

Abstract
We reviewed recent major clinical trials with investigational drugs for the treatment of subjects with neurodegenerative diseases caused by inheritance of gene mutations or associated with genetic risk factors. Specifically, we discussed randomized clinical trials in subjects with Alzheimer’s disease, Huntington’s disease and amyotrophic lateral sclerosis bearing pathogenic gene mutations, and glucocerebrosidase-associated Parkinson’s disease. Learning potential lessons to improve future therapeutic approaches is the aim of this review. Two long-term, controlled trials on three anti-β-amyloid monoclonal antibodies (solanezumab, gantenerumab and crenezumab) in subjects carrying Alzheimer’s disease-linked mutated genes encoding for amyloid precursor protein or presenilin 1 or presenilin 2 failed to show cognitive or functional benefits. A major trial on tominersen, an antisense oligonucleotide designed to reduce the production of the huntingtin protein in subjects with Huntington’s disease, was prematurely interrupted because the drug failed to show higher efficacy than placebo and, at highest doses, led to worsened outcomes. A 28-week trial of tofersen, an antisense oligonucleotide for superoxide dismutase 1 in patients with amyotrophic lateral sclerosis with superoxide dismutase 1 gene mutations failed to show significant beneficial effects but the 1-year open label extension of this study indicated better clinical and functional outcomes in the group with early tofersen therapy. A trial of venglustat, a potent and brain-penetrant glucosylceramide synthase inhibitor, in Parkinson’s disease subjects with heterozygous glucocerebrosidase gene mutations revealed worsened clinical and cognitive performance of patients on the enzyme inhibitor compared to placebo. We concluded that clinical trials in neurodegenerative diseases with a genetic basis should test monoclonal antibodies, antisense oligonucleotides or gene editing directed against the mutated enzyme or the mutated substrate without dramatically affecting physiological wild-type variants.
Key Words: Alzheimer’s disease, amyotrophic lateral sclerosis, amyloid precursor protein, glucocerebrosidase, huntingtin, Huntington’s disease, Parkinson’s disease, presenilin 1, presenilin 2, superoxide dismutase 1
Introduction
Advances in the treatment of more than 4000 known monogenic mutations depend on the development of causal therapies that use the transfer of DNA and/or RNA to modify gene expression in order to correct or compensate for an abnormal phenotype. Biological strategies include the use of patients-derived somatic or induced pluripotent stem cells, gene or mRNA transfer and genome editing (CRISPR/Cas9). Despite the apparent efficacy of these technologies in experimental animal models of hereditary genetic disorders, their successful use in the clinic is a huge challenge (O’Connor and Crystal, 2006). The main biological barriers to all genetic treatment approaches are to deliver and maintain new genetic information, except for genome editing that is stable and genetically transmissible. Overcoming these obstacles requires a full understanding of the molecular basis of the disorder, its mode of inheritance, the type of mutations and genotype-phenotype relationships that result in the disease phenotype. In 2017, the first gene therapy (voretigene neparvovec-rzyl) was approved by the Food and Drug Administration (FDA) for the treatment of inherited retinal dystrophy. In 2019, another gene therapy (onasemnogene abeparvovec-xioi) was approved for spinal muscular atrophy. Both therapies used adeno-associated virus vectors. A number of chimeric antigen receptor T cells genetically engineered to produce an artificial T cell receptor capable of recognizing specific tumor-associated antigens have been approved for treating acute lymphoblastic leukemia (2017), relapsed or refractory large B-cell lymphoma (2017), mantle cell lymphoma (2020) and certain types of large B-cell lymphoma (2021). Genome editing using CRISPR/Cas9 technology has been applied in several genetic disorders including Duchenne muscular dystrophy, hereditary tyrosinemia, cystic fibrosis, β-thalassemia, urea cycle disorders and cataract. To date, the FDA has approved 25 cellular and gene therapy products and received more than 900 applications to investigate gene therapy in clinical trials (US Food and Drug Administration, 2022). Treatment of genetic disorders is also pursued with the use of antisense oligonucleotides, small interfering RNA, RNA aptamers, monoclonal antibodies, enzyme replacement therapies, and selective enzyme inhibitors.
We reviewed recent important results of clinical trials with investigational drugs for the treatment of subjects with neurodegenerative diseases caused by inheritance of gene mutations or associated to genetic risk factors. Specifically, we will discuss major recent trials in subjects with Alzheimer’s disease (AD), Huntington’s disease (HD) and amyotrophic lateral sclerosis (ALS) with genetic origin and glucocerebrosidase (GBA)-associated Parkinson’s disease (PD). Herein, we attempt to learn potential lessons to improve future therapeutic approaches.
Search Strategy and Selection Criteria
We reviewed double-blind, placebo-controlled clinical studies carried out in the last 3 years (2020–2022) in AD, HD, ALS and PD subjects using PubMed, meeting abstracts and ClinicalTrials.gov. Only controlled clinical trials in subjects with disease-related gene mutations were considered. Pilot studies (< 40 subjects) were excluded. We found two publications in AD subjects with APP or PSEN1 or PSEN2 gene mutations, four publications in HD, two publications in ALS subjects with SOD1 gene mutations and three publications in PD subjects with GBA gene mutations. In the final analysis, we discussed three randomized controlled trials on crenezumab, solanezumab and gantenerumab in AD, two trials on tominersen in HD, two trials on toferesen in ALS and three trials on venglusat in PD.
Alzheimer’s Disease
AD is a progressive neurodegenerative disease biologically defined by the presence of β-amyloid (Aβ)-containing extracellular plaques and tau-containing intracellular neurofibrillary tangles. AD represents the most common cause of cognitive impairment in middle-aged and elderly people (Scheltens et al., 2021). As of 2020, approximately 50 million people are living with dementia worldwide, and this number is estimated to increase to 152 million by 2050. Typical clinical features of the disease are early and prominent episodic memory loss, with progressive and variable involvement of other cognitive domains (visuospatial and executive functions, and language). Diagnosis relies on emerging cerebrospinal fluid (CSF) and imaging biomarkers that can detect the key neuropathologic hallmarks of the disease in living people, allowing for the biological characterization of the patients. There are two forms of AD: one is sporadic, generally occurs after 65 years of age and represents the vast majority of cases (> 95%). The other form is familial and is caused by point mutations of the APP gene, which encodes for amyloid precursor protein (APP), or the PSEN1 and PSEN2 genes, which encode for presenilin 1 (PS1) and presenilin 2 (PS2), two enzymes involved in the processing of APP. APP is initially cleaved by the β-secretase at the β-cleavage site and then by the γ-secretase complex within the transmembrane domain. The γ-secretase complex consists of Aph1, nicastrin, presenilin (PS1 and PS2) and Pen-2, which generates the Aβ peptide and the APP intracellular domain. The AD pathogenic alterations are mostly identified in the PSEN1 gene, while mutations in PSEN2 and APP are less frequently observed. No treatments stop or reverse the disease progression, though some may temporarily improve symptoms (Scheltens et al., 2021). Several monoclonal antibodies directed against Aβ have been developed (Panza et al., 2019).
