 1–3 and a transmembrane (TM) domain, forming a structure with nine helices that fold into globular proteins with a relative molecular weight of 21 kDa131. BAX is primarily found as a dormant cytosolic monomer or as an autoinhibited BAX homodimer132.
The inactive BAX monomer structure, determined by nuclear magnetic resonance (NMR), reveals that the α1‒α2 loop partially overlies the α1/α6 trigger site, known as the closed conformation. Additionally, the BH3 helix (α2) is buried in the core of the BAX protein, and the α9 helix is sequestered in the C-terminal canonical site formed by α3‒α5 helices131 (Fig. 2A). Under cellular stress, the physiological BAX agonists BH3-only agonists initiate the BAX activation cascade by binding to the BAX N-terminal trigger site31, leading to the conformational transformation of BAX from a dormant cytosolic monomer into a toxic oligomer14. The conformational activation of BAX occurs through a stepwise mechanism, including displacement of the α1‒α2 loop that otherwise overlies the trigger site (Fig. 2B), exposure of the pro-apoptotic BAX BH3 helix, allosteric release of the C-terminal α9 helix for mitochondrial translocation and insertion32 (Fig. 2C). Subsequently, BH3-only proteins insert their BH3 domain into the unoccupied canonical site, leading to BAX separating into the latch domain (α6‒α8 helixes) and the core domain (α3‒α5 helixes) as well as dislodging its BH3 helix133, 134 (Fig. 2D). Then, the MOM-anchored BAX inserts its BH3 helix into the canonical site of another BAX to form a BH3-in-groove dimer134 (Fig. 2E), which may initiate its oligomerization (Fig. 2F) and permeabilize MOMP. Furthermore, exposed BAX BH3 can target the trigger site of inactive BAX monomers to activate BAX, which is recognized as the BAX autoactivation mechanism32. Importantly, BAX BH3-only proteins that engage the trigger site convert BAX from a closed to an open-loop conformation. In this process, the exposure of the BAX BH3 helix (residues 12–24), a marker of BAX activation, is critical to construct activated homo-oligomerization BAX and membrane disruption, while the allosteric release of the α9 helix is required for mitochondrial translocation.
Figure 2.

The stepwise mechanism of the BAX conformational activation. BH3-only proteins such as BIM BH3 (cyan) engage the trigger site of inactive BAX monomer (gray) (A) (PDB ID: 1F16), which converts BAX from a closed-loop to an open-loop conformation (B) (PDB ID: 2K7W) via the exposure of the BAX BH3 epitope and allosteric release of the C-terminal α9 helix (C). Following this step, the active BAX monomers translocate to the mitochondrial outer membrane (MOM) via its α9 helix. Then BH3-only proteins such as BIM (cyan) bind to the canonical site of MOM BAX (gray), contributing to the formation of core/latch domain swapped dimer (D) (PDB ID: 4ZIE), initiating a series of conformational changes that result in the formation of BH3-in-groove BAX dimer (E) (PDB ID: 4BDU). These symmetrical dimers can form high-order BAX oligomers (F), which execute MOMP to release cytochrome c.
2.2. Physiologic inhibition of BAX
The crystal structure of the cytosolic BAX dimer has been found to demonstrate an autoinhibition mechanism, wherein one protomer's α9 helix engages the trigger site of the second protomer, thereby preventing the N-terminal conformational activation of the first protomer and the displacement of the second protomer's α9 helix132 (Fig. 3A). This mechanism illustrates the ability of cells to regulate BAX activation and BAX-mediated apoptosis through reversible conversion between dimeric and monomeric conformations of BAX.
Figure 3.

(A) A novel BAX autoinhibition mechanism related to the BAX activation pathway. BAX inactive dimer (PDB ID: 4S0O, left); BAX inactive monomer (PDB ID: 1F16, right). (B) The crystal structure of BAX-BH3 helix (cyan) bound to anti-apoptotic protein Bcl-XL (gray) (PDB: 3PL7). (C) Bcl-2 BH4 domain (orange) (PDB ID: 1G5M) engages the BH4-binding domain on BAX (green) to inhibit BAX activation (PDB ID: 1F16). (D) NMR solution structure of vMIA's BAX-binding domain (vMIA-BBD) (light magenta) bound to BAX (gray) (PDB ID: 2LR1). (E) The co-crystal structure of 3C10 (gray) with an inactive form of BAX mutant (P168G) (lime) (PDB ID: 5W5X).
The physiological inhibition of BAX by anti-apoptotic Bcl-2 proteins can be categorized into two mechanisms. These mechanisms involve the neutralization of the exposed BH3-domain of activated BAX with the BH3-binding groove of Bcl-2 anti-apoptotic proteins1 (Fig. 3B) and the binding of the Bcl-2's BH4 domain to the BH4-binding site on BAX135 (Fig. 3C). Structural analysis has revealed that the BH4-binding site is situated in a groove formed by α1, α1‒α2 loop, α2‒α3, and α5‒α6 hairpins. The Bcl-2 BH4 domain directly engages this inhibitory site to block the BH3-triggered conformational activation of BAX (Fig. 3C). Alanine scanning mutagenesis data has identified I19 and L23 of the Bcl-2 BH4 domain as critical residues for stabilizing its binding to BAX135. Furthermore, BH4 domain peptides from Bcl-2/Bcl-XL have been found to inhibit excessive activation of ryanodine receptor (RyR) to block pathological taurolithocholic acid 3-sulfate (TLC-S)-induced) Ca2+ release, thereby protecting pancreatic acinar cells from TLC-S-induced necrosis136. This finding underscores the anti-apoptotic function of the Bcl-2 BH4 domain. Interestingly, the human cytomegalovirus encodes an anti-apoptotic protein, viral mitochondria localized inhibitor of apoptosis (vMIA), which inhibits apoptosis to promote viral infection. NMR structure determination efforts have provided the complex of vMIA's BAX-binding domain (vMIA-BBD) bound to BAX (Fig. 3D). The vMIA-BBD specifically engages a discrete pocket (vMIA site) adjacent to the BH4-binding site and simultaneously stabilizes the α3‒α4 and α5‒α6 hairpins, thereby preventing the conformational changes related to BAX insertion and oligomerization137, 138. Distinct from Bcl-2 anti-apoptotic proteins, which inhibit BAX mitochondrial localization, vMIA promotes the relocation of BAX to mitochondria137. Mutagenesis data has revealed that vMIA-BBD reinforces its binding to BAX making critical polar contacts with BAX's D84 and D86 via its R139 and R146 (Fig. 3D), respectively, as well as specific hydrophobic contacts with the BAX's several hydrophobic residues (V129, P130, L132 and I133) via its L143138. Just as the neutralization of the BH3 domain of pro-apoptotic members with the BH3-binding groove of anti-apoptotic members informed the discovery of BH3 mimetics, the interactions between these natural or peptide BAX antagonists and their corresponding binding sites on BAX offer innovative pharmacologic paradigms for designing BAX antagonists to treat the diseases involving unwanted cell death, resulting in the discovery of BH4 mimetics such as multiple stapled Bcl-2 BH4 domain helices.
Besides, several antibodies have been discovered to directly bind and regulate the anti-apoptotic function of BAX. For instance, the synthetic antibody 3G11 interacts with the inactive form of BAX at its trigger site, forming a stable complex that prevents conformational changes in BAX. The 3G11-mediated BAX inhibition is weakened with the K21E mutant, while it was completely abolished in the presence of the R134 mutant, indicating the significant contribution of R134 to the inhibition compared to K21139. Another antibody 3C10 is shown to specifically contact with the residues 31–45 at the start of the BAX α1‒α2 loop. The complex of 3C10 with an inactive form of BAX mutant (P168G) reveals close contacts between 3C10 and several residues on the α1‒α2 loop (R34, R37, and M38)140 (Fig. 3E). 3C10 blocks the translocation of wild-type BAX to the MOM while activating a mitochondrial form of BAX mutant (S184L) that promotes dimerization and cytochrome c release141. Surprisingly, a point mutation (S184L) at the C-terminal tail of BAX alters the activity of the 3C10 antibody, which binds to the N-terminal trigger site, indicating allosteric communication between the trigger site and the canonical site. Unfortunately, 3C10-mediated BAX regulatory mechanisms are not fully understood due to the unresolved α1‒α2 loop in the 3C10-BAX P16G mutant complex and the absence of a co-crystal structure of 3C10 bound to wild-type BAX. Furthermore, the ability of these antibodies to target intracellular BAX requires further investigation.
Overall, various molecular mechanisms involved in the physiological modulation of BAX have been elucidated, offering numerous opportunities for the development of next-generation apoptosis modulators that directly target BAX.
3. Small-molecule BAX-targeted apoptosis modulators
Contemporary drug discovery strategies of BAX-targeted therapy primarily rely on biochemical screening approaches, especially liposomal release assay139,142. This assay involves encapsulating fluorescent molecules and quenchers in liposomes, and upon BAX activation-mediated permeabilization, the dissociation of these components leads to increased fluorescence intensity. The extent of BAX-mediated permeabilization is accessed by measuring the increase in fluorescence intensity, enabling the quantification of the activation ability of BAX agonists. Direct BAX antagonists are typically identified by evaluating the compound's ability to prevent permeabilization triggered by BH3-only proteins-mediated BAX activation. Various experiments, such as competitive fluorescence polarization assay (FPA), microscale thermophoresis (MST), or isothermal titration calorimetry (ITC), are performed to explore the binding affinity and mechanisms of BAX modulators. Additionally, NMR analysis of chemical shift changes143 upon modulators binding to [15N]BAX, in combination with molecular docking or mutagenesis studies, are further performed to probe the potential binding surfaces and the key modulators-BAX interactions. The cellular biochemical effects of modulators, including BAX activation, cytochrome c release, and apoptosis, are characterized by conventional biochemical approaches such as western blotting, caspase-3/7 assay, and TMRE mitochondrial potential assay.
In addition to elucidating the regulatory mechanism of physiological BAX activation or inhibition, interactions between endogenous ligands such as BH3-only proteins and Bcl-2 anti-apoptotic proteins, as well as small-molecule BAX modulators with BAX, have been extensively investigated to understand how they modulate its pro-apoptotic activity. These studies have identified numerous functional and potentially druggable binding surfaces on BAX, including trigger, S184, canonical and non-canonical (e.g., C126 region), vMIA, and BAI sites (Fig. 4A‒B). Meanwhile, a diversity of BAX direct modulators have been identified, including trigger, S184, canonical and non-canonical sites agonists, vMIA site sensitizers, as well as the trigger, canonical, and BAI sites antagonists (Table 1).
Figure 4.

Overview of a diversity of functional and potential regulatory sites capable of (A) activating (PDB: 2K7W) and (B) inhibiting (PDB: 1F16) BAX pro-apoptotic activity.
Table 1.
Characteristics of small-molecule apoptosis modulators directly targeting BAX.
| Activity | Proposed binding site | Ligand | Structure | Critical contacta | Binding affinitya |
|---|---|---|---|---|---|
| Agonist | Trigger site | BAM7 | 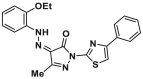 |
K21 | FPA, IC50 = 3.3 μmol/L |
| BTSA1 | 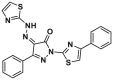 |
K21 | FPA, IC50 = 247 ± 30 nmol/L | ||
| BTSA1.2 | 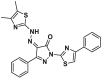 |
K21 | FPA, IC50 = 149 ± 36 nmol/L | ||
| BTC-8 | 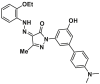 |
K21, E17, D142 | FPA, Ki = 0.8 μmol/L | ||
| 5 | 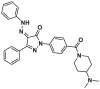 |
K21, D142 | / | ||
| 6 | 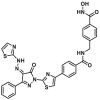 |
K21 | FPA, IC50 = 392.9 ± 18.1 nmol/L | ||
| S184 site | SMBA1 | 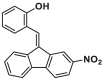 |
S184 | FPA, Ki = 43.3 ± 3.25 nmol/L | |
| CYD-2-11 | 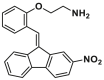 |
S184 | / | ||
| CYD-4-61 | 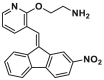 |
S184 | / | ||
| GL0388 | 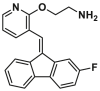 |
S184 | / | ||
| Canonical site | 11 | 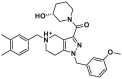 |
/ | / | |
| Non-canonical site | OICR766A | 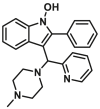 |
C126 | ITC, KD = 255 ± 58 nmol/L | |
| Non-canonical site | Trans-2-hexadecena (t-2-Hex) | 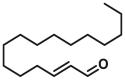 |
C126 | / | |
| Sensitizer | vMIA site | BIF-44 | 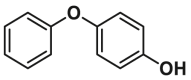 |
/ | ITC, Kd = 37 ± 12 μmol/L |
| Antagonist | Trigger site | Eltrombopag | 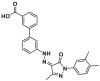 |
R145 | FPA, IC50 = 207 nmol/L MST, KD = 143 nmol/L |
| BAI site | BAI1 | 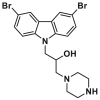 |
V83, L120 | MST, KD = 15 ± 4 μmol/L | |
| BAI2 | 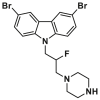 |
V83, L120 | MST, KD = 17.5 ± 4 μmol/L | ||
| Canonical site | MSN-50 | 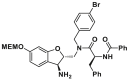 |
/ | / | |
| MSN-125 | 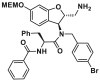 |
/ | / | ||
| DA004 | 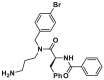 |
/ | / | ||
| Unknown binding site | Bci1 | 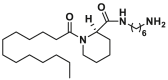 |
/ | / | |
| Bci2 | 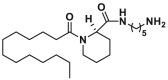 |
/ | / |
“/” means no reported data.
3.1. Small-molecule BAX antagonists
3.1.1. BAX trigger site agonists
The location of the trigger site on the N-terminal surface of BAX was determined using a stapled BIM BH3 helix144. This site is comprised of α1, α6, and α1‒α2 loop and is characterized by a shallow hydrophobic surface consisting of several hydrophobic amino acids (M20, A24, L25, G29, W139, M137, and L141), and a perimeter of polar amino acids (K21, Q28, Q32, E131, and R134)31 (Fig. 5A). This N-terminal trigger site of BAX initiates its direct activation and propagates its autoactivation upon interactions between the exposed BAX BH3 helix of the activated species and the trigger site of dormant monomers31.
