

Abstract
We report our recent development of a conceptually new generation of exceptionally potent non-peptidic HIV-1 protease inhibitors that displayed excellent pharmacological and drug-resistance profiles. Our X-ray structural studies of darunavir and other designed inhibitors from our laboratories led us to create a variety of inhibitors incorporating fused ring polycyclic ethers and aromatic heterocycles to promote hydrogen bonding interactions with the backbone atoms of HIV-1 protease as well as van der Waals interactions with residues in the S2 and S2’ subsites. We have also incorporated specific functionalities to enhance van der Waals interactions in the S1 and S1’ subsites. The combined effects of these structural templates are critical to the inhibitors’ exceptional potency and drug-like properties. We highlight here our molecular design strategies to promote backbone hydrogen bonding interactions to combat drug-resistance and specific design of polycyclic ether templates to mimic peptide-like bonds in the HIV-1 protease active site. Our medicinal chemistry and drug development efforts led to the development of new generation inhibitors significantly improved over darunavir and displaying unprecedented antiviral activity against multidrug-resistant HIV-1 variants.
Graphical Abstract
We describe design and syntheses of a new generation of exceptionally highly potent non-peptidic HIV-1 protease inhibitors.
Introduction
The treatment of Acquired Immunodeficiency Syndrome (AIDS) continues to be a major challenge in medicine.1,2 According to the UNAIDS/WHO’s 2021 report on HIV/AIDS, an estimated 38.4 million people globally are currently living with HIV-1 infection and an estmated 40.1 million people have died from AIDS-related illness since the start of the epidemic.3,4 The advent of highly active antiretroviral therapy (ART) in the mid-1990s, marked the beginning of a new era of treatment for patients with HIV-1 infection and AIDS.5,6 The ART treatment regimen is a combination therapy with HIV-1 protease inhibitor (PI) and reverse transcriptase inhibitor drugs.7,8 ART has significantly reduced morbidity and mortality associated with HIV-1 infection and AIDS. It has also improved the quality of life of patients who have access to these therapies. The use of ART dramatically transformed HIV/AIDS from a fatal disease to a manageable chronic disorder.9,10
Despite the major clinical benefits of ART, initial protease inhibitor-based drugs possessed many detriments including peptide-like features, drug-related side-effects, and poor ADME properties.11,12 The rapid emergence of drug resistance often quickly renders a selected treatment ineffective within a short period of time. Furthermore, HIV-associated neurocognitive disorder (HAND) is affecting nearly fifty percent of the current HIV-positive population.13 These problems seriously complicate the long-term management of HIV-1 infection and prevention of AIDS-related deaths.14,15 Over the years, our laboratories have focused on the design of a conceptually new generation of nonpeptide HIV-1 protease inhibitors that would maintain potent antiviral activity against multidrug-resistant HIV-1 variants and show improved pharmacological properties over existing PI-drugs.16,17 Our research efforts led to the design and discovery of a range of highly potent HIV-1 protease inhibitors that incorporated novel ligands and templates by drawing inspiration from natural products.18,19 One of these protease inhibitors is darunavir (DRV, 1, Figure 1) which displayed drug-like properties and maintained potent antiviral activity against a wide range of highly multidrug-resistant HIV-1 variants.20–22 Darunavir received FDA approval in 2006 for the treatment of patients harboring highly drug-resistant viruses.23,24 Subsequently, it received approval as a first-line therapy for treatment of all HIV/AIDS patients including pediatrics in the current US Department of Health and Human Service (DHHS) guidelines.25,26
Figure 1.

Structure and activity of inhibitors 1 and 2 (PDB: 21EN).
One of the intriguing features of darunavir is the presence of a stereochemically defined and conformationally constrained bicyclic bis-tetrahydrofuran (bis-THF) heterocycle.27,28 The X-ray structure of DRV-bound HIV-1 protease revealed that both oxygens of the bis-THF form very strong hydrogen bonds with the Asp29 and Asp30 backbone amide NHs at the S2 subsite of HIV-1 protease.29,30 The bicyclic ring system of the bis-THF ligand forms van der Waals interactions with residues in the S2 subsite.18,31,32 The P2’ ligand, 4-amino sulfonamide was also specifically chosen to promote hydrogen bonding interactions with the backbone atoms at the S2’ subsite.18,29 The overall structural features of darunavir, its molecular interactions with HIV-1 protease particularly its network of hydrogen bonding interactions with backbone atoms, and its ability to maintain robust antiviral activity with multidrug-resistant HIV-1 variants further stimulated our molecular design of novel scaffolds and templates for further improving inhibitor properties. Herein, we provide highlights of our recent design of new generation HIV-1 protease inhibitors that incorporated unprecedented 6–5-5 ring-fused tetrahydropyrano-furan derivatives as the P2 ligand in combination with alkylaminobenzoxazole and thiazole derivatives as the P2’-ligand. These inhibitors are specifically designed to maximize interactions including van der Waals interactions and hydrogen bonding interactions with the HIV-1 protease backbone atoms to combat drug resistance.
Promoting Backbone Hydrogen Bonding Interactions: Molecular Design Strategy to Combat Drug-Resistance
The underlying principle for our structure-based design of inhibitors to combat drug resistance involves maximizing an inhibitor’s active site interactions, particularly with backbone atoms from the S2 to S2’ subsites.29,33,34 We have critically analyzed and compared various mutant HIV-1 protease X-ray structures with wild-type HIV-1 protease and observed that the active site backbone conformation of these mutant proteases is only distorted minimally compared to wild-type HIV-1 protease.35,36 It is likely that the protease enzyme cannot significantly alter its active site backbone conformation without compromising its catalytic fitness and efficiency required for viral replication. Therefore, our “backbone binding” inhibitor design strategy to combat drug-resistance has evolved to maximize interactions including van der Waals interactions and hydrogen-bonding interactions with the active site of HIV-1 protease.29,34 In principle, such an inhibitor would likely maintain these key interactions including hydrogen-bonds with the backbone atoms of protease mutants. As a consequence, the development of drug-resistant HIV-1 variants would be reduced as altering backbone conformation would lead to impaired proteolysis of the natural polyprotein substrates. Our simplified view is to design an inhibitor like a ‘molecular crab’ that would tightly grip the protein backbone and hold on to the enzyme active site.34 HIV-1 protease is an aspartic acid protease. The active form of HIV-1 protease is a homodimer containing two catalytic aspartic acid residues in the active site. Our model for inhibitor design to combat drug resistance is shown in Figure 2.33 An inhibitor would have a functional group that would mimic the transition-state of the scissile bond cleavage. Among many known transition-state mimetics, we selected (R)-hydroxyethylamine sulfonamide dipeptide isostere with a hydroxyl group with (R)-configuration to bind the catalytic aspartates.28,37 The P1 and P1’ ligands would fill in the hydrophobic S1 and S2 subsites and P2 and P2’ ligands would interact with the S2 and S2’ subsites through robust hydrogen bonding interactions with the backbone atoms as well as van der Waals interactions with the residues.
Figure 2.
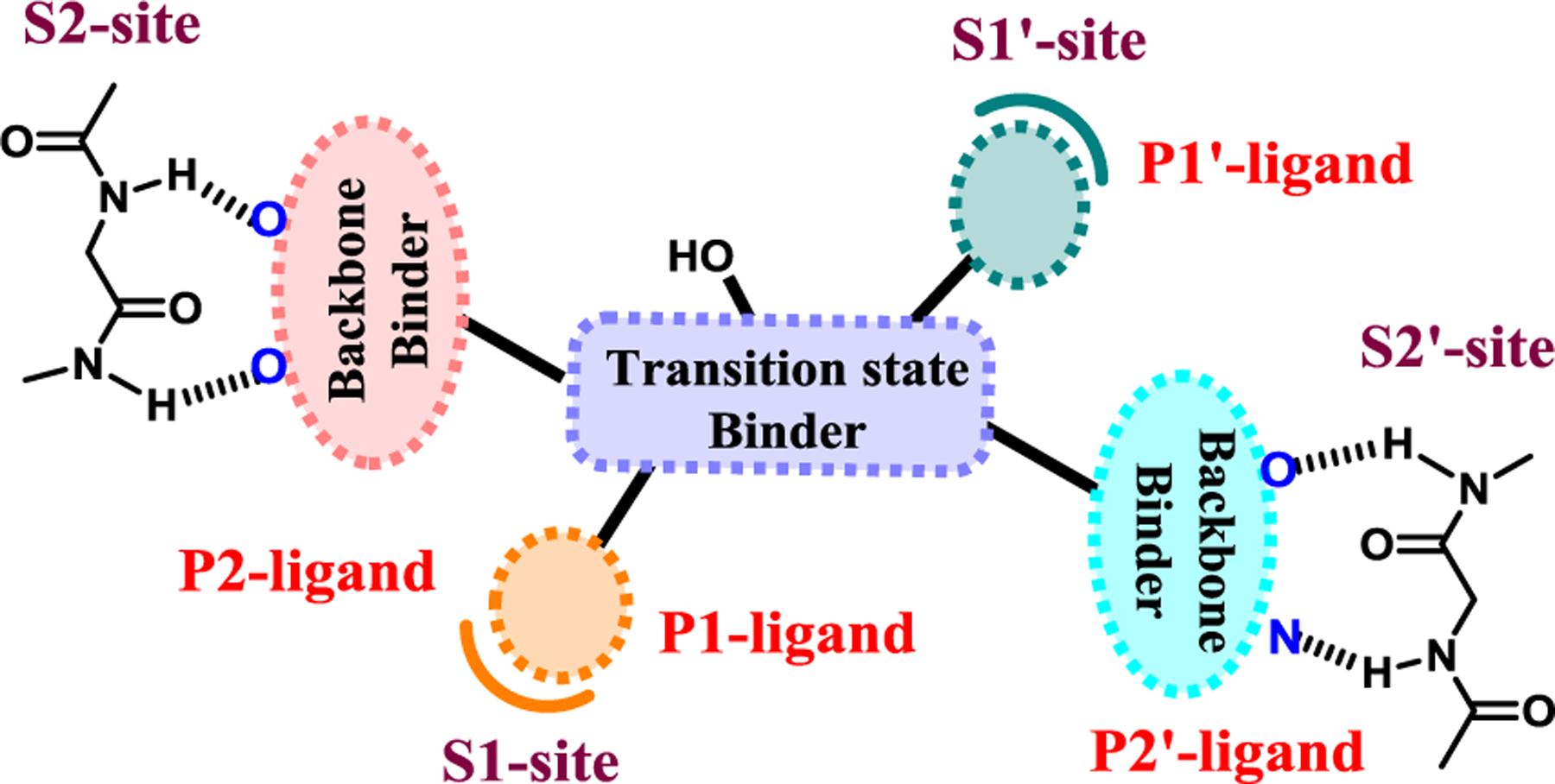
Design models for PIs to combat drug resistance. Promote robust hydrogen bonds through the P2- and P2′-ligands in both the S2 and S2′-subsites. The hydroxyl group to bind to catalytic aspartates mimicking transition-state, and the P1 and P1′-ligands to fill in the S1 and S1′-subsites.
Using our “backbone binding” design strategy, we have recently created a range of exceptionally potent HIV-1 protease inhibitors that incorporated polycyclic ethers as P2 ligands and heterocyclic sulfonamide derivatives as the P2’ ligands. These compounds have shown exceptional antiviral activity against a range of highly multidrug-resistant HIV-1 variants. Through X-ray structural studies and analysis of antiviral activity, we can corroborate our backbone binding molecular design strategy as a powerful design concept to combat drug resistance.19,29,34
Cyclic Ether Templates from Natural products: Inhibitor Design Strategies to Mimic peptide bonds.
Historically, natural products have been important sources for structurally diverse bioactive molecules enabling discovery and development of new drugs.38,39 Fine-tuning and optimization over millions of years of evolution, organisms have biosynthesized a variety of structurally intriguing molecules and molecular templates, specifically custom-made to interact and maintain their activity in biological microenvironments.40 We became motivated to harness some of nature’s molecular templates in our design of new nonpeptide inhibitors, considering the evolutionary fitness and stability of these functionalities. We are particularly intrigued by the cyclic ether and cyclic acetal structural features of numerous bioactive natural products.16,17 As shown in Figure 3, ginkgolides (3), isolated from predinosaur era tree, Ginkgo biloba are used as dietary supplements for human health.41 Laulimalide (4), isolated from marine sponge, exhibits potent anticancer activity.42,43 Azadirachtin (5), isolated from Azadirachts indica A, has shown broad-spectrum insecticide properties.44 Platensimylin, isolated from Streptomyces platensis is a potent broad-spectrum antibiotic.45,46 Eribulin mesylate (7, Halven®), is a semi-synthetic derivative of halichondrin B and is used as an approved anticancer drug.47 As can be seen, these bioactive natural products possess an abundant amount of cyclic ether structural features.
Figure 3.
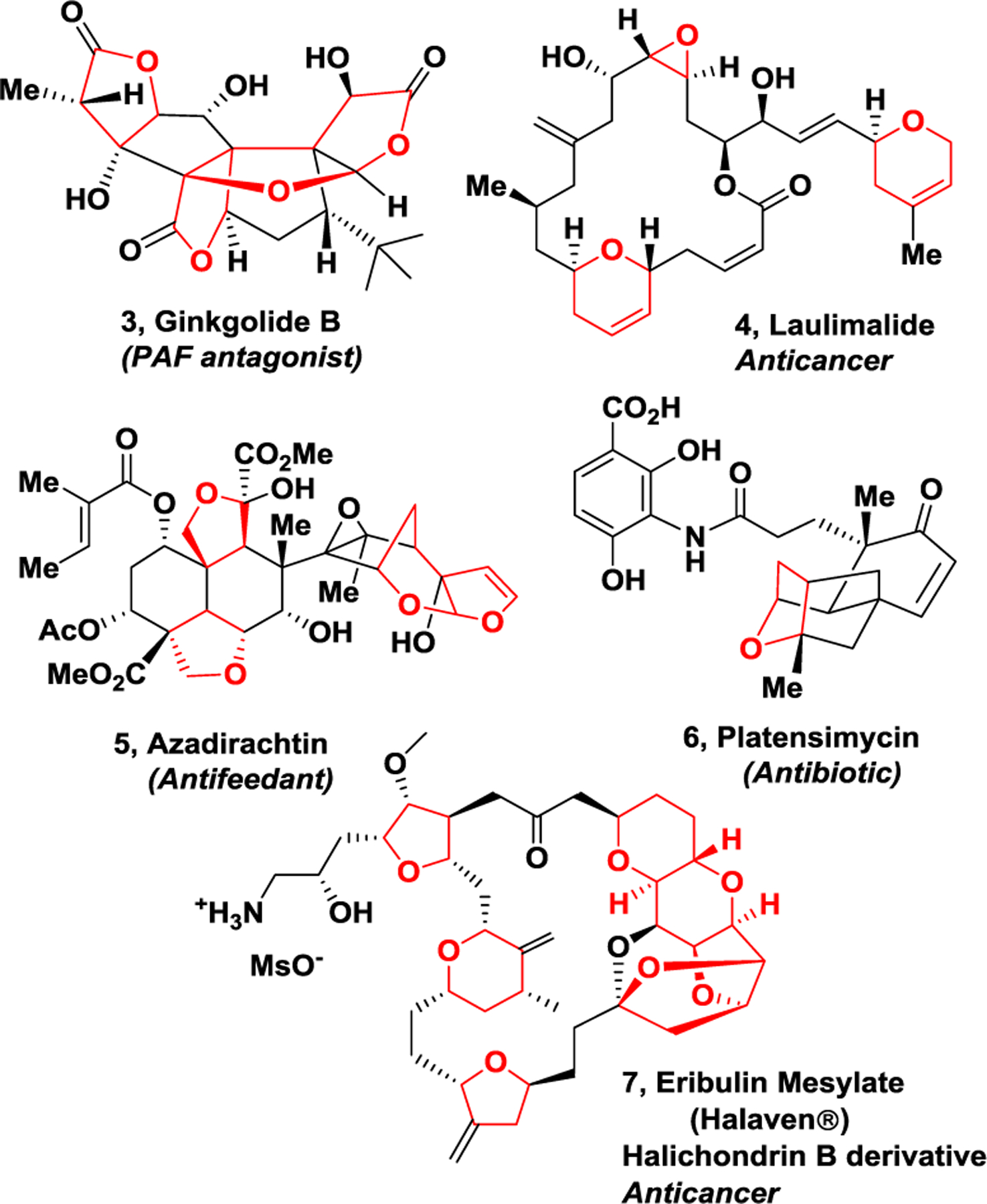
Structure of ginkgolide (3), laulimalide (4), azadirachtin (5), platensimycin (6), and Erlbulin (7) natural products
The cyclic ether functionalities in these molecules are critically important for their biological activity. Presumably, the cyclic ether oxygen is positioned to make up to two hydrogen bonding interactions with target peptide or protein residues in the drug binding site. The side chain substituents around the cyclic ether functionality are likely mimicking the side chains of peptides and proteins. Numerous X-ray structural studies of cyclic ether-derived natural products with target enzymes or receptors revealed that these structural features not only provide overall shape and geometry of the molecules, they also serve as important pharmacophores and make critical interactions with their respective biological target proteins.48,49 An X-ray crystal structure (PDB code: 404H) of laulimide (4) and β-tubulin, with its target protein show these critical interactions.50 As shown in Figure 4, the tetrahydropyran ring oxygen in the macrocycle forms a strong hydrogen bond with the side chain of Asn339.
Figure 4.
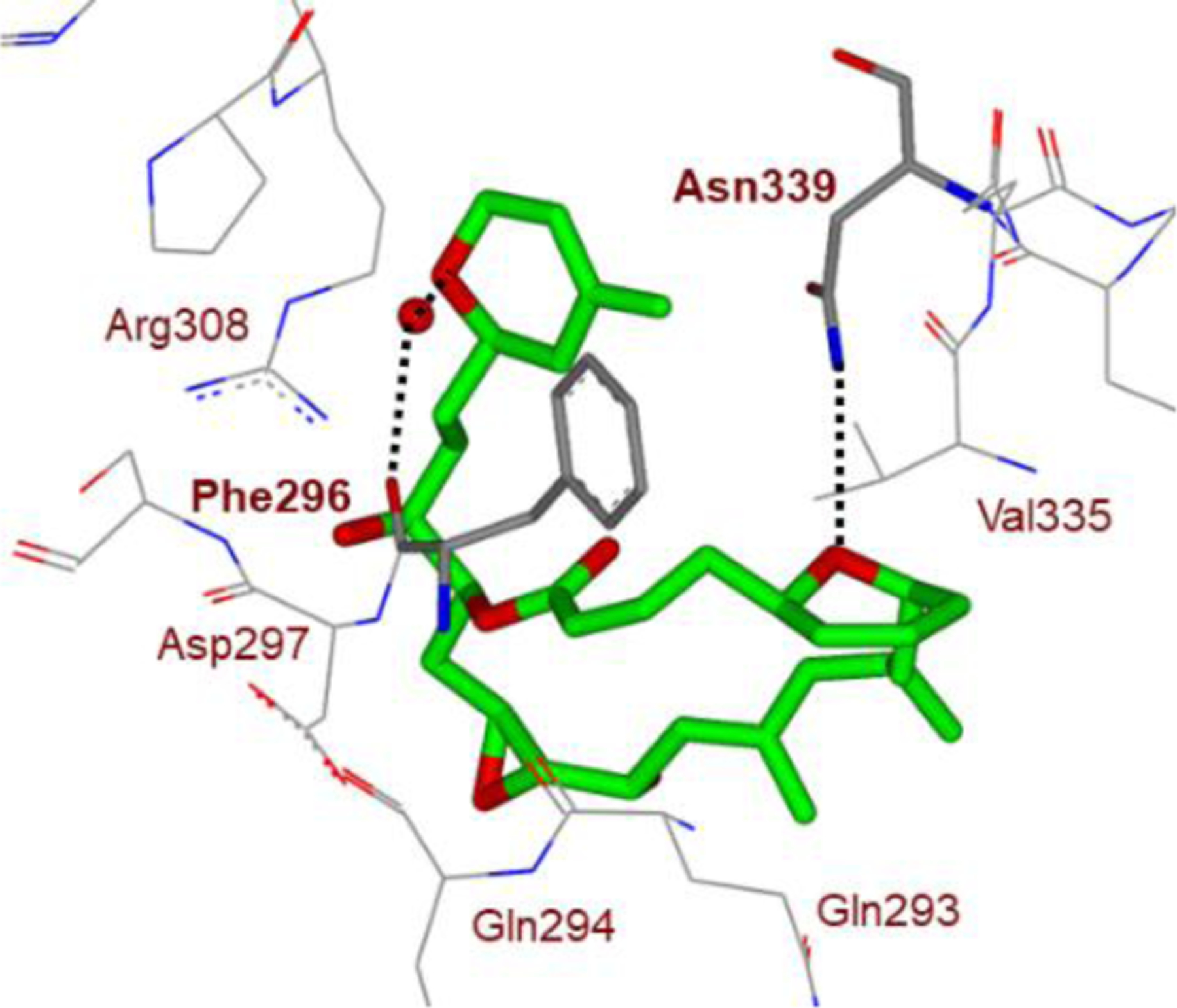
Structure of laulimalide (4, green carbons)-bound to β-tubulin. Hydrogen bonds with THP oxygens are shown with dotted lines.
Also, the oxygen atoms of the dihydropyran side chain form a water-mediated hydrogen bond to the side chain OH of Tyr312 and the main chain backbone carboxyl oxygen of Phe296. These interactions are likely very important for laulimalide’s potent anticancer activity. Similarly, X-ray structural studies of platensimycin (6)-bound bacterial enzyme beta-ketoacyl synthase II (FabF) show that the tetrahydropyranyl oxygen is involved in hydrogen bonding interactions with the Thr270 residue.48 Interestingly, the desoxy derivative showed significantly reduced activity, demonstrating the importance of the ring oxygen.51
The cyclic ether templates, basically may be viewed as peptidomimetic scaffolds. We therefore speculated that incorporation of similar cyclic ether templates in our design of HIV-1 protease inhibitors may lead to nonpeptide derivatives which would alleviate problems inherent to peptide or peptidomimetic based first generation protease inhibitor drugs.52,53 We first systematically explored a variety of cyclic ethers and cyclic sulfone-derived conformationally constrained structural templates to replace peptide-like amide bonds. We then demonstrated that cyclic ether or sulfone-derived heterocycles can mimic peptide and amide bond interactions in the HIV-1 protease active site. This led to the design of a range of highly potent and nonpeptide HIV-1 protease inhibitors with cyclic ether structural features.16,17,27 Our further design optimization then focused on promoting hydrogen bonding interactions with the backbone atoms in the S2 to S2’ subsites of HIV-1 protease.29,34 These design and optimization efforts ultimately culminated the discovery and development of darunavir.18,20,28 In the following section, we briefly highlight the chronology of these design events that led to the development of darunavir; the first approved treatment for patients with drug-resistant HIV.
