

Abstract
Angiotensin II (Ang II) is a potent vasoconstrictor of vascular smooth muscle cells (VSMC) and is implicated in hypertension, but it's role in the regulation of endothelial function is not well known. We and others have previously shown that mechanically activated ion channel, Transient Receptor Potential Vanilloid 4 (TRPV4) mediates flow- and/or receptor-dependent vasodilation via nitric oxide (NO) production in endothelial cells. Ang II was demonstrated to crosstalk with TRPV4 via angiotensin 1 receptor (AT1R) and β-arrestin signaling in epithelial and immortalized cells, however, the role of this crosstalk in endothelial cell function is not fully explored. Ang II treatment significantly downregulated TRPV4 protein expression and TRPV4-mediated Ca2+ influx in human EC without altering TRPV4 mRNA levels. Further, TRPV4-induced eNOS phosphorylation and NO production were significantly reduced in Ang II-treated human EC. Importantly, Ang II infusion in mice revealed that, TRPV4/p-eNOS expression and colocalization was reduced in endothelium in vivo. Finally, Ang II infusion induced vascular remodeling as evidenced by decreased lumen to wall ratio in resistant mesenteric arteries. These findings suggest that Ang II induces endothelial dysfunction and vascular remodeling via downregulation of TRPV4/eNOS pathway and may contribute to hypertension, independent of or in addition to its effect on vascular smooth muscle contraction.
Keywords: Angiotensin II, Ca2+, Endothelial cells, eNOS, TRPV4, Vascular remodeling
Graphical abstract

Highlights
-
•
Ang II treatment reduced TRPV4 protein expression without changing the mRNA levels.
-
•
TRPV4-mediated phosphorylation of eNOS and NO production were reduced in Ang II-treated human EC in vitro.
-
•
Ang II infusion in mice reduced TRPV4 expression in endothelium and lumen to wall ratio in mesenteric arteries in vivo.
-
•
These findings identifies Ang II-TRPV4 axis as a novel target for hypertension.
1. Introduction
Hypertension is one the major factors underlying cardiovascular diseases including heart failure and stroke [1]. The renin-angiotensin system (RAS) is a critical determinant of hypertension in which Ang II is the prominent mediator studied extensively [2]. Ang II has been demonstrated to regulate hypertension by inducing smooth muscle cell contraction and vascular remodeling. Ang II binds to G protein-coupled receptor (GPCR), AT1R, activating Gq/PLC/IP3 mediated Ca2+ influx which induces smooth muscle contraction and vasoconstriction [[3], [4], [5]]. Ca2+ influx is mediated through opening of ion channels which are known to regulate many cellular functions such as proliferation, migration, apoptosis, and differentiation [6]. Among the number of ion channels, TRPV4 channels were demonstrated to crosstalk with Ang II in epithelial cells, immortalized cells, and rat VSMCs. Shukla et al for the first time demonstrated that Ang II-treatment induced ubiquitination and degradation of TRPV4 through AT1R/Gq-dependent activation of β-arrestin pathway [15]. Intriguingly, recent work demonstrated that TRPV4 activation by specific agonist GSK1016790A (GSK1 inhibits AT1R signaling by Ang II [7]. Although, this reciprocal crosstalk between Ang II and TRPV4 is starting to unravel, its role in endothelium or physiological significance is not fully known.
TRPV4 is a mechanosensor expressed in EC and mediates flow- and/or receptor-dependent vasodilation via NO production [8,9]. However, TRPV4 null mice did not display an overt blood pressure phenotype suggesting compensation or alternative role for TRPV4 in hypertension [9]. In fact, recently developed endothelial-specific TRPV4 knockout (TRPV4ECKO) mice demonstrated a mild hypertensive phenotype indicating that while endothelial TRPV4 is important for vasodilation, TRPV4 in other cell types such as VSMC may have a vasoconstrictor function [10]. Indeed, using smooth muscle specific TRPV4KO, it was demonstrated that TRPV4 is expressed in VSMC and its activation triggers vasoconstriction [10]. Despite their implication in hypertension independently, crosstalk between TRPV4-Ang II in EC and the physiological significance in the regulation of hypertension are not fully known.
We have previously demonstrated that TRPV4 is a mechanosensor in EC and regulates their proliferation, migration, and angiogenesis [[11], [12], [13]]. Importantly, we have previously demonstrated that PKCα mediates acetylcholine (ACh)-induced activation of TRPV4-dependent Ca2+ influx in EC that is required for vasodilation [8]. In this study, we investigated if Ang II cross-talks with TRPV4 in EC and its physiological significance using human EC in vitro and Ang II infusion model in vivo.
2. Methods
2.1. Cell Culture
Human microvascular endothelial cells (HMEC-1) were purchased from ATCC (Manassas, VA, United States) and were cultured as previously described (Guarino et al., 2019). Briefly, HMEC-1 were cultured in MCDB-131 media (Corning, United States), supplemented with 10 % FBS, 1 % penicillin-streptomycin, 1 % l-glutamine, 1 % hydrocortisone, and 10 ng/mL human EGF.