Crenezumab is a monoclonal antibody that preferentially binds to oligomeric forms of Aβ (Yang et al., 2019). In June 2022, Genentech announced (Hurdle, 2022) the failure of an AD prevention study on crenezumab in 252 cognitively unimpaired subjects carrying the autosomal dominant form of the disease (Tariot et al., 2018). Participants in the prevention study were carriers of a presenilin 1 mutation (PS1 E280A) and received either crenezumab or placebo for at least 5 years. Compared with placebo, crenezumab did not significantly improve either cognitive or clinical primary or secondary measures. This setback follows the cognitive and clinical failure of another AD prevention study on two other anti-Aβ monoclonal antibodies, solanezumab, a monoclonal antibody selective for soluble monomeric Aβ, and gantenerumab, a monoclonal antibody selective for fibrillar Aβ, in 142 pre-symptomatic subjects carrying PSEN1 or APP autosomal dominant mutations which received monoclonal antibodies or placebo for at least 4 years (Salloway et al., 2021).
The reasons for the failure of crenezumab in subjects with PSEN1 mutations are not clear but two recent studies (CREAD and CREAD2) showed that the drug has no beneficial effects also in subjects with early sporadic AD (Ostrowitzki et al., 2022). In these studies, there was evidence of target engagement by crenezumab since total Aβ42 and Aβ40 levels in plasma and CSF were significantly increased after crenezumab administration. However, there were no significant differences between treatment groups in amyloid PET burden and CSF levels of Aβ oligomers, total tau, phosphorylated tau-181, and other biomarkers studied. Interestingly, at week 53 there was a greater increase in tau PET standardized uptake value ratio in the crenezumab arm compared to placebo. Finally, in a subgroup analysis, crenezumab significantly worsened compared to placebo activities of daily living as measured with the Alzheimer’s Disease Cooperative Study-Activities of Daily Living scale in the subjects with prodromal AD (n = 734) which represented 48% of the studied population. Although crenezumab was designed to target Aβ oligomers (Meilandt et al., 2019), the drug binds also to Aβ monomers, with about 10-fold lower affinity (Ultsch et al., 2016). This means, that in humans the 10-fold selectivity of crenezumab for oligomeric Aβ is not biological relevant and the drug binds to Aβ monomers without affecting oligomeric species. Indeed, in the CREAD studies the baseline CSF concentrations of oligomeric Aβ were very low (2–3 pg/mL) compared to the total CSF Aβ42 levels (570–600 pg/mL). At the end of treatment, the crenezumab and placebo treatments had a negligible effect on Aβ oligomers (–0.93 ± 0.58 pg/mL and –0.22 ± 0.53 pg/mL, respectively), while the effect of the monoclonal antibody on total CSF Aβ42 levels (+281 ± 32 pg/mL) was marked when compared to that of placebo (–1.03 ± 29.70 pg/mL) (Ostrowitzki et al., 2022).
Despite clinical failures of crenezumab, solenezumab and gantenerumab both in familial and sporadic AD, other anti-Aβ monoclonal antibodies targeting aggregated Aβ forms have shown encouraging results. In June 2021, the US Food and Drug Administration accelerated the approval for the anti-Aβ antibody aducanumab for the treatment of early AD (Dunn et al., 2021) after two interrupted studies demonstrated a reduction in Aβ plaques and signals of potential cognitive and clinical efficacy (Budd Haeberlein et al., 2022). Although the FDA decision was widely contested (Nisticò and Borg, 2021), other two potent anti-Aβ monoclonal antibodies, donanemab (Mintun et al., 2021) and lecanemab (Swanson et al., 2021) have shown profound reducing effects in clearing brain amyloid plaques and encouraging results in Phase 2 studies in subjects with sporadic early AD. Importantly, a recent press release by Eisai Co., Ltd. (Tokyo, Japan) and Biogen Inc. (Cambridge, MA, USA) (Hurdle, 2022) have announced positive cognitive and clinical results in a large 18-month study with lecanemab in almost 1800 patients with early AD (CLARITY AD) that could lead to the approval of the drug (Prillaman, 2022). Lecanemab is being also tested in a 4-year prevention study (AHEAD 3–45 study) in 1400 cognitively normal individuals with intermediate and high brain plaque load (Rafii et al., 2022). It has been hypothesized that potent anti-Aβ monoclonal antibodies while dissolving brain Aβ plaques can release monomeric Aβ42 from the plaques and this is reflected in an increase in CSF Aβ42 levels (Imbimbo et al., 2022). The failure of crenezumab, solanezumab and gantenerumab in patients with familial or sporadic AD could be due to their weak effects on brain plaques and thus their inability to increase brain monomeric Aβ42 levels at physiological levels. Indeed, solanezumab studies in sporadic AD patients have shown a significant increase compared to baseline in total CSF Aβ42 levels in drug-treated patients (63–71%) while free monomeric Aβ42 levels decreased by 7–17% (Willis et al., 2018).
Huntington’s Disease
HD is the most common monogenic neurological disorder in the western world, with an estimated prevalence of 5.7 cases per 100,000 persons (Bates et al., 2015). HD is an autosomal dominant progressive neurodegenerative disorder with a cytosine-adenine-guanine (CAG) trinucleotide repeat expansion in the huntingtin gene (HTT) on chromosome 4p. Mutant huntingtin is produced with abnormally long polyglutamine sequences, and it is thought to cause toxic gains of function and protein fragmentation that lead to neuronal dysfunction and death. HD brain pathology mainly includes atrophy of the caudate and putamen. Symptoms generally appear between 30 and 50 years, with an inverse correlation between the number of CAG repeats in the HTT gene and age at onset. Subjects with 40 or more CAG repeats have complete penetrance, while individuals with 36–39 CAG repeats have incomplete penetrance. Generally, symptoms do not manifest in individuals with fewer than 36 CAG repeats. Clinically, HD is characterized by movement disorders (especially chorea), cognitive impairment and neuropsychiatric features. Symptoms begin insidiously, with a slow but relentless deterioration in cognitive and motor function. The average length of survival after clinical onset ranges from 10 to 20 years. Diagnosis of HT is based on genetic testing. There is no cure for HD and full-time care is required in the later stages of the disease. Symptomatic treatments attenuate disease burden and improve quality of life. Tetrabenazine is used to treat movement disorders. Present research is directed to the full understanding of the disease mechanisms, the development of predictive animal models, the clinical testing of drug candidates, and the development of disease-modifying approaches like cell-based therapies for replacing damaged or lost neurons (Ross and Tabrizi, 2011).