Design of Cyclic Ether Containing HIV-1 Protease Inhibitors: From Saquinavir to Darunavir
Saquinavir (8, R031-8959) is one of the first HIV-1 protease inhibitor drugs that was approved by the FDA in 1996 for treatment of patients with HIV/AIDS.54,55 Subsequently, a number of other first-generation inhibitor drugs were approved for use in combination therapy with reverse transcriptase inhibitor drugs.19,56,57 These combination therapies with HIV-1 protease inhibitors resulted in a dramatic reduction of HIV/AIDS related deaths among patients who had access to these therapies.9,10 However, therapeutic efficacy of first-generation PI drugs was limited due to peptide-like features and poor ADME properties of these drugs. The emergence of drug-resistant HIV-1 strains made the therapies ineffective. Subsequent development addressing these issues led to second-generation PI drugs, such as lopinavir, atazanevir, and tipranavir.37,56 However, the emergence of highly multidrug-resistant HIV-1 variants among infected patients became a significant limitation.
In our effort to utilize structure-based design to develop next generation nonpeptide PIs effective against drug-resistant HIV-1 variants, we first explored saquinavir active site interactions with a stereochemically-defined cyclic ether and cyclic sulfone-derived heterocycles in an effort to replace the peptidic bonds of saquinavir. These endeavors resulted in the design and discovery of a variety of potent nonpeptide PIs.58–61
Further optimization of cyclic ether templates followed based upon the X-ray structures of saquinavir-bound HIV-1 protease and X-ray structures of early lead inhibitors with HIV-1 protease. The saquinavir-bound HIV-1 protease X-ray structure revealed that the carboxyl oxygen of the P2-asparagine ligand and the amide carboxyl oxygen of P3-quinaldic amide form two strong hydrogen bonds with the backbone amide NHs of Asp30 and Asp29 in the S2 and S3 subsites (Figure 5).54,62 Based upon the X-ray structures, we then designed a fused bicyclic hexahydrofuro[2,3-b]furan to replace both amide carbonyl bonds in the saquinavir structure.27,61,62 As shown in Figure 4, inhibitor 9, with a stereochemically defined (3R,3aS,6aR)-bis-tetrahydrofuranyl urethane displayed very potent HIV-1 protease inhibitory activity as well as antiviral activity. Indeed, our X-ray structural studies of PI-9-bound HIV-1 protease and comparison of X-ray structure of saquinavir-bound protease revealed that the fused cyclic ether template of the bis-THF ligand oxygens formed strong hydrogen bonds with Asp29 and Asp30 backbone amide NHs in the active site.61,62 The bicyclic bis-THF core also makes extensive van der Waals interactions in the S2 subsite. In essence, the bis-THF ligand effectively replaced two amide bonds and the bulky P3–10π-aromatic quinaldic moiety of saquinavir. Through our detailed structure-activity relationship studies, we also demonstrated that ring stereochemistry, position of ring oxygens, ring size of bis-THF in PI9, are optimal for effective binding interactions in the active site.27,61
Figure 5.
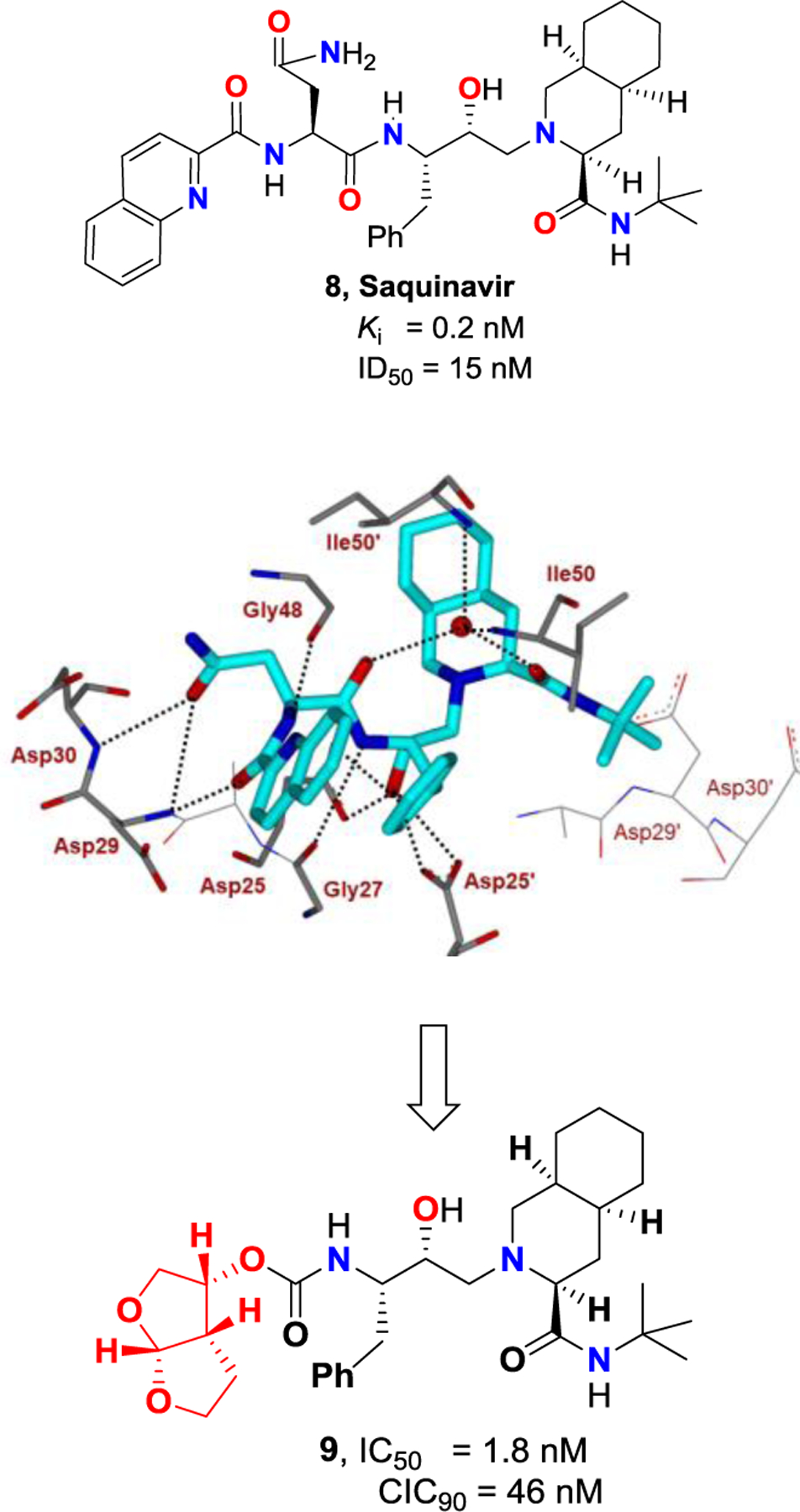
Structure and activity of inhibitors 8 and 9 (PDB: 1HXB).
Following our discovery of the cyclic ether derived nonpeptide bis-THF heterocycle as a highly effective P2 ligand for HIV-1 protease inhibitors, we then focused our efforts to design PIs that would make extensive backbone hydrogen bonding interactions in both S2 to S2’ subsites.29,34 Since the active enzyme, HIV-1 protease is a homodimer, we also planned to promote extensive van der Waals interactions throughout the active site. Towards these objectives, we first designed PIs incorporating (R)-(hydroxyethyl)sulfonamide-derived transition-state binder with a p-methoxybenzene sulfonamide as the P2’ ligand, with the plan to have hydrogen binding interactions in the S2’-site through the methoxyl oxygen.62,63 This optimization resulted in PI 10 (Figure 6, later named as TMC-126), which exhibited exceptional HIV-1 protease inhibitory activity as well as antiviral activity. Enzyme inhibitory activity of inhibitor 10 against mutant proteases resistant to a majority of first-generation approved PIs is shown in Table 1.64,65 As can be seen, protease inhibitory activity of 10 is less than 100 pM and changes of activity were not more than 5-fold. Table 2 shows antiviral activity of inhibitor 10 against a panel of highly multi-PI-resistant HIV-1 strains isolated from patients. Antiviral activity of PI 10 did not change much against these multidrug-resistant HIV-1 variants whereas, other approved PIs exhibited a high level of drug resistance.63,66 The X-ray structural analysis of 10-bound HIV-1 protease revealed that both the bis-THF ligand and the 4-methoxy sulfonamide P2’-ligand formed extensive hydrogen bonding and van der Waals interactions with the backbone atoms in the S2 and S2’ subsites.32,37,67 As shown in Figure 6, both lone pairs of top tetrahydrofuranyl oxygen atom of bis-THF ligand form two hydrogen bonds with Asp30 and Asp29 backbone amide NHs. The bottom oxygen however forms a single hydrogen bond with Asp29 backbone amide NH in the S2 subsite. Also, both P1 and P1’ ligands fill in the hydrophobic S1 and S1’ subsites effectively. Inhibitor 10 showed a high genetic barrier to drug resistance acquisition. Also, in vitro selection of HIV-1 strains resistant to inhibitor 10 was difficult to attain.66 The extraordinary antiviral activities of 10 against multidrug-resistant HIV-variants as well as its durability towards the development of drug-resistance can be attributed to the extensive interactions of 10 with the protease, particularly the formation of a network of hydrogen bonding interactions with the backbone atoms from the S2 to S2′-subsites.29,34
Figure 6.
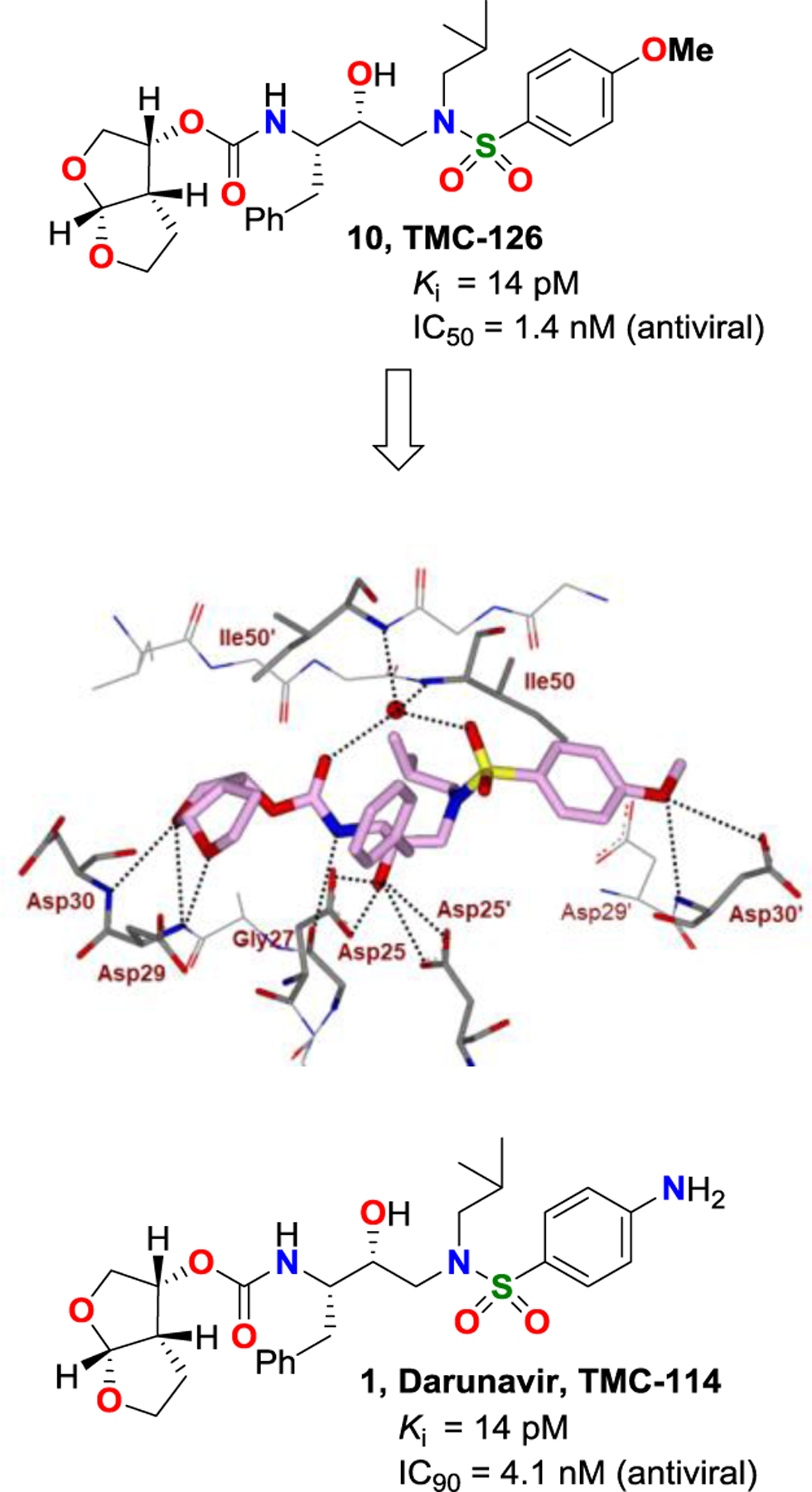
Structure and activity of inhibitors 1 and 10. X-ray structure of 10-bound HIV-1 protease (PDB: 317E).
Table 1.
Inhibitory potency of 10 against wild-type and mutant proteases
| Enzyme | Ki (pM) | Ki wt/Ki mut | Vitalitya |
|---|---|---|---|
| WT | 14 | 1 | 1 |
| D30N | <5 | 0.33 | 0.3 |
| V32I | 8 | 0.57 | 0.5 |
| I84V | 40 | 2.85 | 1 |
| V32I/I84V | 70 | 5 | 0.7 |
| M46F/V82A | <5 | 0.33 | 0.1 |
| G48V/L90M | <5 | 0.33 | 0.1 |
| V82F/I84V | 7 | 0.5 | 0.1 |
| V82T/I84V | 22 | 1.57 | 0.1 |
| V32I/K45I/F53L/A71V/I84V/L89M | 31 | 2.2 | 0.1 |
| V32I/L33F/K45I/F53L/A71V/I84V | 46 | 3.3 | 0.1 |
| 20R/36I/54V/71V/82T | 31 | 2.2 | 0.1 |
The vitality is a measure of the enzymatic fitness of a mutant protease in presence of an inhibitor, here 10 (determined as (Ki*kcat/Km)mut/(Ki*kcat/Km)wild).
Table 2.
Antiviral activity of 10 against HIV-1 isolated from previously treated HIV-infected patients
| Virus[a] | IC50 μM (fold change) | |||||
|---|---|---|---|---|---|---|
| RTV | IDV | SQV | NFV | APV | 10 | |
| WT | 0.044 (1) | 0.013 (1) | 0.010 (1) | 0.023 (1) | 0.025 (1) | 0.0007 (1) |
| 1 | >1 (>23) | >1 (>77) | 0.27 (27) | >1 (>43) | 0.27 (11) | 0.004 (6) |
| 2 | >1 (>23) | 0.49 (38) | 0.037 (4) | 0.33 (14) | 0.28 (11) | 0.0013 (2) |
| 3 | >1 (>23) | 0.49 (38) | 0.036 (4) | >1 (>43) | 0.26 (10) | 0.001 (1) |
| 4 | >1 (>23) | 0.21 (16) | 0.033 (3) | 0.09 (4) | 0.31 (12) | 0.0016 (2) |
| 5 | >1 (>23) | >1 (>77) | 0.31 (31) | 0.41 (18) | 0.67 (27) | 0.0024 (3) |
| 6 | >1 (>23) | 0.30 (23) | 0.19 (19) | >1 (>43) | 0.16 (6) | 0.0005 (1) |
| 7 | >1 (>23) | >1 (>77) | 0.12 (12) | >1 (>43) | 0.49 (20) | 0.0055 (8) |
| 8 | >1 (>23) | 0.55 (42) | 0.042 (4) | >1 (>43) | 0.15 (6) | 0.001 (1) |
Amino acid substitutions identified in the protease-encoding regions of viruses compared to the consensus sequence cited from the Los Alamos database. See reference 57 for details.
We have designed and synthesized a variety of other very potent PIs that promote the formation of a network of hydrogen bonds like PI 10. Further optimization of ligand binding as well as pharmacokinetic properties ultimately led to the design and development of inhibitor 1 (Darunavir, TMC-114) with a bis-THF P2 ligand and a 4-aminobenzenesulfonamide as the P2’ ligand. Darunavir exhibited exceptionally potent protease inhibitory and antiviral activity.28,62,63 Also, it displayed unprecedented broad-spectrum antiviral activity against a wide range of highly multidrug-resistant HIV-1 variants.21,22 As shown in Table 3, darunavir exhibited very potent antiviral activity (IC50 = 0.003–0.029 μM) against a series of HIV-1 strains, which were selected for their resistance against saquinavir (SQV), amprenavir (APV), indinavir (IDV), nelfinavir (NFV), and ritonavir (RTV) after exposure of the virus to increasing concentrations of these PIs. Also, darunavir maintained excellent antiviral activity against a panel of highly multi-PI- resistant HIV-1 variants isolated from AIDS patients who previously failed anti-HIV regimens after receiving other anti-HIV drugs.21,22 Darunavir received an accelerated FDA approval in June, 2006, for patients harboring drug-resistant HIV. Then in 2008, its approval was extended to all HIV/AIDS patients including pediatrics.16 Darunavir emerged as a first-line therapy and it is often preferred for HIV/AIDS patients who did not receive antiretroviral drugs previously due to its tolerance and a high genetic barrier to the development of drug-resistant viruses compared to other HIV-1 protease inhibitor drugs.15,16 The unique structural design of darunavir, its molecular interactions with the protease active site, and resilience against multidrug-resistant HIV-1 variants have stimulated significant research interest regarding its mechanism of action.28,68 Using intermolecular fluorescence resonance energy transfer (FRET)-based HIV expression assay, we have shown that darunavir blocks HIV-1 protease dimerization.69,70 Therefore, darunavir’s unique dual modes of action which include (1) inhibition of the catalytic protease dimer and (2) inhibition of protease monomer dimerization to form the active enzyme, may be responsible for its robust drug-resistance profiles and high genetic barrier to the development of drug-resistant HIV-1 variants.20,28,68
Table 3.
Activity of darunavir against HIV-1 clinical isolates in PHA-PBMC infected cells[a]
| IC50 values (μM) | ||||||
|---|---|---|---|---|---|---|
| Virus | SQV | APV | IDV | NFV | RTV | DRV (1) |
| WT | 0.010 | 0.023 | 0.018 | 0.019 | 0.027 | 0.003 |
| 1 | 0.004 | 0.011 | 0.018 | 0.033 | 0.032 | 0.003 |
| 2 | 0.23 (23) | 0.39 | >1 (>56) | 0.54 (28) | >1 (>37) | 0.004 (1) |
| 3 | 0.30 (30) | 0.34 | >1 (>56) | >1 (>53) | >1 (>37) | 0.02 (7) |
| 4 | 0.35 (35) | 0.75 (33) | >1 (>56) | >1 (>53) | >1 (>37) | 0.029 (10) |
| 5 | 0.14 (14) | 0.16 (7) | >1 (>56) | 0.36 (19) | >1 (>37) | 0.004 (1) |
| 6 | 0.31 (31) | 0.34 (15) | >1 (>56) | >1 (>53) | >1 (>37) | 0.013 (4) |
| 7 | 0.037 (4) | 0.28 (12) | >1 (>56) | 0.44 (23) | >1 (>37) | 0.003 (1) |
| 9 | 0.029 (3) | 0.25 (11) | 0.39 (22) | 0.32 (17) | 0.44 (16) | 0.004 (1) |
Amino acid substitutions identified in the protease-encoding regions of viruses compared to the consensus sequence cited from the Los Alamos database. See reference 13 for details.
We have determined a high resolution X-ray crystal structure of darunavir-bound HIV-1 protease (Figure 1). These structures show that darunavir makes extensive interactions including a network of hydrogen bonds throughout the HIV-1 protease active site. Both oxygens of the P2 bis-THF ligand formed two very strong hydrogen bonds with the Asp29 and Asp30 backbone amide NHs in the S2 subsite.30,31,71 The P2’-aminosulfonamide’s amine functionality formed hydrogen bonds with the Asp30’ backbone NH as well as with the side chain carboxylic acid in the S2’-subsite. Both P1 phenylmethyl group and the P1’ isobutyl moiety filled in the hydrophobic pockets in S1 and S1’ subsites. Furthermore, a tetra-coordinated structural water molecule forms hydrogen bonds with both NH atoms of flap residues Ile50/50’ and the inhibitor’s P2 urethane carbonyl and one of the P2’-sulfonamide (SO2) oxygens.72,73 Presumably, these ligand-binding site interactions are responsible for darunavir’s unprecedented antiviral activity and drug-resistance profiles.29,34
Following darunavir’s development, utilizing our SAR and critical molecular insights from active site interactions, we have designed a variety of other bicyclic and polycyclic ether templates to enhance the backbone-binding interactions, as well as to improve van der Waals interactions in the active site. These endeavours resulted in a series of exceptionally potent HIV-1 protease inhibitors that exhibited improved antiviral activity against multidrug-resistant HIV-1 variants compared to darunavir and other approved PI drugs. Figure 7 depicts structure and potency of a selected number of representative inhibitors (11-16).74–81 A detailed account of their design, synthesis, biological evaluation and structural studies have been reported and reviewed by us.19 Of particular importance, we desgined and incorporated stereochemically defined and conformationally constrained cyclic-ether structural features in the P2 ligand to interact specifically with active site.
Figure 7.