2.2. Western blot analysis
Cells were serum starved and treated with and without Ang II (200 nM) for 48 h. After, cells were lysed in RIPA buffer containing 1× protease and phosphatase inhibitor cocktails (Millipore Sigma and Roche, Basel, Switzerland). Lysates were loaded into precast polyacrylamide gels (Bio-Rad) for electrophoresis. Gels were transferred onto a PVDF membrane and blocked in 5 % milk powder in tris-buffered saline (TBS) with 0.1 % Tween-20. Membranes were incubated overnight at 4 °C with primary antibodies: TRPV4 (1:300; Alomone Labs, Jerusalem, Israel); eNOS-phospho Ser1177 (1:1000; BD Bioscience, New Jersey); eNOS (1:1000; Cell Signaling Technology, USA) and GAPDH (1:5000; Cell Signaling Technology, USA). After incubation, membranes were washed 3 times with 1× TBST for 5 min each, followed by 1 h incubation at room temperature in appropriate secondary antibody, anti-rabbit or anti-mouse (1:5000; Cell Signaling Technology, USA) conjugated with horseradish peroxidase (Cell Signaling Technology, USA). Signals were detected with western blot imager (Azure 500 Fluorescent Western Blot Imager, Dublin, United States), and developed with an immobilon ECL ultra-western HRP substrate (Darmstadt, Germany) or Azure biosystem chemiluminescent substrates (Dublin, CA, United States). Quantification was performed using Image J software (NIH).
2.3. Quantitative PCR (qPCR)
RNA was extracted from HMEC-1 treated with and without Ang II (200 nM/48 h) by using the RNeasy mini kit (Qiagen, Hilden, Germany) and quantified with a BioTek micro plate reader. Revert aid first strand cDNA synthesis kit (Thermo Fisher Scientific, USA) was used to synthesize cDNA, and fast SYBR green master mix (Thermo Fisher Scientific, USA) was used for qPCR on Real-Time PCR Systems (Applied Biosystems, StepOnePlus). The following real-time primers were obtained from Integrated DNA Technologies (Coralville, IA, United States): hGAPDH (forward-5′-TGC ACC ACC AAC TGC TTA GC-3′, reverse: 5′-GGC ATG GAC TGT GGT CAT GAG −3′) and hTRPV4 (forward-5′-TCA CTC TCA CCG CCT ACT ACC A-3′, reverse: 5′-CCC AGT GAA GAG CGT AAT GAC C-3′). Gene expression was normalized to GAPDH and ΔΔCt values were expressed as a fold change.
2.4. Calcium imaging
HMEC-1 were cultured as described previously, on MatTek glass bottom dishes (MatTek, Ashland, MA, United States). After treatment with Ang II as described above, cells were loaded with Fluo-4/AM (4 μM) for 25 min and were washed in previously described Ca2+ media [8,14]. Ca2+ imaging was performed on an Olympus FluoView 300 microscope (Olympus, Shinjuku, Tokyo, Japan)/Leica SP5 confocal microscope after stimulation with the TRPV4 agonist, GSK1016790A (100 nM).
2.5. Immunofluorescence of phospho-endothelial nitric oxide synthase
HMEC-1 were cultured on glass cover slips in six-well plates until they were ∼ 70–80 % confluent. After this, the complete media was replaced with serum free media, left for overnight, followed by treatment with and without Ang II (200 nM for 48 h). Cells were then washed with PBS (3 times) and fixed for 20 min at room temperature in PBS containing 4 % paraformaldehyde. Cells were then rinsed and permeabilized with 0.25 % Triton X-100 in PBS and blocked with serum-containing media for 20 min. Afterwards, cells were incubated with the primary phospho-endothelial nitric oxide synthase (p-eNOS) antibody (1:100; BD Bioscience, New Jersey) at room temperature for 1 h. The cells were then washed with PBS and incubated with Alexa Fluor-conjugated secondary antibodies (1:500). Cells were mounted on glass slides using Fluoromount-G™ mounting medium containing DAPI (Invitrogen, USA). Images were obtained using multiphoton microscope (Leica Microsystems) with a 40× objective and processed using Image J software (NIH).
2.6. NO measurement
HMEC-1 were seeded on glass cover slips in six-well plates at the appropriate densities. At ∼70–80 % confluency, complete media was replaced with serum free media overnight, and cells were treated with and without Ang II (200 nM/48 h), followed by stimulation with known TRPV4 activator GSK-1016790A(100 nM/30 min) and incubation with 10 μM diaminofluorescein-FM diacetate (DAF-FM DA; NO-sensitive fluorescent dye) without phenol red at 37 °C in 5 % CO2 for 30 min. Cells were then washed with PBS (3 times) and fixed for 20 min at room temperature in PBS containing 4 % paraformaldehyde. Cells were then washed with PBS (3 times) and mounted on glass slides using Fluoromount-G™ mounting medium containing DAPI (Invitrogen, USA). Images were obtained using multiphoton microscope (Leica Microsystems) with a 40× objective and processed using Image J software (NIH).
2.7. Animals and angiotensin II infusion
3–5 months old male mice (C57BL/6 J) were used in this study. All experiments were performed according to the guidelines and approval of the Institutional Animal Care and Use Committee of The University of Toledo. Mice were housed in a temperature-controlled room with a 12:12 h light-dark cycle and maintained with access to food and water ad libitum. Mice were divided into two groups, control (n = 8) and experimental (Ang II) (n = 8) group. Control mice received saline, while the experimental group received 0.2 μg/kg of Ang II daily by the intraperitoneal (i.p.) route for a period of 28 days. On day 29, mice were euthanized, aorta, and mesenteric arteries were collected.