Figure 5.

(A) Structural features of the trigger site (PDB: 2K7W). (B) The NMR-derived structure of BIM-BH3 helix (cyan) bound to BAX (gray) (PDB: 2K7W). (C) The iterative structural optimization of BAX agonists bearing pyrazol-3-one core. (D) The design strategy of HDAC-BAX hybrids.
During cellular stress, the natural BAX trigger site agonists BH3-only proteins induce the conversion of the α1‒α2 loop from a closed to an open configuration, leading to the initiation of BAX activation and exposure of the trigger site. Due to this loop opening, BH3-only proteins utilize their BH3 domain to engage the trigger site and propagate subsequent allosteric conformational changes. The crystal structure of BIM-BH3, in complex with the trigger site of BAX, exhibits several stabilizing ionic interactions between the trigger-site residues and charged residues of the BH3 domains31. BIM-BH3 makes critical contact with K21 via the anionic carboxylate of BIM D138 and forms hydrogen bonds with BAX D142 and E146 via the anionic carboxylate of BIM D145145 (Fig. 5B). Further mutagenesis studies suggest that the critical hydrogen bonds between BAX K21 and BIM-BH3 or other BAX trigger site agonists determine their capability to induce BAX activation. Molecular dynamics simulations suggest that BIM-BH3 may promote the displacement of the α1‒α2 loop at the trigger site via the spatial repulsion between its two bulky tyrosine residues (Y162/Y163) and the α1‒α2 loop145. Following the ‘opening’ of the α1‒α2 loop, a cryptic pocket called the trigger bottom pocket is exposed and consists of residues L27, Q28, I31, Q32, A35, V121, A124, L125, P130, I133, and M137. Through extensive molecular dynamics simulations, Feng et al. found that trigger site activators BAM7 and BTSA1 can bind to the trigger bottom pocket and increase the conformational flexibility involving the α1‒α2 loop, which is a critical event in BAX activation146.
Owing to the challenge of generating stable, recombinant, monomeric BAX proteins and the dynamic context in which compounds engaging the trigger site cause conformational changes in BAX, conventional screening methods are less amenable to discovering BAX trigger site agonists. Through in silico screening based on the topography of the trigger site, Gavathiotis et al. have identified a series of BAX activator molecules (BAMs). These BAMs were then evaluated using an original competitive fluorescence polarization assay (FPA), resulting in the discovery of the first gain-of-function direct BAX activator, BAM7 (1, BAX, IC50 = 3.31 μmol/L)147 (Fig. 5C). Subsequent NMR and molecular docking studies demonstrate that BAM7 selectively binds to the N-terminal trigger site and forms a hydrogen bond with BAX K21 via the pyrazolone carbonyl, promoting characteristic structural changes and ultimately inducing BAX functional activation to trigger apoptosis.
Through structural optimization of the phenylhydrazono and phenylthiazole of BAM7, Stornaiuolo et al. have identified a series of BAX targeting compounds (BTCs) where BTC-8 (2, BAX, IC50 = 0.8 μmol/L) is the most potent small molecule binding to the BAX trigger site148 (Fig. 5C). BTC-8 differs significantly from BAM7 by replacing the phenylthiazole moiety with the biphenyl moiety, which contains an exocyclic basic group on the terminal phenyl ring. Molecular docking analysis reveals that BTC-8 reinforces its binding to the trigger site through critical contacts with D142 via the exocyclic basic group as well as a hydrogen bond with E17 via the hydroxyl group. In a murine LLC1 tumor model, a single dose of BTC-8 (1 mg/kg) administered intraperitoneally exhibits excellent anti-tumor effects from highly size-reduced tumor lesions to decreased tumor burden and lung area in mice through on-target BAX activation, without causing significant toxicity to normal tissues. In vitro studies have shown that BTC-8-induced BAX activation is responsible for anti-proliferative effects, cell cycle arrest, and apoptosis in a panel of glioblastoma (GBM) cells, as well as effectively blocking self-renewal and inducing apoptosis in glioma stem cells (GSCs) while sparing normal cells such as mesenchymal stem cells (MSCs) and human lymphocytes149. Furthermore, BTC-8 and the alkylating agent Temozolomide (TMZ) (50 nmol/L BTC-8 + 50 μmol/L TMZ) have demonstrated additive anti-proliferative effects on both GBM cells and GSCs.
The lead optimization of BAM7 results in the identification of a series of analogs. Among these, compound BTSA1 (3, BAX trigger site activator 1) exhibits the most binding affinity to the trigger site97, with an IC50 value of 250 nmol/L, in comparison to FITC-BIM SAHB (BAX, IC50 = 280 nmol/L) and BAM7 (Fig. 5C). Molecular docking, driven by the NMR analysis of [15N]BAX after BTSA1 titration, reveals that BTSA1 engages in the crevice of the trigger site formed by residues M20, K21, A24, L25, Q28, G138, L141 and R145. The strongest interaction was observed between the pyrazolone carbonyl and BAX K21, which is recognized as a key interaction of BAX agonists. Notably, the thiazole group lies adjacent to a presumed hinge site for the α1‒α2 loop that opens upon initiation of the conformation activation. Moreover, the bulky phenyl group attached to the pyrazolone core may act to mimic the bulky tyrosine (BIM Y162, Y163) and contribute to the ‘opening’ of the α1‒α2 loop, which may account for the enhanced binding affinity of BTSA1. Furthermore, a battery of biochemical analyses confirms that BTSA1 specifically engages the trigger site of BAX to induce the exposure of the pro-apoptotic BH3 domain in a dose-dependent manner, leading to BAX membrane translocation, BAX-mediated membrane permeabilization, and ultimately the release of cytochrome c from mitochondria. BTSA1 triggers substantial and rapid BAX-mediated apoptosis in human acute myeloid leukemia (AML) cell lines, which is regulated by the expression of anti-apoptotic Bcl-2 proteins and the cytosolic conformation of BAX. The in vivo pharmacokinetic studies of BTSA1 in mice demonstrate that it has a long half-life (t1/2 = 15 h) in mouse plasma and favorable oral bioavailability (F = 51%) at a dose of 10 mg/kg. Treatment with a single intraperitoneal dose of BTSA1 (10 mg/kg, qod) leads to anti-leukemic effects, ranging from reduced infiltration of human leukemia cells in blood to increased host survival in human AML xenografts. Additionally, BTSA1 has no obvious toxic effects on healthy hematopoietic cells or other tissues at a dose of 15 mg/kg.
Despite the potent in vivo anti-leukemic efficacy and favorable pharmacokinetic profiles of BTSA1, it has the potential to generate the reactive and toxic metabolite aminothiazole owing to its poor metabolic stability. Gavathiotis et al. discovered a new analog, BTSA1.2 (4, BAX, IC50 = 149 ± 36 nmol/L) by installing the two methyl groups to the thiazole group of BTSA1 (Fig. 5C), which provides significantly improved binding affinity to BAX and metabolic stability in mouse liver microsomes150. Moreover, BTSA1.2 exhibits favorable pharmacokinetic properties including a long half-life (t1/2 = 14 h) in mouse plasma, favorable oral bioavailability (F = 49.2%), and significant plasma exposure (AUC = ∼100 μmol h/L). Compared to BTSA1, BTSA1.2 is well tolerated as demonstrated by the lack of toxicity to healthy hematopoiesis and other tissues in vivo even at a higher dose (200 mg/kg, p.o.). Accumulating evidence demonstrates that the inhibition of activated BAX by overexpressed Bcl-XL in solid tumors is responsible for resistance to direct and indirect therapeutic BAX activation, highlighting the therapeutical potential of combining BAX agonists with Bcl-XL inhibitors to overcome apoptosis resistance in solid tumors by enhancing apoptotic priming via inhibition of the anti-apoptotic blockade. A panel of resistant solid tumor cell lines is sensitive to the combination of BTSA1.2 and clinical Bcl-XL/Bcl-2 inhibitor Navitoclax, with strong synergistic effects. Combined treatment of BTSA1.2 and Navitoclax by oral administration daily exhibits strong anti-tumor effects from tumor growth inhibition to tumor regression in resistant SW480 colorectal xenografts (BTSA1.2 200 mg/kg + Navitoclax 100 mg/kg) compared to single agent treatments, as well as effectively in patient-derived xenograft models from COLO-1 and COLO-2 tumors (BTSA1.2 200 mg/kg + Navitoclax 50 mg/kg), while it is well tolerated and less toxic to platelets.
Our group discovered a series of pyrazolone derivatives by structural optimization of the trigger site activator BTSA1. Among them, compound 5 induces BAX conformational activation and exhibits improved anti-tumor efficacy in A549 cells with favorable in vitro stability and CYPs profiles151. Numerous studies reveal that HDAC inhibitors exert their anti-tumor effects in part by increasing the expression of pro-apoptotic proteins (e.g., BIM, BID, and BAX) and decreasing the expression of anti-apoptotic proteins (e.g., Bcl-2, Bcl-XL, and Bcl-W) to reactivate apoptosis152, 153, thus providing a rationale for combinations with BAX direct activators. Feng et al. demonstrated that combined treatment of BAX activator BTSA1 and HDAC inhibitors Vorinostat (SAHA) in Hela cells have moderate synergistic anti-proliferative activity and enhanced BAX-dependent apoptosis (SAHA, IC50 = 2.65 ± 0.01 μmol/L; BTSA1, IC50 = 11.47 ± 0.20 μmol/L; BTSA1 + SAHA, IC50 = 0.81 ± 0.01 μmol/L)154. Based on this finding, they converted the dual treatment of BTSA1 and SAHA into HDAC-BAX multiple hybrids by installing a zinc-binding group (ZBG) and a linker at the solvent-exposed phenylthiazole moiety. Further biological investigations indicate that compound 6 emerges as the most effective HDAC‒BAX multiple hybrids154 (Fig. 5D), which has good BAX affinity (BAX, IC50 = 392.9 ± 18.1 nmol/L) and HDAC inhibitory activity (HDACs, IC50 = 62.8 ± 2.1 nmol/L). Compound 6 exhibits significantly increased anti-proliferative activities against Hela cells (IC50 = 0.86 ± 0.04 μmol/L) compared to single agent treatments by several mechanisms, including enhancing BAX-mediated apoptosis with activation and upregulation of BAX, altering chromatin remodeling with HDACs inhibition. This finding provides a new paradigm for direct BAX activators in combination with agents indirectly targeting the Bcl-2 protein family (such as proteasome inhibitors or HDACs inhibitors) or for developing multi-target drugs based on BAX to treat resistant solid tumors.
3.1.2. BAX S184 site agonists
Emerging evidence indicates that the phosphorylation of BAX at S184 controls its pro-apoptotic function. Xin et al. demonstrated that nicotine inactivates the pro-apoptotic activity of BAX through AKT-mediated phosphorylation of S184, resulting in increased apoptotic resistance in lung cancer cells155. Kale et al. found that excessive activation of AKT pathway-mediated S184 phosphorylation is strongly related to poor prognosis and therapeutic resistance (e.g., BH3 mimetics and chemotherapy) in a range of malignancies, especially ovarian cancer cells86. In contrast, protein phosphatase 2A (PP2A), a physiological BAX phosphorylase, restores the pro-apoptotic activity of BAX through dephosphorylation of S184156. Mutagenesis analysis indicates that BAX S184 is the major functional switch controlling its pro-apoptotic activity157. S184 phosphorylation of BAX exerts two survival mechanisms by blocking BAX conformational activation through the inhibition of BAX mitochondrial insertion and inactivation of BAX binding to BH3-only proteins to disarm its pro-apoptotic function. Overall, the therapeutic targeting of the S184 site to modulate the phosphorylation status of BAX S184 provides a promising opportunity to discover BAX agonists.
The S184 site refers to a sub-region of the canonical groove centered on S184. It is located in the C-terminal tail of BAX, which is strongly related to BAX translocation and insertion into the mitochondrial outer membrane (MOM)131. The binding pocket is formed by the α9 helix and joint between α4 and α5 helices consisting of residues D98, S184, S101, D102, N106, R109, F176, V180 and A183 (Fig. 6A).
Figure 6.

(A) Structural features of the S184 site (PDB: 2K7W). (B) The iterative structural optimization of BAX agonists targeting the S184 site.
Through a combined computational and BAX-dependent cytotoxicity screening, several small molecule BAX agonists (SMBAs) have been identified, where compound SMBA1 (7, BAX, Ki = 43.3 ± 3.25 nmol/L) bearing a fluorene core specifically binds to BAX with the most potent binding affinity158 (Fig. 6B). Mechanistically, SMBA1 engages the S184 site to inhibit BAX phosphorylation, leading to BAX mitochondrial insertion and release of cytochrome c, thereby promoting BAX-dependent apoptosis in A549 cells. In A549 lung cancer xenografts, SMBA1 (2–60 mg/kg, i.p., qd) suppresses apoptosis in a dose-dependent manner from the conversion of inactive BAX to the activated form, without significant toxic effects on surrounding normal tissues at therapeutically effective doses. In addition, SMBA1 strongly suppresses GBM cell growth through the activation of BAX-mediated apoptosis in vitro and in vivo with relatively low toxicity159.