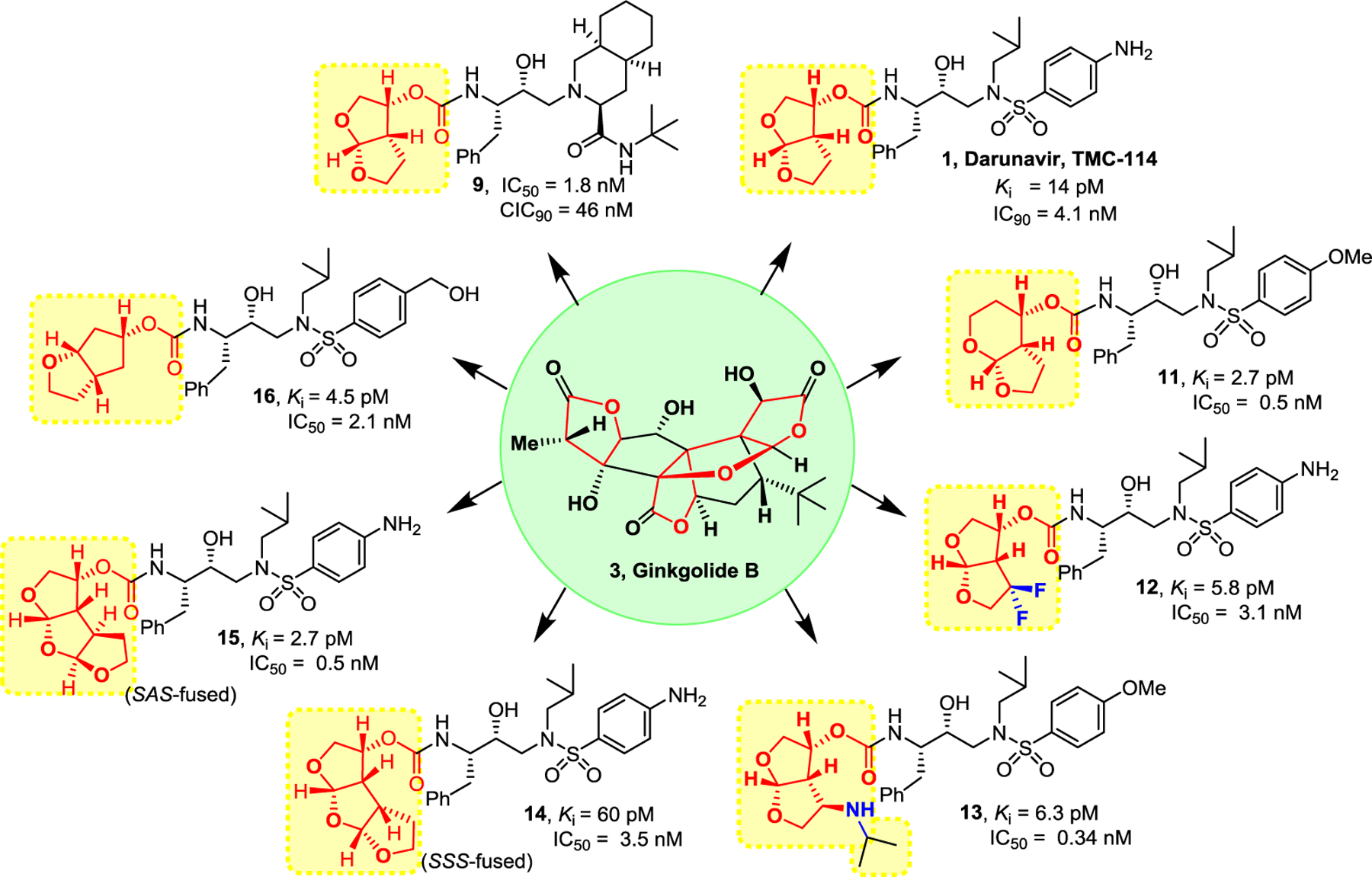
Design and synthesis of polycyclic-ether containing highly potent HIV-1 protease inhibitors.
Design of Crown-THF-based New Generation of HIV-1 Protease Inhibitors
In our post darunavir development, we set out to design a structurally more conformationally constrained and preorganized ligand that would enhance overall backbone hydrogen bonding and van der Waals interactions in the protease active site.29,34 The X-ray structure of darunavir-bound HIV-1 protease revealed that the bis-THF P2 ligand can be modified to enhance ligand-binding site interactions.30,31 In the context of our design of bicyclic 6–5-fused acetal-derived inhibitor 11, we showed that the hexahydrofuropyranol heterocycle has made better alignment with the backbone amide NHs in the S2-subsite over the bis-THF ligand.74 Presumably, the extra methylene group in the new tetrahydropyranyl tetrahydrofuran-fused (Tp-THF) ligand may have contributed toward improved van der Waals interactions. Inhibitor 11 displayed enhanced enzyme inhibitory and antiviral activity over the corresponding bis-THF-derived inhibitor 10. In addition, inhibitor 11 exhibited impressive antiviral activity against a panel of highly multidrug-resistant HIV-1 variants, comparable to darunavir and its methoxy derivative 10.74,82 Indeed, our X-ray structural analysis of 11-bound HIV-1 protease and 10-bound HIV-1 protease complexes show that both oxygens of the Tp-THF ligand of inhibitor 11 promoted stronger hydrogen bond formation with the backbone NHs of Asp29 and Asp30.74
Based upon the above critical ligand-binding site interactions, we then speculated that a bicyclic 7–5-fused acetal-derived P2 ligand in inhibitor structure 17 (Figure 8) may further improve hydrogen bonding interactions in the active site, however, due to conformational instability of the seven-membered ring cycle, we planned to incorporate a methylene bridge to further stabilize the conformation. This hypothesis led to the design of a conformationally restricted crown-like tetrahydropyrano-tetrahydrofuran (crown-THF or Crn-THF) ligand shown in inhibitor structure 18. The ligand has three extra methylene groups compared to bis-THF ligand in darunavir and three methylene groups are expected to engage in van der Waals interactions with nearby residues in the active site. This Crn-THF ligand is a relatively larger P2 ligand than the bis-THF ligand of darunavir, and we speculated that it may confer better adaptability to protease mutation.
Figure 8.
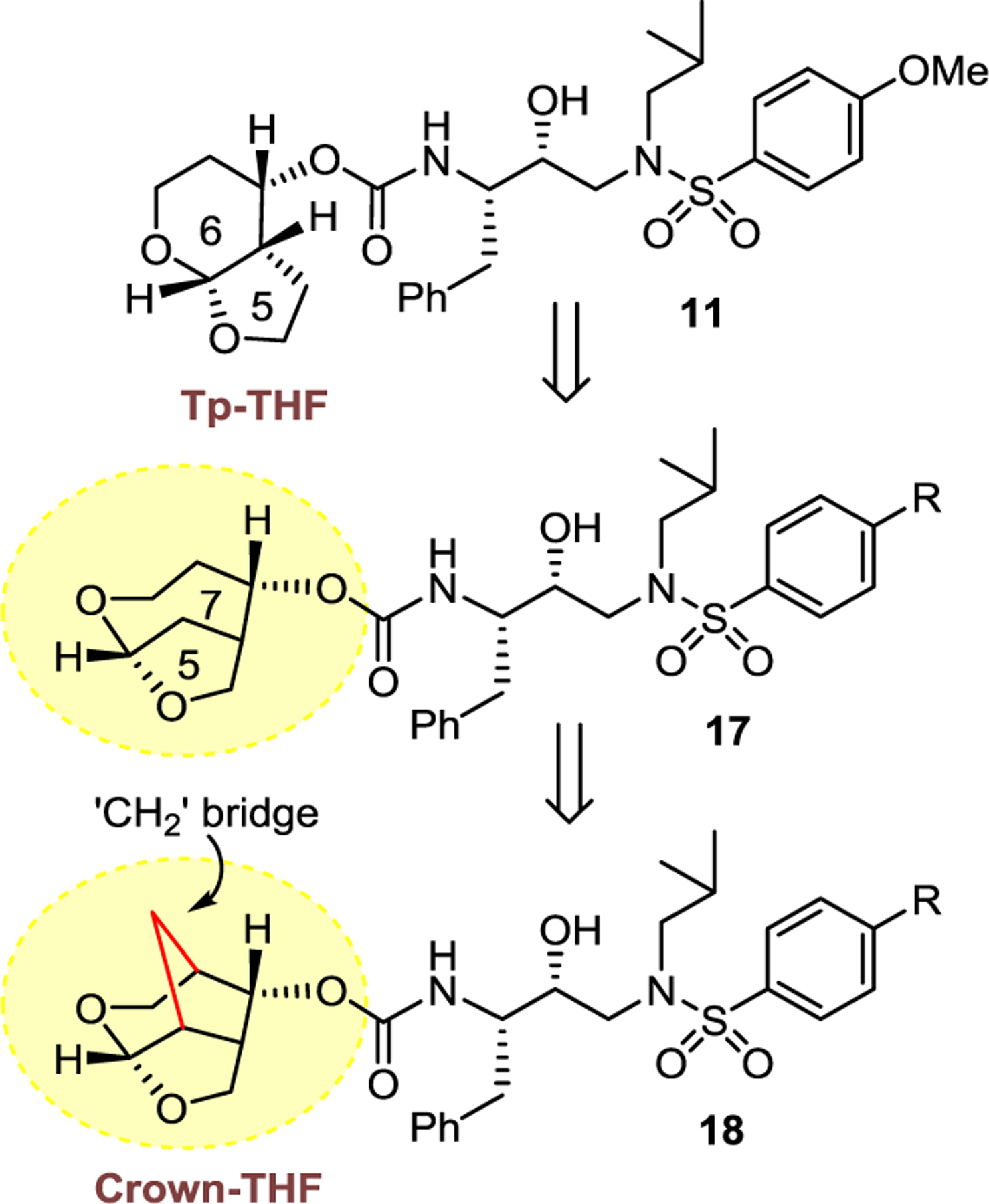
Design of Crown-THF P2 ligand
Our examination of the preliminary models of endo-crn-THF and exo-crn-THF-derived inhibitors indicated that the acetal oxygens in the endo-derivative are more favorably positioned to form hydrogen bonds with Asp30 and Asp29 amide NHs. The tricyclic core of endo-crn-THF ligand also appeared to fill the hydrophobic pocket in the S2 site more effectively than the exo isomer. As can be seen in Figure 9, inhibitor 19 with an endo-Crn-THF P2 ligand and a 4-methoxybenzenesulfonamide as the P2′-ligand, displayed an enzyme inhibitory Ki of 14 pM compared to inhibitor 20 with an exo-crn-THF ligand (Ki 10.8 nM).83,84 Inhibitor 21, with a p-aminobenzenesulfonamide as the P2′-ligand, exhibited a comparable Ki value to compound 19. Both inhibitors 19 and 21 with an endo-crn-THF P2 ligand exhibited very potent antiviral activity comparable to darunavir and inhibitor 10. Inhibitor 20 with an exo-crn-THF displayed no measurable antiviral activity.
Figure 9.
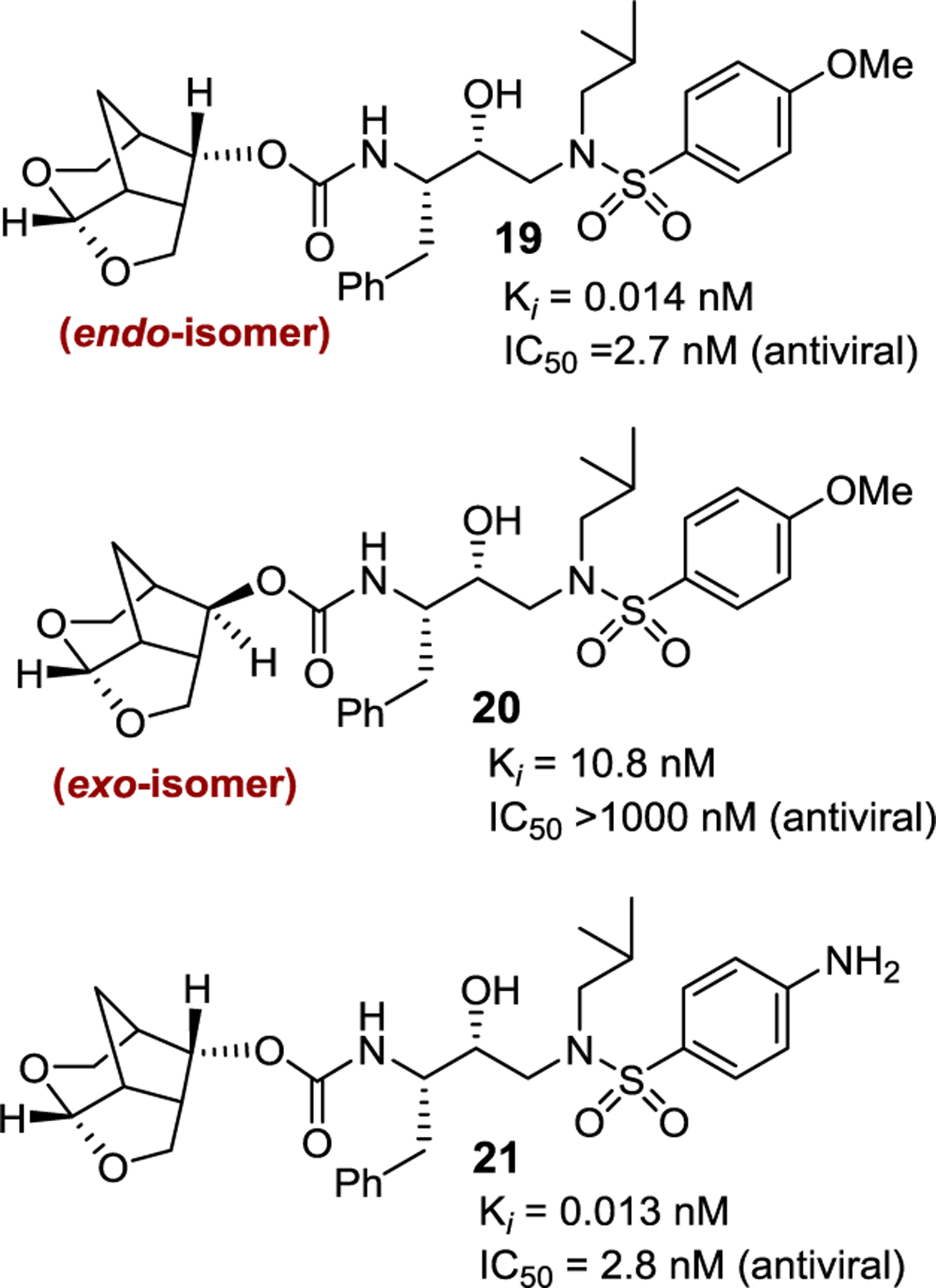
Structure and activity of inhibitor 19–21.
We determined a high resolution X-ray crystal structure of inhibitor 19-bound HIV-1 protease.83,85 As shown in Figure 10, both acetal oxygens of the tricyclic crown scaffold interact with backbone residues and the side chain Asp30 carboxylate group in S2 subsite, comparable to the interactions of darunavir. This Crn-THF ligand is conformationally restricted and larger than the P2 bis-THF ligand of darunavir. It displaced residues 45’–48’ by up to 0.8 Å, and resulted in an enlarged inhibitor-binding cavity. The Crn-THF ligand is also involved in significantly enhanced van der Waals interactions with Ile47, Val32, and Leu76 residues in the S2-site compared to bis-THF ligand of darunavir.
Figure 10.
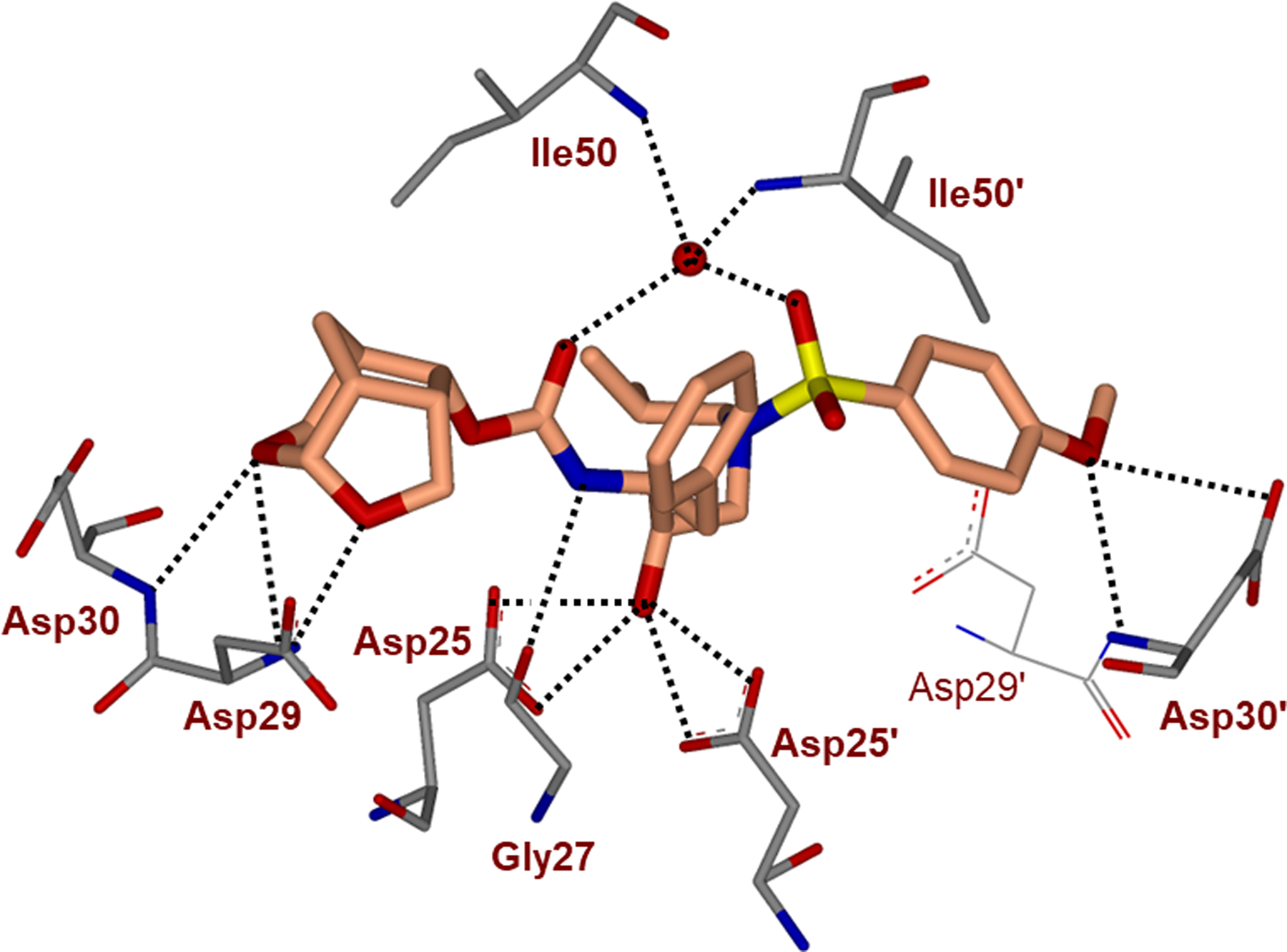
Inhibitor 19-bound X-ray structure of HIV-1 protease (PDB code: 5ULT). All hydrogen bonds are indicated by dotted lines.
For improving overall ligand-binding interactions of inhibitors, we then planned to replace P2’-ligand aminobenzene sulfonamide ligand of darunavir with other aromatic heterocycles that would promote enhanced hydrogen bonding interactions as well as to improve hydrophobic contacts in the S2’ subsite. In particular, we planned to incorporate various alkylaminobenzothiazole and benzoxazole hetereocycles in combination with the crown-THF P2 ligand for this new class of HIV-1 protease inhibitors. We selected these heterocycles as they are structural features of numerous medicinally important compounds with drug-like properties.86,87 Among various alkylamino groups examined by us, aminocyclopropylbenzo-thiazole (Abt) sulfonamide as the P2’ ligand showed the optimum results. As shown in Figure 11, replacement of the 4- aminosulfonamide P2’ ligand of inhibitor 21, with an aminocyclopropylbenzothiazole ligand, resulted in inhibitor 22, which displayed a slight reduction of enzyme Ki value, however there is a nearly 10-fold improvement of antiviral activity over inhibitor 21.83 In comparison, inhibitor 23 resulting from substitution of darunavir’s 4-aminosulfonamide P2’ ligand with an aminocyclopropylbenzothiazole ligand showed only slight improvement in antiviral activity over darunavir. Inhibitors 21, 22 and 23 were evaluated against DRV-resistant HIV-1 variants. Inhibitor 22 exhibited very potent antiviral activity against all HIV-1 variants examined compared to inhibitors 21, 23, and several FDA approved PI drugs.83,84
Figure 11.
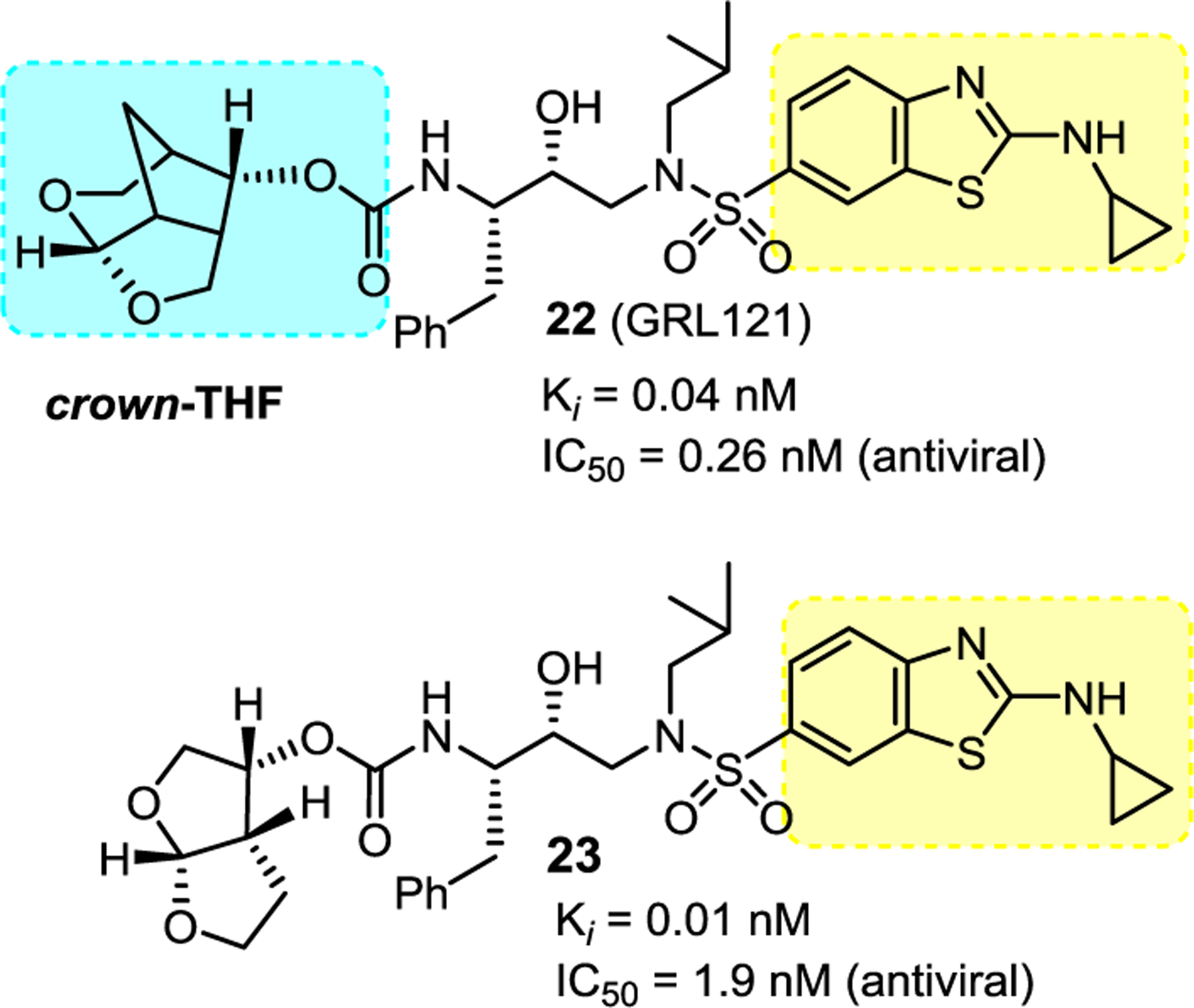
Structure and activity of inhibitor 22–23.