Tominersen is an antisense oligonucleotide (ASO) designed to reduce the production of the huntingtin protein, which is believed to be the cause of HD. Optimism around the drug soared after a 28-week Phase I/II trial (NCT02519036) in 46 HD patients, because tominersen significantly reduced levels of mutant huntingtin in the CSF (Tabrizi et al., 2019). In March 2021, Roche prematurely interrupted a 2-year, Phase III trial (NCT03761849) on tominersen in 899 patients with HD following a planned review of the data that indicated that the potential benefits of the drug did not outweigh its risks (Kwon, 2021). The study tested two tominersen dose regimens: 120 mg given either every 8 weeks or every 16 weeks. Roche reported that after 69 weeks, participants on the highest tominersen dose regimen (120 mg per 8 weeks) experienced a more marked motor function and cognitive decline than those in the placebo group. Participants in the lowest dose regimen (120 mg per 16 weeks) did not show overall benefit compared with those receiving placebo. Participants in both tominersen-treated groups also experienced larger increases in brain ventricle volumes than did those who received a placebo. Tominersen suppressed the production of the mutant form of huntingtin but also of the healthy wild-type, and this could have caused neurotoxicity. One week after the interruption of the tominersen trial, Wave Life Sciences revealed that it would discontinue the development of two of its ASOs that were in Phase I/II clinical development in HD (Kwon, 2021).
Amyotrophic Lateral Sclerosis
ALS is the most common form of motor neuron disease (Hardiman et al., 2017). ALS may initially occur in any body segment (bulbar, cervical, thoracic, or lumbosacral), manifested as upper motor neuron (spasticity, slowness of movement, increased tendon reflexes) or lower motor neuron (weakness, reduced reflexes, muscle atrophy, fasciculations, and cramps) symptoms or signs. Approximately 70% of patients develop spinal-onset disease, mainly characterized by asymmetric limb weakness, while around 25% of patients suffer from bulbar-onset disease, mainly characterized by dysarthria and dysphagia. Up to 50% of ALS patients present with cognitive impairment during the course of disease, and 13% of them suffer from concomitant behavioral variant frontotemporal dementia. The global incidence of ALS is 2–3 per 100,000 individuals, with a lifetime risk of ALS in western countries of 1 in 350 for men and 1 in 400 for women. The disease can affect people at any age, but usually starts around 60 years or at 50 years in inherited cases. The average survival is 2–4 years and death is usually due to respiratory failure. Most cases of ALS (90–95%) are classified as sporadic ALS with no known cause. The remaining 5–10% of cases have a genetic cause and these are classified as familial ALS. About 2% of ALS cases are linked to mutations of SOD1, an antioxidant enzyme whose activity is preserved in most mutant forms. Mutant SOD1 protein forms toxic intracellular aggregates. In sporadic ALS, cytoplasmic aggregates of wild-type SOD1 are also observed. It is believed that misfolded forms of mutant SOD1 can cause misfolding and aggregation of wild-type SOD1 in adjacent neurons in a prion-like propagation fashion. There are no disease-modifying treatments for ALS. The available symptomatic treatments are aimed to attenuate clinical burden. Riluzole may prolong median survival by 2–3 months. Non-invasive ventilation may prolong survival and improve quality of life. Mechanical ventilation can prolong survival without affecting disease progression (Brown and Al-Chalabi, 2017).
Tofersen is an ASO that can reduce SOD1 protein synthesis via modulating the degradation of SOD1 messenger RNA. In a dose-ranging Phase I/II study on tofersen in 50 patients with ALS due to SOD1 mutations, the highest drug dose of 100 mg given intrathecally over a period of 12 weeks decreased significantly, compared to baseline, CSF SOD1 levels by 33% (Miller et al., 2020). In June 2022, Biogen announced encouraging clinical results from the 12-month open label extension phase of a 28-week, double-blind, placebo-controlled study on tofersen in patients with ALS associated with SOD1 mutations (Kos et al., 2022). The original 28-week, double-blind, placebo-controlled Phase III study evaluated the effects of tofersen 100 mg per day in 108 patients, in which 72 received tofersen (faster progression predicted in 39) and 36 received placebo (faster progression predicted in 21). In faster-progressors, tofersen triggered greater reductions than placebo in concentrations of SOD1 in CSF (–29% vs. +16%) and of neurofilament light chains (a marker of neuronal damage) in plasma (–60% vs. +20%). In the faster-progression subgroup (primary analysis), the change to week 28 in the Revised Amyotrophic Lateral Sclerosis Functional Rating Scale (ALSFRS-R) score was –6.98 with tofersen and –8.14 with placebo (difference, 1.2 points; 95% confidence interval [CI], –3.2 to 5.5; P = 0.97) (Miller et al., 2022). Ninety-five participants entered the open label extension phase of the study with a median exposure to tofersen of approximately 20 months. The 12-month open label extension data made a comparison between early initiation of tofersen and delayed initiation of tofersen (6 months later). Over 12 months in the overall study population, the group with early therapy showed better clinical outcomes as measured by the improvement in the ALSFRS-R (difference of 3.5 points; 95% CI: 0.4 to 6.7), in the respiratory function as measured by slow vital capacity (difference of 9.2 percent-predicted; 95% CI: 1.7 to 16.6) and in the muscle strength as measured by the handheld dynamometry megascore (difference of 0.28; 95% CI: 0.05 to 0.52). Early survival data suggested a lower risk of death or pulmonary ventilation (hazard ratio: 0.36; 95% CI: 0.14 to 0.94) and death (hazard ratio: 0.27; 95% CI: 0.08 to 0.89) with earlier initiation of tofersen. The 12-month results indicated sustained reductions in total SOD1 protein and neurofilament light chains (Miller et al., 2022).