A structural-based lead optimization strategy turns out into a series of SMBA1 derivatives by introducing various O-alkylamino side chains tethered to the hydroxyl group to access deeper S184 binding pocket, where analogs CYD-2-11 (8, BAX, Ki = 34.1 ± 8.54 nmol/L) with a 2-aminoethyl into the pyridyl ring has slightly increased binding to BAX compared to SMBA1 (BAX, Ki = 43.3 ± 3.25 nmol/L)84 (Fig. 6B). CYD-2-11 (40 mg/kg, i.p.) effectively suppresses lung cancer growth via the induction of BAX-mediated apoptosis, including cell/patient-derived xenografts and genetically engineered mouse models (GEMMs) with the KRASG12D mutant. Li et al. found that enhanced AKT-mediated S184 phosphorylation of BAX was responsible for the resistance to the mTOR inhibitor RAD00184. Therefore, CYD-2-11 in combination with the mTOR inhibitor RAD001 (CYD-2-11 40 mg/kg + RAD001 1 mg/kg, i.p., qd) exhibits potent anti-tumor effects against lung cancer growth with or without acquired resistance in vitro and in vivo. Further replacing the upper phenyl ring of CYD-2-11 with the pyridyl ring affords the analog CYD-4-61 (9, MDA-MB-231, IC50 = 0.07 ± 0.03 μmol/L; MCF-7, IC50 = 0.06 ± 0.03 μmol/L)160 (Fig. 6B), which displays significantly increased anti-proliferative effects against breast cancer cells compared to CYD-2-11 (MDA-MB-231, IC50 = 3.22 ± 0.49 μmol/ L; MCF-7, IC50 = 3.81 ± 0.76 μmol/L) and SMBA1 (MDA-MB-231, IC50 = 5.6 ± 0.69 μmol/L; MCF-7, IC50 = 8.3 ± 1.44 μmol/L). Molecular docking analysis suggests that CYD-4-61 reinforces its binding to the S184 site through hydrogen bonding with D102 via the terminal amino group and a hydrogen bond with R107 via the nitrogen atom of the pyridine ring. A battery of structural, cellular, and in vivo studies demonstrate that CYD-4-61 (2.5 mg/kg, i.p., qd, TGI = 55%) exhibits stronger anti-tumor activity against breast cancer than SMBA1 (40 mg/kg, i.p., qd, TGI = 51%) by inhibiting BAX phosphorylation and eventually leading to apoptosis. However, poor cell selectivity over normal cells (SI = 1.6/1.8) and body weight loss in tumor models with CYD-4-61 hamper further preclinical studies. To reduce the toxicity of CYD-4-61, Liu et al. tried to replace the toxicophore nitro group with various bioisosteres, where compound GL0388 (10, Fig. 6B) with 2-fluorine displays significantly reduced toxicity and less anti-proliferative activity (MDA-MB-231, IC50 = 0.07 ± 0.03 μmol/L; MCF-7, IC50 = 0.06 ± 0.03 μmol/L; SI = 5.8/10.7)161. GL0388 (20 mpk, i.p., qod) significantly suppresses human triple-negative breast cancer xenografts in association with the activated apoptosis pathway but showed slight body weight loss at therapeutically effective doses.
Overall, therapeutic targeting the S184 site to promote BAX-mediated apoptosis alone or in combination with other established therapies exhibits remarkable anti-tumor efficacy for several BAX-expressing solid tumors in vitro and in vivo, providing a new paradigm for pharmacologic modulation of BAX activation. Future investigations should focus on enhancing cell selectivity to enlarge the therapeutic window of S184 site activators with a great balance between anti-tumor potency and drug-likeness.
3.1.3. BAX canonical site agonists
The canonical site refers to the BH3-binding groove constructed by α2‒α5 helixes and is relatively long and deep compared to the trigger site. The cytosolic BAX protein remains inactive, potentially as a result of its transmembrane α9 helix occupying the canonical site131. Previous findings have demonstrated that interactions between the BH3 domain of BH3-only proteins and the canonical site of MOM-associated BAX can cause additional conformational changes, including its separation into the latch domain and core domain and dislodgement of its BH3 domain, ultimately nucleating its oligomerization to activate MOMP.
Based on these findings, Zhao et al. proposed a new paradigm for BAX activation by competitively engaging the BH3-binding groove to promote its insertion and oligomerization. Through in-silico screening of the canonical site, Zhao et al. identified a small molecule 11 (ZINC 14750348) with a molecular weight of 489.3 Da (Fig. 7A) that promotes BAX insertion into the MOM, leading to BAX-dependent apoptosis in vitro162. In a murine LLC tumor model, a single dose of 11 (40 mg/kg, i.p., qd) represses tumor growth via apoptosis. Unfortunately, sufficient biophysical, structural, and biochemical evidence is lacking to confirm the binding characteristics of compound 11. Consequently, the rationale and efficacy of pharmacologically targeting the canonical site to directly activate BAX remains uncertain. Further investigations should focus on elucidating the basic molecular mechanisms responsible for 11-induced BAX activation (especially for the binding site), which seems to be applied for the development of novel BAX activators targeting the canonical site or other potentially druggable binding sites.
Figure 7.

(A) BAX agonist 11 engages the canonical site to promote BAX insertion into the MOM and oligomerization (PDB: 1F16). (B) BAX agonist 12 (OICR766A) targets an unrecognized binding site to activate BAX, which requires the presence of C126 (PDB: 2K7W). (C) The chemical reaction of the lipid electrophile trans-2-hexadecenal (t-2-hex) with BAX C126. (D) BAX agonist 13 (BIF-44) specifically binds to the vMIA site and allosterically mobilizes the key regions implicated in BAX activation, thereby sensitizing BH3-only proteins-triggered conformational activation of BAX (PDB: 2K7W).
3.1.4. BAX non-canonical site agonists
Liposome- or mitochondria-based permeabilization assays can quantify membrane permeabilization, which probes the capacity for compound-induced BAX activation and is used extensively for screening BAX direct agonists139. By using the liposome or mitochondria-based permeabilization assays, Brahmbhatt et al. identified several BAX direct agonists with different structural characteristics, where compound OICR766A (12, liposome, EC50 = 0.1 μmol/L; mitochondria, EC50 = 0.1 μmol/L) exhibits the most potent effects on BAX activation163 (Fig. 7B). Intriguingly, OICR766A binds directly to BAX protein (ITC, BAX, KD = 255 ± 58 nmol/L) leading to promoting BAX oligomerization in solution, which is poorly inhibited by anti-apoptotic protein Bcl-XL. Mutagenesis studies reveal that the presence of C126 is essential for OICR766A (ITC, BAXC126A, KD > 20 μmol/L) to BAX and OICR766A-induced conformational changes in BAX. Cellular studies demonstrate that OICR766A triggers BAX oligomerization and membrane localization to activate MOMP, ultimately leading to BAX/BAK-dependent apoptosis. Intriguingly, OICR766A specifically targets an unknown binding site and activates BAX via a mechanism distinct from that of trigger site agonists.
Chipuk et al. found that the specific binding of the sphingolipid product trans-2-hexadecenal (t-2-hex) to BAX might lower the thermodynamic constraints on the conformational changes of BAX (Fig. 7C), which may simultaneously trigger BAX oligomerization and facilitate BH3-only protein-induced BAX activation164. The addition of sphingolipid inhibitors blocks BAX conformational changes and cytochrome c release, suggesting that the sphingolipid milieu is essential for inducing functional BAX activation and apoptosis. Although seminal findings elucidate the molecular mechanism by which sphingolipid metabolism cooperates with the Bcl-2 family to regulate apoptosis, minimal structural data shows that t-2-Hex interacts with the BAX protein. Interestingly, Cohen et al. found that t-2-Hex directly and covalently modified C126 of monomeric BAX, resulting in the conformational perturbation of the α5/α6 hairpin adjacent to C126 and influencing key regions related to BAX initiation (α1‒α2 loop, and α2) and translocation (α9), ultimately promoting the conformational activation of BAX and BAX-dependent apoptosis165. These findings shed light on why t-2-Hex enhances BH3-triggered BAX-dependent apoptosis in a C126-dependent manner. The explicit mechanism of the covalent lipidation of BAXC126 to activate BAX provides a rational theoretical basis for the development of covalent BAX agonists targeting the C126 region.
In recent advancements, scholars have made notable progress in comprehending how BAX is activated, shedding light on the crucial role of C126 in BAX oligomerization and BAX-induced cell death. This opens up new possibilities for activating BAX by targeting the C126 region either covalently or noncovalently. It is imperative for forthcoming research to further investigate the binding properties of OICR766A and the essential attributes of the C126 region. This in-depth exploration will enable the translation of these structural revelations into potent BAX agonists.
3.2. Small-molecule BAX sensitizers
The vMIA site is composed of residues of the flexible loops between α1 and α2 (E44‒D48), α3‒α4 (D84‒D86), α5‒α6 (C126‒T127), α9 helixes (L181, A183) and C terminus (I187‒G192). Emerging evidence suggests that the vMIA protein engagement stabilizes the α3/α4 and α5/α6 hairpins, effectively thereby suppressing the conformational activation of BAX137, 138. This discovery opens up possibilities for the development of potent BAX modulators that can compete with the vMIA protein.
By utilizing a saturation transfer difference (STD) NMR-based screening strategy in combination with liposomal release assay, Gavathiotis et al. identified various BAX-interacting fragments (BIFs), where BIF-44 (13, ITC, BAX, Kd = 37 ± 12 μmol/L) exhibited the potent sensitizer profile rather than the direct activator profile166 (Fig. 7D). Interestingly, BIF-44 competes with physiological BAX inhibitors, such as vMIA and the Bcl-2 BH4 domain, for BAX interaction, suggesting that BIF-44 may rely on another mechanism to reduce the apoptosis threshold. Further structural and mechanistic analyses reveal that BIF-44 specifically binds to the vMIA site and allosterically mobilizes the key regions implicated in BAX initiation (α1‒α2 loop) and oligomerization (BAX BH3 helix), thereby enhancing BH3-mediated conformational activation of BAX. The underlying mechanisms of BIF-44-triggered allosteric sensitization of inactive BAX provide novel structural insights into BAX modulation and provide an opportunity to develop BAX sensitizers by targeting BAX-inhibitory motifs. Although both vMIA and BIF-44 target the vMIA site and form similar contacts with D84, the key interactions governing vMIA site-mediated activation or inhibition remain unclear138,166. Taken together, investigations into the modulation of the vMIA site by these modulators highlight the allosteric communications that occur between the vMIA site and the trigger site, as well as the vMIA site and the canonical site.
3.3. Small-molecule BAX antagonists
Preventing aberrant cell death has been extensively investigated as a therapeutical strategy for a host of diseases such as neurodegeneration and heart failure42,115. Distinct from BAX agonists or sensitizers that promote or induce the conformation activation of BAX, BAX antagonists mainly stabilize its inactive conformation or prevent the formation of high-order oligomers. Based on their binding sites, current small-molecule BAX antagonists can be divided into four categories, BAX trigger site antagonists, BAX BAI site antagonists, BAX canonical site antagonists, and BAX antagonists bound to unrecognized sites. BAX trigger or BAI site antagonists directly bind to cytosolic BAX monomer and simultaneously induce allosteric conformational changes to stabilize its inactive conformation, thereby suppressing the pro-apoptotic activity of BAX. Being different from these antagonists that inhibited conformational activation of BAX, BAX canonical site antagonists inhibit the BAX oligomerization, and BAX antagonists with an unknown binding site block the BAX channel activity, thereby preventing BAX-mediated MOMP and cell death.
3.3.1. BAX trigger site antagonists
In an attempt to identify small-molecule BAX modulators with attractive drug-like properties, Gavathiotis et al. initially built a pharmacophore expansion and scaffold similarity search strategy based on the reported small-molecule BAX trigger site agonists. Subsequently, they conducted a screening of a library containing FDA-approved small molecule drugs and successfully discovered the first BAX trigger site antagonist, Eltrombopag (14, Fig. 8A), which is a thrombopoietin receptor agonist used in the clinical treatment of chronic immune thrombocytopenia145. Mechanically, compound 14 competitively engages the trigger site of cytosolic BAX (BAX, IC50 = 207 nmol/L), impeding the conformation changes of the α1‒α2 loop triggered by the trigger site agonists or BH3-only proteins. Simultaneously, it promotes discrete conformational changes in α4, α6, and α7/α4‒α5 loop to stabilize the interaction between α9 helix and the canonical site (Fig. 8B), consequently stabilizing the inactive conformation of BAX and inhibiting BAX's pro-apoptotic activity. Collectively, HSQC-NMR analysis, molecular docking, and mutagenesis studies demonstrate that 14 reinforces its binding to the trigger site of inactive BAX through a salt bridge with R145 via the anionic carboxylate coupled with a hydrogen bond between the pyrazolone carbonyl and R134. To further explore the key ionic interactions between R145 and the carboxylate, Gavathiotis et al. converted 14 into its corresponding methyl ester, which displays a significantly decreased binding affinity (BAX, IC50 > 5 μmol/L). Importantly, the significantly decreased inhibition activity of 14 with R145E mutant (IC50 = 6.8 μmol/L) compared other mutants (K21E, IC50 = 3.1 μmol/L; R134E, EC50 = 3.8 μmol/L) or WT (IC50 = 2.7 μmol/L) suggests the critical interactions between the 14's anionic carboxylate and R145 is essential for 14-mediated BAX inhibition, which is consistent with the R134 contributed to antibody 3G11-mediated BAX inhibition139. These findings highlight that the specific contacts with R134 should be considered in the design of BAX trigger site antagonists.
Figure 8.

(A) The trigger site antagonist 14 (Eltrombopag) as well as BAI site antagonists 15 (BAI1), 16, 17 (BAI2), and 18 block the conformational activation of BAX. (B) Structural features of the trigger site and BAI site on inactive BAX (PDB: 1F16).
3.3.2. BAX BAI site antagonists
The BAI site is located at the junction of the C terminus of α3, the α3‒α4 loop, and the residues of α5 and α6 helixes. It is structurally characterized by a hydrophobic pocket consisting of multiple hydrophobic amino acids (I80, A81, V83, T85, P88, V91, V95, F116, L120, W139, and L185), along with a few peripheral polar amino acids (D84 and K123)167 (Fig. 8B).
Through in vitro inhibition of cytochrome c release assay triggered by BH3-only proteins, Bombrun et al. identified a series of 3,6-dibromocarbazole piperazine derivatives as small-molecule cytochrome c release inhibitors, where compound 15 (77 ± 7%@5 μmol/L), 16 (61 ± 5%@5 μmol/L), 17 (41 ± 2%@5 μmol/L) and 18 (61 ± 2%@5 μmol/L) exhibit relatively powerful inhibition activities168. These compounds show sub-micromolar IC50 values of BAX channel activities (15: 0.52 ± 0.21 μmol/L, 16: 0.37 ± 0.12 μmol/L, 17: 0.68 ± 0.22 μmol/L, 18: 0.68 ± 0.09 μmol/L, Fig. 8A), suggesting they inhibit cytochrome c release via the blockage of BAX channel. To explore the location of identified cytochrome c release inhibitors, Bombrun et al. installed the fluorescent moieties BODIPY 493/503 methyl bromide or 5-iodoacetamido-fluorescein into the compound 15 to generate two fluorescent probes. The fluorescent probes are found to be colocalized with the mitochondrial marker Mitotraker, suggesting compound 15 inhibits cytochrome c release via interacting with BAX to block BAX channel activity168. These studies reveal the mechanistic links between the 15-mediated inhibition of cytochrome c release and the blockage of BAX channel activity, while its accurate molecular mechanism remains unclear.