2.8. Immunofluorescence
Aorta sections (5 μm) were incubated with the following primary antibodies: mouse anti-eNOS-phospho Ser1177 (1:200; BD Bioscience, New Jersey), mouse anti-CD31 (1:50; Invitrogen, Carlsbad, CA, USA), and anti-TRPV4 (1:50; Alomone Labs, Jerusalem, Israel). After the samples were washed with PBS, the tissue sections were incubated with appropriate secondary antibodies coupled to Alexa Fluor-488 or Alexa Fluor-594 (Invitrogen, USA) and mounted using Fluoromount-G™ mounting medium containing DAPI. Slides were imaged with 40× objective using Leica SP5 multiphoton microscope and processed using NIH ImageJ software. Paraffin sections of mesenteric arteries were exposed to antigen retrieval, stained with TRPV4 and CD31 antibodies and imaged as described above. For TRPV4 internalization, cells were transfected with TRPV4-EGFP [11], serum starved and stimulated with Ang II (200 nM/2 h), fixed and imaged with 63× objective using Leica SP5 multiphoton microscope and processed using NIH ImageJ software.
2.9. Mesenteric isolation and histological analysis
A second or third branch of the mesenteric arterial bed of 130 to 280 μm in lumen diameter and about 3 mm in length was carefully dissected out and was used for histological examination. Mesenteric artery morphometric analysis was performed with ImageJ software.
2.10. Statistical analysis
Statistical analyses were carried out using GraphPad Prism 8 statistical software. Analysis was performed using one-way ANOVA with Tukey's multiple comparisons test and student's t-test (unpaired, nonparametric) with Mann-Whitney test was used. All values reported are the Mean ± SEM. A value of *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, was considered significant.
3. Results
3.1. Angiotensin II treatment downregulates TRPV4 protein expression and TRPV4- mediated calcium influx without altering TRPV4 mRNA levels in HMECs
To determine if Ang II cross-talks with TRPV4 in human EC, we first measured the TRPV4 protein expression after Ang II treatment (200 nM for 48 h). Western blot analysis revealed that Ang II treatment significantly (p ≤ 0.05) downregulated TRPV4 protein expression in human EC (Fig. 1A and B). Next, we measured the TRPV4 mRNA levels. qPCR analysis showed that there was no significant difference in TRPV4 mRNA in control and Ang II treated human EC (Fig. 1C). Further, we found that Ang II treatment induced internalization of TRPV4 as evidenced by disappearance of TRPV4-EGFP from the plasma membrane, with distinct puncta like appearance in the cell indicating targeting to intracellular vesicles (Supplemental Fig. 1).
Fig. 1.
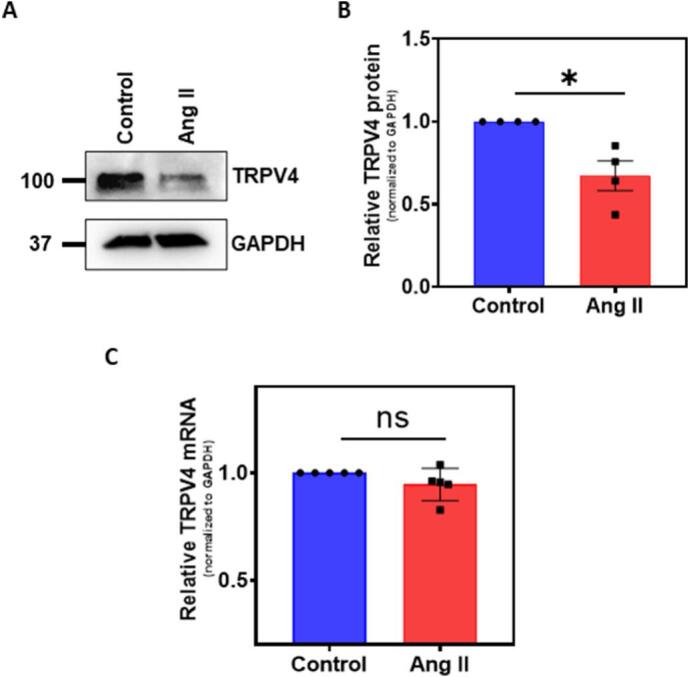
Angiotensin II treatment downregulates TRPV4 protein expression in HMECs. Cells were treated with Ang II (200 nM for 48 h) and the expression of TRPV4 was assessed using western blot and quantitative PCR (qPCR). A) Western blots showing TRPV4 protein expression. B) Relative TRPV4 expression levels in control and Ang II treated HMEC. GAPDH was used as loading control and for normalization (n = 4 biological replicates/group). C) qPCR analysis of TRPV4 mRNA levels in control and Ang II treated HMEC. (n = 5 biological replicates/group). All data points represented are the mean ± SEM, Statistical significance (unpaired t-test; nonparametric, Mann-Whitney test) is indicated by * p ≤ 0.05.