Parkinson’s Disease
PD is the second-most common neurodegenerative disorder affecting 2–3% of the population aged 65 years or above (Poewe et al., 2017). The disease is mainly characterized by the progressive loss of dopaminergic neurons in the substantia nigra pars compacta. Histologically, the disease is characterized by the presence of intra-neuronal Lewy body inclusions containing α-synuclein aggregates. Core clinical features of PD include bradykinesia, rigidity, resting tremor and postural instability. A spectrum of non-motor symptoms has also been described, including cognitive impairment, autonomic dysfunction, disorders of sleep, neuropsychiatric symptoms and hyposmia; some of these symptoms can precede by years the onset of classic motor symptoms. PD is diagnosed clinically, but neuroimaging techniques such as dopamine transporter SPECT are helpful in differential diagnosis distinguishing between essential tremor and tremor from parkinsonian syndromes. Pharmacological treatment of PD is based on dopamine replacement therapy to improve disease symptoms. Levodopa is the gold standard and it is administered with a levodopa decarboxylase inhibitor (carbidopa) to reduce its peripheral breakdown and to improve efficacy. Other available drugs include dopamine agonists, catechol-O-methyltransferase inhibitors, monoamine oxidase type B inhibitors, an N-methyl-D-aspartate antagonist, and anticholinergic medications. Innovative surgical therapies such as deep brain stimulations may be used when motor symptoms persist despite optimal medical management and for those developing intractable levodopa-related motor complications (Poewe et al., 2017). α-Synuclein is a small cytoplasmic protein that can misfold and form aggregated polymers, which are a major component of Lewy bodies and Lewy neurites. Rare genetic mutations of the SNCA gene encoding α-synuclein lead to autosomal dominantly inherited PD (Singleton et al., 2003). α-Synuclein is highly expressed in the brain and its physiological roles include vesicular transport and neurotransmitter release, including the release of dopamine (Bridi and Hirth, 2018). A proposed mechanism for the progressive nature of PD is the propagation of the misfolded oligomeric α-synuclein from neuron to neuron in a prion-like fashion (Volpicelli-Daley and Brundin, 2018). Targeting intra- and extra-neuronal α-synuclein aggregates has been proposed as a disease-modifying treatment of PD (Fields et al., 2019). Mutations in the GBA gene, encoding the lysosomal enzyme glucocerebrosidase (GBA), increase the risk of developing PD (Migdalska-Richards and Schapira, 2016). There are 7–10% of patients with PD carrying a GBA mutation and presenting with reduced GBA activity, earlier disease onset, and more rapid cognitive decline. It is believed that GBA loss of function triggers accumulation of its substrate, glucosylceramide (GL-1), which accelerates the formation of toxic α-synuclein oligomers (Sardi et al., 2018). High plasma levels of GL-1 are associated with impaired cognition in PD patients and have been proposed as a marker of toxic α-synuclein aggregates in the brain (Lerche et al., 2021). In animal models of synucleinopathy, the use of a brain-penetrant glucosylceramide synthase inhibitor reduced α-synuclein aggregation in the hippocampus (Sardi et al., 2017).
Venglustat is a potent, CNS-penetrant, glucosylceramide synthase inhibitor that reduces GL-1 production. In a 2-week, Phase I study involving 55 healthy individuals, venglustat was found to be safe and well tolerated and dose-dependently reduced plasma GL-1 levels (Peterschmitt et al., 2021a). A Phase IIa study in 29 PD patients with GBA mutations confirmed the ability of venglustat to dose-dependently inhibit GL-1 levels in plasma and also in CSF (Peterschmitt et al., 2022). A 1-year, Phase II study (MOVES-PD) randomized 221 participants with PD and heterozygous GBA mutations to a 1-year regimen of once-daily venglustat or placebo, plus 2 years of follow-up (Peterschmitt, 2021b). The primary endpoint was the Unified Parkinson’s Disease Rating Scale (UPRDS) Part II and III; secondary outcomes were the PD Cognitive Rating Scale, UPRDS all parts, and Hoehn and Yahr score. In March 2021, Sanofi announced the negative results of the MOVES-PD study with the primary endpoint of the MDS-UPDRS Part II and III worsening more with venglustat than placebo. A similar trend was seen for the MDS-UPRDS total score and for the PD Cognitive Rating Scale. Target engagement was deemed successful. There was an approximately 75% reduction in glucosylceramide GL-1 levels in plasma and CSF within 2 weeks after administration and the levels remained within normal limits during the study. A decrease in α-synuclein in CSF was also documented in patients receiving venglustat (Peterschmitt et al., 2021b).
Lessons for Potential Future Approaches
The encouraging clinical results obtained in the open label extension phase on the study of tofersen in patients with ALS and SOD1 mutations may be informative for other CNS degenerative conditions with genetic basis. SOD1 genetic variants are generally determined by heterozygous mutations inherited in an autosomal dominant manner. These mutations involve one allele, leaving partial production of the wild-type SOD1 by the normal allele still possible. Mutated SOD1 tends to be a little more unstable than the wild-type, therefore patients usually have more wild-type SOD1 than mutated SOD1. It is believed that the toxicity of mutant SOD1 might arise from an initial misfolding reducing nuclear protection from the active enzyme (Sau et al., 2007). In the tofersen trial in ALS, a rational approach has been adopted: a drug (ASO) decreased the levels of the toxic mutated enzyme (mSOD1) but did not affect either the substrate of the enzyme (superoxide) or the metabolite (hydrogen peroxide) produced by the enzyme. In the 28-week, double-blind, placebo-controlled trial, tofersen dose-dependently lowered SOD1 levels in CSF with a 36% average decrease compared baseline on the 100-mg dose vs. 3% decrease with placebo (Miller et al., 2022). No signals of toxicity were detected in this study. Compared to placebo, the 100-mg dose showed positive trends with less decline in the ALSFRS-R score (–1.19 vs. –5.63 points), the percentage of predicted slow vital capacity (–7.08 vs. –14.46 percentage points) and the handheld dynamometry megascore (–0.03 vs. –0.26 points). Compared to baseline, at day 85 the concentrations of neurofilament light chains in plasma and CSF were largely unchanged in the placebo group, whereas they were decreased in the 100-mg group. The results of the 12-month open label extension confirmed the positive clinical and biological trends in favor of the 100-mg dose observed in the initial 28-week double-placebo, controlled trial. The delayed-start design of the study suggests that tofersen may have disease-modifying properties although this hypothesis needs to be confirmed in a larger cohort study.
The clinical failure of the 69-week, Phase II/III trial of tominersen in HD is not a complete surprise. The results of the previous 4-week, Phase I/II study have evidenced neurotoxicity signals. Mean brain ventricular volume showed dose-dependent and time-dependent increases on days 113 and 197, i.e. 4 and 16 weeks after the last intrathecal administration (day 85). Elevations of the concentration of neurofilament light protein in CSF occurred in some patients in the 90-mg and 120-mg cohorts at day 113 or day 141, again 4 and 8 weeks after cessation of the regimen (Tabrizi et al., 2019). These long-term neurotoxicity signals might reveal structural detrimental effects of the drug of the brain. The dose-dependent neurotoxicity signals were again observed in the Phase III study and led to the premature interruption of the trial. Several factors could have contributed to tominersen’s failure. The drug suppresses production of the healthy, as well as the mutant form of huntingtin, and this could have caused problems. In the Phase I/II study we know that tominersen caused dose-dependent decreases compared to baseline in the CSF concentration of mutant huntingtin in CSF at the 4-week post-dose sampling point of –20%, –25%, –28%, –42%, and –38% in the 10-, 30-, 60-, 90-, and 120-mg dose groups, respectively. Unfortunately, we do not know the extent of the drug’s inhibition of physiological wild-type huntingtin.