The liposomal release assay is also used to screen small molecules inhibiting the truncated form of BID (tBID)-triggered BAX-mediated MOMP. Gavathiotis et al. discovered that the above-mentioned carbazole-based compounds 15 and 17 dose-dependently inhibit a range of BAX activators (tBID, BIM SAHB, BAM7) or heat-triggered BAX-mediated MOMP with low micromolar IC50 values (2–4 μmol/L). These inhibitory effects on BAX are independent of both the activators and their concentration, indicating direct inhibition activities on BAX, suggesting their direct inhibition activities on BAX167. Compounds 15 and 17 were called BAX activation inhibitor 1 (BAI1) or 2 (BAI2) in their published articles (Fig. 8A), respectively. Ligand detected 1H-NMR and microscale thermophoresis (MST) experiments demonstrate BAI1 and BAI2 directly binds to BAX with dissociation constants of 15.0 ± 4 μmol/L and 17.5 ± 4 μmol/L, respectively. SiteMap (Schrödinger, LLC, 2017) guided by HSQC NMR analysis molecular docking and mutagenesis studies confirms a relatively deep pocket formed by α3, α4, α5, and α6 helixes. BAI2 and BAI2 engage this pocket called the BAI site to inactivate BAX. Interestingly, these compounds effectively block the essential conformational activation of BAX (α1‒α2 loop) induced by BH3 agonists, rather than impeding the binding of the activators to the trigger site, indicating an allosteric mechanism of BAX inhibition. Additionally, BAI1 selectively inhibits BAX-medicated cell death induced by BH3 agonists or trigger site agonist BTSA1. Taken together, the above target validation and mechanism studies of these carbazole-based compounds reveal an unrecognized regulatory pocket, providing insights into the rational design and development of BAX allosteric antagonists for the treatment of diseases associated with aberrant BAX-dependent cell death.
Encouraged by BAI1's specific inhibition effects on BAX-dependent cell death, Gavathiotis and co-workers subsequently evaluated its potential therapeutic advantages in the context of pathological cell death. Currently, anthracycline doxorubicin (DOX), a widely used chemotherapy, causes severe and dose-dependent cardiomyopathy169, 170 related to both apoptosis and necrosis171. Preliminary mechanism studies confirm that BAX-dependent apoptosis and necrosis are implicated in doxorubicin-elicited cardiomyopathy and BAI1 effectively abrogates cardiomyocyte apoptosis or necrosis induced by diverse stimuli (staurosporine, prolonged hypoxia, hypoxia/reoxygenation or doxorubicin) in vitro, raising the possibility that BAI1 protected against doxorubicin-induced cardiomyopathy172. Intriguingly, combination therapy (DOX 2 mg/kg + BAI1 2 mg/kg, i.p.) provides significant cardiac protection without the loss of doxorubicin's anti-tumor potency in breast cancer LM2 and acute myelocytic leukemia MLL-AF9 cell-derived xenograft (CDX) models as well as breast cancer patient-derived xenograft (PDX) models. Consist with higher expression of BAX in tumor cells than in the heart, BH3 profiling reveals a significant apoptotic threshold in tumorous versus heart tissue isolated from PDX models, suggesting the fundamental difference of BAX levels accounted for the selectivity of BAI1 in cardioprotection without the loss of cancer treatment. This study demonstrates that therapeutical inhibition of BAX with small molecules effectively overcomes doxorubicin-induced cardiomyopathy via dual inhibition of apoptosis and necrosis in vitro and in vivo, offering an alternative strategy for the development of cardioprotection agents for treating BAX-dependent cardiomyopathies resulting from multiple cancer therapies.
3.3.3. BAX canonical site antagonists
Utilizing a MOMP-mimicking liposome dye-release assay, Niu and colleagues identified two molecules MSN-50 (19, IC50 = 2–4 μmol/L) and MSN-125 (20, IC50 = 6–8 μmol/L) dose-dependently inhibits BAX-mediated membrane permeabilization (MP) (Fig. 9A). Interestingly, both of them prevent BAX- and BAK-mediated MOMP, thereby inhibiting apoptosis in human colon cancer HCT-116 cells or BMK cells and protecting primary cortical neurons from glutamate excitotoxicity172. Chemical crosslinking analysis reveals that MSN compounds disrupt the interaction between T56 (α2) and R94 (α4′), partially interaction between L59 (α2) and M79 (α3′), thereby partially interfering with BH3-in-groove dimer interfaces (Fig. 9A). These data suggest that MSN compounds might bind to the canonical site to prevent the formation of correct BAX dimer, resulting in the inhibition of BAX oligomerization and MOMP. The molecular mechanism of these compounds inhibiting BAX demonstrates that the correct formation of BAX symmetric dimers and high-order oligomers is necessary for triggering MOMP. Retaining the pharmacophore common of MSN compounds to afford compound DAN004 (21), which shares a similar molecular mechanism of MSN compounds and exhibits enhanced MP-inhibiting activity (IC50 = ∼0.7 μmol/L)172 (Fig. 9A). However, DAN004's marked toxicity hinders its further studies. The discovery of MSN compounds provides chemical tools for the investigation of the molecular mechanism of BAX oligomerization and might serve as a hit for the development of BAX/BAK dual antagonists to treat a wide range of diseases in the context of aberrant cell death.
Figure 9.

(A) Several proposed canonical site antagonists 19 (MSN-50), 20 (MSN-125), and 21 (DAN004) inhibit BAX oligomerization via interfering with the BH3-grove dimer (PDB: 4BDU), as probed by chemical crosslinking studies. (B) BAX channel blockers 22 (Bci1) and 23 (Bci2) with an unrecognized binding site block the BAX channel activity to prevent BAX-mediated apoptosis.
3.3.4. BAX antagonists bound to an unrecognized site
Through high throughput screening of low molecular weight compound library via the liposome channel assay, Antonsson and colleagues identified two BAX selective channel blockers Bci1 (22, BAX channel, IC50 = 0.81 ± 0.22 μmol/L) and Bci2 (23, BAX channel, IC50 = 0.89 ± 0.29 μmol/L) featuring a C12 lipid-like tail173 (Fig. 9B). Both of them dose-dependently prevent BAX-mediated cytochrome c release from isolated mitochondria without effects on the conformational activation of BAX or its membrane translocation, insertion, or oligomerization. In a gerbil brain ischemia model, Bci1 or Bci2 (30 mg/kg, i.p.) exhibits powerful neuroprotection, decreasing hippocampal damage by 45% or 55%, respectively. The significantly increased cytosolic cytochrome c levels are observed in cytosolic cytochrome c levels, suggesting their neuroprotective effect is closely related to the blockage of the BAX channel activity in vivo. This finding highlights the significance of BAX channel activity in apoptosis and offers an alternative efficient target to treat ischemic injuries.
4. Concluding remarks and future perspectives
The clinical validation of targeting apoptotic pathways has been demonstrated with the approval of the selective anti-apoptotic protein Bcl-2 inhibitor Venetoclax for the treatment of hematologic malignancies. The dysregulation of the pro-apoptotic protein BAX is implicated in various diseases involving pathologic cell survival (e.g., cancer, autoimmunity) and cell death (e.g., neurodegeneration, and heart failure). Hence, the pharmacological modulation of the pro-apoptotic activity of BAX with small molecules offers a promising paradigm to block aberrant cell death or restore cell death for curing a host of diseases characterized by dysregulated cell death.
Over the last decade, studies elucidating the molecular mechanisms that contribute to the physiological modulation of BAX have been booming, including BH3-triggered BAX activation, inactivation of BAX by S184 phosphorylation, and suppression of BAX conformational activation by protein (e.g., Bcl-2 BH4 and vMIA) or peptide (e.g., antibodies 3G11 and 3C10) antagonists engagement. These investigations have revealed a diversity of physiologic binding sites capable of modulating BAX pro-apoptotic activity, including the trigger, S184, canonical, BH4-binding, and vMIA sites. Biochemical screening approaches (e.g., liposomal release assay) in combination with NMR analysis identified multiple small-molecule BAX modulators, offering a series of pharmacologic binding sites that modulate BAX-mediated cell death, such as the BAI site and C126 region. Identification and characterization of these potentially druggable have prompted the development of BAX-selective targeting modulators, which are divided into three subgroups: BAX agonists capable of activating BAX directly, such as trigger site, S184 site, canonical site, and non-canonical site agonists; BAX sensitizers capable of sensitizing the function of BAX, such as vMIA site sensitizers; and BAX antagonists capable of preventing BAX-mediated cell death, such as trigger site, BAI site and canonical site antagonists. Intriguingly, some of the binding surfaces on BAX, such as the trigger, canonical, and vMIA sites, might act as activation or inhibition dependent on the key interactions with small molecules, peptides, or proteins. For example, the interactions between agonists and BAX K21 are crucial for BAX activation, while the contacts between antagonists and BAX R145 are important for BAX inhibition.
Direct BAX activation with small molecules offers an innovative strategy for cancer treatment with the therapeutical potential to overcome the apoptosis resistance of diverse approved therapeutic agents such as chemotherapies, BH3-mimetics, and mTOR inhibitors in a wide range of cancers. Intriguingly, the combination of Bcl-2/Bcl-XL inhibitor Navitoclax and BAX trigger site agonist BTSA1.2 synergistically suppresses apoptosis-resistant solid/hematologic tumor cells, CDX, and PDX models while sparing healthy tissues. BAX S184 site agonists CYD-2-11 not only potently suppress CDX and PDX models, as well as GEMMs with the KRASG12D mutant, but also reverse the resistance of mTOR inhibitors in vitro and in vivo. Conversely, inhibition of BAX with small molecules such as BAX BAI site antagonist BAI1 effectively mitigates the chemotherapies-induced side effects without the loss of their anti-tumor potency in both CDX and PDX models, offering a rational strategy to improve cancer chemotherapies. Ischemic stroke, the majority of stroke (∼85%)174, is partially attributed to unwanted apoptosis of neurons within the penumbra of the infarct120. However, the limited treatment options and extremally short therapeutical window (<3 h) of current treatment lead to its massive mortality and morbidity175. Intriguingly, Bci compounds significantly protect neurons that decrease hippocampal damage via the blockage of BAX channel activity in a gerbil brain ischemia model. Similarly, MSN compounds exhibit remarkable neuroprotection by preventing both BAX- and BAK-mediated cell death in vitro and in vivo. These findings strongly proof that therapeutical inhibiting BAX-mediated cell death is effective in attenuating the effects of neuronal cell death stimuli, providing an opportunity to treat ischemic stroke as well as degenerative diseases by drugging BAX directly.
Great advances have been made in the discovery of various BAX-targeted apoptosis modulators. However, the development of BAX therapeutics as clinical drugs faces several challenges. Firstly, the lack of co-crystal structures of BAX with identified small molecule modulators not only becomes a potential barrier to a deeper understanding of the pharmacological modulation of BAX but also hinders the rational design of potent BAX-targeted apoptosis modulators by structure-based approaches. Secondly, most of the current BAX modulators have not advanced to clinical trials owing to their narrow therapeutic window or poor drug-like properties, necessitating a balance between the potency and safety profile of these compounds. Despite BAX agonists may theoretically share a therapeutic window similar to current BH3 mimetics in cancer, it remains uncertain whether current BAX agonists can be safely transitioned to preclinical studies or clinically used drugs, especially given the poor cell/tissue selectivity of S184 site agonists. Notably, the relatively superior BAX trigger site activators BTSA1 and its analogs still possess poor to moderate anti-tumor potency (hematologic tumor cell lines, IC50 < 3 μmol/L; solid tumor cell lines, IC50 > 10 μmol/L) and metabolic instability146, and the generation of the reactive and toxic metabolite aminothiazole will hamper their further development. The potency and ADMET properties of the identified leads require further optimization. The development of small-molecule BAX antagonists is still in its early lead discovery stages. Current reported BAX antagonists merely provide proof-of-concept for the feasibility of direct BAX inhibition in the context of pathologic cell death and serve as potent pharmacological tools to probe the consequences of direct BAX inhibition under physiological and pathological conditions. There is an urgent need for the structural optimization of potency and drug-like properties of these tools or lead compounds to discover more powerful clinical candidates and conduct deeper evaluations of the therapeutical value and safety of pharmacologically BAX modulation under pathological conditions.
Besides, BAX is implicated in multiple forms of non-apoptotic cell death cell death by influencing several intracellular organelles’ membrane permeability. In contrast to apoptosis, the key step of mitochondrial-dependent necrosis is the opening of the mitochondrial permeability transition pore (MPTP) in the inner mitochondrial membrane, which promotes the loss of the inner membrane potential, swelling, and eventual rupture of the organelle membrane26,176. Overexpression of BAX in tumor cells promotes mitochondrial permeability transition (MPT), consequently disrupting the integrity of the plasma membrane to induce necrosis177, 178, 179. Emerging evidence indicates that BAX drives mitochondrial fusion180 and MPTP-dependent Ca2+ release181 to sensitize the opening of MPTP, ultimately promoting necrosis. Additionally, BAX plays an important role in autophagy via its effects on lysosomal membrane permeability. In their monomeric state, they increase the lysosomal membrane permeability to promote autophagy without being activated182. Moreover, necroptosis, a hybrid of apoptosis and necrosis, is dependent on BAX/BAK oligomerization. During the necroptotic cell death process, necroptotic stimuli-induced mixed-lineage kinase-domain-like (MLKL) protein activation leads to the downregulation of pro-apoptotic protein Mcl-1 that in turn results in BAX/BAK oligomerization, ultimately promoting MOMP and reinforcing MPTP opening183. While the stepwise regulated mechanism of BAX conformational activation for apoptosis is well-defined, the regulated mechanisms of BAX-mediated non-apoptotic cell death are less clear.