We determined the X-ray crystal structure of inhibitor 22-bound wild-type HIV-1 protease and the important interactions are shown in Figure 12. In particular, the P2′ ligand’s benzothiazole amine nitrogen also makes strong hydrogen bonds with the backbone NH of Asp30′ and the cyclopropyl amine nitrogen forms an additional hydrogen bond with the side chain carboxylate of Asp30′. The Abt ligand makes more hydrophobic contacts with the protease than the 4-aminobenzenesulfonamide ligand of DRV. Also, both oxygens of the crn-THF ligand formed strong hydrogen bonds with backbone amide NHs of Asp29 and Asp30. The tricyclic ligand scaffold is involved in van der Waals interactions with the HIV-1 protease.83 Presumably, these extensive molecular interactions are responsible for observed superior antiviral profiles of inhibitor 22 against a panel of highly multidrug-resistant HIV-1 variants.83,84
Figure 12.
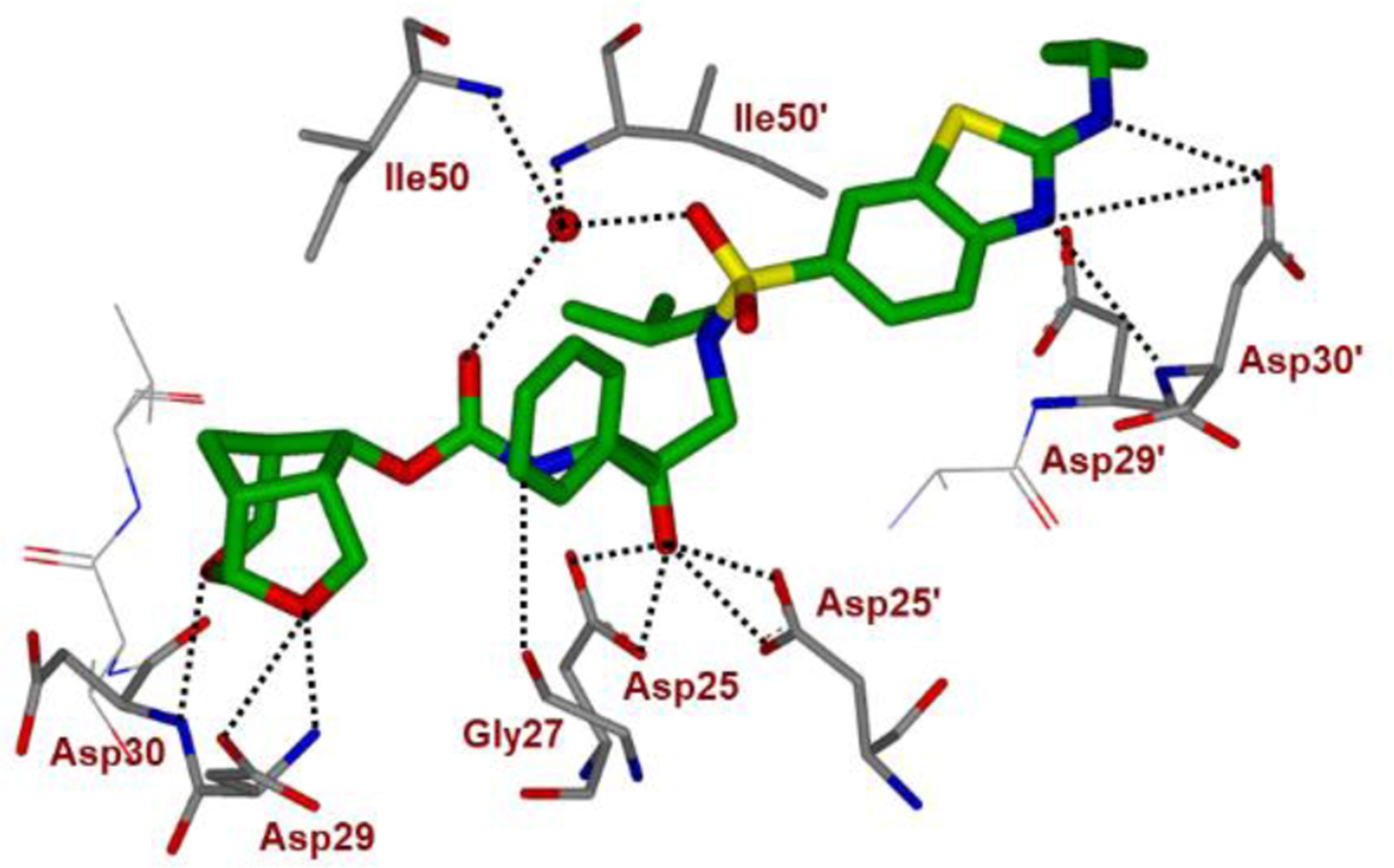
Inhibitor 22-bound X-ray structure of HIV-1 protease (PDB code: 5TYR). All hydrogen bonds are indicated by dotted lines.
Based upon our detailed structural analysis of inhibitor 22-bound HIV-1 protease, we then planned to further optimize the inhibitor-protease van der Waals hydrogen ligand-binding site interactions of inhibitor 22 to improve drug properties. Our overall plans were to preserve all backbone bonding interactions of 22 with HIV-1 protease and interactions in the active site. We planned to incorporate fluorine atoms in the P1-ligand to improve van der Waals contact in the S1-subsite. Our particular preference for fluorine atoms is due to fact that fluorines are often used to increase lipophilicity and help improve drug penetration in the CNS.88,89 As shown in Figure 13, our incorporation of a 3,5-difluorobenzyl moiety as the P1 ligand in 22 resulted in exceptionally potent inhibitor 2, displaying a protease inhibitory Ki value of 14 pM and antiviral IC50 value of 0.017 nM, a more than 150-fold potency enhancement over darunavir. Inhibitor 2 is over 15-fold more potent than inhibitor 22 with no fluorine, showing the importance of bis-fluorines on the P1-ligand.90,91 We also investigated activity of monofluoro derivatives. Inhibitor 24 with a 3-fluorophenylmethyl as the P1-ligand displayed a protease inhibitory Ki value of 0.3 pM and an antiviral IC50 of 0.055 nM. Inhibitor 25 with a 4-fluorophenylmethyl as the P1 ligand exhibited a reduction in both protease inhibitory and antiviral activity over its 3-fluoro derivative.90,92 We also investigated various alkylaminobenzoxazole derivatives as the P2’ ligand in place of P2’ ligand of inhibitor 2. Inhibitor 26 with an isopropylaminobenzoxazole displayed the best results, showing a Ki value 5 pM and antiviral IC50 of 0.055 nM.93
Figure 13.
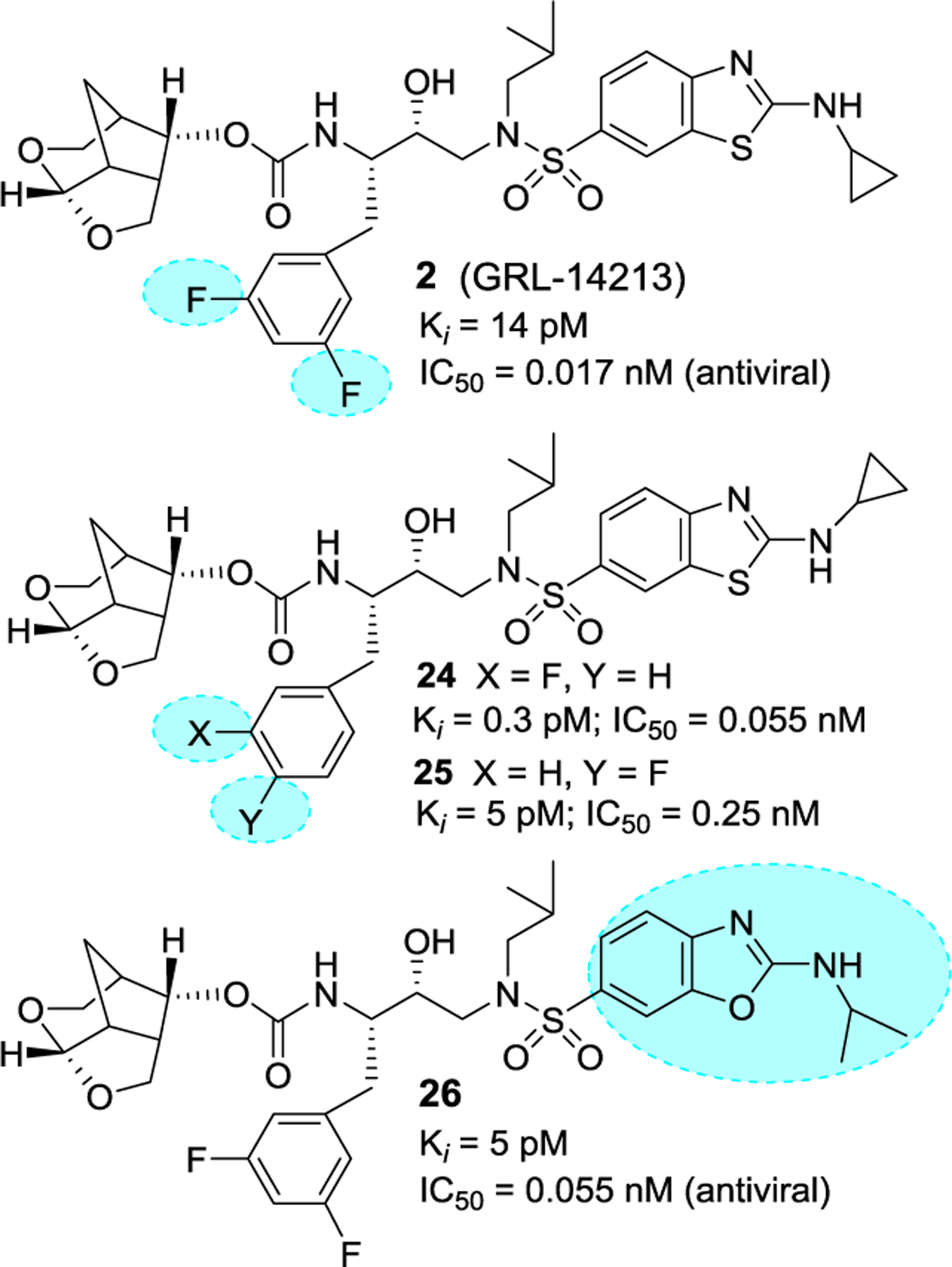
Structure and activity of inhibitors 2 and 24–26.
We evaluated inhibitor 2 and inhibitor 22 without fluorine against darunavir-resistant HIV-1 variants and compared antiviral IC50 values against approved PIs. These DRV-resistant variants HIV-1DRVRs are highly resistant to clinically used PIs including DRV and RT inhibitors (NRTIs) such as tenofovir. As shown in Table 4, both LPV and ATV failed to block the replication of DRV-resistant HIV-1 variants. Inhibitor 22 with no fluorine very effectively blocked the replication of all three highly DRV-resistant HIV-1 variants.90,91 However, inhibitor 2 with two-fluorines on the P1 ligand displayed exceptional antiviral activity in low picomolar concentration against DRV-resistant HIV-1 variants selected in the presence of DRV over 20 viral passages.94,95 Inhibitor 2 maintained IC50 values of 1.2 nM against HIV-1DRVR51, the most multi-PI/NRTI-resistant HIV-1 variants. In comparison, darunavir displayed >1000-fold increase in its IC50 values (IC50 2.5 μM). Furthermore, inhibitor 2 exerted very potent antiviral activity against the HIV-2 variants and showed much improved cytotoxicity profile with a selectivity index (CC50/IC50) as high as 2,473,684 compared to DRV (41,562).90,91
Table 4.
Antiviral activity of inhibitors 2 and 22 against highly DRV-resistant HIV-1 variants.
| Mean IC50 in nM ± SD (fold-change) | |||||
|---|---|---|---|---|---|
| LPV | ATV | DRV | 22 (GRL-121) | 2 (GRL-142) | |
| cHIVnl4-3WT | 13 | 4.0 | 3.2 | 0.26 | 0.019 |
| HIVDRVRP20 | >1000 (>77) | 450 (113) | 51 (16) | 0.075 (0.3) | 0.0024 (0.1) |
| HIVDRVRP30 | >1000 (>77) | >1000 (>250) | 220 (79) | 1.9 (7) | 0.023 (1) |
| HIVDRVRP51 | >1000 (>77) | >1000 (>250) | 2500 (781) | 32 (279) | 1.2 (63) |
Numbers in parentheses represent fold changes in IC50s for each isolate compared to the IC50s for wild-type cHIVNL4-3WT. All assays were conducted in triplicate, and the data shown represent mean values (±1 standard deviation) derived from the results of three independent experiments.
Inhibitor 22 was then evaluated against a variety of HIV-1 variants that had been selected in vitro with other FDA-approved PI drugs. These variants acquired multiple amino acid substitutions in the protease of the virus resulting in viral resistance to each PI drug. As shown in Table 5, two widely used PIs LPV and ATV failed to block these variants effectively compared to DRV. Inhibitor 22 without fluorine, maintained very potent antiviral activity against all resistant HIV-1 variants (IC50 values ranging from 1.8 pM to 0.13 nM). Inhibitor 2 with bis-fluorines displayed exceptional antiviral activity against all seven highly P1-resistant HIV-1 variants, showing antiviral IC50 values, ranging from 8.5 fM to 0.015 nM.90,91
Table 5.
Antiviral activity of 2 and 22 against highly PI-resistant HIV-1 variants.
| Mean IC50 in nM ± SD (fold-change) | ||||||
|---|---|---|---|---|---|---|
| Virus Species | LPV | ATV | DRV | 22 (GRL-121) | 2 (GRL-142) | |
| cHIVnl4–3WT | 13 ± 2 | 4.0 ± 2.3 | 3.2 ± 0.7 | 0.26 ± 0.05 | 0.019 ± 0.017 | |
| HIVSQV-5μM | >1000 (>77) | 430 ± 20 (108) | 17 ± 7 (5) | 0.026 ± 0.01 (0.1) | 0.00018 ± 0.00003 (0.009) | |
| HIVAPV-5μM | 280 ± 15 (22) | 3.0 ± 1.0 (1) | 39 ± 16 (12) | 0.13 ± 0.08 (0.5) | 0.0000085 ± 0.000008 (0.0004) | |
| HIVLPV-5μM | >1000 (>77) | 46 ± 10 (12) | 280 ± 50 (86) | 0.0018 ± 0.001 (0.007) | 0.000019 ± 0.0000014 (0.0001) | |
| HIVIDV-5μM | 250 ± 15 (19) | 56 ± 9 (14) | 37 ± 8 (12) | 0.0092 ± 0.016 (0.04) | 0.00018 ± 0.00028 (0.009) | |
| HIVNFV-5μM | 37 ± 3 (3) | 12 ± 2 (3) | 7.7 ± 3 (2) | 0.048 ± 0.02 (0.2) | 0.00024 ± 0.00026 (0.01) | |
| HIVATV-5μM | 310 ± 20 (24) | >1000 (>250) | 25 ± 1 (8) | 0.092 ± 0.09 (0.4) | 0.015 ± 0.004 (0.8) | |
| HIVTPV-15μM | >1000 (>77) | >1000 (>250) | 40 ± 3 (13) | 0.063 ± 0.02 (0.2) | 0.00024 ± 0.00007 (0.01) | |
invitroHIVPIR, in vitro PI-selected HIV-1 variants;
rCLHIV, recombinant clinical HIV-1 variants. Numbers in parentheses represent fold changes in IC50s for each isolate compared to the IC50s for wild-type cHIVNL4–3WT. All assays were conducted in triplicate, and the data shown represent mean values (±1 standard deviation) derived from the results of three independent experiments. DOI: https://doi.org/10.7554/eLife.28020.005
We assessed genetic barrier to the emergence of HIV-1 variants resistant to inhibitors 2 and 22 and compared with other approved PI drugs. We aimed to select HIV-1 variants resistant to ATV and DRV and inhibitors 2 and 22 by propagating CHIVNL4–3WT using increasing concentrations of each inhibitor. As shown in Figure 14 (top panel), HIV-1 became resistant to ATV and started replicating in the presence of 5 μM by week 36. There were 7-amino acid substitutions in the protease encoding region.91 However, the emergence of DRV-resistant variants were considerably delayed at concentration above 0.1 μM DRV at week 36. In comparison, CHIVNL4–3WT failed to spread in the presence of 0.01 μM concentration of 22 or in the presence of 0.003 μM concentrations of 2, even after 36 weeks, indicating that HIV-1 failed to select any meaningful substitutions in the PR encoding region of the virus. The effect of inhibitors 2 and 22 against a mixture of 11 multi-PI-resistant clinical HIV-1 isolates (HIV11MIX) were also evaluated and the results are shown in Figure 14 (bottom panel). As can be seen, the virus quickly became highly resistant to both ATV and DRV however, both inhibitors 2 and 22 very effectively blocked the propagation of HIV11MIX in the presence of >0.07 nM inhibitor 22 or >0.017 nM concentration of inhibitor 2. The results indicate that inhibitor 2 displayed an extremely high genetic barrier to resistance.91
Figure 14.
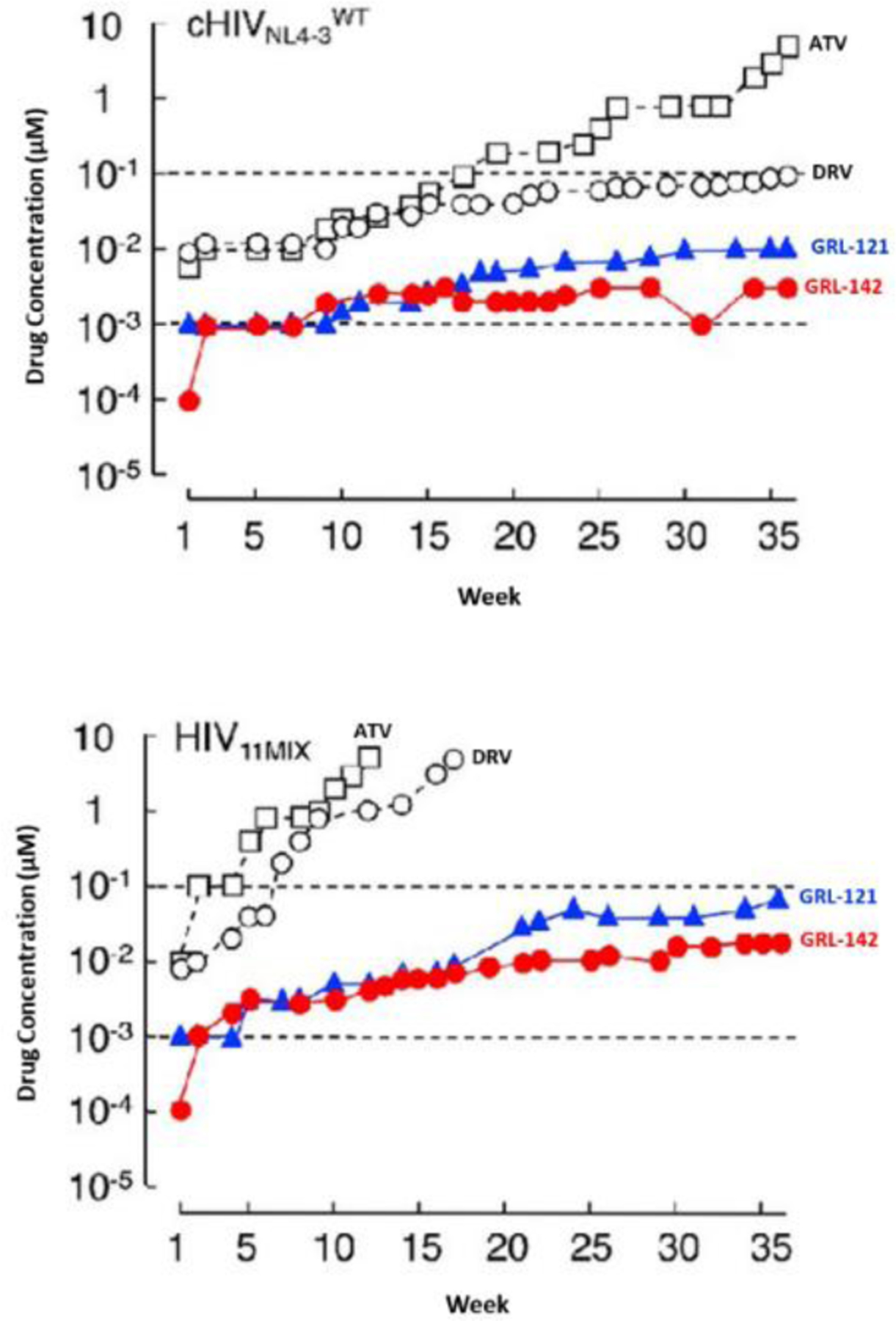
Inhibitor 2 shows high genetic barrier to the development of HIV-1 variants in vitro. Top panel A, against HIVNL4–3; bottom panel B, against a mixture of 11 multi-PI-resistant HIV-1 isolates (HIV11MIX). DOI: https://doi.org/10.7554/eLife.28020.005
It appears that inhibitor 2, like DRV, exhibits a dual mechanism of action, (i) inhibition of HIV-1 protease dimerization and (ii) inhibition of proteolytic activity of HIV-1 protease. Dimerization of HIV-1 protease subunits is an essential process for the acquisition of proteolytic activity of HIV-1 protease. We previously demonstrated that DRV and TPV inhibit the dimerization of HIV-1 protease monomers by using the FRET-based HIV-1 expression assay.69,70 Other approved PIs do not block dimerization of HIV-1 protease subunits. We have now observed that inhibitor 2 blocks protease dimerization very potently, at 0.1 nM concentration, which were significantly lower than DRV. It improved by at least a factor of 1000-fold (thousand) compared to DRV.91 The data suggests that inhibitor 2 could be developed as a monotherapy.
Inhibitor 2 is more lipophilic than inhibitor 22 or DRV. Inhibitor 2 shows favorable penetration into the brain of rats. When DRV and compound 2 were perorally administered to rats (n = 2) at a dose of 5 mg/kg plus RTV (8.33 mg/kg), the Cmax was achieved around 90 and 360 min after the administration, respectively. The concentrations of DRV and inhibitor 2 in plasma, CSF, and brain were determined in 15 and 90 min for DRV and 60 and 360 min for inhibitor 2 after the administration. Drug concentrations of 2 in the brain were 7.24 ± 9.65 and 32.6 ± 1.4 nM in 60 and 360 min, respectively. The latter concentration (32.6 nM) represents ~1,882 fold greater than the IC50 value and ~114 fold greater than the IC95 value of inhibitor 2. The data strongly suggest that inhibitor 2 would potently block the infection and replication of HIV-1 in the brain.90,91 This may serve to prevent or treat HAND complication.