The apparent clinical success of tofersen in ALS patients with mutated SOD1 suggests that we may treat patients with autosomal dominant familial AD with an ASO or a monoclonal antibody specific for the mutated enzyme (mPSEN1 or mPSEN2) or mutated APP (mAPP) rather than targeting the metabolite of wild-type APP (Aβ). It has been shown that pathogenic PSEN1 mutations may inhibit also wild-type presenilin function, suppressing γ-secretase-dependent cleavage of APP and Notch. Surprisingly, mutant PSEN1 could stimulate production of Aβ42 by wild-type PSEN1 while decreasing its production of Aβ40 (Heilig et al., 2013). Thus, targeting mutant PSEN1 gene in patients with inherited autosomal dominant AD appears a rational approach. Indeed, recent studies have showed that CRISPR-Cas9 gene editing technology is able to selectively disrupt PSEN1 mutations leading to an autosomal dominant form of early-onset AD and counteract the AD-associated phenotype (Konstantinidis et al., 2022).
The anti-α-synuclein therapeutic approach in PD started in 2003 when Braak et al. (2003), after examining the brains of patients with PD and individuals with incidental Lewy pathology, proposed a unifying theory according to which α-synuclein pathology progressed through different stages and propagated in different brain areas of affected individuals. This neuropathology study made the role of α-synuclein central in the development of neurodegenerative Parkinsonism. Almost 20 years later, two Phase II trials of humanized monoclonal anti-α-synuclein antibodies in PD have been reported: one with cinpanemab, which binds to the N-terminal end of α-synuclein, and prasinezumab the C-terminal. The SPARK trial tested intravenous monthly administrations of cinpanemab (250, 1250, or 3500 mg) vs. placebo for 1 year (Lang et al., 2022) whereas the PASADENA trial tested intravenous monthly administrations of prasinezumab (1500 mg or 4500 mg doses) vs. placebo for 1 year (Pagano et al., 2022). In both trials, the primary endpoint was the total Movement Disorders Society-Unified Parkinson Disease Rating Scale (MDS-UPDRS) score at 52 weeks. There was no dose-response change or separation from placebo with either approach. Unfortunately, CSF biomarkers, including α-synuclein, were not measured. Thus, target engagement could not be verified. Nevertheless, both monoclonal antibodies were shown to markedly bind to aggregated α-synuclein in preclinical studies. In Phase 1 studies, prasinezumab dose-dependently reduced from baseline free serum α-synuclein levels in subjects with PD (Jankovic et al., 2018). In a Phase 1 study involving healthy volunteers and participants with PD, cinpanemab–α-synuclein complexes were found in the plasma suggesting dose-dependent biologic activity in subjects with PD (Brys et al., 2019). The negative SPARK and PASADENA studies indicate that the general reduction of a physiological protein like α-synuclein in patients with sporadic PD does not produce clinical benefits. These negative results mimic the negative outcome of the indirect approach of lowering α-synuclein levels with venglustat in PD subjects with heterozygous GBA mutations. It is important to point out that in general in neurologic gene defects, the mutations are in heterozygous form and therefore both the mutated form of the protein and the wild-type form are present in the organism. In these cases, a drug targeting the mutated protein may also affect the wild-type physiological form. When the mutation is in homozygous form, only the mutated form of the protein exists, generally with important clinical consequences. On the other hand, there are also dominant mutations, which cause negative clinical effects even in heterozygous conditions. Ideally, drugs targeting a specific mutation should be initially tested in homozygous subjects to have a higher chance to target the pathological protein. This is the strategy that is being pursued in AD. A 1-year, open label, Phase 1 study (NCT03634007) in 15 AD APOE ε4/ε4 carriers with evidence of brain amyloid accumulation is presently assessing the safety and tolerability of intracisternal AAV-mediated delivery of a human APOE2 cDNA sequence (AAVrh.10hAPOE2).
In conclusion, the results of recent clinical trials in neurodegenerative diseases with a genetic basis suggest that selective monoclonal antibodies, antisense oligonucleotides or gene editing should be directed against the mutated enzyme or the mutated substrate without dramatically affecting physiological wild-type variants (Figure 1).
Figure 1.
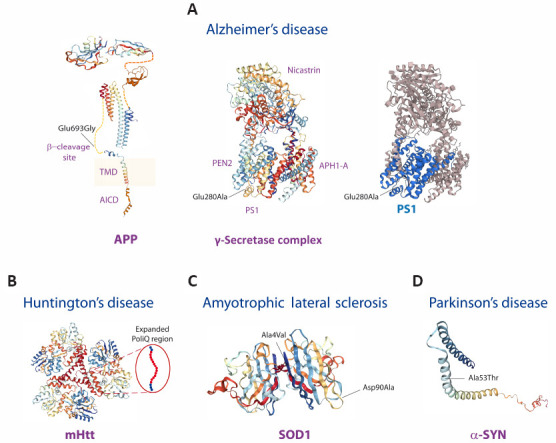
The 3D structures of amyloid precursor protein (APP), presenilin 1 (PS1), huntingtin (Htt), superoxide dismutase (SOD1), and α-synuclein (α-SYN) proteins are shown.
Examples of protein mutations underlying familial dominant forms of Alzheimer’s disease (A), Huntington’s disease (B), amyotrophic lateral sclerosis (C), and Parkinson’s disease (D) are shown (for structure rendering see Additional file 1 (144.6KB, pdf) ). (A) In Alzheimer’s disease, there are hundreds of point missense mutations of APP, PS1 and PS2 proteins. As example, the Glu693Gly mutation of APP and the Glu280Ala mutation of PS1 are shown. APP is initially cleaved by the β-secretase at the β-cleavage site and then by the γ-secretase complex within the transmembrane domain (TMD). The γ-secretase complex is formed by the sequential assembly of Aph1, nicastrin, presenilin (PS1 and PS2) and Pen-2 and generates the Aβ peptide and the APP intracellular domain. Most of the AD pathogenic alterations are identified in the PSEN1 gene, while mutations in PSEN2 and APP are less frequently observed. (B) In Huntington’s disease, the genetic expansion of the CAG triplet repeat of the HTT gene leads to extra glutamines in the poly-Q region near the N-terminus of the Htt protein. (C) In amyotrophic lateral sclerosis, more than 180 SOD1 variants exist. As example, the Ala4Val and Asp90Ala mutations of SOD1 are shown. The Ala4Val mutation is the most common SOD1-ALS mutation and it is localized within the dimer interface region and is frequent in the North American population and it is associated with an early onset, and short duration of the disease. The Asp90Ala mutation is diffused in the Scandinavia and it has a longer post-presentation survival. (D) In Parkinson’s disease, there are several α-synuclein mutations. As example, the Ala53Thr mutation at the α-synuclein N-terminus is shown. This missense mutation is associated with an aggressive early onset familial Parkinson’s disease form.