Future efforts should focus on characterizing the high–resolution complex structures of modulators with BAX, which is favorable for building a clear understanding of the activation or inhibition determinants and facilitating the discovery of more powerful BAX modulators. It is crucial to identify the most effective pharmacological targets among the binding sites or conformations of BAX. Accumulating evidence suggests that trigger site agonists exhibit broad anti-tumor potency in vitro and in vivo with well-characterized mechanisms of action and excellent pharmacokinetic profiles, suggesting that the trigger site may be a more potent regulatory site for the development of BAX therapeutics in cancer. The therapeutical potential of BAX/BAK dual agonists or antagonists is worth exploring in the diseases of dysregulated cell death, such as MSN compounds as BAX/BAK dual inhibitors for the treatment of degenerative diseases. Moreover, future investigations should also pay attention to the precisely regulated mechanisms of BAX-mediated non-apoptotic cell death. For diseases related to multiple forms of dysregulated cell death (e.g., heart failure), pharmacological manipulation of BAX may provide a rate-limiting therapeutical strategy, such as the use of BAX antagonist BAI compounds for the treatment of doxorubicin-induced cardiomyopathy.
Carefully and effectively manipulating BAX with small molecules presents blueprints for developing next-generation apoptosis modulators and represents a pressing and worthy challenge for medicinal chemistry studies. Through continued dedication, we are confident that it is feasible to leverage advanced medicinal chemistry, and structural and mechanistic insights to develop clinically-viable BAX therapeutics for the treatment of diseases characterized by uncontrolled cell death.
Author contributions
Zhenwei Zhang reviewed the literature, drew the figures, and drafted the manuscript. Linghui Hou, Dan Liu, and Shenglin Luan proofread the structures, figures, and tables. Min Huang provided supervision and revised the manuscript. Linxiang Zhao conceived the review topic and supervised the whole process. All of the authors have read and approved the final manuscript.
Conflicts of interest
The authors declare no conflicts of interest.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Grant No. 82173687, 82204218), Liaoning Revitalization Talents Program (XLYC1902089, China), Natural Science Foundation of Liaoning Province (2020-BS-155, China), General Scientific Research Projects of Department of Education in Liaoning Province (LJKQZ2021032, LJKZ0930, China), and Excellent Youth Talent Support Program of Shenyang Pharmaceutical University (YQ202304, China).
Footnotes
Peer review under the responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Contributor Information
Min Huang, Email: huangcrazye@sina.com.
Linxiang Zhao, Email: linxiang.zhao@vip.sina.com.
References
- 1.Czabotar P.E., Lessene G., Strasser A., Adams J.M. Control of apoptosis by the Bcl-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol. 2014;15:49–63. doi: 10.1038/nrm3722. [DOI] [PubMed] [Google Scholar]
- 2.Oltvai Z.N., Milliman C.L., Korsmeyer S.J. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell. 1993;74:609–619. doi: 10.1016/0092-8674(93)90509-o. [DOI] [PubMed] [Google Scholar]
- 3.Kiefer M.C., Brauer M.J., Powers V.C., Wu J.J., Umansky S.R., Tomei L.D., et al. Modulation of apoptosis by the widely distributed Bcl-2 homologue Bak. Nature. 1995;374:736–739. doi: 10.1038/374736a0. [DOI] [PubMed] [Google Scholar]
- 4.Hsu S.Y., Kaipia A., McGee E., Lomeli M., Hsueh A.J. Bok is a pro-apoptotic Bcl-2 protein with restricted expression in reproductive tissues and heterodimerizes with selective anti-apoptotic Bcl-2 family members. Proc Natl Acad Sci U S A. 1997;94:12401–12406. doi: 10.1073/pnas.94.23.12401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tsujimoto Y., Finger L.R., Yunis J., Nowell P.C., Croce C.M. Cloning of the chromosome breakpoint of neoplastic B cells with the t(14;18) chromosome translocation. Science. 1984;226:1097–1099. doi: 10.1126/science.6093263. [DOI] [PubMed] [Google Scholar]
- 6.Boise L.H., González-García M., Postema C.E., Ding L., Lindsten T., Turka L.A., et al. bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell. 1993;74:597–608. doi: 10.1016/0092-8674(93)90508-n. [DOI] [PubMed] [Google Scholar]
- 7.Lin E.Y., Orlofsky A., Berger M.S., Prystowsky M.B. Characterization of A1, a novel hemopoietic-specific early-response gene with sequence similarity to bcl-2. J Immunol. 1993;151:1979–1988. [PubMed] [Google Scholar]
- 8.Kozopas K.M., Yang T., Buchan H.L., Zhou P., Craig R.W. MCL1, a gene expressed in programmed myeloid cell differentiation, has sequence similarity to BCL2. Proc Natl Acad Sci U S A. 1993;90:3516–3520. doi: 10.1073/pnas.90.8.3516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gibson L., Holmgreen S.P., Huang D.C., Bernard O., Copeland N.G., Jenkins N.A., et al. bcl-w, a novel member of the bcl-2 family, promotes cell survival. Oncogene. 1996;13:665–675. [PubMed] [Google Scholar]
- 10.Ke N., Godzik A., Reed J.C. Bcl-B, a novel Bcl-2 family member that differentially binds and regulates Bax and Bak. J Biol Chem. 2001;276:12481–12484. doi: 10.1074/jbc.C000871200. [DOI] [PubMed] [Google Scholar]
- 11.Zhang H., Holzgreve W., De Geyter C. Bcl2-L-10, a novel anti-apoptotic member of the Bcl-2 family, blocks apoptosis in the mitochondria death pathway but not in the death receptor pathway. Hum Mol Genet. 2001;10:2329–2339. doi: 10.1093/hmg/10.21.2329. [DOI] [PubMed] [Google Scholar]
- 12.Aouacheria A., Arnaud E., Venet S., Lalle P., Gouy M., Rigal D., et al. Nrh, a human homologue of Nr-13 associates with Bcl-Xs and is an inhibitor of apoptosis. Oncogene. 2001;20:5846–5855. doi: 10.1038/sj.onc.1204740. [DOI] [PubMed] [Google Scholar]
- 13.Letai A., Bassik M.C., Walensky L.D., Sorcinelli M.D., Weiler S., Korsmeyer S.J. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell. 2002;2:183–192. doi: 10.1016/s1535-6108(02)00127-7. [DOI] [PubMed] [Google Scholar]
- 14.Wang K., Yin X.M., Chao D.T., Milliman C.L., Korsmeyer S.J. BID: a novel BH3 domain-only death agonist. Genes Dev. 1996;10:2859–2869. doi: 10.1101/gad.10.22.2859. [DOI] [PubMed] [Google Scholar]
- 15.O'Connor L., Strasser A., O'Reilly L.A., Hausmann G., Adams J.M., Cory S., et al. Bim: a novel member of the Bcl-2 family that promotes apoptosis. EMBO J. 1998;17:384–395. doi: 10.1093/emboj/17.2.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nakano K., Vousden K.H. PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell. 2001;7:683–694. doi: 10.1016/s1097-2765(01)00214-3. [DOI] [PubMed] [Google Scholar]
- 17.Oda E., Ohki R., Murasawa H., Nemoto J., Shibue T., Yamashita T., et al. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science. 2000;288:1053–1058. doi: 10.1126/science.288.5468.1053. [DOI] [PubMed] [Google Scholar]
- 18.Yang E., Zha J., Jockel J., Boise L.H., Thompson C.B., Korsmeyer S.J. Bad, a heterodimeric partner for Bcl-XL and Bcl-2, displaces Bax and promotes cell death. Cell. 1995;80:285–291. doi: 10.1016/0092-8674(95)90411-5. [DOI] [PubMed] [Google Scholar]
- 19.Inohara N., Ding L., Chen S., Núñez G. Harakiri, a novel regulator of cell death, encodes a protein that activates apoptosis and interacts selectively with survival-promoting proteins Bcl-2 and Bcl-XL. EMBO J. 1997;16:1686–1694. doi: 10.1093/emboj/16.7.1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Willis S.N., Fletcher J.I., Kaufmann T., van Delft M.F., Chen L., Czabotar P.E., et al. Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science. 2007;315:856–859. doi: 10.1126/science.1133289. [DOI] [PubMed] [Google Scholar]
- 21.Willis S.N., Chen L., Dewson G., Wei A., Naik E., Fletcher J.I., et al. Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev. 2005;19:1294–1305. doi: 10.1101/gad.1304105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Simmons M.J., Fan G., Zong W.X., Degenhardt K., White E., Gélinas C. Bfl-1/A1 functions, similar to Mcl-1, as a selective tBid and Bak antagonist. Oncogene. 2008;27:1421–1428. doi: 10.1038/sj.onc.1210771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kuwana T., Bouchier-Hayes L., Chipuk J.E., Bonzon C., Sullivan B.A., Green D.R., et al. BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol Cell. 2005;17:525–535. doi: 10.1016/j.molcel.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 24.Chen L., Willis S.N., Wei A., Smith B.J., Fletcher J.I., Hinds M.G., et al. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell. 2005;17:393–403. doi: 10.1016/j.molcel.2004.12.030. [DOI] [PubMed] [Google Scholar]
- 25.Certo M., Del Gaizo Moore V., Nishino M., Wei G., Korsmeyer S., Armstrong S.A., et al. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell. 2006;9:351–365. doi: 10.1016/j.ccr.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 26.Tait S.W., Green D.R. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol. 2010;11:621–632. doi: 10.1038/nrm2952. [DOI] [PubMed] [Google Scholar]
- 27.Green D.R., Kroemer G. The pathophysiology of mitochondrial cell death. Science. 2004;305:626–629. doi: 10.1126/science.1099320. [DOI] [PubMed] [Google Scholar]
- 28.Yang J., Liu X., Bhalla K., Kim C.N., Ibrado A.M., Cai J., et al. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science. 1997;275:1129–1132. doi: 10.1126/science.275.5303.1129. [DOI] [PubMed] [Google Scholar]
- 29.Kharbanda S., Pandey P., Schofield L., Israels S., Roncinske R., Yoshida K., et al. Role for Bcl-XL as an inhibitor of cytosolic cytochrome c accumulation in DNA damage-induced apoptosis. Proc Natl Acad Sci U S A. 1997;94:6939–6942. doi: 10.1073/pnas.94.13.6939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nijhawan D., Fang M., Traer E., Zhong Q., Gao W., Du F., et al. Elimination of Mcl-1 is required for the initiation of apoptosis following ultraviolet irradiation. Genes Dev. 2003;17:1475–1486. doi: 10.1101/gad.1093903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gavathiotis E., Suzuki M., Davis M.L., Pitter K., Bird G.H., Katz S.G., et al. BAX activation is initiated at a novel interaction site. Nature. 2008;455:1076–1081. doi: 10.1038/nature07396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gavathiotis E., Reyna D.E., Davis M.L., Bird G.H., Walensky L.D. BH3-triggered structural reorganization drives the activation of proapoptotic BAX. Mol Cell. 2010;40:481–492. doi: 10.1016/j.molcel.2010.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brouwer J.M., Westphal D., Dewson G., Robin A.Y., Uren R.T., Bartolo R., et al. Bak core and latch domains separate during activation, and freed core domains form symmetric homodimers. Mol Cell. 2014;55:938–946. doi: 10.1016/j.molcel.2014.07.016. [DOI] [PubMed] [Google Scholar]
- 34.Moldoveanu T., Grace C.R., Llambi F., Nourse A., Fitzgerald P., Gehring K., et al. BID-induced structural changes in BAK promote apoptosis. Nat Struct Mol Biol. 2013;20:589–597. doi: 10.1038/nsmb.2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dewson G., Kratina T., Czabotar P., Day C.L., Adams J.M., Kluck R.M. Bak activation for apoptosis involves oligomerization of dimers via their alpha6 helices. Mol Cell. 2009;36:696–703. doi: 10.1016/j.molcel.2009.11.008. [DOI] [PubMed] [Google Scholar]
- 36.Walensky L.D. Direct BAKtivation. Nat Struct Mol Biol. 2013;20:536–538. doi: 10.1038/nsmb.2579. [DOI] [PubMed] [Google Scholar]
- 37.Singh R., Letai A., Sarosiek K. Regulation of apoptosis in health and disease: the balancing act of Bcl-2 family proteins. Nat Rev Mol Cell Biol. 2019;20:175–193. doi: 10.1038/s41580-018-0089-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ren D., Tu H.C., Kim H., Wang G.X., Bean G.R., Takeuchi O., et al. BID, BIM, and PUMA are essential for activation of the BAX- and BAK-dependent cell death program. Science. 2010;330:1390–1393. doi: 10.1126/science.1190217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang K., Gross A., Waksman G., Korsmeyer S.J. Mutagenesis of the BH3 domain of BAX identifies residues critical for dimerization and killing. Mol Cell Biol. 1998;18:6083–6089. doi: 10.1128/mcb.18.10.6083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fletcher J.I., Meusburger S., Hawkins C.J., Riglar D.T., Lee E.F., Fairlie W.D., et al. Apoptosis is triggered when prosurvival Bcl-2 proteins cannot restrain Bax. Proc Natl Acad Sci U S A. 2008;105:18081–18087. doi: 10.1073/pnas.0808691105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen H.C., Kanai M., Inoue-Yamauchi A., Tu H.C., Huang Y., Ren D., et al. An interconnected hierarchical model of cell death regulation by the BCL-2 family. Nat Cell Biol. 2015;17:1270–1281. doi: 10.1038/ncb3236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vitale I., Pietrocola F., Guilbaud E., Aaronson S.A., Abrams J.M., Adam D., et al. Apoptotic cell death in disease-current understanding of the NCCD 2023. Cell Death Differ. 2023;30:1097–1154. doi: 10.1038/s41418-023-01153-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Delbridge A.R.D., Grabow S., Strasser A., Vaux D.L. Thirty years of Bcl-2: translating cell death discoveries into novel cancer therapies. Nat Rev Cancer. 2016;16:99–109. doi: 10.1038/nrc.2015.17. [DOI] [PubMed] [Google Scholar]