Next, to determine the functional consequence of TRPV4 downregulation, we measured TRPV4-mediated Ca2+ influx in control and Ang II treated human EC. Stimulation with TRPV4 specific agonist, GSK-1016790A (GSK1; 100 nM) induced robust Ca2+ influx that was significantly (p ≤ 0.01) abolished when human EC were pretreated with Ang II (200 nM/48 h) (Fig. 2A-C), suggesting that Ang II treatment results in functional downregulation of TRPV4 in human EC.
Fig. 2.
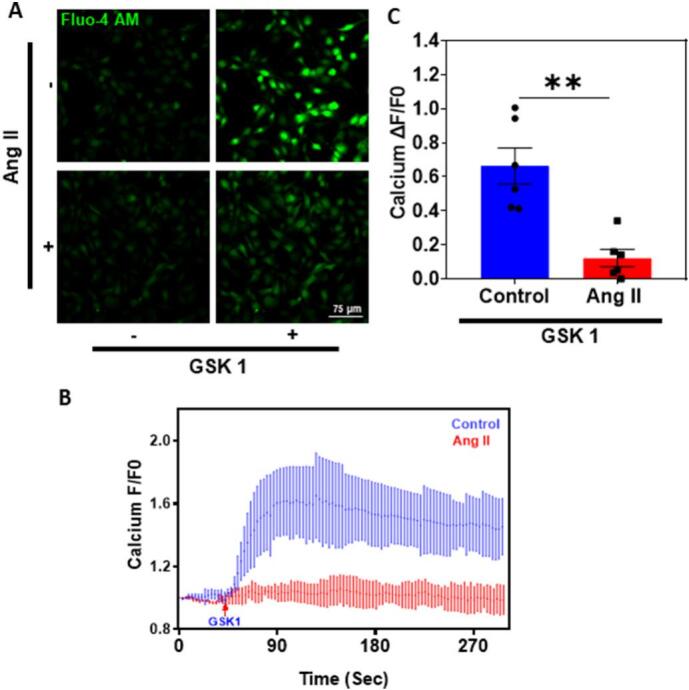
Angiotensin II treatment attenuates TRPV4 mediated calcium influx in HMECs. A) Representative fluorescence images of cytosolic Ca2+ influx in response to TRPV4 agonist GSK1016790A (GSK1) (n = 3 biological replicates/group, representative images are from ∼5 fields/group/experiment). (Magnification: 40×; Scale bar: 75 μm). B) Representative traces showing relative changes in cytosolic Ca2+ in response to GSK1 in the fluo-4/AM-loaded HMECs treated with (n = 3 biological replicates/group, data points represent an average of 128 cells/group) and without Ang II (200 nM/48 h) (n = 3 biological replicates/group, data points represent an average of 143 cells/group). The arrow represents the time when the cells were stimulated with GSK1. C) Quantitative analysis of cytosolic Ca2+ influx induced by GSK1 in HMEC (n = 3 biological replicates/group). All data points represented are the mean ± SEM. Statistical significance (unpaired t-test, nonparametric, Mann-Whitney test) is indicated by * p ≤ 0.01.
3.2. TRPV4 mediated eNOS phosphorylation and NO production are inhibited by Angiotensin II
We next investigated if Ang II mediated TRPV4 downregulation modulates endothelial function. To achieve this, we measured eNOS phosphorylation as well as NO production in the human EC using phospho-specific antibodies against eNOS Ser1177 and DAF-FM DA, respectively. Immunofluorescence analysis revealed that cells stimulated with GSK1 showed robust staining of phospho-eNOS, which was inhibited by pretreatment of the cells with Ang II (Fig. 3A). Quantification of the fluorescence intensity revealed significant reduction in eNOS phosphorylation (Fig. 3B). Western blot analysis also showed increased phosphorylation of eNOS in response to GSK1, which was inhibited by Ang II treatment (Fig. 3C and D). We then measured NO generation, where cells stimulated with GSK1 showed significant NO production, which was inhibited by pretreatment of the cells with Ang II (Fig. 4A and B).
Fig. 3.