We determined a high resolution X-ray crystal structure of inhibitor 2-bound wild-type HIV-1 protease and the important interactions are shown in Figure 14. Inhibitor 2 retains all key hydrogen bonds observed between DRV and the backbone atoms of HIV-1 protease. The P2’ thiazole nitrogen of 2 forms a hydrogen bond interaction with the backbone amide of Asp 30′ similar to the interaction of 4-aminobenzene of DRV. The P2′ amino group forms a strong hydrogen bond interaction with the Asp 30′ side chain. Interestingly, the cyclopropyl group bulges out and forms N-H…O interactions with the side chain of Asp 30′. Both oxygen atoms in the Crn-THF form strong hydrogen bonds with the backbone amide NHs of Asp29 and Asp30. In addition, the Crn-THF ligand has formed enhanced van der Waals interactions in the S2 subsite compared to the bis-THF ligand of darunavir.90,91
As shown in Figure 16, both fluorine atoms on the P1 ligand of inhibitor 2 form important halogen bond interactions in the protease active site. One of the fluorine atoms interacts with the tips of both flaps by forming a strong polar interaction with the backbone NH group of Ile50 (C-F…H-N) and an orthogonal multipolar interaction (C-F…C-O) with the backbone carbonyl ofGly49.[41,42] This fluorine makes hydrophobic interactions with Gly48, Ile50 and Pro81′. The second fluorine atom forms multipolar interactions with the guanidium group of Arg8′, stabilizing the dimer of HIV-1 protease by a critical inter-subunit ion pair. This fluorine atom also has van der Waals contact with Val82′. Both P1 and P2′ ligands in inhibitor 2 considerably contributed additional interactions, improving the favorable inhibitor-HIV-1 protease interactions in comparison to DRV. These new ligand-binding site interactions are likely to account for the improved activity of inhibitor 2.
Figure 16.
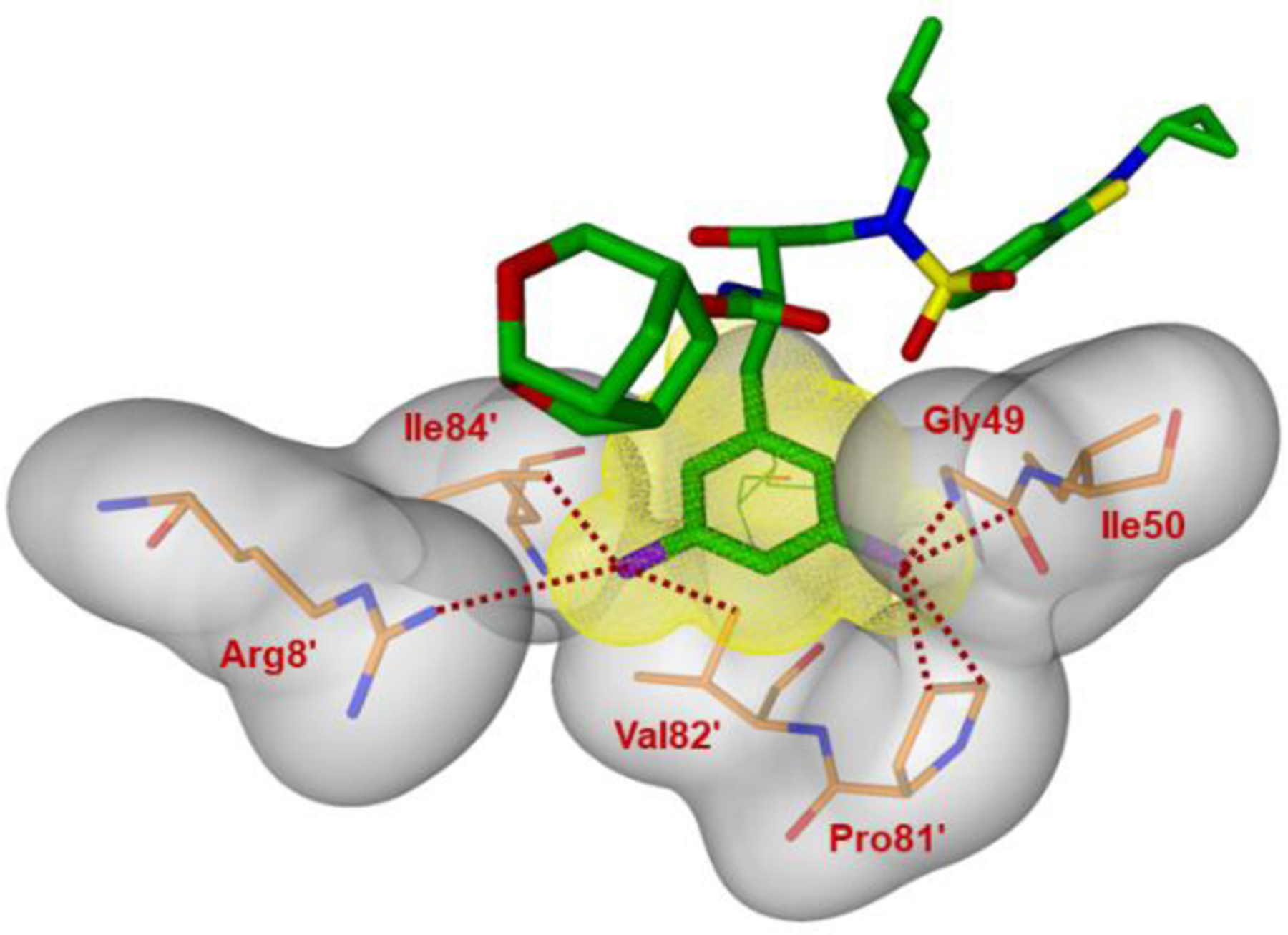
Side view of the S1 subsite. The protein surface is shown in transparent gray. The van der Waals surface for the fluorinated P1-ligand for inhibitor 2 is shown in orange wire mesh and van der Walls contacts are shown by dotted lines.
Synthesis of Crown-THF-derived HIV-1 Protease Inhibitor 2 (GRL-142)
Our synthetic strategy for inhibitor 2 is shown in Scheme 1. Inhibitor 2 was obtained by alkoxycarbonylation of ligand alcohol 27 and hydroxyethylaminosulfonamide amine derivative 28.96,97 Stereochemically defined tricyclic ligand alcohol 27 was synthesized from 3-hydroxybicyclo[2.2.1]hept-5-ene derivative 29. We planned to synthesize this intermediate via an enantioselective Diels-Alder reaction. The fluorinated isosteric amines were synthesized from azido diol 30 utilizing Sharpless asymmetric epoxidation98,99 of the corresponding fluorinated allylic alcohol, followed by regioselective epoxide opening as described by us previously.100
Scheme 1.

Synthetic strategies for the preparation of inhibitor 2
The synthesis of tricyclic crown-THF ligand alcohol 27 in optically active form is shown in Scheme 2. As described previously, we utilized a chiral oxazaborolidinium cation catalyzed Diels–Alder reaction developed by Corey and Mukherjee.101,102 The Diels-Alder reaction of readily available ethyl vinyl boronate 31103 and cyclopentadiene afforded the corresponding cycloadduct, which was oxidized with H2O2 to furnish optically active alcohol 29 in excellent yield and in excellent optical purity (98% ee). Alcohol 29 was then converted to bicyclic acetal 33 in a four-steps to provide bicyclic acetal 33 (2:1 mixture). The lactol mixture was exposed to TFA to provide tricyclic exo-alcohol 34 in good yield. This exo-alcohol was then converted to endo-alcohol 27 by oxidation followed by reduction of the resulting ketone.104 Crown-THF ligand alcohol, (3S,3aS,5R,7aS,8S)-hexahydro-4H-3,5-methanofuro[2,3-b]-pyran-8-ol (27) was obtained in good yield. This overall route is efficient and provided access to both endo- and exo-ligand alcohols in optically active form (>99% ee).
Scheme 2.
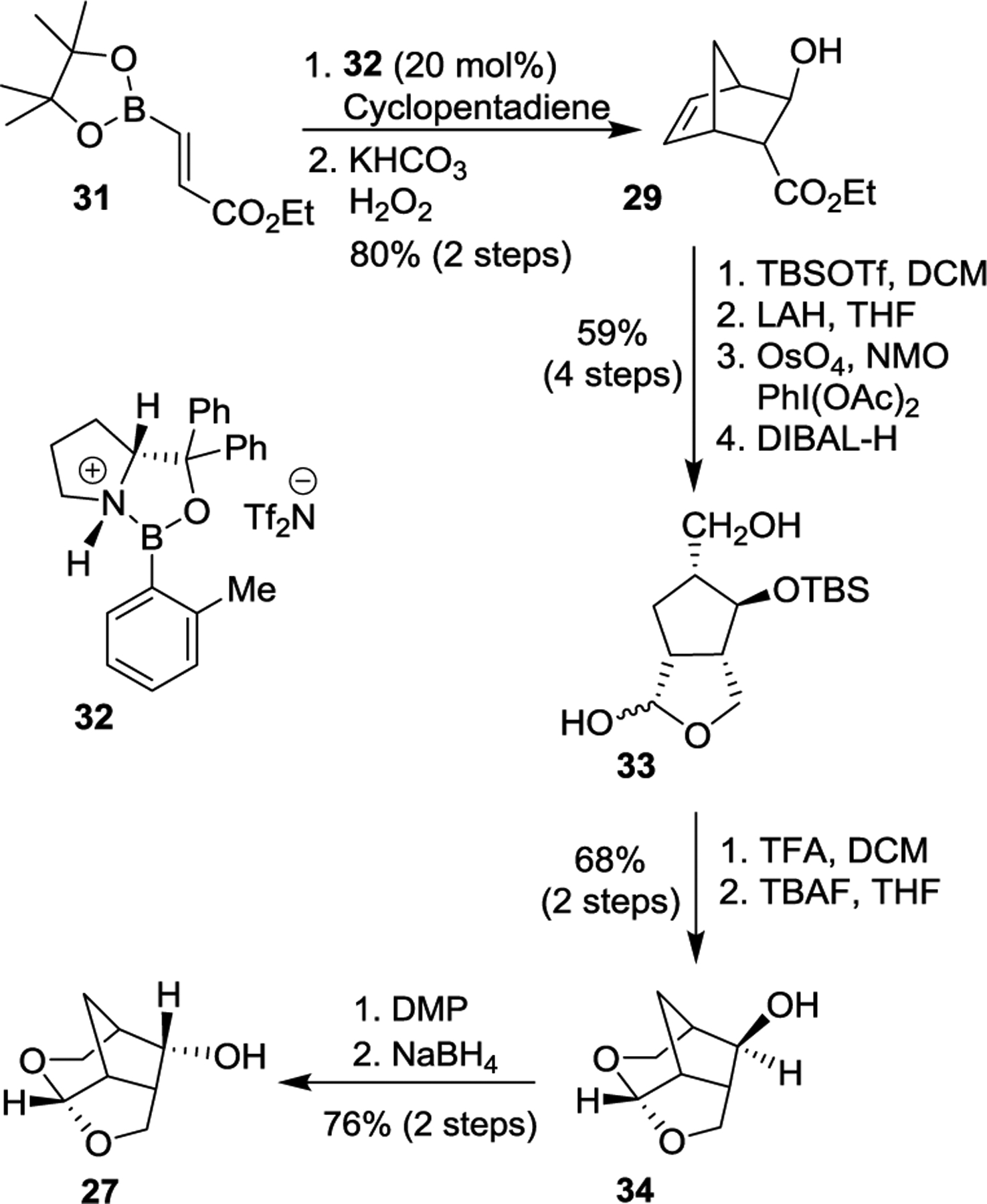
Synthesis of Crn-THF ligand 27.
Our synthesis of the 3,5-difluorobenzene epoxide derivative for the synthesis of inhibitor 2 is shown in Scheme 2. Commercially available 1-bromo-3,5-difluorobenzene 35 was converted to its 3,5-difluorophenyl magnesium bromide and this Grignard reagent was reacted with butadiene monoxide to provide allylic alcohol 16.61 Sharpless asymmetric epoxidation of 16 with (−)-diethyl-D-tartrate resulted in the corresponding epoxide which was reacted with TMSN3 and Ti(OiPr)4 to afford the azido diol 37.98,99,105 Diol 37 was then converted to epoxide 38 as described previously.106,107
Epoxide 38 was then converted to inhibitor 2 as shown in Scheme 4. Reaction of 38 with isobutylamine provided azidoalcohol 39. This was then reacted with sulfonyl chloride 40 to provide the sulfonamide, which was converted to Boc amine derivative 41. Oxidation of the sulfide provided the sulfone, which was then reacted with cyclopropylamine followed by deprotection of Boc with TFA to furnish aminoalcohol 42. Crown-THF ligand alcohol 27 was converted to activated carbonate 43 which reacted with amine 42 to furnish inhibitor 2 in good overall yield.90,91
Scheme 4.
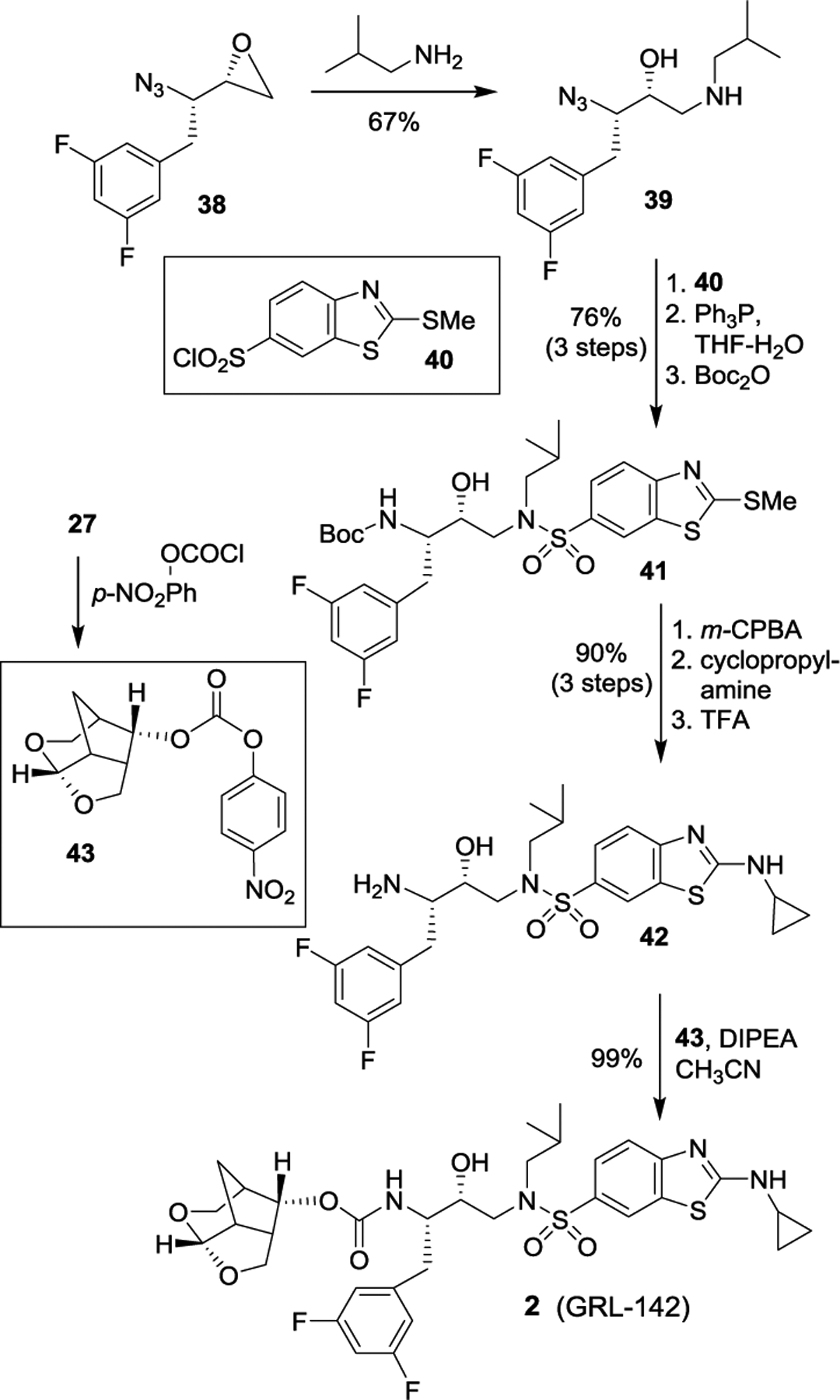
Synthesis of protease inhibitor 2.
Synthesis of Fused Tricyclic Polyether-derived HIV-1 Protease Inhibitors
Recently, we have designed, synthesized and evaluated another new class of HIV-1 protease inhibitors containing stereochemically defined fused tricyclic polyethers as the P2 ligands and a variety of sulfonamide derivatives as the P2’ ligands.108 As described previously, based upon the X-ray crystal structure of DRV (1)-bound HIV-1 protease and its methoxy derivative 10-bound HIV-1 protease, we have designed hexahydrofuropyranol-derived urethane as the P2-ligand in inhibitor 11, which exhibited enhanced activity over inhibitor 10.74 Inhibitor 11 maintained potent antiviral activity against drug-resistant HIV-1 strains similar to darunavir and its methoxy derivative 10. In an effort to promote further backbone interactions in the S2 subsite with the bicyclic acetal functionality, we speculated that a methylene group would be more effective. As shown in Figure 17, a hydroxymethyl urethane in inhibitor structure 44 would be optimum for effective contact with the backbone amide NHs. However, we planned to incorporate a methylene bridge to promote stablity as shown in inhibitor structure 45. An inhibitor model shows an increase of the dihedral angle of the acetal template and better alignment with Asp29 and Asp30 backbone amide NHs. The resulting preorganized and conformationally constrained octahydro-2H-1,7-dioxacyclopenta[cd]indene incorporates three extra methylene groups over bis-THF ligand in DRV. This ligand is expected to make more favorable van der Waals interactions since these methylene groups are located in the hydrophobic space surrounding Ile47, Val32, Leu76, and Ile50′ residues in the S2 subsite. We presumed that the umbrella-like tetrahydropyranofuran (Umb-THF) may show better adaptability to protease mutation. We planned to optimize ligand-binding site interations of inhibitors with an Umb-THF P2 ligand in combination with a fluorinated phenylmethyl P1 ligand and benzothiazole derivatives to improve activity and drug resistance profiles.
Figure 17.

Design of Umb-THF P2 ligand
We have synthesized a series of inhibitors incorporating Umb-THF as the P2 ligand in combination with other P1 and P2’ ligands.108 All inhibitors containing the Umb-THF as the P2 ligand provided very potent activity. As can be seen in Figure 18, inhibitor 46 with an (2aS,2a1S,4R,4aS,7aS)-octahydro-2H-1,7-dioxacyclopentaindenyl urethane as the P2 ligand with a 4-methoxybenzenesulfonamide as the P2′-ligand, displayed an enzyme inhibitory Ki of 8 pM and antiviral IC50 value of 9 nM. Interestingly inhibitor 47 with an enantiomeric ligand also showed very potent activity however, both inhibitors displayed reduced antiviral activity compared to inhibitor 10 or 11. We have also investigated the effect of alternative fused tetrahydrofurano-pyran rings in inhibitor 48. This inhibitor showed a reduction in enzyme and antiviral activity compared to inhibitor 46.108
Figure 18.
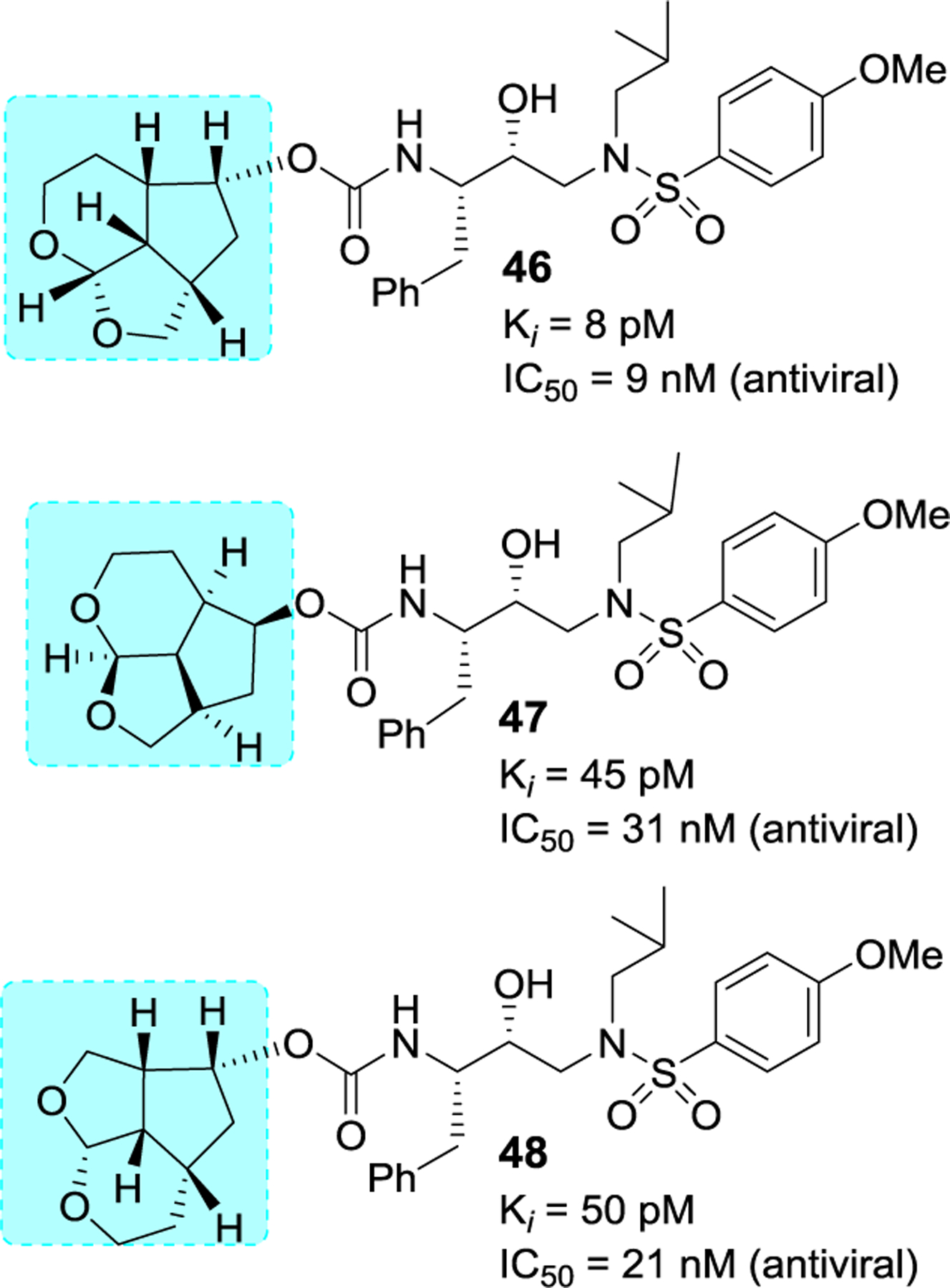
Structure and activity of inhibitors 46–48.