- 44.Mihalyova J., Jelinek T., Growkova K., Hrdinka M., Simicek M., Hajek R. Venetoclax: a new wave in hematooncology. Exp Hematol. 2018;61:10–25. doi: 10.1016/j.exphem.2018.02.002. [DOI] [PubMed] [Google Scholar]
- 45.Carneiro B.A., El-Deiry W.S. Targeting apoptosis in cancer therapy. Nat Rev Clin Oncol. 2020;17:395–417. doi: 10.1038/s41571-020-0341-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Heinicke U., Haydn T., Kehr S., Vogler M., Fulda S. Bcl-2 selective inhibitor ABT-199 primes rhabdomyosarcoma cells to histone deacetylase inhibitor-induced apoptosis. Oncogene. 2018;37:5325–5339. doi: 10.1038/s41388-018-0212-5. [DOI] [PubMed] [Google Scholar]
- 47.Faber A.C., Corcoran R.B., Ebi H., Sequist L.V., Waltman B.A., Chung E., et al. BIM expression in treatment-naive cancers predicts responsiveness to kinase inhibitors. Cancer Discov. 2011;1:352–365. doi: 10.1158/2159-8290.CD-11-0106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Oltersdorf T., Elmore S.W., Shoemaker A.R., Armstrong R.C., Augeri D.J., Belli B.A., et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 49.Tse C., Shoemaker A.R., Adickes J., Anderson M.G., Chen J., Jin S., et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008;68:3421–3428. doi: 10.1158/0008-5472.CAN-07-5836. [DOI] [PubMed] [Google Scholar]
- 50.Souers A.J., Leverson J.D., Boghaert E.R., Ackler S.L., Catron N.D., Chen J., et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med. 2013;19:202–208. doi: 10.1038/nm.3048. [DOI] [PubMed] [Google Scholar]
- 51.Cerella C., Dicato M., Diederich M. BH3 mimetics in AML therapy: death and beyond? Trends Pharmacol Sci. 2020;41:793–814. doi: 10.1016/j.tips.2020.09.004. [DOI] [PubMed] [Google Scholar]
- 52.Deng J. How to unleash mitochondrial apoptotic blockades to kill cancers? Acta Pharm Sin B. 2017;7:18–26. doi: 10.1016/j.apsb.2016.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wu D., Li Y., Zheng L., Xiao H., Ouyang L., Wang G., et al. Small molecules targeting protein‒protein interactions for cancer therapy. Acta Pharm Sin B. 2023;13:4060–4088. doi: 10.1016/j.apsb.2023.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Roberts A.W., Seymour J.F., Brown J.R., Wierda W.G., Kipps T.J., Khaw S.L., et al. Substantial susceptibility of chronic lymphocytic leukemia to Bcl-2 inhibition: results of a phase I study of navitoclax in patients with relapsed or refractory disease. J Clin Oncol. 2012;30:488–496. doi: 10.1200/JCO.2011.34.7898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kipps T.J., Eradat H., Grosicki S., Catalano J., Cosolo W., Dyagil I.S., et al. A phase 2 study of the BH3 mimetic Bcl-2 inhibitor navitoclax (ABT-263) with or without rituximab, in previously untreated B-cell chronic lymphocytic leukemia. Leuk Lymphoma. 2015;56:2826–2833. doi: 10.3109/10428194.2015.1030638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tron A.E., Belmonte M.A., Adam A., Aquila B.M., Boise L.H., Chiarparin E., et al. Discovery of Mcl-1-specific inhibitor AZD5991 and preclinical activity in multiple myeloma and acute myeloid leukemia. Nat Commun. 2018;9:5341. doi: 10.1038/s41467-018-07551-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Caenepeel S., Brown S.P., Belmontes B., Moody G., Keegan K.S., Chui D., et al. AMG 176, a selective Mcl-1 inhibitor, is effective in hematologic cancer models alone and in combination with established therapies. Cancer Discov. 2018;8:1582–1597. doi: 10.1158/2159-8290.CD-18-0387. [DOI] [PubMed] [Google Scholar]
- 58.Szlavik Z., Csekei M., Paczal A., Szabo Z.B., Sipos S., Radics G., et al. Discovery of S64315, a potent and selective Mcl-1 inhibitor. J Med Chem. 2020;63:13762–13795. doi: 10.1021/acs.jmedchem.0c01234. [DOI] [PubMed] [Google Scholar]
- 59.Choudhary G.S., Al-Harbi S., Mazumder S., Hill B.T., Smith M.R., Bodo J., et al. Mcl-1 and Bcl-XL-dependent resistance to the Bcl-2 inhibitor ABT-199 can be overcome by preventing PI3K/AKT/mTOR activation in lymphoid malignancies. Cell Death Dis. 2015;6 doi: 10.1038/cddis.2014.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang Q., Riley-Gillis B., Han L., Jia Y., Lodi A., Zhang H., et al. Activation of RAS/MAPK pathway confers Mcl-1 mediated acquired resistance to Bcl-2 inhibitor venetoclax in acute myeloid leukemia. Signal Transduct Targeted Ther. 2022;7:51. doi: 10.1038/s41392-021-00870-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang Z., Bai L., Hou L., Deng H., Luan S., Liu D., et al. Trends in targeting Bcl-2 anti-apoptotic proteins for cancer treatment. Eur J Med Chem. 2022;232 doi: 10.1016/j.ejmech.2022.114184. [DOI] [PubMed] [Google Scholar]
- 62.Blombery P., Thompson E.R., Nguyen T., Birkinshaw R.W., Gong J.N., Chen X., et al. Multiple BCL2 mutations cooccurring with Gly101Val emerge in chronic lymphocytic leukemia progression on venetoclax. Blood. 2020;135:773–777. doi: 10.1182/blood.2019004205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tausch E., Close W., Dolnik A., Bloehdorn J., Chyla B., Bullinger L., et al. Venetoclax resistance and acquired BCL2 mutations in chronic lymphocytic leukemia. Haematologica. 2019;104:e434–e437. doi: 10.3324/haematol.2019.222588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lin K.H., Winter P.S., Xie A., Roth C., Martz C.A., Stein E.M., et al. Targeting Mcl-1/Bcl-XL forestalls the acquisition of resistance to ABT-199 in acute myeloid leukemia. Sci Rep. 2016;6 doi: 10.1038/srep27696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kaefer A., Yang J., Noertersheuser P., Mensing S., Humerickhouse R., Awni W., et al. Mechanism-based pharmacokinetic/pharmacodynamic meta-analysis of navitoclax (ABT-263) induced thrombocytopenia. Cancer Chemother Pharmacol. 2014;74:593–602. doi: 10.1007/s00280-014-2530-9. [DOI] [PubMed] [Google Scholar]
- 66.Wang X., Bathina M., Lynch J., Koss B., Calabrese C., Frase S., et al. Deletion of MCL-1 causes lethal cardiac failure and mitochondrial dysfunction. Genes Dev. 2013;27:1351–1364. doi: 10.1101/gad.215855.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Papatzimas J.W., Gorobets E., Maity R., Muniyat M.I., MacCallum J.L., Neri P., et al. From inhibition to degradation: targeting the antiapoptotic protein myeloid cell leukemia 1 (Mcl-1) J Med Chem. 2019;62:5522–5540. doi: 10.1021/acs.jmedchem.9b00455. [DOI] [PubMed] [Google Scholar]
- 68.Rasmussen M.L., Taneja N., Neininger A.C., Wang L., Robertson G.L., Riffle S.N., et al. Mcl-1 inhibition by selective BH3 mimetics disrupts mitochondrial dynamics causing loss of viability and functionality of human cardiomyocytes. iScience. 2020;23 doi: 10.1016/j.isci.2020.101015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Romanov-Michailidis F., Hsiao C.C., Urner L.M., Jerhaoui S., Surkyn M., Miller B., et al. Discovery of an oral, beyond-rule-of-five Mcl-1 protein‒protein interaction modulator with the potential of treating hematological malignancies. J Med Chem. 2023;66:6122–6148. doi: 10.1021/acs.jmedchem.2c01953. [DOI] [PubMed] [Google Scholar]
- 70.Bai L., Chen J., Liu L., McEachern D., Aguilar A., Zhou H., et al. 338 BM-1252 (APG-1252): a potent dual specific Bcl-2/Bcl-xL inhibitor that achieves complete tumor regression with minimal platelet toxicity. Eur J Cancer. 2014;50:109–110. [Google Scholar]
- 71.Patterson C.M., Balachander S.B., Grant I., Pop-Damkov P., Kelly B., McCoull W., et al. Design and optimisation of dendrimer-conjugated Bcl-2/xL inhibitor, AZD0466, with improved therapeutic index for cancer therapy. Commun Biol. 2021;4:112. doi: 10.1038/s42003-020-01631-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Khan S., Zhang X., Lv D., Zhang Q., He Y., Zhang P., et al. Bcl-XL PROTAC degrader achieves safe and potent antitumor activity. Nat Med. 2019;25:1938–1947. doi: 10.1038/s41591-019-0668-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang Z., Li H., Gou L., Li W., Wang Y. Antibody‒drug conjugates: recent advances in payloads. Acta Pharm Sin B. 2023;13:4025–4059. doi: 10.1016/j.apsb.2023.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Casara P., Davidson J., Claperon A., Le Toumelin-Braizat G., Vogler M., Bruno A., et al. S55746 is a novel orally active Bcl-2 selective and potent inhibitor that impairs hematological tumor growth. Oncotarget. 2018;9:20075–20088. doi: 10.18632/oncotarget.24744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Le Gouill S., Wermke M., Morschhauser F., Lim S.T., Salles G., Kloos I., et al. A new Bcl-2 inhibitor (S55746/BCL201) as monotherapy in patients with relapsed or refractory non-Hodgkin lymphoma: preliminary results of the first-in-human trial. Hematol Oncol. 2017;35:47–48. [Google Scholar]
- 76.Diepstraten S.T., Anderson M.A., Czabotar P.E., Lessene G., Strasser A., Kelly G.L. The manipulation of apoptosis for cancer therapy using BH3-mimetic drugs. Nat Rev Cancer. 2022;22:45–64. doi: 10.1038/s41568-021-00407-4. [DOI] [PubMed] [Google Scholar]
- 77.Hata A.N., Engelman J.A., Faber A.C. The Bcl-2 family: key mediators of the apoptotic response to targeted anticancer therapeutics. Cancer Discov. 2015;5:475–487. doi: 10.1158/2159-8290.CD-15-0011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bose P., Gandhi V., Konopleva M. Pathways and mechanisms of venetoclax resistance. Leuk Lymphoma. 2017;58:1–17. doi: 10.1080/10428194.2017.1283032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Liu Z., Ding Y., Ye N., Wild C., Chen H., Zhou J. Direct activation of Bax protein for cancer therapy. Med Res Rev. 2016;36:313–341. doi: 10.1002/med.21379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jansson A., Sun X.F. Bax expression decreases significantly from primary tumor to metastasis in colorectal cancer. J Clin Oncol. 2002;20:811–816. doi: 10.1200/JCO.2002.20.3.811. [DOI] [PubMed] [Google Scholar]
- 81.Milas I., Komaki R., Hachiya T., Bubb R.S., Ro J.Y., Langford L., et al. Epidermal growth factor receptor, cyclooxygenase-2, and BAX expression in the primary non-small cell lung cancer and brain metastases. Clin Cancer Res. 2003;9:1070–1076. [PubMed] [Google Scholar]
- 82.Baltaziak M., Duraj E., Koda M., Wincewicz A., Musiatowicz M., Kanczuga-Koda L., et al. Expression of Bcl-XL, Bax, and p53 in primary tumors and lymph node metastases in oral squamous cell carcinoma. Ann N Y Acad Sci. 2006;1090:18–25. doi: 10.1196/annals.1378.002. [DOI] [PubMed] [Google Scholar]
- 83.Bush J.A., Li G. The role of Bcl-2 family members in the progression of cutaneous melanoma. Clin Exp Metastasis. 2003;20:531–539. doi: 10.1023/a:1025874502181. [DOI] [PubMed] [Google Scholar]
- 84.Li R., Ding C., Zhang J., Xie M., Park D., Ding Y., et al. Modulation of Bax and mTOR for cancer therapeutics. Cancer Res. 2017;77:3001–3012. doi: 10.1158/0008-5472.CAN-16-2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gardai S.J., Hildeman D.A., Frankel S.K., Whitlock B.B., Frasch S.C., Borregaard N., et al. Phosphorylation of Bax Ser184 by Akt regulates its activity and apoptosis in neutrophils. J Biol Chem. 2004;279:21085–21095. doi: 10.1074/jbc.M400063200. [DOI] [PubMed] [Google Scholar]
- 86.Kale J., Kutuk O., Brito G.C., Andrews T.S., Leber B., Letai A., et al. Phosphorylation switches Bax from promoting to inhibiting apoptosis thereby increasing drug resistance. EMBO Rep. 2018;19:e45235. doi: 10.15252/embr.201745235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Song J.Y., Ryu S.H., Cho Y.M., Kim Y.S., Lee B.M., Lee S.W., et al. Wip1 suppresses apoptotic cell death through direct dephosphorylation of BAX in response to γ-radiation. Cell Death Dis. 2013;4:e744. doi: 10.1038/cddis.2013.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Perego P., Giarola M., Righetti S.C., Supino R., Caserini C., Delia D., et al. Association between cisplatin resistance and mutation of p53 gene and reduced bax expression in ovarian carcinoma cell systems. Cancer Res. 1996;56:556–562. [PubMed] [Google Scholar]