Additional file:
Additional file 1 (144.6KB, pdf) : Structure rendering.
Footnotes
Author contributions: BPI, CI and RN designed and wrote the manuscript. VT helped to search the literature and prepared the figure. All authors have read and approved the final manuscript.
Conflicts of interest: BPI is an employee at Chiesi Farmaceutici. He is listed among the inventors of a number of Chiesi Farmaceutici’s patents of anti-Alzheimer drugs. Viviana Triaca, Camillo Imbimbo and Robert Nisticò have no conflict of interest to declare.
Availability of data and materials: All data generated or analyzed during this study are included in this published article and its supplementary information files.
C-Editors: Zhao M, Liu WJ, Wang L; T-Editor: Jia Y
References
- 1.Bates GP, Dorsey R, Gusella JF, Hayden MR, Kay C, Leavitt BR, Nance M, Ross CA, Scahill RI, Wetzel R, Wild EJ, Tabrizi SJ. Huntington disease. Nat Rev Dis Primers. (2015);1:15005. doi: 10.1038/nrdp.2015.5. [DOI] [PubMed] [Google Scholar]
- 2.Braak H, Del Tredici K, R¨ub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging. (2003);24:197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- 3.Bridi JC, Hirth F. Mechanisms of α-synuclein induced synaptopathy in Parkinson's disease. Front Neurosci. (2018);12:80. doi: 10.3389/fnins.2018.00080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brys M, Fanning L, Hung S, Ellenbogen A, Penner N, Yang M, Welch M, Koenig E, David E, Fox T, Makh S, Aldred J, Goodman I, Pepinsky B, Liu Y, Graham D, Weihofen A, Cedarbaum JM. Randomized phase I clinical trial of anti-α-synuclein antibody BIIB054. Mov Disord. (2019);34:1154–1163. doi: 10.1002/mds.27738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brown RH, Al-Chalabi A. Amyotrophic lateral sclerosis. N Engl J Med. (2017);377:162–172. doi: 10.1056/NEJMra1603471. [DOI] [PubMed] [Google Scholar]
- 6.Budd Haeberlein S, Aisen PS, Barkhof F, Chalkias S, Chen T, Cohen S, Dent G, Hansson O, Harrison K, von Hehn C, Iwatsubo T, Mallinckrodt C, Mummery CJ, Muralidharan KK, Nestorov I, Nisenbaum L, Rajagovindan R, Skordos L, Tian Y, van Dyck CH, et al. Two randomized phase 3 studies of aducanumab in early Alzheimer's disease. J Prev Alzheimers Dis. (2022);9:197–210. doi: 10.14283/jpad.2022.30. [DOI] [PubMed] [Google Scholar]
- 7.Dunn B, Stein P, Cavazzoni P. Approval of aducanumab for Alzheimer disease—the FDA's perspective. JAMA Intern Med. (2021);181:1276–1278. doi: 10.1001/jamainternmed.2021.4607. [DOI] [PubMed] [Google Scholar]
- 8.Fields CR, Bengoa-Vergniory N, Wade-Martins R. Targeting alpha-synuclein as a therapy for Parkinson's disease. Front Mol Neurosci. (2019);12:299. doi: 10.3389/fnmol.2019.00299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hardiman O, Al-Chalabi A, Chio A, Corr EM, Logroscino G, Robberecht W, Shaw PJ, Simmons Z, van den Berg LH. Amyotrophic lateral sclerosis. Nat Rev Dis Primers. (2017);3:17071. doi: 10.1038/nrdp.2017.71. [DOI] [PubMed] [Google Scholar]
- 10.Heilig EA, Gutti U, Tai T, Shen J, Kelleher RJ., 3rd Trans-dominant negative effects of pathogenic PSEN1 mutations on γ-secretase activity and Aβproduction. J Neurosci. (2013);33:11606–11617. doi: 10.1523/JNEUROSCI.0954-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hurdle J. Genentech provides update on Alzheimer's prevention initiative study evaluating crenezumab in autosomal dominant alzheimer's disease. (2022). [Accessed June 16, 2022]. https://www.businesswire.com/news/home/20220616005106/en/Genentech-Provides-Updateon-Alzheimer%E2%80%99s-Prevention-Initiative-Study-Evaluating-Crenezumab-in-Autosomal-Dominant-Alzheimer%E2%80%99s-Disease.