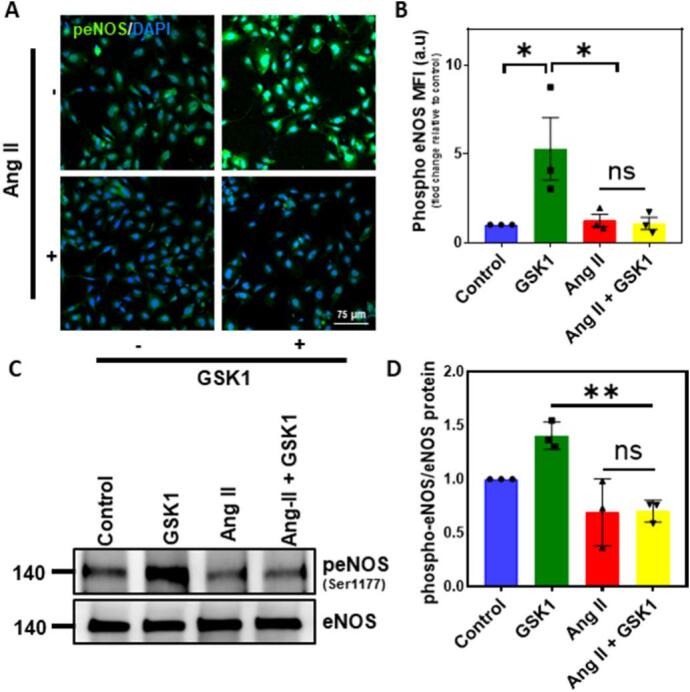
TRPV4 mediated eNOS phosphorylation is inhibited by Angiotensin II. HMEC were treated with Ang II and/or GSK1 and the expression of phospho-eNOS (S1177) was determined by immunofluorescence and western blot. A) Representative confocal fluorescence images of human EC stained with phospho-eNOS (Ser1177) (n = 3 biological replicates/group, representative images are from 5 fields/group/experiment). (Magnification: 40×, scale bar: 75 μm). B) Quantification of mean fluorescence intensity (MFI) of phospho-eNOS in HMEC (n = 3 biological replicates/group, data points represent average of 5 fields/group/experiment). C) Western blots showing phospho-eNOS expression (n = 3 biological replicates/group). D) Relative expression of phospho-eNOS. Total eNOS was used as a loading control and for normalization (n = 3 biological replicates/group). All data points represented are the mean ± SEM. ANOVA with Tukey's multiple comparisons test was performed. Statistical significance is indicated by * p ≤ 0.05, ** p ≤ 0.01.
Fig. 4.
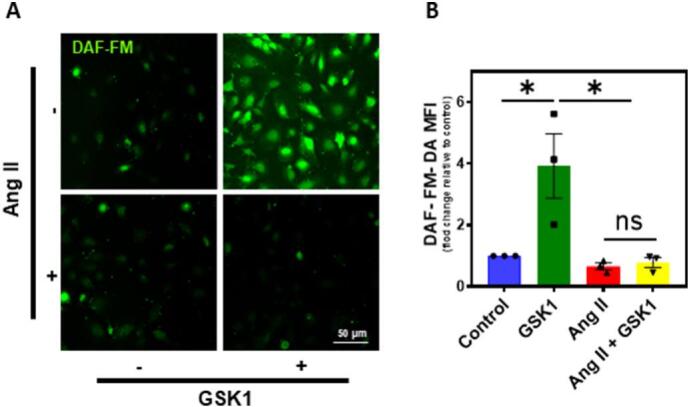
TRPV4 mediated NO production is inhibited by Angiotensin II. HMEC were treated with Ang II and/or GSK1 and NO levels were determined by loading with DAF-FM. A) Representative images of HMEC showing DAF fluorescence indicative of NO levels. (n = 3 biological replicates/group, representative images are from 5 fields/group/experiment). (Magnification: 40×, scale bar: 50 μm). B) Quantification of DAF fluorescence in HMEC (n = 3 biological replicates/group, data points represent an average of 5 fields/group/experiment). All data points represented are the mean ± SEM from at least three independent experiments. ANOVA with Tukey's multiple comparisons test was performed. Statistical significance is indicated by * p ≤ 0.05.
3.3. Angiotensin II infusion downregulates TRPV4/p-eNOS expression in endothelium in vivo and induces vascular remodeling
To investigate if Ang II induces endothelial dysfunction in vivo via downregulation of TRPV4/eNOS pathway, we infused Ang II for 28 days. Immunohistochemical analysis of aortic cross sections from vehicle-treated mice showed robust TRPV4 staining in the endothelium (red; Fig. 5A) which is co-localized (yellow) with p-eNOS (green Fig. 5A). As a control, we found that TRPV4 was co-localized with endothelial marker CD31 (Supplemental Fig. 2A). In contrast, TRPV4 and p-eNOS expression and co-localization was significantly attenuated in the aorta of Ang II-infused mice (green; Fig. 5A). Similarly, we found co-localization of TRPV4 with CD31 in the endothelium of control mesenteric arteries which was significantly reduced in Ang II-treated arteries (Supplemental Fig. 2B).
Fig. 5.