- 89.Fresquet V., Rieger M., Carolis C., García-Barchino M.J., Martinez-Climent J.A. Acquired mutations in Bcl-2 family proteins conferring resistance to the BH3 mimetic ABT-199 in lymphoma. Blood. 2014;123:4111–4119. doi: 10.1182/blood-2014-03-560284. [DOI] [PubMed] [Google Scholar]
- 90.Aguilar-Santelises M., Rottenberg M.E., Lewin N., Mellstedt H., Jondal M. Bcl-2, Bax and p53 expression in B-CLL in relation to in vitro survival and clinical progression. Int J Cancer. 1996;69:114–119. doi: 10.1002/(SICI)1097-0215(19960422)69:2<114::AID-IJC8>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 91.Molica S., Dattilo A., Giulino C., Levato D., Levato L. Increased Bcl-2/Bax ratio in B-cell chronic lymphocytic leukemia is associated with a progressive pattern of disease. Haematologica. 1998;83:1122–1124. [PubMed] [Google Scholar]
- 92.Bannerji R., Kitada S., Flinn I.W., Pearson M., Young D., Reed J.C., et al. Apoptotic-regulatory and complement-protecting protein expression in chronic lymphocytic leukemia: relationship to in vivo rituximab resistance. J Clin Oncol. 2003;21:1466–1471. doi: 10.1200/JCO.2003.06.012. [DOI] [PubMed] [Google Scholar]
- 93.Starczynski J., Pepper C., Pratt G., Hooper L., Thomas A., Milligan D., et al. Common polymorphism G(-248)A in the promoter region of the bax gene results in significantly shorter survival in patients with chronic lymphocytic leukemia once treatment is initiated. J Clin Oncol. 2005;23:1514–1521. doi: 10.1200/JCO.2005.02.192. [DOI] [PubMed] [Google Scholar]
- 94.Manoochehri M., Karbasi A., Bandehpour M., Kazemi B. Down-regulation of BAX gene during carcinogenesis and acquisition of resistance to 5-FU in colorectal cancer. Pathol Oncol Res. 2014;20:301–307. doi: 10.1007/s12253-013-9695-0. [DOI] [PubMed] [Google Scholar]
- 95.Agrawal S.G., Liu F.T., Wiseman C., Shirali S., Liu H., Lillington D., et al. Increased proteasomal degradation of Bax is a common feature of poor prognosis chronic lymphocytic leukemia. Blood. 2008;111:2790–2796. doi: 10.1182/blood-2007-10-110460. [DOI] [PubMed] [Google Scholar]
- 96.Shi L., Chen J., Yang J., Pan T., Zhang S., Wang Z. MiR-21 protected human glioblastoma U87MG cells from chemotherapeutic drug temozolomide induced apoptosis by decreasing Bax/Bcl-2 ratio and caspase-3 activity. Brain Res. 2010;1352:255–264. doi: 10.1016/j.brainres.2010.07.009. [DOI] [PubMed] [Google Scholar]
- 97.Sharifi S., Barar J., Hejazi M.S., Samadi N. Roles of the Bcl-2/Bax ratio, caspase-8 and 9 in resistance of breast cancer cells to paclitaxel. Asian Pac J Cancer Prev APJCP. 2014;15:8617–8622. doi: 10.7314/apjcp.2014.15.20.8617. [DOI] [PubMed] [Google Scholar]
- 98.Pluta P., Smolewski P., Pluta A., Cebula-Obrzut B., Wierzbowska A., Nejc D., et al. Significance of Bax expression in breast cancer patients. Pol Przegl Chir. 2011;83:549–553. doi: 10.2478/v10035-011-0087-4. [DOI] [PubMed] [Google Scholar]
- 99.Bukholm I.R., Bukholm G., Nesland J.M. Reduced expression of both Bax and Bcl-2 is independently associated with lymph node metastasis in human breast carcinomas. Apmis. 2002;110:214–220. doi: 10.1034/j.1600-0463.2002.100303.x. [DOI] [PubMed] [Google Scholar]
- 100.Krajewski S., Blomqvist C., Franssila K., Krajewska M., Wasenius V.M., Niskanen E., et al. Reduced expression of proapoptotic gene BAX is associated with poor response rates to combination chemotherapy and shorter survival in women with metastatic breast adenocarcinoma. Cancer Res. 1995;55:4471–4478. [PubMed] [Google Scholar]
- 101.Reyna D.E., Garner T.P., Lopez A., Kopp F., Choudhary G.S., Sridharan A., et al. Direct activation of BAX by BTSA1 overcomes apoptosis resistance in acute myeloid leukemia. Cancer Cell. 2017;32:490–505 e10. doi: 10.1016/j.ccell.2017.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Walensky L.D. Targeting BAX to drug death directly. Nat Chem Biol. 2019;15:657–665. doi: 10.1038/s41589-019-0306-6. [DOI] [PubMed] [Google Scholar]
- 103.Spitz A.Z., Gavathiotis E. Physiological and pharmacological modulation of BAX. Trends Pharmacol Sci. 2022;43:206–220. doi: 10.1016/j.tips.2021.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Marsden V.S., Strasser A. Control of apoptosis in the immune system: bcl-2, BH3-only proteins and more. Annu Rev Immunol. 2003;21:71–105. doi: 10.1146/annurev.immunol.21.120601.141029. [DOI] [PubMed] [Google Scholar]
- 105.Strasser A., Whittingham S., Vaux D.L., Bath M.L., Adams J.M., Cory S., et al. Enforced BCL2 expression in B-lymphoid cells prolongs antibody responses and elicits autoimmune disease. Proc Natl Acad Sci U S A. 1991;88:8661–8665. doi: 10.1073/pnas.88.19.8661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bouillet P., Metcalf D., Huang D.C., Tarlinton D.M., Kay T.W., Köntgen F., et al. Proapoptotic Bcl-2 relative Bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science. 1999;286:1735–1738. doi: 10.1126/science.286.5445.1735. [DOI] [PubMed] [Google Scholar]
- 107.Mason K.D., Lin A., Robb L., Josefsson E.C., Henley K.J., Gray D.H., et al. Proapoptotic Bak and Bax guard against fatal systemic and organ-specific autoimmune disease. Proc Natl Acad Sci U S A. 2013;110:2599–2604. doi: 10.1073/pnas.1215097110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Rathmell J.C., Lindsten T., Zong W.X., Cinalli R.M., Thompson C.B. Deficiency in Bak and Bax perturbs thymic selection and lymphoid homeostasis. Nat Immunol. 2002;3:932–939. doi: 10.1038/ni834. [DOI] [PubMed] [Google Scholar]
- 109.Bardwell P.D., Gu J., McCarthy D., Wallace C., Bryant S., Goess C., et al. The Bcl-2 family antagonist ABT-737 significantly inhibits multiple animal models of autoimmunity. J Immunol. 2009;182:7482–7489. doi: 10.4049/jimmunol.0802813. [DOI] [PubMed] [Google Scholar]
- 110.Manzur M., Fleming P., Huang D.C., Degli-Esposti M.A., Andoniou C.E. Virally mediated inhibition of Bax in leukocytes promotes dissemination of murine cytomegalovirus. Cell Death Differ. 2009;16:312–320. doi: 10.1038/cdd.2008.152. [DOI] [PubMed] [Google Scholar]
- 111.Norris K.L., Youle R.J. Cytomegalovirus proteins vMIA and m38.5 link mitochondrial morphogenesis to Bcl-2 family proteins. J Virol. 2008;82:6232–6243. doi: 10.1128/JVI.02710-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Arnoult D., Skaletskaya A., Estaquier J., Dufour C., Goldmacher V.S. The murine cytomegalovirus cell death suppressor m38.5 binds Bax and blocks Bax-mediated mitochondrial outer membrane permeabilization. Apoptosis. 2008;13:1100–1110. doi: 10.1007/s10495-008-0245-2. [DOI] [PubMed] [Google Scholar]
- 113.Jurak I., Schumacher U., Simic H., Voigt S., Brune W. Murine cytomegalovirus m38.5 protein inhibits Bax-mediated cell death. J Virol. 2008;82:4812–4822. doi: 10.1128/JVI.02570-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Erekat N.S. Apoptosis and its therapeutic implications in neurodegenerative diseases. Clin Anat. 2022;35:65–78. doi: 10.1002/ca.23792. [DOI] [PubMed] [Google Scholar]
- 115.Hyman B.T., Yuan J. Apoptotic and non-apoptotic roles of caspases in neuronal physiology and pathophysiology. Nat Rev Neurosci. 2012;13:395–406. doi: 10.1038/nrn3228. [DOI] [PubMed] [Google Scholar]
- 116.Hollville E., Romero S.E., Deshmukh M. Apoptotic cell death regulation in neurons. FEBS J. 2019;286:3276–3298. doi: 10.1111/febs.14970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Dhapola R., Hota S.S., Sarma P., Bhattacharyya A., Medhi B., Reddy D.H. Recent advances in molecular pathways and therapeutic implications targeting neuroinflammation for Alzheimer's disease. Inflammopharmacology. 2021;29:1669–1681. doi: 10.1007/s10787-021-00889-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kumari S., Dhapola R., Reddy D.H. Apoptosis in Alzheimer's disease: insight into the signaling pathways and therapeutic avenues. Apoptosis. 2023;28:943–957. doi: 10.1007/s10495-023-01848-y. [DOI] [PubMed] [Google Scholar]
- 119.Gould T.W., Buss R.R., Vinsant S., Prevette D., Sun W., Knudson C.M., et al. Complete dissociation of motor neuron death from motor dysfunction by Bax deletion in a mouse model of ALS. J Neurosci. 2006;26:8774–8786. doi: 10.1523/JNEUROSCI.2315-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Gibson M.E., Han B.H., Choi J., Knudson C.M., Korsmeyer S.J., Parsadanian M., et al. BAX contributes to apoptotic-like death following neonatal hypoxia-ischemia: evidence for distinct apoptosis pathways. Mol Med. 2001;7:644–655. [PMC free article] [PubMed] [Google Scholar]
- 121.Girgenrath M., Dominov J.A., Kostek C.A., Miller J.B. Inhibition of apoptosis improves outcome in a model of congenital muscular dystrophy. J Clin Invest. 2004;114:1635–1639. doi: 10.1172/JCI22928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Tehranian R., Rose M.E., Vagni V., Pickrell A.M., Griffith R.P., Liu H., et al. Disruption of Bax protein prevents neuronal cell death but produces cognitive impairment in mice following traumatic brain injury. J Neurotrauma. 2008;25:755–767. doi: 10.1089/neu.2007.0441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Dong H., Fazzaro A., Xiang C., Korsmeyer S.J., Jacquin M.F., McDonald J.W. Enhanced oligodendrocyte survival after spinal cord injury in Bax-deficient mice and mice with delayed Wallerian degeneration. J Neurosci. 2003;23:8682–8691. doi: 10.1523/JNEUROSCI.23-25-08682.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.D'Orsi B., Kilbride S.M., Chen G., Perez Alvarez S., Bonner H.P., Pfeiffer S., et al. Bax regulates neuronal Ca2+ homeostasis. J Neurosci. 2015;35:1706–1722. doi: 10.1523/JNEUROSCI.2453-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hochhauser E., Cheporko Y., Yasovich N., Pinchas L., Offen D., Barhum Y., et al. Bax deficiency reduces infarct size and improves long-term function after myocardial infarction. Cell Biochem Biophys. 2007;47:11–20. doi: 10.1385/cbb:47:1:11. [DOI] [PubMed] [Google Scholar]
- 126.Amgalan D., Garner T.P., Pekson R., Jia X.F., Yanamandala M., Paulino V., et al. A small-molecule allosteric inhibitor of BAX protects against doxorubicin-induced cardiomyopathy. Nat Can (Ott) 2020;1:315–328. doi: 10.1038/s43018-020-0039-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wei Q., Dong G., Chen J.K., Ramesh G., Dong Z. Bax and Bak have critical roles in ischemic acute kidney injury in global and proximal tubule-specific knockout mouse models. Kidney Int. 2013;84:138–148. doi: 10.1038/ki.2013.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Jang H.S., Padanilam B.J. Simultaneous deletion of Bax and Bak is required to prevent apoptosis and interstitial fibrosis in obstructive nephropathy. Am J Physiol Ren Physiol. 2015;309:F540–F550. doi: 10.1152/ajprenal.00170.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Mei S., Li L., Wei Q., Hao J., Su Y., Mei C., et al. Double knockout of Bax and Bak from kidney proximal tubules reduces unilateral urethral obstruction associated apoptosis and renal interstitial fibrosis. Sci Rep. 2017;7 doi: 10.1038/srep44892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Pogmore J.P., Uehling D., Andrews D.W. Pharmacological targeting of executioner proteins: controlling life and death. J Med Chem. 2021;64:5276–5290. doi: 10.1021/acs.jmedchem.0c02200. [DOI] [PubMed] [Google Scholar]
- 131.Suzuki M., Youle R.J., Tjandra N. Structure of Bax: coregulation of dimer formation and intracellular localization. Cell. 2000;103:645–654. doi: 10.1016/s0092-8674(00)00167-7. [DOI] [PubMed] [Google Scholar]
- 132.Garner T.P., Reyna D.E., Priyadarshi A., Chen H.C., Li S., Wu Y., et al. An autoinhibited dimeric form of BAX regulates the BAX activation pathway. Mol Cell. 2016;64:431. doi: 10.1016/j.molcel.2016.10.005. [DOI] [PubMed] [Google Scholar]
- 133.Robin A.Y., Krishna Kumar K., Westphal D., Wardak A.Z., Thompson G.V., Dewson G., et al. Crystal structure of Bax bound to the BH3 peptide of Bim identifies important contacts for interaction. Cell Death Dis. 2015;6 doi: 10.1038/cddis.2015.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Czabotar P.E., Westphal D., Dewson G., Ma S., Hockings C., Fairlie W.D., et al. Bax crystal structures reveal how BH3 domains activate Bax and nucleate its oligomerization to induce apoptosis. Cell. 2013;152:519–531. doi: 10.1016/j.cell.2012.12.031. [DOI] [PubMed] [Google Scholar]