We then examined aminocyclopropylbenzothiazole (Abt) sulfonamide as the P2’ ligand. As shown in Figure 19, both inhibitors 49 and 50 with the enantiomeric Umb-ligand as the P2 ligand and Abt as the P2’ ligand displayed significant improvement in antiviral activity compared to inhibitors 46 and 47 with 4-methoxybenzenesulfonamide as the P2′-ligand. Then, incorporation of a 3,5-difluorobenzyl moeity as the P1 ligand in inhibitors 49 and 50 provided inhibitors 51 and 52 with enantiomeric P2 ligands. Both inhibitors exhibited exceptionally potent activity, displaying antiviral IC50 values of 23 pM for inhibitor 51 and 27 pM for inhibitor 52, comparable to Crown-THF-derived inhibitor 2.
Figure 19.
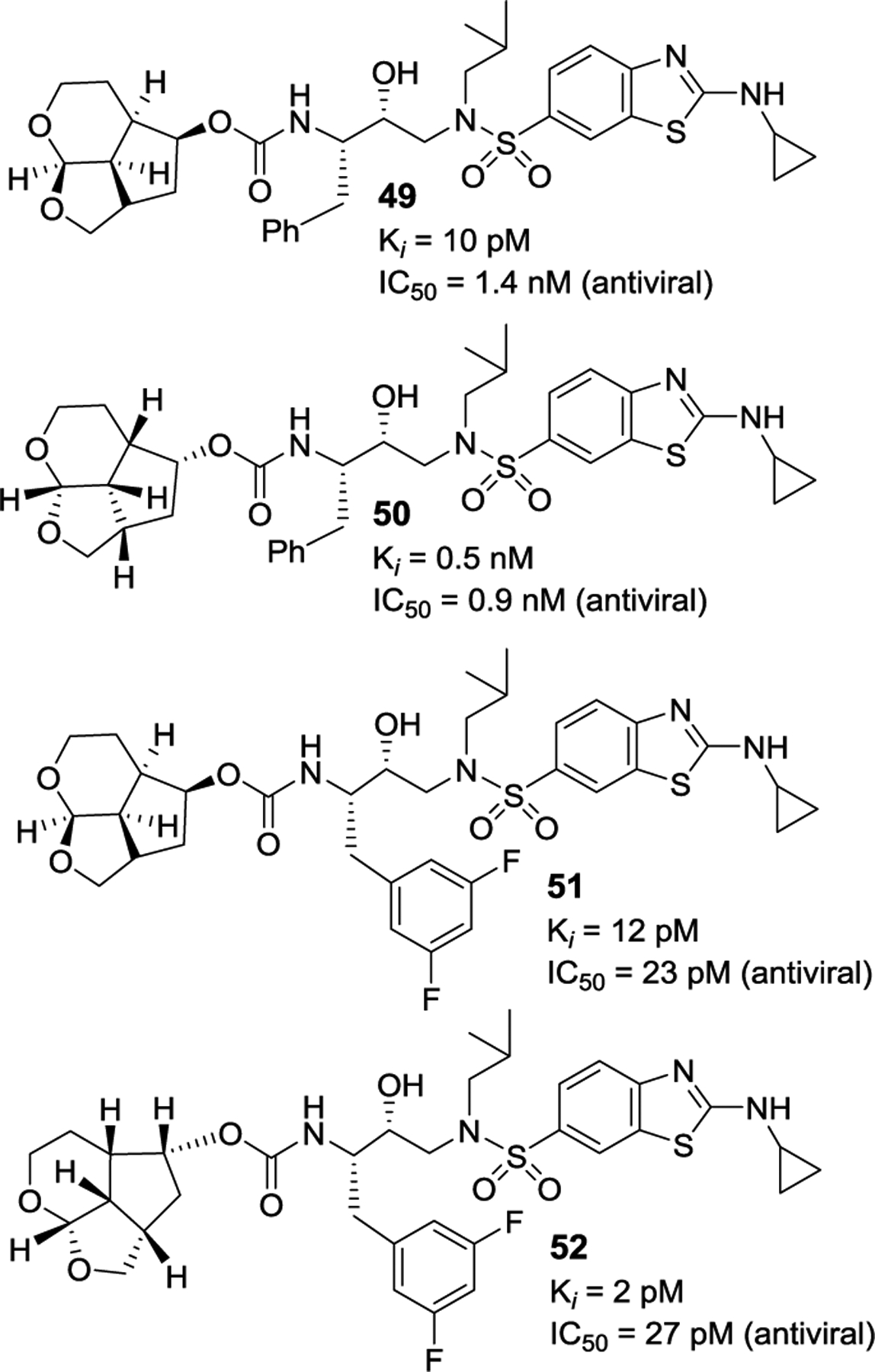
Structure and activity of inhibitors 49–52.
We evaluated the antiviral activity of both potent Umb-THF-derived inhibitors 51 and 52 against a panel of HIV-1 variants that had been selected in vitro with three widely used FDA-approved PI drugs as described previously.109,110 The results are shown in Table 6. As can be seen, both inhibitors 51 and 52 maintained exceptional activity against all three HIV-1 variants with EC50 values ranging from 0.0003 to 0.002 nM. Essentially, both inhibitors did not show any loss in antiviral activity compared to wild-type HIVNL4–3.
Table 6.
Comparison of the antiviral activity of 51 and 52 and other PIs against highly PI-resistant HIV-1 variants.
| EC50 (nM)a | ||||||
|---|---|---|---|---|---|---|
| Virusa | LPV | APV | ATV | DRV | 51 | 52 |
| HIV-1NL4–3 | 0.032 | 0.087 | 0.0033 | 0.003 | 0.002 | 0.0012 |
| HIV-1ATVR5μM | >1 (>31) | >1 (>11) | >1 (>303) | 0.024 (8) | 0.0015 (1) | 0.0006 (0.3) |
| HIV-1LPVR5μM | >1 (>31) | 0.19 (2) | 0.029 (9) | 0.026 (9) | 0.002 (2) | 0.002 (1) |
| HIV-1APVR5μM | 0.39 (12) | >1 (>11) | 0.07 (21) | 0.2 (67) | 0.00032 (0.3) | 0.0003 (0.2) |
The EC50 (50% effective concentration) values were determined by using MT-4 cells as target cells. MT-4 cells (105/mL) were exposed to 100 TCID50s of each HIV-1, and the inhibition of p24 Gag protein production by each drug was used as an endpoint. All assays were conducted in duplicate, and the data shown represent mean values (± S.D.) derived from the results of two independent experiments.
We determined high resolution X-ray crystal structures of both inhibitors 51- and 52-bound HIV-1 protease. All key inhibitor-protease interactions are shown in Figure 20. The Umb-THF P2 ligand forms stronger hydrogen bonds with backbone atoms in the S2-site. Also, both enantiomeric ligands make enhanced van der Waals interactions in S2-site compared to the bis-THF ligand of DRV. The acetal oxygens are suitably positioned and formed strong hydrogen bonds with backbone NHs of Asp29 and Asp30. The Umb-THF ligands form significant van der Waals interactions with the side chain atoms of Ile50, Ile47, Val32, and Ile84 in the S2 subsite. The bulging P2 ligand in 52 is nicely accommodated by shifting the Gly48 carbonyl group in flap region into an alternate conformation. Interestingly, the enantiomeric Umb-THF ligand in inhibitor 51 rotated towards the CD1 atom of Ile50 to form a van der Waals contact. These interactions are extensive and they contributed towards the exceptional potency and superior drug resistance properties of inhibitors 51 and 52.108
Figure 20A.
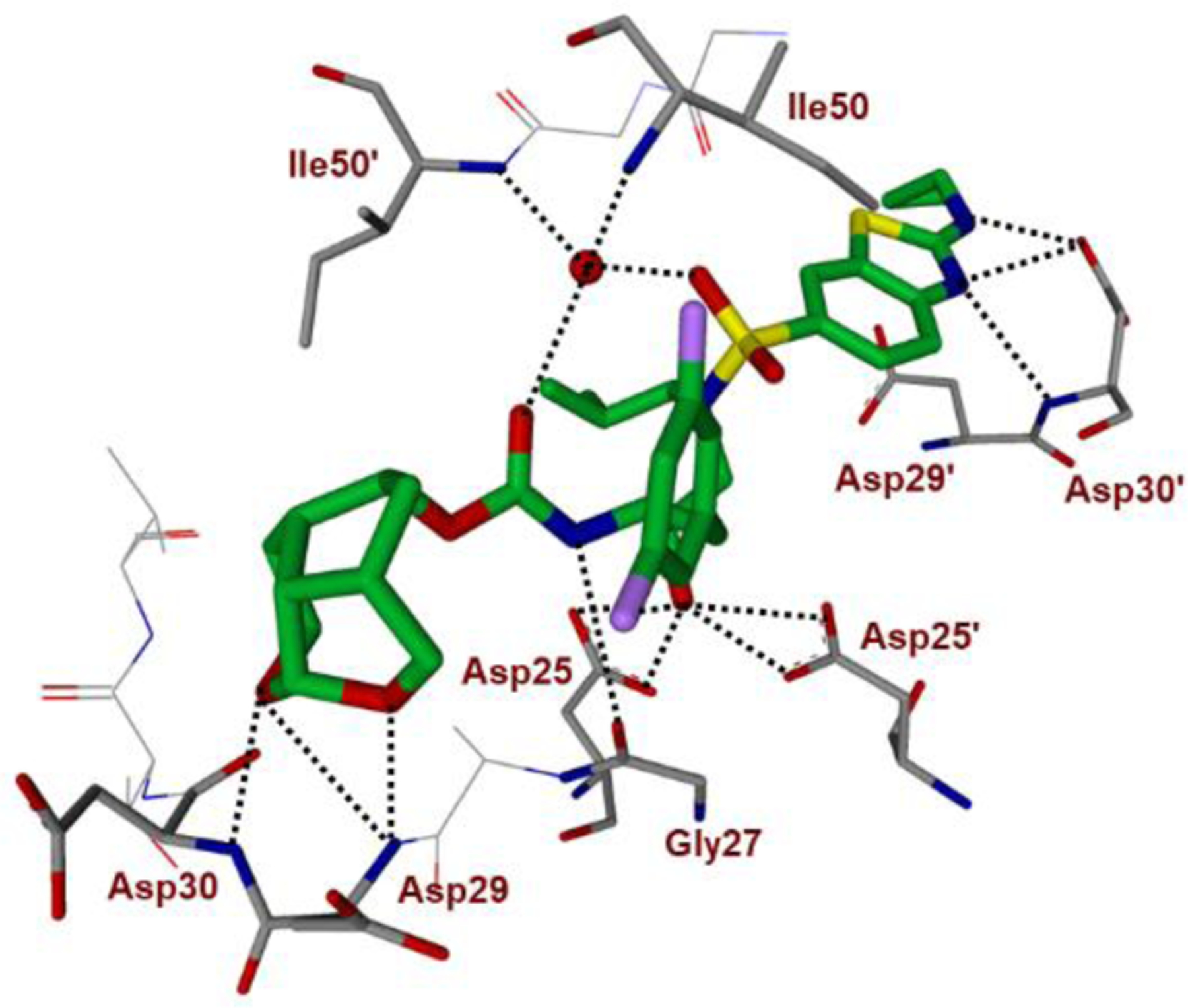
Inhibitor 52-bound HIV-1 protease X-ray structure is shown (PDB code: 6CDJ). The inhibitor carbon atoms are shown in green and hydrogen bonds are shown by black dotted lines.
Our synthesis of optically active Umb-THF ligand alcohol is shown in Scheme 5. Dihydropyran was converted to bromo ether 13 using NBS and propargyl alcohol. Dehydrobromination of bromo ether provided ene-yne derivative 14.111 Pauson-Khand cyclization112,113 of 14 with CO2(CO)8 in hexane furnished tricyclic enone 15. Transfer hydrogenation followed by reduction of the resulting ketone furnished racemic ligand alcohol 16. Lipase catalyzed enzymatic resolution114,115 of racemic alcohol 16 afforded optically active alcohol (+)-16 and its acetic derivative 17. Saponification of 17 provided optically active ligand alcohol (−)-16.
Scheme 5.
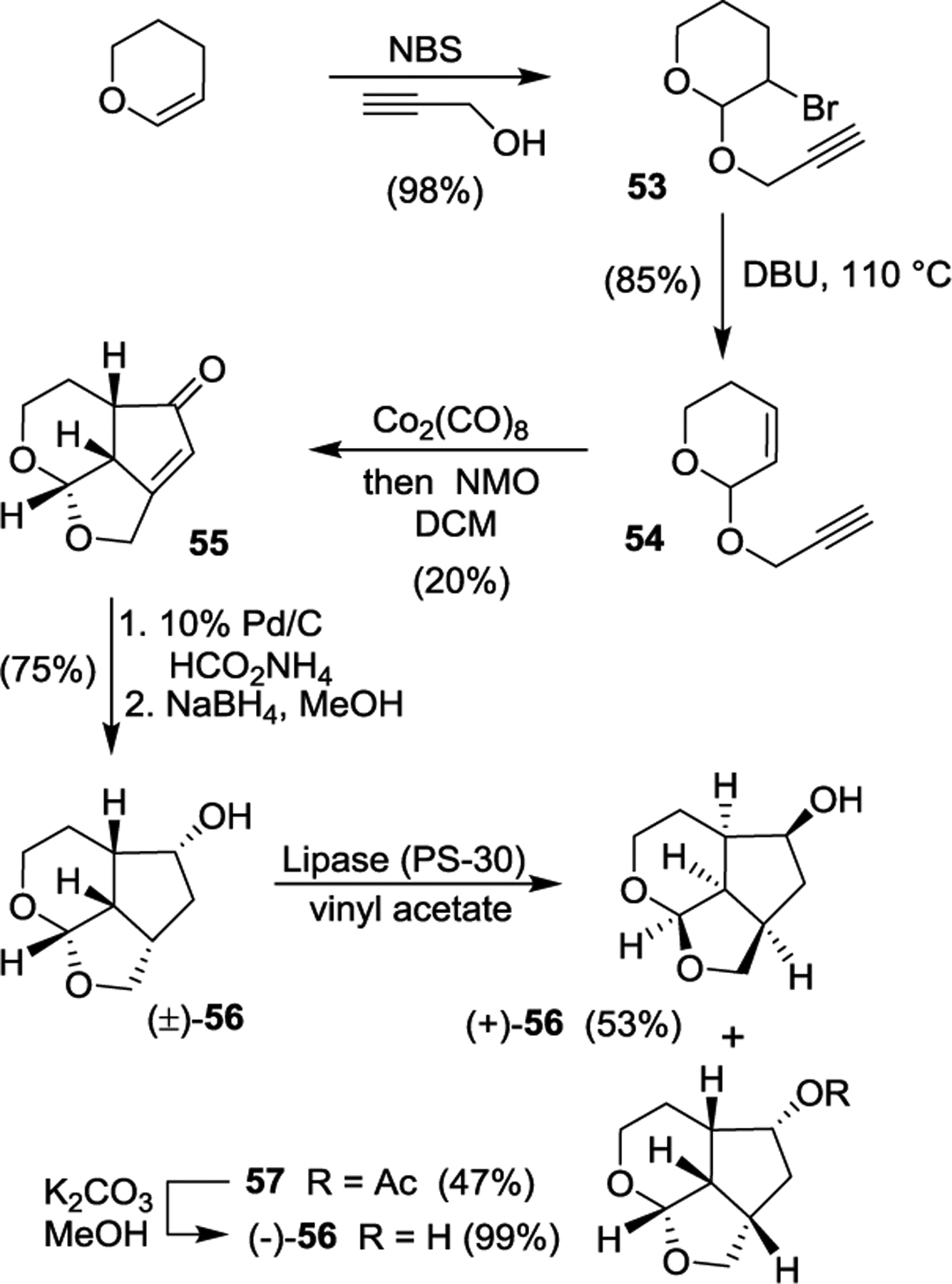
Synthesis of optically active ligand alcohol 16.
Above optically active ligand alcohols were converted to their respective mixed activated carbonate derivatives 18 and 19. Reaction of these carbonates with amine 42 containing fluorines in the P1 ligand, furnished inhibitors 51 and 52, respectively. The overall route involving Pauson-Khand cyclization as a key step provided convenient access to various Umb-THF ligands for SAR and optimization work.108 Our preliminary results show that the combination of the fused tricylic Umb-THF as the P2 ligands, a cyclopropylaminobenzothiazole derivative as the P2’-ligand, and a 3,5-difluorophenylmethyl as the P1 ligand resulted in exceptionally potent inhibitors with marked drug resistance profiles. Further optimization of the ligand-binding site interactions based upon the X-ray structures of inhibitor-HIV-1 protease complexes may lead to inhibitors with improved properties.
Recent development of related HIV-1 protease inhibitors from other laboratories.
The design, synthesis, and optimization of protease inhibitors for clinical development continues to be a very active area of research. Darunavir, the last approved protease inhibitor drug, sets a high standard for next generation protease inhibitors. Several laboratories recently reported further structural modification of darunavir scaffold to improve potency and drug-resistance properties. We will report here new and relevant inhibitors since 2016 that are not reported in previous reviews.19,57
Based upon X-ray structures of darunavir-bound HIV-1 protease (PDB ID: 4HLA), we have shown that incorporation of carboxylic acid, carboxamide or stereochemically defined hydroxyl alkyl groups on the P2’-ligand of darunavir led to enhanced hydrogen bonding interactions in the S2’-subsite.116 As shown in Figure 21, compounds 60-62 exhibited very potent protease inhibitory activity, however, they displayed significantly reduced antiviral activity possibly due to efflux properties of these derivatives.117,118 X-ray structural studies of 60-bound HIV-1 protease documented extensive hydrogen bonding interactions in the S2’-site.117 Raines and co-workers reported the design of boronic acid based derivatives and showed extraordinary enhancement of HIV-1 protease inhibitory activity.119 Representative derivatives 63 and 64 showed femtomolar Ki values, however antiviral activity was reduced significantly. X-ray structural studies showed similar enhanced hydrogen bonding interactions as the carboxylic acid derivative 60. Interestingly, inhibitor 65, containing crown-THF ligands as the P2 ligand and boronic acid as the P2’ ligand showed improved antiviral activity.118 We have examined antiviral activity of inhibitors 64 and 65 against DRV-resistant HIV-1 variants and the results were compared against darunavir and our recent highly potent inhibitor 2 as shown in Table 7.118 All boronic acid and carboxylic acid-derived inhibitors exhibited significantly reduced antiviral activity against the highly DRV-resistant HIV-1 variants.
Figure 21.
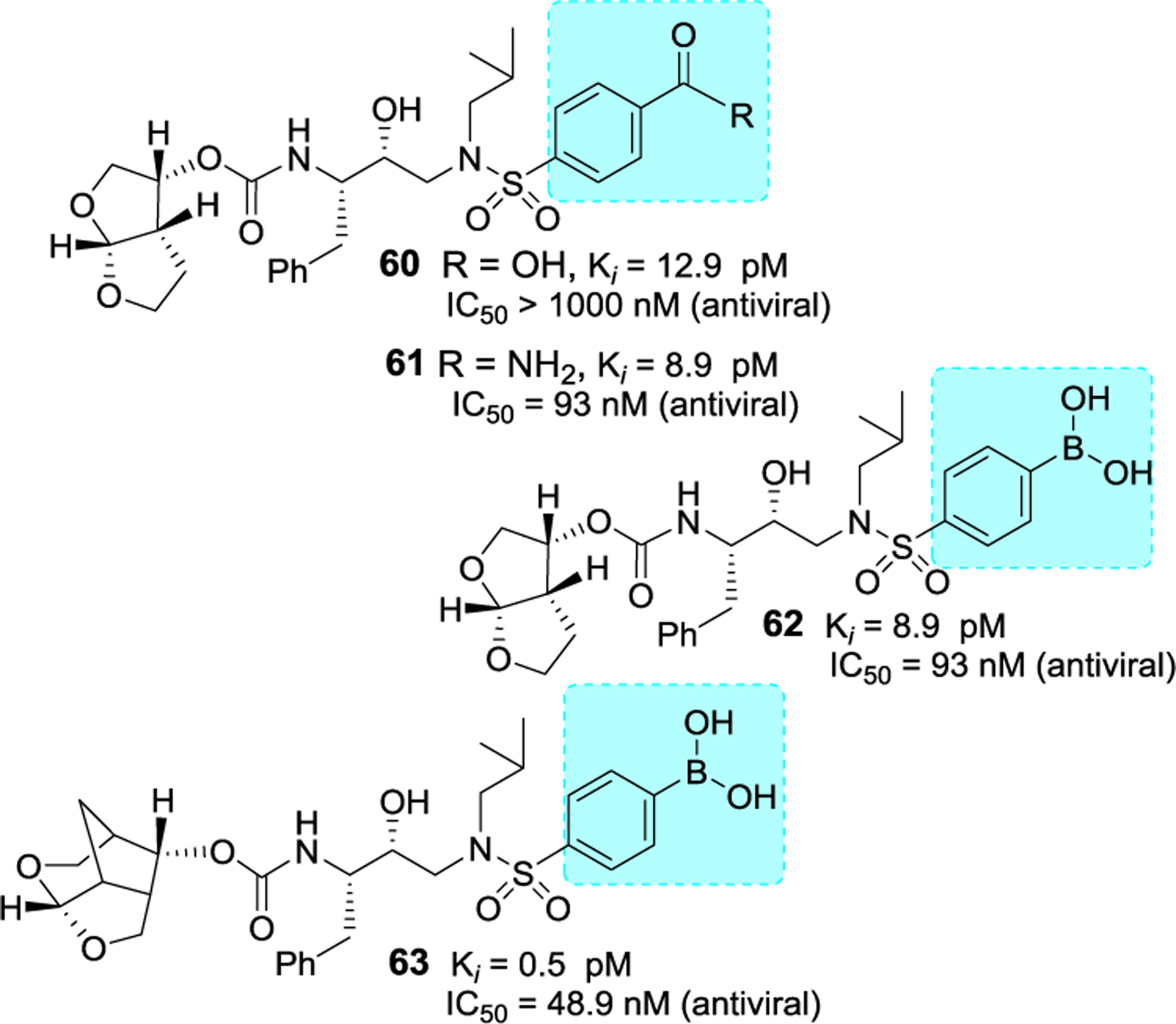
Structure and activity of inhibitors 60–53.
Table 7.
Antiviral activity of inhibitors against highly DRV-resistant HIV-1 variants.
| Mean IC50 in nM ± SD (fold-change) | |||||
|---|---|---|---|---|---|
| LPV | 1 | 62 | 63 | 2 (GRL-142) | |
| cHIVnl4-3WT | 13 ± 2 | 3.2 | 48.9 | 37.7 | 0.017 |
| HIVDRVRP20 | >1000 (>77) | 525.8 (164) | 219.3 (4) | 71.2 (2) | 0.0024 |
| HIVDRVRP30 | >1000 (>77) | 601 (>187) | 946.4 (19) | 532.3 (14) | 0.14 |
| HIVDRVRP51 | >1000 (>77) | 5429.7 (>1000) | >1000 | >1000 | 1.3 |
Numbers in parentheses represent fold changes in IC50s for each isolate compared to the IC50s for wild-type cHIVNL4-3WT. All assays were conducted in triplicate.