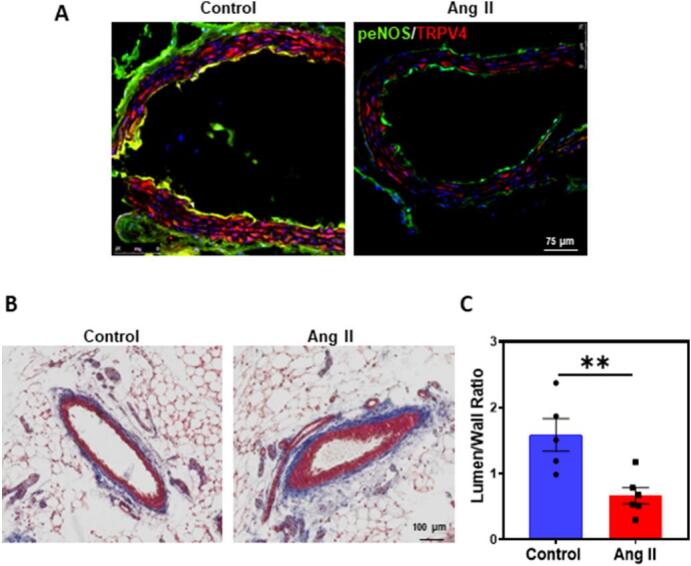
Angiotensin II infusion reduces TRPV4/p-eNOS expression in aortic endothelium and vascular remodeling in mesenteric vessels in vivo. A) Representative confocal immunofluorescence images of aorta (cross-section) depicting the p-eNOS/TRPV4 staining in endotheium with and without Ang II infusion (Red: TRPV4; Green: p-eNOS) (n = 5 (control) and n = 3 (Ang II) biological replicates/group). (Magnification: 40×, scale bar: 75 μm). B) Representative images of mesenteric (cross-section) arteries showing the vessel diameter. C) Quantification of mean lumen/wall ratio in mesenteric vessels (n = 5 (control) and n = 6 (Ang II) biological replicates/ group). (Magnification: 20×; Scale bar:100um). All data points represented are the mean ± SEM. Statistical significance (unpaired t-test; nonparametric, Mann-Whitney test) is indicated by ** p ≤ 0.01. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Finally, to determine the pathophysiological significance of TRPV4/eNOS/NO downregulation by Ang II, we measured lumen to wall ratio in histological sections of mesenteric arteries. We found that lumen to wall area ratio was significantly reduced in mesenteric arteries of Ang II-infused mice compared to vehicle controls (Fig. 5B and C).
4. Discussion
In the present study we demonstrate that Ang II treatment downregulates TRPV4 and induces endothelial dysfunction and remodeling of resistant arteries via downregulation of TRPV4/eNOS/NO pathway. We and others have previously demonstrated that TRPV4 is required for both ACh-mediated (GPCR) and flow-dependent vasodilation through NO production [8,9]. However, the impact of a vasoconstrictor GPCR such as Ang II on TRPV4 in EC is not well known. Ang II typically induces vasoconstriction in VSMC via AT1R/Gq signaling [5]. Interestingly, Ang II treatment demonstrated to downregulate TRPV4 through β-arrestin dependent ubiquitination in epithelial and rat smooth muscle cells [7,15,16]. Conversely, TRPV4 activation appear to downregulate Ang II/AT1R signaling in epithelial cells through calcineurin pathway [7]. However, neither the functional significance of this Ang II-TRPV4 crosstalk nor its existence in endothelial cells is not fully known. Here, we demonstrate that chronic Ang II treatment reduces TRPV4 protein expression without altering TRPV4 mRNA levels in human EC. Our results are in line with the previous findings in other cells that Ang II degrades TRPV4 targeting it to ubiquitination/proteosome pathway via Gq/β-arrestin [7]. In fact, we found that Ang II treatment for 2 h internalizes TRPV4 as evidenced by disappearance of TRPV4-EGFP from the plasma membrane, with distinct puncta like appearance in the cell indicating targeting to intracellular vesicles (Supplemental Fig. 1; Fig. 6). However, in contrast to these results, it has been reported that Ang II treatment does not affect TRPV4 protein expression in the mesenteric arteries and human coronary artery endothelial cells (HCAECs) overexpressing TRPV4-GFP. The possible reasons for the difference between these two studies could be experimental conditions such as 1) the duration of Ang II infusion, 14 days Vs 28 days 2) TRPV4 expression was measured in HCAECs overexpressing TRPV4-GFP but not in EC expressing endogenous TRPV4 and 3) Ang II treatment duration 24 h Vs 48 h. Although there is a difference in the duration and dose of Ang II treatment between these two studies, Ang II treatment, in fact, significantly reduced TRPV4-mediated calcium influx in HCAECs in the previous study. Based on these findings, Nishijima et al. suggested that Ang II may alter TRPV4 function or signaling but not it's expression [17]. Importantly, our results uncover the functional consequence of this crosstalk demonstrating that Ang II treatment reduces TRPV4-mediated Ca2+ influx, eNOS phosphorylation, and NO production leading to endothelial dysfunction. Although NO is the major mediator of TRPV4 facilitated vasodilation, TRPV4 channels can also induce vasodilation through the activation of intermediate (IK)- and small (SK)-conductance channels and dependent hyperpolarization of VSMC in some resistant arteries [18,19]. In fact, Ang II treatment was demonstrated to reduce TRPV4 mediated vasodilation in isolated cerebral (parenchymal) arteries which predominantly use TRPV4-EDHF pathway for vasodilation [20]. Therefore, it is plausible that TRPV4 mediates vasodilation through NO or EDHF in different vascular beds and that Ang II-mediated TRPV4 downregulation impairs vasodilation in both kind of vessels.
Fig. 6.

Angiotensin II induces endothelial dysfunction via downregulation of TRPV4/eNOS/NO signaling axis. In normal endothelium, TRPV4 activation leads to calcium influx in EC which in turn results in the phosphorylation of eNOS (endothelial nitric oxide synthase) at S1177and NO (nitric oxide) production leading to vasodilation. However, infusion of Ang II in mice may cause internalization and downregulation of TRPV4 (via β-arrestin) and mediated calcium influx, eNOS phosphorylation and NO production resulting in decreased vasodilation (Created with Biorendor.com).