- 135.Barclay L.A., Wales T.E., Garner T.P., Wachter F., Lee S., Guerra R.M., et al. Inhibition of pro-apoptotic BAX by a noncanonical interaction mechanism. Mol Cell. 2015;57:873–886. doi: 10.1016/j.molcel.2015.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Vervliet T., Gerasimenko J.V., Ferdek P.E., Jakubowska M.A., Petersen O.H., Gerasimenko O.V., et al. BH4 domain peptides derived from Bcl-2/Bcl-XL as novel tools against acute pancreatitis. Cell Death Dis. 2018;4:58. doi: 10.1038/s41420-018-0054-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Arnoult D., Bartle L.M., Skaletskaya A., Poncet D., Zamzami N., Park P.U., et al. Cytomegalovirus cell death suppressor vMIA blocks Bax- but not Bak-mediated apoptosis by binding and sequestering Bax at mitochondria. Proc Natl Acad Sci U S A. 2004;101:7988–7993. doi: 10.1073/pnas.0401897101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Ma J., Edlich F., Bermejo G.A., Norris K.L., Youle R.J., Tjandra N. Structural mechanism of Bax inhibition by cytomegalovirus protein vMIA. Proc Natl Acad Sci U S A. 2012;109:20901–20906. doi: 10.1073/pnas.1217094110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Uchime O., Dai Z., Biris N., Lee D., Sidhu S.S., Li S., et al. Synthetic antibodies inhibit Bcl-2-associated X Protein (BAX) through blockade of the N-terminal activation site. J Biol Chem. 2016;291:89–102. doi: 10.1074/jbc.M115.680918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Robin A.Y., Iyer S., Birkinshaw R.W., Sandow J., Wardak A., Luo C.S., et al. Ensemble properties of Bax determine its function. Structure. 2018;26:1346–1359.e5. doi: 10.1016/j.str.2018.07.006. [DOI] [PubMed] [Google Scholar]
- 141.Iyer S., Anwari K., Alsop A.E., Yuen W.S., Huang D.C., Carroll J., et al. Identification of an activation site in Bak and mitochondrial Bax triggered by antibodies. Nat Commun. 2016;7 doi: 10.1038/ncomms11734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Lovell J.F., Billen L.P., Bindner S., Shamas-Din A., Fradin C., Leber B., et al. Membrane binding by tBid initiates an ordered series of events culminating in membrane permeabilization by Bax. Cell. 2008;135:1074–1084. doi: 10.1016/j.cell.2008.11.010. [DOI] [PubMed] [Google Scholar]
- 143.Shuker S.B., Hajduk P.J., Meadows R.P., Fesik S.W. Discovering high-affinity ligands for proteins: SAR by NMR. Science. 1996;274:1531–1534. doi: 10.1126/science.274.5292.1531. [DOI] [PubMed] [Google Scholar]
- 144.Walensky L.D., Kung A.L., Escher I., Malia T.J., Barbuto S., Wright R.D., et al. Activation of apoptosis in vivo by a hydrocarbon-stapled BH3 helix. Science. 2004;305:1466–1470. doi: 10.1126/science.1099191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Spitz A.Z., Zacharioudakis E., Reyna D.E., Garner T.P., Gavathiotis E. Eltrombopag directly inhibits BAX and prevents cell death. Nat Commun. 2021;12:1134. doi: 10.1038/s41467-021-21224-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Feng G., Zhang X., Li Y., Wang R. Analysis of the binding sites on BAX and the mechanism of BAX activators through extensive molecular dynamics simulations. J Chem Inf Model. 2022;62:5208–5222. doi: 10.1021/acs.jcim.0c01420. [DOI] [PubMed] [Google Scholar]
- 147.Gavathiotis E., Reyna D.E., Bellairs J.A., Leshchiner E.S., Walensky L.D. Direct and selective small-molecule activation of proapoptotic BAX. Nat Chem Biol. 2012;8:639–645. doi: 10.1038/nchembio.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Stornaiuolo M., La Regina G., Passacantilli S., Grassia G., Coluccia A., La Pietra V., et al. Structure-based lead optimization and biological evaluation of BAX direct activators as novel potential anticancer agents. J Med Chem. 2015;58:2135–2148. doi: 10.1021/jm501123r. [DOI] [PubMed] [Google Scholar]
- 149.Daniele S., Pietrobono D., Costa B., Giustiniano M., La Pietra V., Giacomelli C., et al. Bax activation blocks self-renewal and induces apoptosis of human glioblastoma stem cells. ACS Chem Neurosci. 2018;9:85–99. doi: 10.1021/acschemneuro.7b00023. [DOI] [PubMed] [Google Scholar]
- 150.Lopez A., Reyna D.E., Gitego N., Kopp F., Zhou H., Miranda-Roman M.A., et al. Co-targeting of BAX and Bcl-XL proteins broadly overcomes resistance to apoptosis in cancer. Nat Commun. 2022;13:1199. doi: 10.1038/s41467-022-28741-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Zhang Z., Zhao S., Pei J., Hou L., Luan S., Deng H., et al. Optimization of BAX trigger site activator BTSA1 with improved antitumor potency and in vitro ADMET properties. Eur J Med Chem. 2023;248 doi: 10.1016/j.ejmech.2022.115076. [DOI] [PubMed] [Google Scholar]
- 152.Zhao B., He T. Chidamide, a histone deacetylase inhibitor, functions as a tumor inhibitor by modulating the ratio of Bax/Bcl-2 and P21 in pancreatic cancer. Oncol Rep. 2015;33:304–310. doi: 10.3892/or.2014.3595. [DOI] [PubMed] [Google Scholar]
- 153.Wang J., Kim T.H., Ahn M.Y., Lee J., Jung J.H., Choi W.S., et al. Sirtinol, a class III HDAC inhibitor, induces apoptotic and autophagic cell death in MCF-7 human breast cancer cells. Int J Oncol. 2012;41:1101–1109. doi: 10.3892/ijo.2012.1534. [DOI] [PubMed] [Google Scholar]
- 154.Liang T., Zhou Y., Elhassan R.M., Hou X., Yang X., Fang H. HDAC-Bax multiple ligands enhance Bax-dependent apoptosis in HeLa Cells. J Med Chem. 2020;63:12083–12099. doi: 10.1021/acs.jmedchem.0c01454. [DOI] [PubMed] [Google Scholar]
- 155.Xin M., Deng X. Nicotine inactivation of the proapoptotic function of Bax through phosphorylation. J Biol Chem. 2005;280:10781–10789. doi: 10.1074/jbc.M500084200. [DOI] [PubMed] [Google Scholar]
- 156.Xin M., Deng X. Protein phosphatase 2A enhances the proapoptotic function of Bax through dephosphorylation. J Biol Chem. 2006;281:18859–18867. doi: 10.1074/jbc.M512543200. [DOI] [PubMed] [Google Scholar]
- 157.Wang Q., Sun S.Y., Khuri F., Curran W.J., Deng X. Mono- or double-site phosphorylation distinctly regulates the proapoptotic function of Bax. PLoS One. 2010;5 doi: 10.1371/journal.pone.0013393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Xin M., Li R., Xie M., Park D., Owonikoko T.K., Sica G.L., et al. Small-molecule Bax agonists for cancer therapy. Nat Commun. 2014;5:4935. doi: 10.1038/ncomms5935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Fan S., Xu Q., Wang L., Wan Y., Qiu S. SMBA1, a Bax activator, Induces cell cycle arrest and apoptosis in malignant glioma cells. Pharmacology. 2020;105:164–172. doi: 10.1159/000500292. [DOI] [PubMed] [Google Scholar]
- 160.Liu G., Yin T., Kim H., Ding C., Yu Z., Wang H., et al. Structure‒activity relationship studies on Bax activator SMBA1 for the treatment of ER-positive and triple-negative breast cancer. Eur J Med Chem. 2019;178:589–605. doi: 10.1016/j.ejmech.2019.06.004. [DOI] [PubMed] [Google Scholar]
- 161.Liu G., Kim H., Wang P., Fricke D.R., Chen H., Wang T., et al. Further lead optimization on Bax activators: design, synthesis and pharmacological evaluation of 2-fluoro-fluorene derivatives for the treatment of breast cancer. Eur J Med Chem. 2021;219 doi: 10.1016/j.ejmech.2021.113427. [DOI] [PubMed] [Google Scholar]
- 162.Zhao G., Zhu Y., Eno C.O., Liu Y., Deleeuw L., Burlison J.A., et al. Activation of the proapoptotic Bcl-2 protein Bax by a small molecule induces tumor cell apoptosis. Mol Cell Biol. 2014;34:1198–1207. doi: 10.1128/MCB.00996-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Brahmbhatt H., Uehling D., Al-Awar R., Leber B., Andrews D. Small molecules reveal an alternative mechanism of Bax activation. Biochem J. 2016;473:1073–1083. doi: 10.1042/BCJ20160118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Chipuk J.E., McStay G.P., Bharti A., Kuwana T., Clarke C.J., Siskind L.J., et al. Sphingolipid metabolism cooperates with BAK and BAX to promote the mitochondrial pathway of apoptosis. Cell. 2012;148:988–1000. doi: 10.1016/j.cell.2012.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Cohen D.T., Wales T.E., McHenry M.W., Engen J.R., Walensky L.D. Site-dependent cysteine lipidation potentiates the activation of proapoptotic BAX. Cell Rep. 2020;30:3229–3239.e6. doi: 10.1016/j.celrep.2020.02.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Pritz J.R., Wachter F., Lee S., Luccarelli J., Wales T.E., Cohen D.T., et al. Allosteric sensitization of proapoptotic BAX. Nat Chem Biol. 2017;13:961–967. doi: 10.1038/nchembio.2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Garner T.P., Amgalan D., Reyna D.E., Li S., Kitsis R.N., Gavathiotis E. Small-molecule allosteric inhibitors of BAX. Nat Chem Biol. 2019;15:322–330. doi: 10.1038/s41589-018-0223-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Bombrun A., Gerber P., Casi G., Terradillos O., Antonsson B., Halazy S. 3,6-Dibromocarbazole piperazine derivatives of 2-propanol as first inhibitors of cytochrome c release via Bax channel modulation. J Med Chem. 2003;46:4365–4368. doi: 10.1021/jm034107j. [DOI] [PubMed] [Google Scholar]
- 169.Lefrak E.A., Pitha J., Rosenheim S., Gottlieb J.A. A clinicopathologic analysis of adriamycin cardiotoxicity. Cancer. 1973;32:302–314. doi: 10.1002/1097-0142(197308)32:2<302::aid-cncr2820320205>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 170.Middleman E., Luce J., Frei E., 3rd Clinical trials with adriamycin. Cancer. 1971;28:844–850. doi: 10.1002/1097-0142(1971)28:4<844::aid-cncr2820280407>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 171.Renu K., Abilash V.G., Tirupathi Pichiah P.B., Arunachalam S. Molecular mechanism of doxorubicin-induced cardiomyopathy‒an update. Eur J Pharmacol. 2018;818:241–253. doi: 10.1016/j.ejphar.2017.10.043. [DOI] [PubMed] [Google Scholar]
- 172.Niu X., Brahmbhatt H., Mergenthaler P., Zhang Z., Sang J., Daude M., et al. A small-molecule inhibitor of Bax and Bak oligomerization prevents genotoxic cell death and promotes neuroprotection. Cell Chem Biol. 2017;24:493–506.e5. doi: 10.1016/j.chembiol.2017.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Hetz C., Vitte P.A., Bombrun A., Rostovtseva T.K., Montessuit S., Hiver A., et al. Bax channel inhibitors prevent mitochondrion-mediated apoptosis and protect neurons in a model of global brain ischemia. J Biol Chem. 2005;280:42960–42970. doi: 10.1074/jbc.M505843200. [DOI] [PubMed] [Google Scholar]
- 174.Tsao C.W., Aday A.W., Almarzooq Z.I., Alonso A., Beaton A.Z., Bittencourt M.S., et al. Heart disease and stroke statistics-2022 update: a report from the American Heart Association. Circulation. 2022;145:e153–e639. doi: 10.1161/CIR.0000000000001052. [DOI] [PubMed] [Google Scholar]
- 175.Fugate J.E., Rabinstein A.A. Absolute and relative contraindications to IV rt-PA for acute ischemic stroke. Neurohospitalist. 2015;5:110–121. doi: 10.1177/1941874415578532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Halestrap A.P. What is the mitochondrial permeability transition pore? J Mol Cell Cardiol. 2009;46:821–831. doi: 10.1016/j.yjmcc.2009.02.021. [DOI] [PubMed] [Google Scholar]
- 177.Pastorino J.G., Chen S.T., Tafani M., Snyder J.W., Farber J.L. The overexpression of Bax produces cell death upon induction of the mitochondrial permeability transition. J Biol Chem. 1998;273:7770–7775. doi: 10.1074/jbc.273.13.7770. [DOI] [PubMed] [Google Scholar]
- 178.Shinoura N., Yoshida Y., Asai A., Kirino T., Hamada H. Relative level of expression of Bax and Bcl-XL determines the cellular fate of apoptosis/necrosis induced by the overexpression of Bax. Oncogene. 1999;18:5703–5713. doi: 10.1038/sj.onc.1202966. [DOI] [PubMed] [Google Scholar]
- 179.Zamzami N., Hirsch T., Dallaporta B., Petit P.X., Kroemer G. Mitochondrial implication in accidental and programmed cell death: apoptosis and necrosis. J Bioenerg Biomembr. 1997;29:185–193. doi: 10.1023/a:1022694131572. [DOI] [PubMed] [Google Scholar]
- 180.Whelan R.S., Konstantinidis K., Wei A.C., Chen Y., Reyna D.E., Jha S., et al. Bax regulates primary necrosis through mitochondrial dynamics. Proc Natl Acad Sci U S A. 2012;109:6566–6571. doi: 10.1073/pnas.1201608109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Karch J., Kwong J.Q., Burr A.R., Sargent M.A., Elrod J.W., Peixoto P.M., et al. Bax and Bak function as the outer membrane component of the mitochondrial permeability pore in regulating necrotic cell death in mice. Elife. 2013;2 doi: 10.7554/eLife.00772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Karch J., Schips T.G., Maliken B.D., Brody M.J., Sargent M.A., Kanisicak O., et al. Autophagic cell death is dependent on lysosomal membrane permeability through Bax and Bak. Elife. 2017;6 doi: 10.7554/eLife.30543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Karch J., Kanisicak O., Brody M.J., Sargent M.A., Michael D.M., Molkentin J.D. Necroptosis interfaces with MOMP and the MPTP in mediating cell death. PLoS One. 2015;10 doi: 10.1371/journal.pone.0130520. [DOI] [PMC free article] [PubMed] [Google Scholar]