- 12.Imbimbo BP, Ippati S, Imbimbo C, Balducci C. Should we lower or raise levels of amyloid-βin the brains of Alzheimer patients? Pharmacol Res. (2022);183:106390. doi: 10.1016/j.phrs.2022.106390. [DOI] [PubMed] [Google Scholar]
- 13.Jankovic J, Goodman I, Safirstein B, Marmon TK, Schenk DB, Koller M, Zago W, Ness DK, Griffith SG, Grundman M, Soto J, Ostrowitzki S, Boess FG, Martin-Facklam M, Quinn JF, Isaacson SH, Omidvar O, Ellenbogen A, Kinney GG. Safety and tolerability of multiple ascending doses of PRX002/RG7935, an anti-α-synuclein monoclonal antibody, in patients with Parkinson disease:a randomized clinical trial. JAMA Neurol. (2018);75:1206–1214. doi: 10.1001/jamaneurol.2018.1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kalia LV, Lang AE. Parkinson's disease. Lancet. (2015);386:896–912. doi: 10.1016/S0140-6736(14)61393-3. [DOI] [PubMed] [Google Scholar]
- 15.Konstantinidis E, Molisak A, Perrin F, Streubel-Gallasch L, Fayad S, Kim DY, Petri K, Aryee MJ, Aguilar X, György B, Giedraitis V, Joung JK, Pattanayak V, Essand M, Erlandsson A, Berezovska O, Ingelsson M. CRISPR-Cas9 treatment partially restores amyloid-β42/40 in human fibroblasts with the Alzheimer's disease PSEN 1 M146L mutation. Mol Ther Nucleic Acids. (2022);28:450–461. doi: 10.1016/j.omtn.2022.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kwon D. Failure of genetic therapies for Huntington's devastates community. Nature. (2021);593:180. doi: 10.1038/d41586-021-01177-7. [DOI] [PubMed] [Google Scholar]
- 17.Lang AE, Siderowf AD, Macklin EA, Poewe W, Brooks DJ, Fernandez HH, Rascol O, Giladi N, Stocchi F, Tanner CM, Postuma RB, Simon DK, Tolosa E, Mollenhauer B, Cedarbaum JM, Fraser K, Xiao J, Evans KC, Graham DL, Sapir I, et al. Trial of Cinpanemab in early Parkinson's disease. N Engl J Med. (2022);387:408–420. doi: 10.1056/NEJMoa2203395. [DOI] [PubMed] [Google Scholar]
- 18.Lerche S, Schulte C, Wurster I, Machetanz G, Roeben B, Zimmermann M, Deuschle C, Hauser AK, Bohringer J, Krageloh-Mann I, Waniek K, Lachmann I, Petterson XT, Chiang R, Park H, Wang B, Liepelt-Scarfone I, Maetzler W, Galasko D, Scherzer CR, et al. The mutation matters: CSF profiles of GCase, sphingolipids, alpha-synuclein in PDGBA. Mov Disord. (2021);36:1216–1228. doi: 10.1002/mds.28472. [DOI] [PubMed] [Google Scholar]
- 19.Meilandt WJ, Maloney JA, Imperio J, Lalehzadeh G, Earr T, Crowell S, Bainbridge TW, Lu Y, Ernst JA, Fuji RN, Atwal JK. Characterization of the selective in vitro and in vivo binding properties of crenezumab to oligomeric Aβ. Alzheimers Res Ther. (2019);11:97. doi: 10.1186/s13195-019-0553-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Migdalska-Richards A, Schapira A. The relationship between glucocerebrosidase mutations and Parkinson disease. J Neurochem. (2016);139:77–90. doi: 10.1111/jnc.13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miller T, Cudkowicz M, Shaw PJ, Andersen PM, Atassi N, Bucelli RC, Genge A, Glass J, Ladha S, Ludolph AL, Maragakis NJ, McDermott CJ, Pestronk A, Ravits J, Salachas F, Trudell R, Van Damme P, Zinman L, Bennett CF, Lane R, et al. Phase 1-2 trial of antisense oligonucleotide tofersen for SOD1 ALS. N Engl J Med. (2020);383:109–119. doi: 10.1056/NEJMoa2003715. [DOI] [PubMed] [Google Scholar]
- 22.Miller TM, Cudkowicz ME, Genge A, Shaw PJ, Sobue G, Bucelli RC, Chiò A, Van Damme P, Ludolph AC, Glass JD, Andrews JA, Babu S, Benatar M, McDermott CJ, Cochrane T, Chary S, Chew S, Zhu H, Wu F, Nestorov I, et al. Trial of antisense oligonucleotide tofersen for SOD1 ALS. N Engl J Med. (2022);387:1099–1110. doi: 10.1056/NEJMoa2204705. [DOI] [PubMed] [Google Scholar]
- 23.Mintun MA, Lo AC, Duggan Evans C, Wessels AM, Ardayfio PA, Andersen SW, Shcherbinin S, Sparks J, Sims JR, Brys M, Apostolova LG, Salloway SP, Skovronsky DM. Donanemab in early Alzheimer's disease. N Engl J Med. (2021);384:1691–1704. doi: 10.1056/NEJMoa2100708. [DOI] [PubMed] [Google Scholar]
- 24.Nisticò R, Borg JJ. Aducanumab for Alzheimer's disease: A regulatory perspective. Pharmacol Res. (2021);171:105754. doi: 10.1016/j.phrs.2021.105754. [DOI] [PubMed] [Google Scholar]
- 25.O'Connor TP, Crystal RG. Genetic medicines:treatment strategies for hereditary disorders. Nat Rev Genet. (2006);7:261–276. doi: 10.1038/nrg1829. [DOI] [PubMed] [Google Scholar]
- 26.Ostrowitzki S, Bittner T, Sink KM, Mackey H, Rabe C, Honig LS, Cassetta E, Woodward M, Boada M, van Dyck CH, Grimmer T, Selkoe DJ, Schneider A, Blondeau K, Hu N, Quartino A, Clayton D, Dolton M, Dang Y, Ostaszewski B, et al. Evaluating the safety and efficacy of crenezumab vs. placebo in adults with early Alzheimer disease:two phase 3 randomized placebo-controlled trials. JAMA Neurol. (2022) doi: 10.1001/jamaneurol.2022.2909. doi:10.1001/jamaneurol.2022.2909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pagano G, Taylor KI, Anzures-Cabrera J, Marchesi M, Simuni T, Marek K, Postuma RB, Pavese N, Stocchi F, Azulay JP, Mollenhauer B, López-Manzanares L, Russell DS, Boyd JT, Nicholas AP, Luquin MR, Hauser RA, Gasser T, Poewe W, Ricci B, et al. Trial of prasinezumab in early-stage Parkinson's disease. N Engl J Med. (2022);387:421–432. doi: 10.1056/NEJMoa2202867. [DOI] [PubMed] [Google Scholar]
- 28.Panza F, Lozupone M, Seripa D, Imbimbo BP. Amyloid-βimmunotherapy for alzheimer disease: Is it now a long shot? Ann Neurol. (2019);85:303–315. doi: 10.1002/ana.25410. [DOI] [PubMed] [Google Scholar]
- 29.Peterschmitt MJ, Crawford NPS, Gaemers SJM, Ji AJ, Sharma J, Pham TT. Pharmacokinetics, pharmacodynamics, safety, and tolerability of oral venglustat in healthy volunteers. Clin Pharmacol Drug Dev. (2021a);10:86–98. doi: 10.1002/cpdd.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Peterschmitt JM. Venglustat in Parkinson's disease patients with a GBA mutation:results from part 2 of the Phase 2 MOVES-PD trial. The 15th International Virtual Conference for Alzheimer's &Parkinson's Diseases. (2021b). [Accessed November 16, 2022]. https://cslide.ctimeetingtech.com/adpd21/attendee/confcal/persons/P.