Our study also provide evidence that TRPV4 expression is significantly reduced in vascular endothelium in vivo after infusion of Ang II. Further, we show that endothelial p-eNOS expression and its colocalization with TRPV4 is reduced in vivo. Ang II treatment did not reduce CD31 expression confirming the specific downregulation of TRPV4 and p-eNOS in the endothelium, after receiving Ang II for 28 days. However, our results suggest that this downregulation of TRPV4/eNOS pathway appears to induce endothelial dysfunction as manifested by increased remodeling (decreased lumen/wall ratio) of the mesenteric arteries. NO not only mediates vasodilation but also demonstrated to inhibit VSMC proliferation and migration [21]. Our findings that TRPV4-mediated NO production is reduced in Ang II-treated EC, suggest that increased wall thickness of the arteries could be due to VSMC proliferation triggered by low levels of NO in these vessels. Although we have not measured TRPV4 expression, VSMC contraction, and/or phenotype switching in response to Ang II treatment, immunofluorescence revealed that TRPV4 expression is intact in the smooth muscle area suggesting that Ang II treatment may not affect TRPV4 expression in VSMC. Hence, the vascular remodeling observed in the present study may be due to inhibition of vasodilation as well as increase in vasoconstriction by Ang II working on endothelial and smooth muscle cells, respectively. This is further complicated by the fact that smooth muscle TRPV4 is predominantly implicated in vasoconstriction, though two pools of TRPV4 that exerts opposite effects on vascular tone were identified in VSMC, which needs to be investigated further [22]. In summary, our results demonstrate that Ang II, in addition to its role in VSMC, can contribute to hypertension by inducing endothelial dysfunction via downregulation of TRPV4/eNOS/NO pathway (Fig. 6).
4.1. Limitations
The major limitation of this study is that blood pressure was not measured before or after Ang II infusion in mice. However, we do not expect any change in the blood pressure as TRPV4 appears to have contrasting roles in endothelium and VSMC [23]. We used only male mice for the remodeling studies which needs to be investigated in female mice to confirm if there is sex/gender difference in Ang II induced endothelial dysfunction and vascular remodeling.
4.2. Perspectives
Ang II has been implicated as a designated vasoconstrictor majorly through its action on VSMC via Gq/Rho/Rho kinase pathway. Although Ang II was demonstrated to inhibit TRPV4-dependent vasodilation in cerebral parenchymal arteries and mesenteric arteries, the molecular mechanisms are not known. Our findings that Ang II induce endothelial dysfunction and vascular remodeling via downregulation of TRPV4 and dependent eNOS/NO pathway uncovered hitherto VSMC independent role for Ang II in the regulation of vascular tone. This, coupled with the recent findings that TRPV4 activation inhibits Ang II signaling [7], indicates that TRPV4 acts as an endogenous regulator of Ang II and identifies Ang II-TRPV4 crosstalk as a novel target for the treatment of hypertension.
CRediT authorship contribution statement
CKT., NK., and VK., conceptualized and designed the study. NK, VK, and KD performed research, analyzed the data, and edited the manuscript. S.P. provided materials and edited the manuscript. CKT secured funding, interpreted, edited, and wrote the manuscript. The co–first authorship and the order were determined according to the time spent on this project.
Grant support
This work was supported by the National Institutes of Health (NIH) (R01HL119705, R01HL148585, and R15CA202847; American Heart Association (AHA-TPA-971237) to CKT and R01AI144115; SP).
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jmccpl.2023.100055.
Appendix A. Supplementary data
Supplementary figures
Data availability
The datasets presented in the current study are available from the corresponding author on reasonable request.
References
- 1.Tackling G., Borhade M.B. StatPearls; Treasure Island (FL): 2023. Hypertensive heart disease. [Google Scholar]
- 2.Su C., Xue J., Ye C., Chen A. Role of the central renin-angiotensin system in hypertension (review) Int J Mol Med. 2021;47(6) doi: 10.3892/ijmm.2021.4928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lu Y., Sun X., Peng L., Jiang W., Li W., Yuan H., et al. Angiotensin II-induced vascular remodeling and hypertension involves cathepsin L/V- MEK/ERK mediated mechanism. Int J Cardiol. 2020;298:98–106. doi: 10.1016/j.ijcard.2019.09.070. [DOI] [PubMed] [Google Scholar]
- 4.Schiffrin E.L. Vascular remodeling in hypertension: mechanisms and treatment. Hypertension. 2012;59(2):367–374. doi: 10.1161/HYPERTENSIONAHA.111.187021. [DOI] [PubMed] [Google Scholar]
- 5.Griendling K.K., Ushio-Fukai M., Lassegue B., Alexander R.W. Angiotensin II signaling in vascular smooth muscle. New concepts. Hypertension. 1997;29(1 Pt 2):366–373. doi: 10.1161/01.hyp.29.1.366. [DOI] [PubMed] [Google Scholar]