Besides carboxylic acid and boronic acid functionalities, we and others have explored stereochemically defined hydroxyl groups to make enhanced hydrogen bonding interactions in the S2’ site. The effect of these functionalities on the antiviral activity was evaluated. As shown in Figure 22, darunavir derivative 64 with a (R)-hydroxyl group on the P2’ ligand showed comparable in vitro activity as darunavir. Interestingly, the derivative 65 with (S)-isomer showed much reduction in potency.27,116 Rusere and co-workers have also investigated the effect of various polar groups on the P2’ ligand in combination with other P1’ ligands.120 As shown in Figure 23, inhibitors 66–69 have shown very potent protease inhibitory activity against drug-resistant variants 184V and I50V/A71V. The X-ray structure of inhibitor 67-bound HIV-1 protease showed extensive interactions in the S2’-subsite as shown in Figure 24.120
Figure 22.
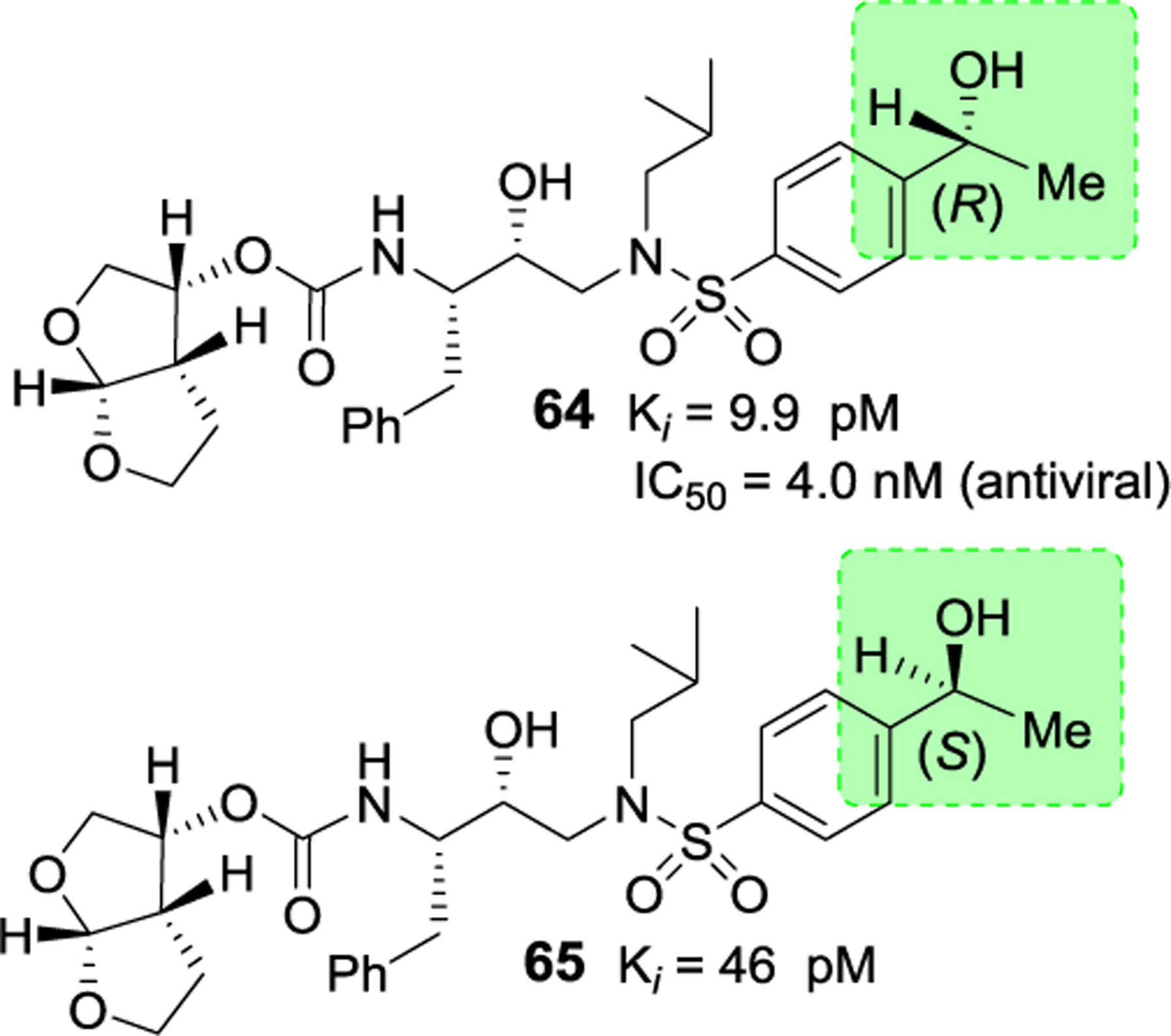
Structure and activity of inhibitors 64 and 65.
Figure 23.
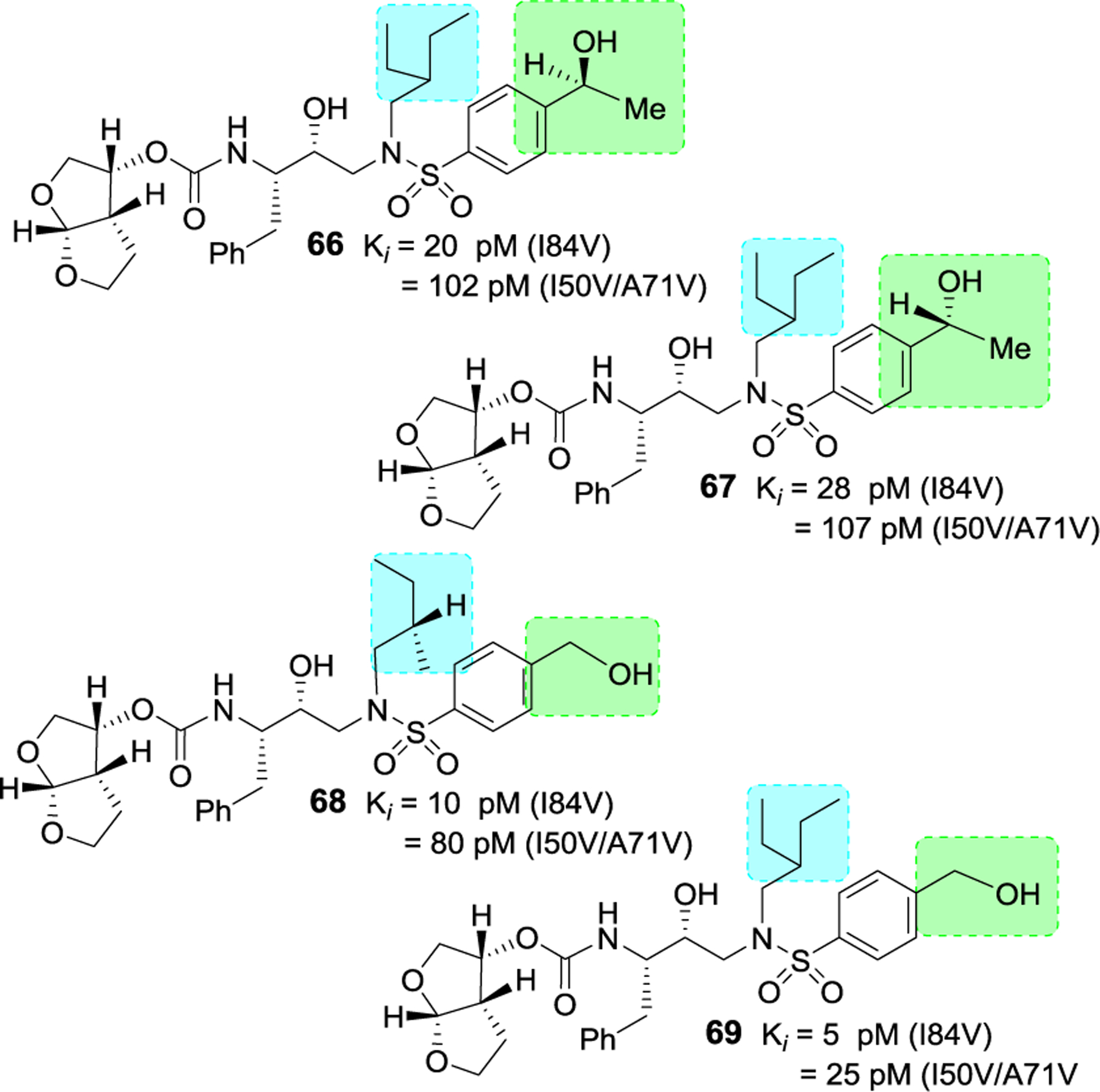
Structure and activity of inhibitors 66–69.
Figure 24.
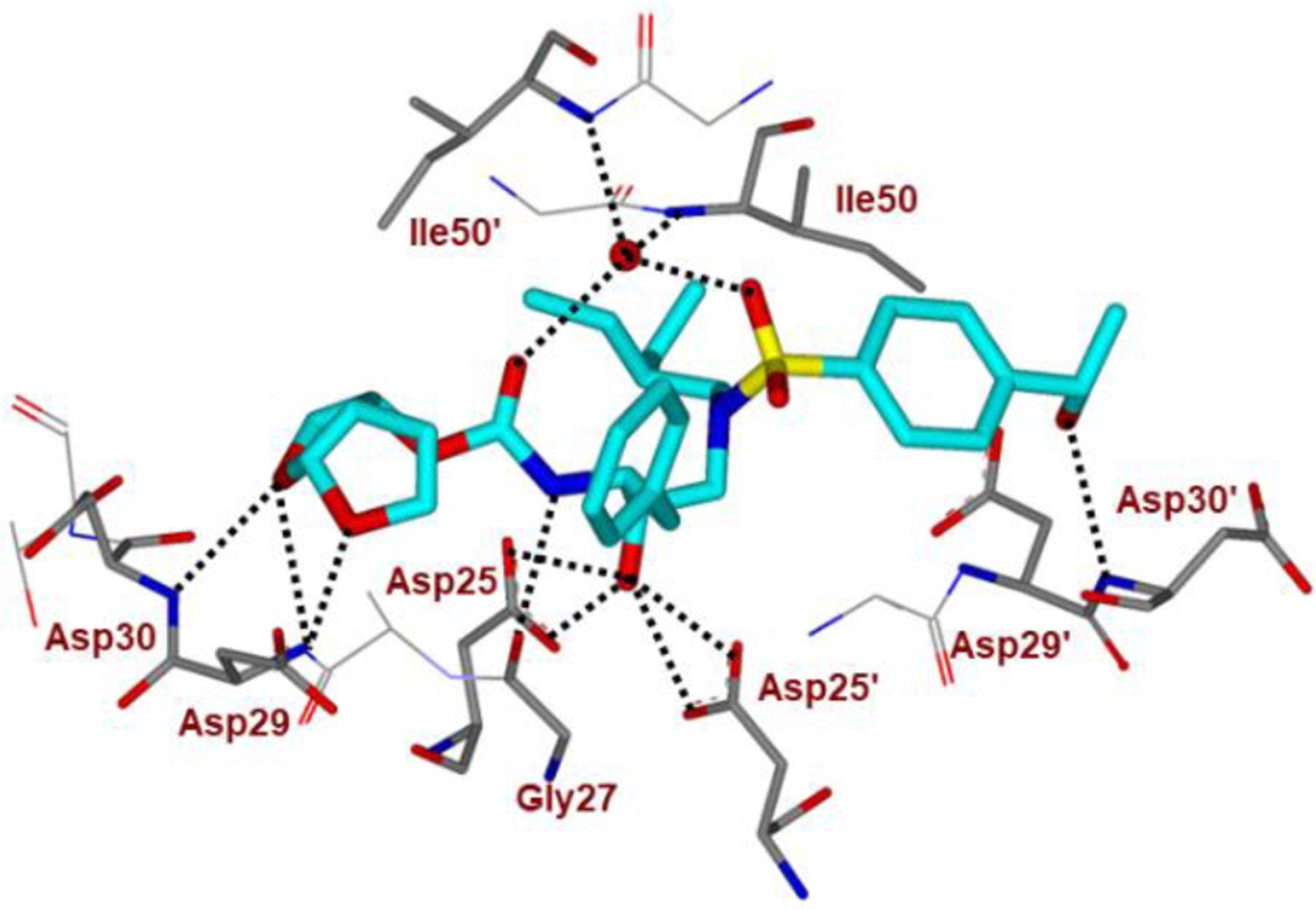
Inhibitor 67-bound HIV-1 protease X-ray structure is shown (PDB code: 6OXV). The inhibitor carbon atoms are shown in cyan and hydrogen bonds are shown by black dotted lines.
Rusere and co-workers have investigated protease inhibitors incorporating bis-tetrahydrofuran as the P2 or P2’ ligand in psuedosymmetric dipeptide isosteres related to lopinavir.121,122 The authors’ structure-activity relationship studies revealed that bis-THF carbamate can be incorporated in both sides of the isostere without affecting much of enzyme inhibitory activity. Representative inhibitors 70–73 in Figure 25 showed very potent Ki values against wild-type as well as protease mutants I84V and I50V/A71V. However, antiviral activity of these derivatives is not improved over darunavir or lopinavir. X-ray structural studies have indicated strong hydrogen bonding interactions of the bis-THF ligand with backbone amide NHs Asp29 and Asp30 as well as Asp29’ and Asp30’ NH similar to interactions of bis-THF ligand in the S2 subsite of darunavir.121
Figure 25.
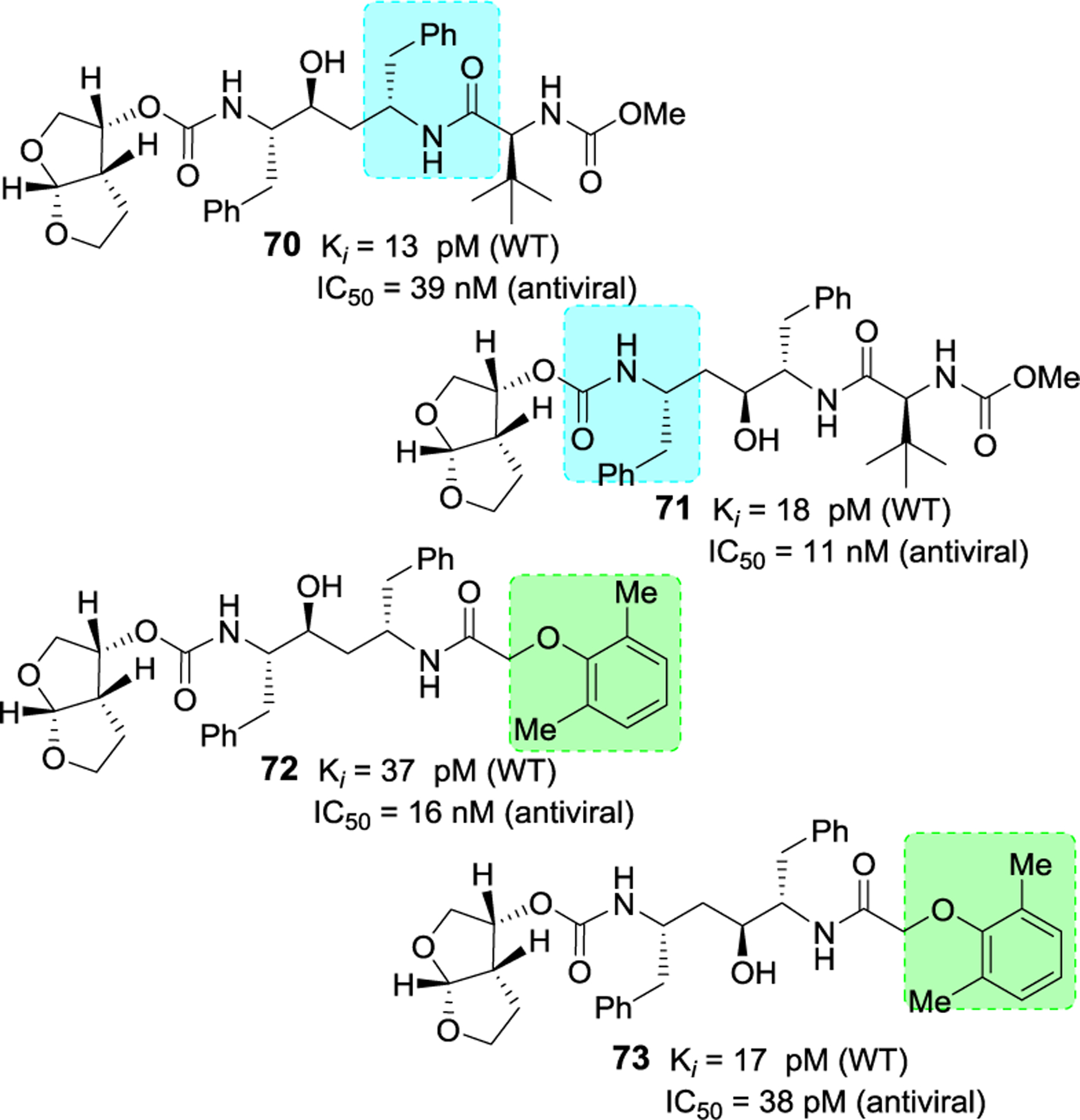
Structure and activity of inhibitors 70–73.
Bai and co-workers have reported design, synthesis, and structure-activity relationship studies of HIV-1 protease inhibitors incorporating stereochemically defined aminotetrahydrofuran-derived tertiary amine-amide as the P2’ ligand in hydroxysulfonamide isostere scaffold.123,124 Many amide derivatives were explored and a number of tertiary amide derived inhibitors have shown low nanomolar to subnanomolar HIV-1 protease inhibitory activity. Among representative examples 74–77 in Figure 26, inhibitor 77 with N(S-tetrahydrofuran)-N(2-methoxyethyl)acetamide P2 ligand in combination with 3,4-methylenedioxybenzene sulfonamide P2’ ligand exhibited best protease inhibitory activity (IC50 0.35 nM). These derivatives showed low cytotoxicity. However, antiviral activity of these compounds was not reported.123
Figure 26.
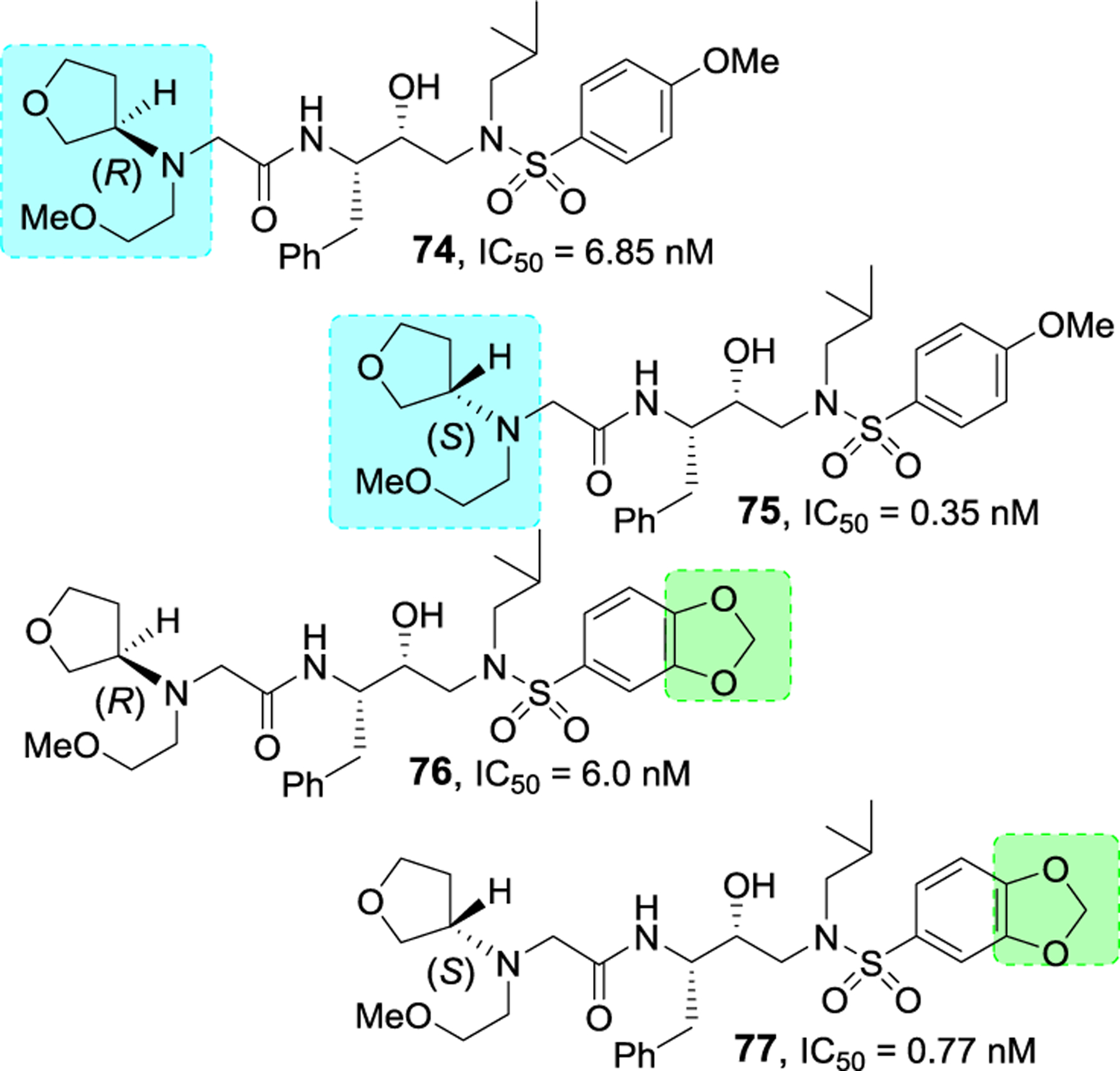
Structure and activity of inhibitors 74–77.
Bungard and co-workers reported the design and synthesis of a potent series of HIV-1 protease inhibitors containing a piperazine sulfonamide core.125,126 The design of these inhibitors was based upon the work of pyrrolidine based inhibitor 78 (Figure 27), reported by Coburn and co-workers.127 Presumably, the pyrrolidine NH binds to catalytic Asp25 and Asp25’ in the active site. A truncated derivative 79 containing a morpholine was then designed. This compound weakly inhibited HIV-1 protease (11% inhibition at 1μM), however, structural studies of inhibitor-bound HIV-1 protease showed key interactions with catalytic Asp25 and Asp25’ in the active site. Also, the two phenyl groups occupied the S1 and S3 regions in the protease active site.
Figure 27.