- 31.Peterschmitt MJ, Saiki H, Hatano T, Gasser T, Isaacson SH, Gaemers SJM, Minini P, Saubadu S, Sharma J, Walbillic S, Alcalay RN, Cutter G, Hattori N, Höglinger GU, Marek K, Schapira AHV, Scherzer CR, Simuni T, Giladi N, Sardi SP, et al. Safety, pharmacokinetics, and pharmacodynamics of oral venglustat in patients with Parkinson's disease and a GBA mutation:results from part 1 of the randomized, double-blinded, placebo-controlled MOVES-PD trial. J Parkinsons Dis. (2022);12:557–570. doi: 10.3233/JPD-212714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Poewe W, Seppi K, Tanner CM, Halliday GM, Brundin P, Volkmann J, Schrag AE, Lang AE. Parkinson disease. Nat Rev Dis Primers. (2017);3:17013. doi: 10.1038/nrdp.2017.13. [DOI] [PubMed] [Google Scholar]
- 33.Prillaman M. Alzheimer's drug slows mental decline in trial - but is it a breakthrough? Nature. (2022);610:15–16. doi: 10.1038/d41586-022-03081-0. [DOI] [PubMed] [Google Scholar]
- 34.Rafii MS, Sperling RA, Donohue MC, Zhou J, Roberts C, Irizarry MC, Dhadda S, Sethuraman G, Kramer LD, Swanson CJ, Li D, Krause S, Rissman RA, Walter S, Raman R, Johnson KA, Aisen PS. The AHEAD 3-45 Study: Design of a prevention trial for Alzheimer's disease. Alzheimers Dement. doi:10.1002/alz.12748. (2022) doi: 10.1002/alz.12748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ross CA, Tabrizi SJ. Huntington's disease:from molecular pathogenesis to clinical treatment. Lancet Neurol. (2011);10:83–98. doi: 10.1016/S1474-4422(10)70245-3. [DOI] [PubMed] [Google Scholar]
- 36.Salloway S, Farlow M, McDade E, Clifford DB, Wang G, Llibre-Guerra JJ, Hitchcock JM, Mills SL, Santacruz AM, Aschenbrenner AJ, Hassenstab J, Benzinger TLS, Gordon BA, Fagan AM, Coalier KA, Cruchaga C, Goate AA, Perrin RJ, Xiong C, Li Y, et al. A trial of gantenerumab or solanezumab in dominantly inherited Alzheimer's disease. Nat Med. (2021);27:1187–1196. doi: 10.1038/s41591-021-01369-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sardi SP, Viel C, Clarke J, Treleaven CM, Richards AM, Park H, Olszewski MA, Dodge JC, Marshall J, Makino E, Wang B, Sidman RL, Cheng SH, Shihabuddin LS. Glucosylceramide synthase inhibition alleviates aberrations in synucleinopathy models. Proc Natl Acad Sci U S A. (2017);114:2699–2704. doi: 10.1073/pnas.1616152114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sardi SP, Cedarbaum JM, Brundin P. Targeted therapies for Parkinson's disease:from genetics to the clinic. Mov Disord. (2018);33:684–696. doi: 10.1002/mds.27414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sau D, De Biasi S, Vitellaro-Zuccarello L, Riso P, Guarnieri S, Porrini M, Simeoni S, Crippa V, Onesto E, Palazzolo I, Rusmini P, Bolzoni E, Bendotti C, Poletti A. Mutation of SOD1 in ALS:a gain of a loss of function. Hum Mol Genet. (2007);16:1604–1618. doi: 10.1093/hmg/ddm110. [DOI] [PubMed] [Google Scholar]
- 40.Scheltens P, De Strooper B, Kivipelto M, Holstege H, Chételat G, Teunissen CE, Cummings J, van der Flier WM. Alzheimer's disease. Lancet. (2021);397:1577–1590. doi: 10.1016/S0140-6736(20)32205-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, et al. Alpha-synuclein locus triplication causes Parkinson's disease. Science. (2003);302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- 42.Swanson CJ, Zhang Y, Dhadda S, Wang J, Kaplow J, Lai RYK, Lannfelt L, Bradley H, Rabe M, Koyama A, Reyderman L, Berry DA, Berry S, Gordon R, Kramer LD, Cummings JL. A randomized, double-blind, phase 2b proof-of-concept clinical trial in early Alzheimer's disease with lecanemab, an anti-Aβprotofibril antibody. Alzheimers Res Ther. (2021);13:80. doi: 10.1186/s13195-021-00813-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tabrizi SJ, Leavitt BR, Landwehrmeyer GB, Wild EJ, Saft C, Barker RA, Blair NF, Craufurd D, Priller J, Rickards H, Rosser A, Kordasiewicz HB, Czech C, Swayze EE, Norris DA, Baumann T, Gerlach I, Schobel SA, Paz E, Smith AV, et al. Targeting huntingtin expression in patients with Huntington's disease. N Engl J Med. (2019);380:2307–2316. doi: 10.1056/NEJMoa1900907. [DOI] [PubMed] [Google Scholar]
- 44.Tariot PN, Lopera F, Langbaum JB, Thomas RG, Hendrix S, Schneider LS, Rios-Romenets S, Giraldo M, Acosta N, Tobon C, Ramos C, Espinosa A, Cho W, Ward M, Clayton D, Friesenhahn M, Mackey H, Honigberg L, Sanabria Bohorquez S, Chen K, et al. The Alzheimer's Prevention Initiative Autosomal-Dominant Alzheimer's Disease Trial: A study of crenezumab versus placebo in preclinical PSEN1 E280A mutation carriers to evaluate efficacy and safety in the treatment of autosomal-dominant Alzheimer's disease, including a placebo-treated noncarrier cohort. Alzheimers Dement. (2018);4:150–160. doi: 10.1016/j.trci.2018.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ultsch M, Li B, Maurer T, Mathieu M, Adolfsson O, Muhs A, Pfeifer A, Pihlgren M, Bainbridge TW, Reichelt M, Ernst JA, Eigenbrot C, Fuh G, Atwal JK, Watts RJ, Wang W. Structure of crenezumab complex with Aβshows loss of β-hairpin. Sci Rep. (2016);6:39374. doi: 10.1038/srep39374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.US Food and Drug Administration (2022) Approved Cellular and Gene Therapy Products. [Accessed September 19, 2022]. https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/approved-cellular-and-gene-therapy-products.
- 47.Volpicelli-Daley L, Brundin P. Prion-like propagation of pathology in Parkinson disease. Handb Clin Neurol. (2018);153:321–335. doi: 10.1016/B978-0-444-63945-5.00017-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Willis BA, Sundell K, Lachno DR, Ferguson-Sells LR, Case MG, Holdridge K, DeMattos RB, Raskin J, Siemers ER, Dean RA. Central pharmacodynamic activity of solanezumab in mild Alzheimer's disease dementia. Alzheimers Dement. (2018);4:652–660. doi: 10.1016/j.trci.2018.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang T, Dang Y, Ostaszewski B, Mengel D, Steffen V, Rabe C, Bittner T, Walsh DM, Selkoe DJ. Target engagement in an Alzheimer trial: Crenezumab lowers amyloid βoligomers in cerebrospinal fluid. Ann Neurol. (2019);86:215–224. doi: 10.1002/ana.25513. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.