- 6.Canales Coutino B., Mayor R. Mechanosensitive ion channels in cell migration. Cells Dev. 2021;166 doi: 10.1016/j.cdev.2021.203683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zaccor N.W., Sumner C.J., Snyder S.H. The nonselective cation channel TRPV4 inhibits angiotensin II receptors. J Biol Chem. 2020;295(29):9986–9997. doi: 10.1074/jbc.RA120.014325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Adapala R.K., Talasila P.K., Bratz I.N., Zhang D.X., Suzuki M., Meszaros J.G., et al. PKCalpha mediates acetylcholine-induced activation of TRPV4-dependent calcium influx in endothelial cells. Am J Physiol Heart Circ Physiol. 2011;301(3):H757–H765. doi: 10.1152/ajpheart.00142.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang D.X., Mendoza S.A., Bubolz A.H., Mizuno A., Ge Z.D., Li R., et al. Transient receptor potential vanilloid type 4-deficient mice exhibit impaired endothelium-dependent relaxation induced by acetylcholine in vitro and in vivo. Hypertension. 2009;53(3):532–538. doi: 10.1161/HYPERTENSIONAHA.108.127100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ottolini M., Hong K., Cope E.L., Daneva Z., DeLalio L.J., Sokolowski J.D., et al. Local Peroxynitrite impairs endothelial transient receptor potential Vanilloid 4 channels and elevates blood pressure in obesity. Circulation. 2020;141(16):1318–1333. doi: 10.1161/CIRCULATIONAHA.119.043385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Adapala R.K., Thoppil R.J., Ghosh K., Cappelli H.C., Dudley A.C., Paruchuri S., et al. Activation of mechanosensitive ion channel TRPV4 normalizes tumor vasculature and improves cancer therapy. Oncogene. 2016;35(3):314–322. doi: 10.1038/onc.2015.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kanugula A.K., Adapala R.K., Jamaiyar A., Lenkey N., Guarino B.D., Liedtke W., et al. Endothelial TRPV4 channels prevent tumor growth and metastasis via modulation of tumor angiogenesis and vascular integrity. Angiogenesis. 2021;24(3):647–656. doi: 10.1007/s10456-021-09775-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thodeti C.K., Matthews B., Ravi A., Mammoto A., Ghosh K., Bracha A.L., et al. TRPV4 channels mediate cyclic strain-induced endothelial cell reorientation through integrin-to-integrin signaling. Circ Res. 2009;104(9):1123–1130. doi: 10.1161/CIRCRESAHA.108.192930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Adapala R.K., Katari V., Kanugula A.K., Ohanyan V., Paruchuri S., Thodeti C.K. Deletion of endothelial TRPV4 protects heart from pressure overload-induced hypertrophy. Hypertension. 2023;80(11):2345–2356. doi: 10.1161/HYPERTENSIONAHA.123.21528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shukla A.K., Kim J., Ahn S., Xiao K., Shenoy S.K., Liedtke W., et al. Arresting a transient receptor potential (TRP) channel: beta-arrestin 1 mediates ubiquitination and functional down-regulation of TRPV4. J Biol Chem. 2010;285(39):30115–30125. doi: 10.1074/jbc.M110.141549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ma Z., Viswanathan G., Sellig M., Jassal C., Choi I., Garikipati A., et al. beta-Arrestin-mediated angiotensin II type 1 receptor activation promotes pulmonary vascular remodeling in pulmonary hypertension. JACC Basic Transl Sci. 2021;6(11):854–869. doi: 10.1016/j.jacbts.2021.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nishijima Y., Zheng X., Lund H., Suzuki M., Mattson D.L., Zhang D.X. Characterization of blood pressure and endothelial function in TRPV4-deficient mice with l-NAME- and angiotensin II-induced hypertension. Physiol Rep. 2014;2(1) doi: 10.1002/phy2.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sonkusare S.K., Bonev A.D., Ledoux J., Liedtke W., Kotlikoff M.I., Heppner T.J., et al. Elementary Ca2+ signals through endothelial TRPV4 channels regulate vascular function. Science. 2012;336(6081):597–601. doi: 10.1126/science.1216283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Filosa J.A., Yao X., Rath G. TRPV4 and the regulation of vascular tone. J Cardiovasc Pharmacol. 2013;61(2):113–119. doi: 10.1097/FJC.0b013e318279ba42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marrelli S.P., O’Neil R G., Brown R.C., Bryan R.M., Jr. PLA2 and TRPV4 channels regulate endothelial calcium in cerebral arteries. Am J Physiol Heart Circ Physiol. 2007;292(3):H1390–H1397. doi: 10.1152/ajpheart.01006.2006. [DOI] [PubMed] [Google Scholar]
- 21.Jeremy J.Y., Rowe D., Emsley A.M., Newby A.C. Nitric oxide and the proliferation of vascular smooth muscle cells. Cardiovasc Res. 1999;43(3):580–594. doi: 10.1016/s0008-6363(99)00171-6. [DOI] [PubMed] [Google Scholar]
- 22.Liu L., Guo M., Lv X., Wang Z., Yang J., Li Y., et al. Role of transient receptor potential Vanilloid 4 in vascular function. Front Mol Biosci. 2021;8 doi: 10.3389/fmolb.2021.677661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen Y.L., Daneva Z., Kuppusamy M., Ottolini M., Baker T.M., Klimentova E., et al. Novel smooth muscle Ca(2+)-signaling nanodomains in blood pressure regulation. Circulation. 2022;146(7):548–564. doi: 10.1161/CIRCULATIONAHA.121.058607. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary figures
Data Availability Statement
The datasets presented in the current study are available from the corresponding author on reasonable request.