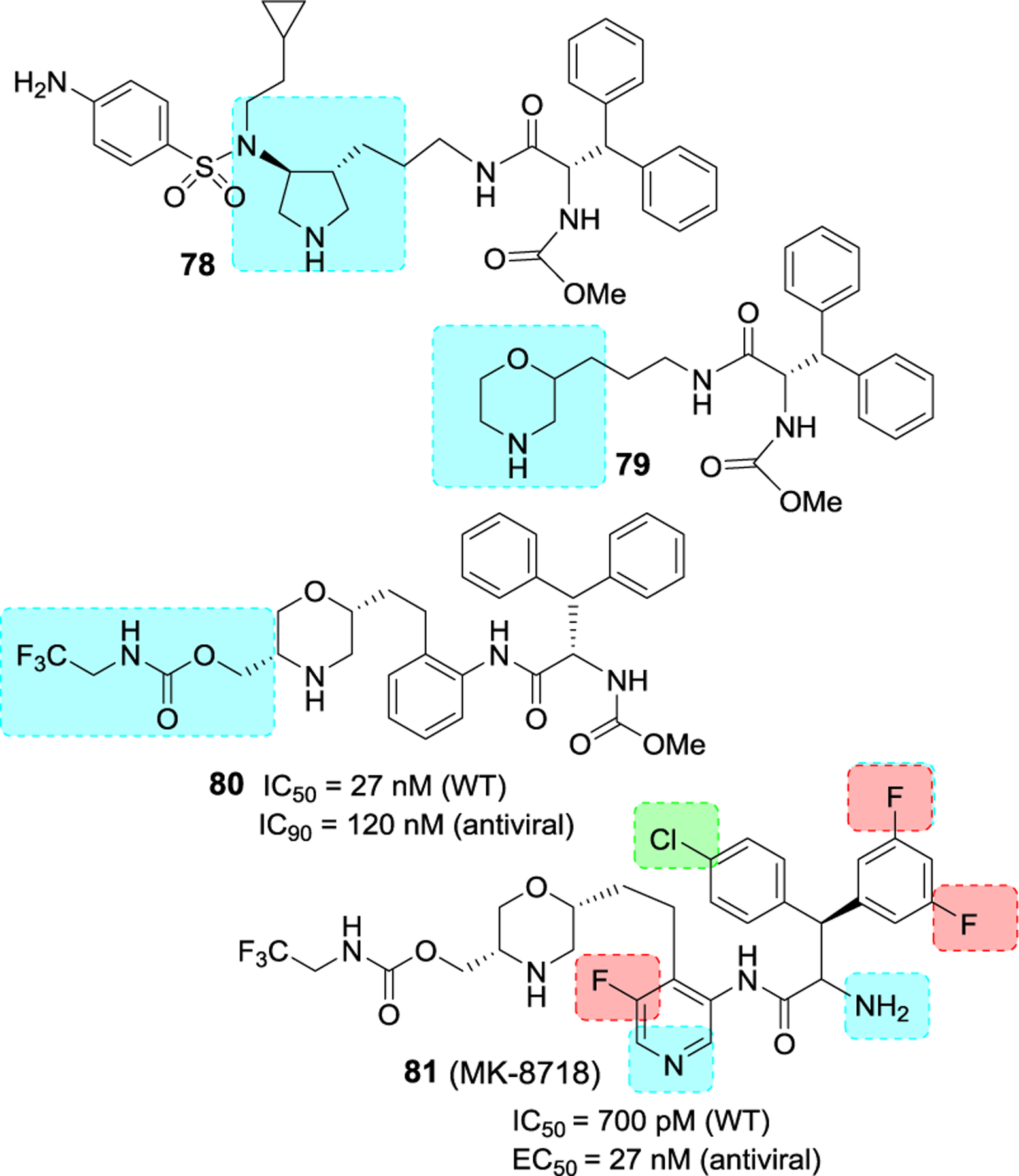
Structure and activity of inhibitors 78–81.
Subsequently, the X-ray structure-based optimization of substituents and incorporation of functionalities were carried out. In inhibitor 80, an aniline moiety was incorporated on the side chain and a methyl carbamate functionality was introduced on the morpholine ring. Compound 80 displayed low nanomolar HIV-1 protease inhibitory activity and exhibited antiviral IC95 value of 120 nM. Rat pharmacokinetic studies showed oral favorable bioavailability for compound 80 (5 mpk, F = 19%). Further optimization of P1, P1’, P2 and P3 ligands resulted in Inhibitor 81 (MK 8718) with improved antiviral and pharmacokinetic properties.125
Further design based upon the structural information of inhibitor 81-bound HIV-1 protease led to the development of very potent inhibitors containing piperazine sulfonamide core. As shown in Figure 28, inhibitor 82 exhibited very potent enzyme inhibitory activity (IC50 12 pM) and antiviral activity (EC50 2.8 nM), comparable to darunavir and atazanevir. The X-ray structure of a related inhibitor 83-bound HIV-1 protease was determined.125 As shown in Figure 29, the structure revealed unique modes of interaction with catalytic aspartates as well as interactions of inhibitor’s sulfonamide group with Ile50 and Ile50’ located in the flaps of HIV-1 protease. The piperazine NH is involved in hydrogen bonding interactions with catalytic Asp25 and Asp25’ in the active site.
Figure 28.
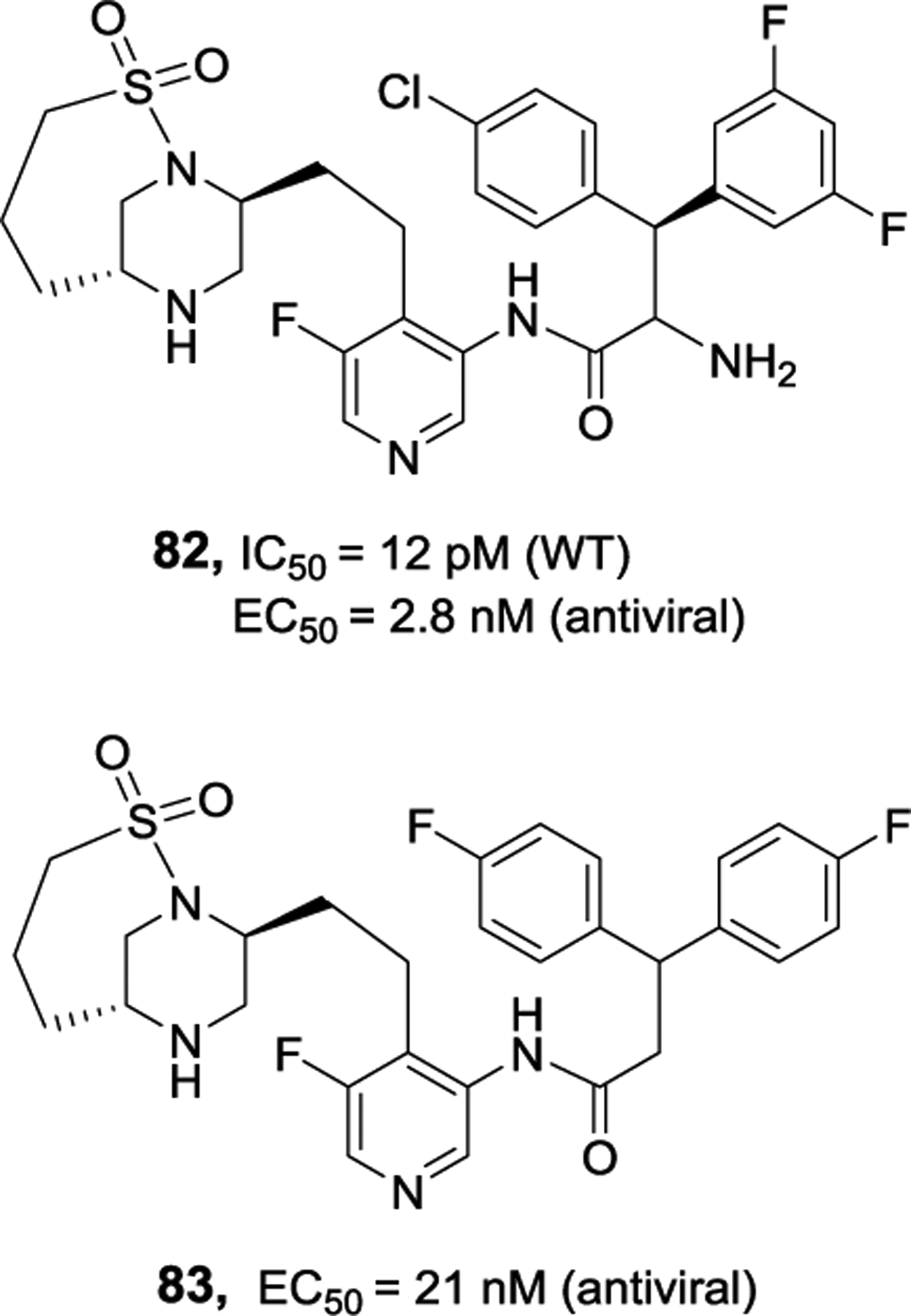
Structure and activity of inhibitors 82 and 83.
Figure 29.
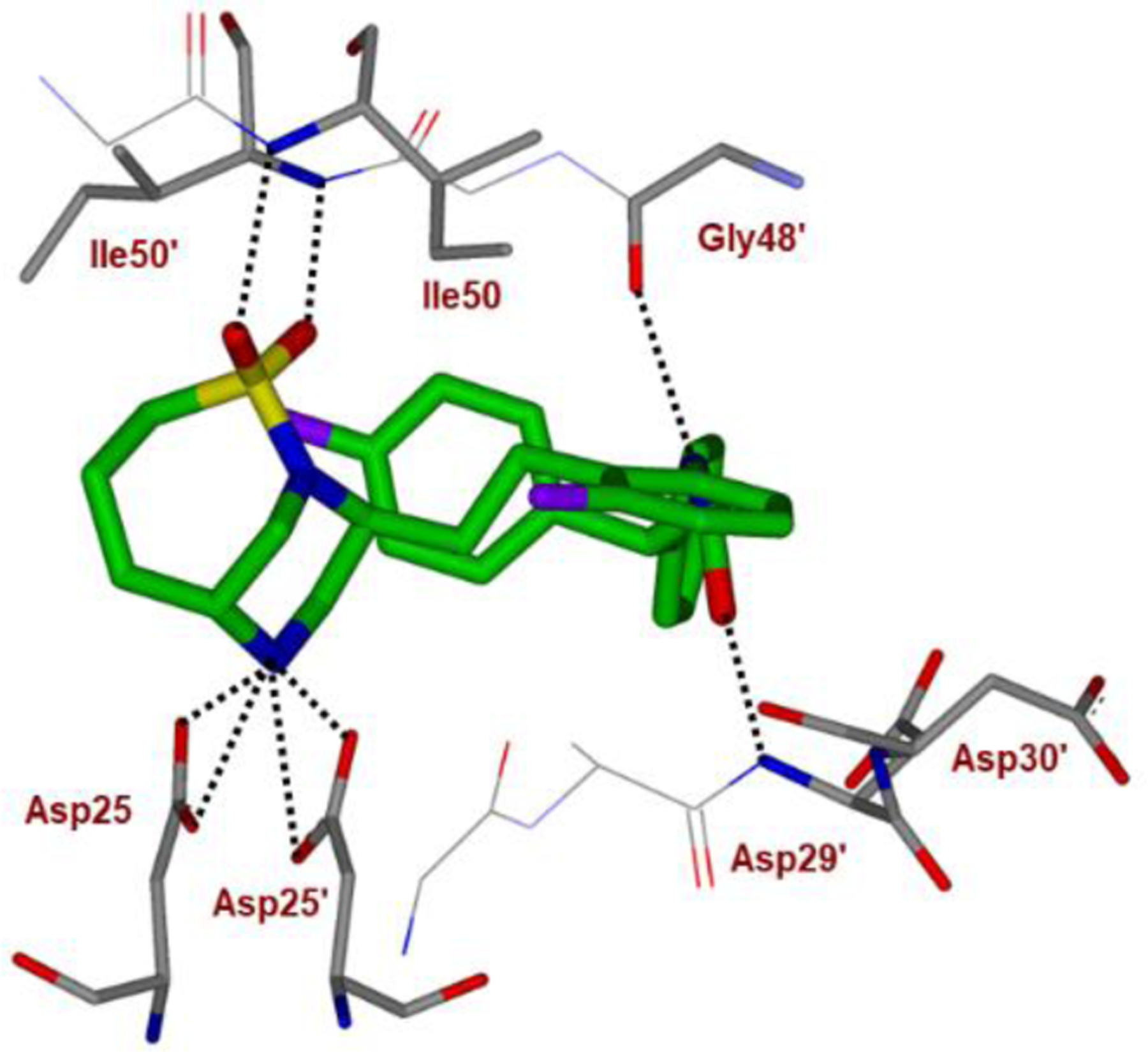
Inhibitor 83-bound HIV-1 protease X-ray structure is shown (PDB code: 6B3H). The inhibitor carbon atoms are shown in green and hydrogen bonds are shown by black dotted lines.
Conclusion
HIV-1 protease inhibitors continue to play an important role in the current treatment and management of patients with HIV-1 infection and AIDS. The last approved HIV-1 protease inhibitor drug is darunavir, which has become widely used first-line therapy for treatment of HIV/AIDS patients. Darunavir based therapy is particularly effective for treatment of patients harboring multidrug-resistant HIV-1 variants that do not respond to existing ART regimens. Darunavir is conceptually different from other approved protease inhibitor drugs. Daruanvir exhibits unprecedented dual mechanism of action, (1) it inhibits catalytically active dimeric HIV-1 protease; (2) it prevents dimerization of protease monomers into the active enzyme. During darunavir design, we incorporated a stereochemically defined bis-tetrahydrofuran heterocycle as the P2 ligand in combination with a p-aminobenezenesulfonamide as the P2’ ligand. These structural features are specifically designed to make extensive hydrogen bonding interactions from S2 and S2’ subsites of HIV-1 protease. The ‘backbone binding’ interactions of darunavir on both sides of the dimeric protease as well as the polyether structural features of the bis-THF ligand are critical to its extraordinary antiviral activity against multidrug-resistant HIV-1 strains.
Since our discovery of darunavir, we have set our focus on the design and development of a new generation of protease inhibitors which can show robust drug-resistant profiles and maintain potency against darunavir and other PI-resistant viruses. Also, we planned to design inhibitors that can cross the blood-brain barrier to inhibit HIV-1 replication in the CNS and reduce developing HAND. In this review, we provided highlights of our recent development of exceptionally potent new generation HIV-1 protease inhibitors that incorporated a host of new polyether-like conformationally constrained polycyclic ligands such as, crown-THF, Umb-THF as P2 ligands; aminobenzothiazole and aminobenzooxazole-derived P2’ ligands, and fluorinated phenylmethyl derivatives as the P1 ligand to make enhanced backbone binding as well as van der Waals interactions in the active site. Among these inhibitors, inhibitor GRL-142 and related derivatives are particularly very intriguing as these compounds display significantly better antiviral activity, drug-resistance profiles, and drug-like properties compared to darunavir. We feature design principles for cyclic polyether-derived nonpeptide ligands motivated by bioactive natural products, backbone binding design strategies to combat drug resistance, in-depth biological studies including in vitro antiviral studies against a range of MDR variants, and X-ray structural studies of inhibitor-protease complex to gain structural insights into activity and properties. We hope this review will further stimulate design and discovery efforts toward improved treatment against HIV infection and AIDS.
Figure 15.
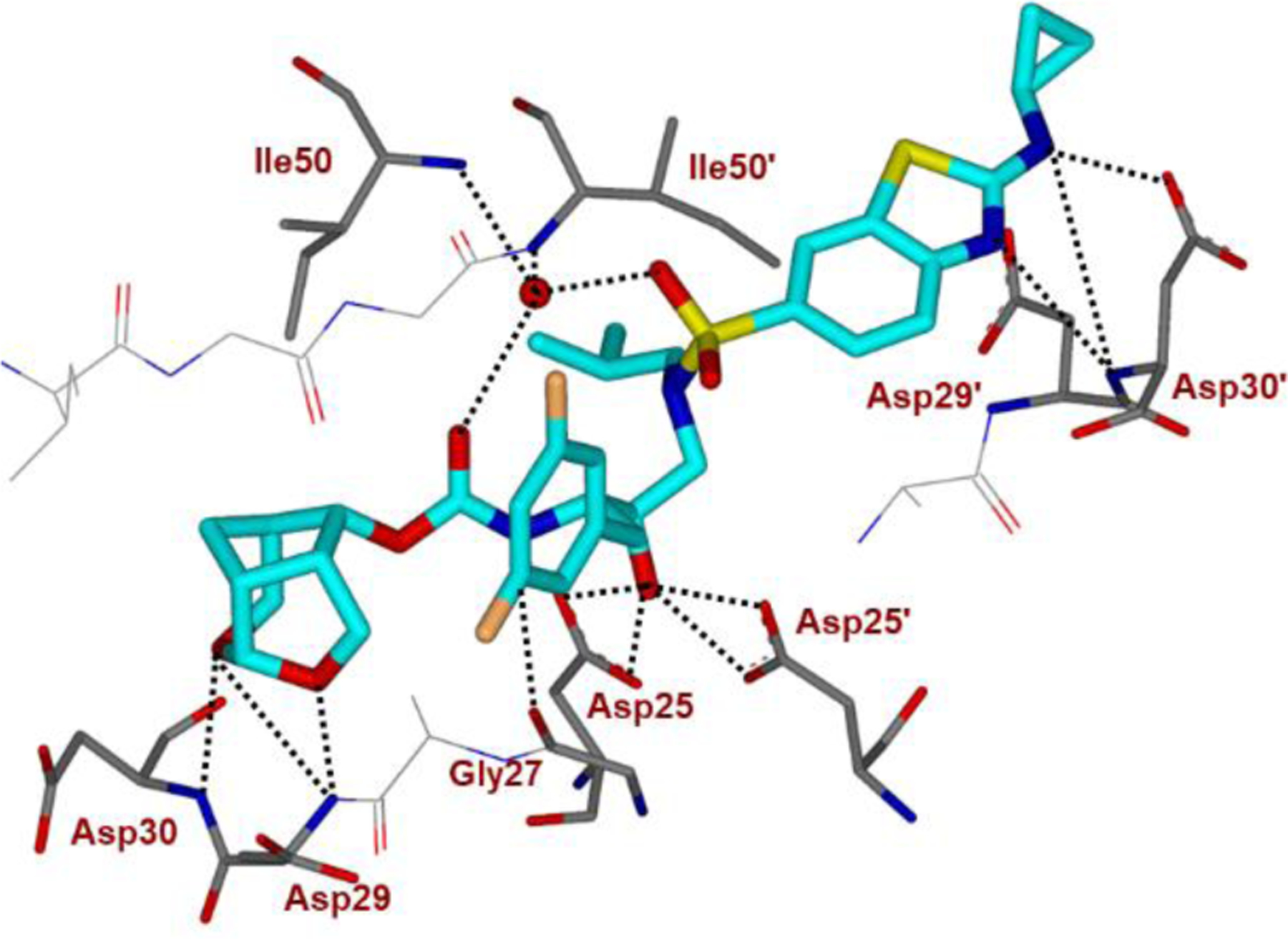
Inhibitor 2-bound X-ray structure of HIV-1 protease. The major orientation of the inhibitor is shown. The inhibitor carbon atoms are shown in green, water molecules are red spheres, and the hydrogen bonds are indicated by dotted lines (PDB ID: 6BZ2).
Figure 20B.
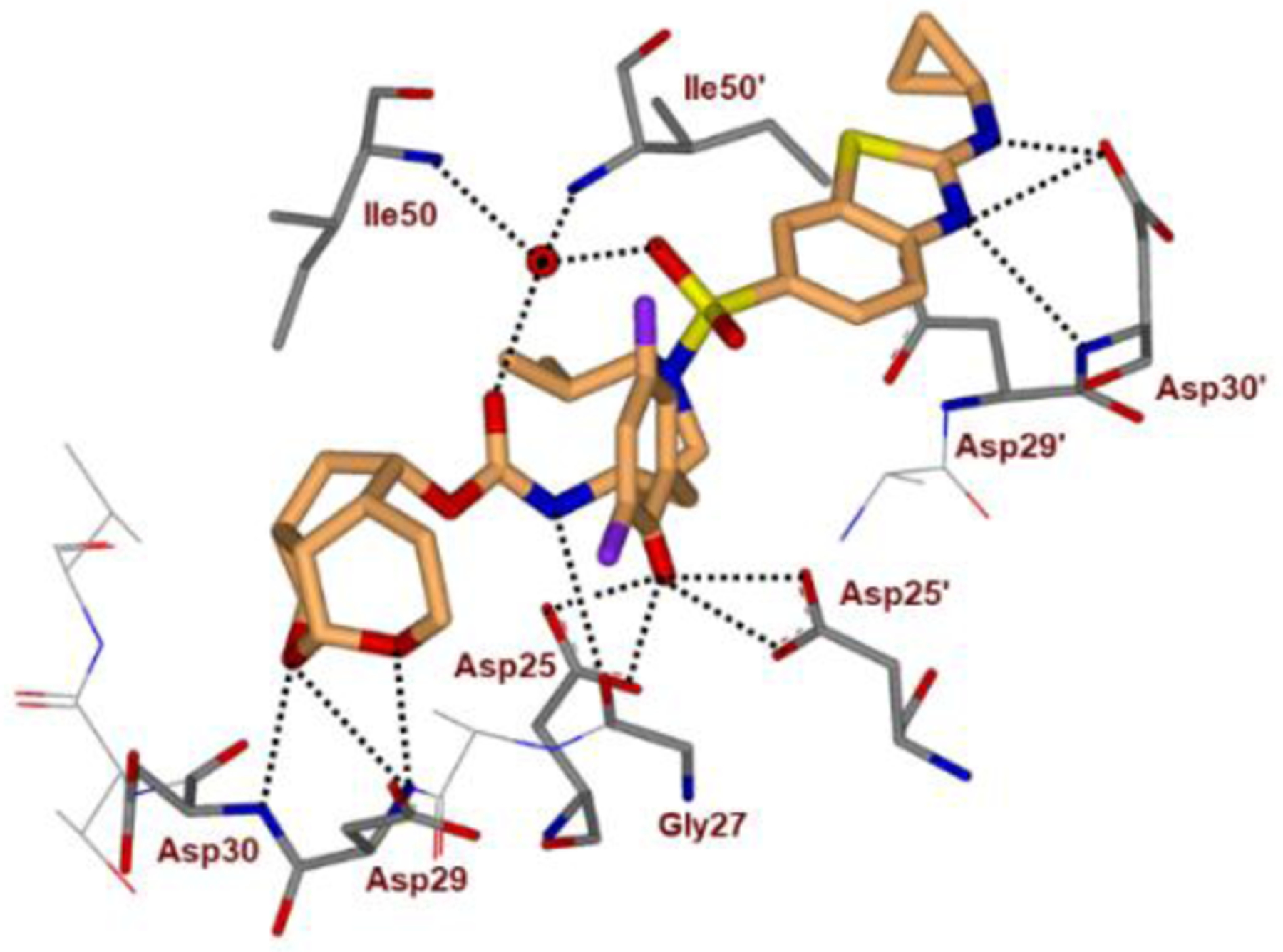
Inhibitor 51-bound HIV-1 protease X-ray structure is shown (PDB code: 6CDL). The inhibitor carbon atoms are shown in cyan and hydrogen bonds are shown by black dotted lines.
Scheme 3.
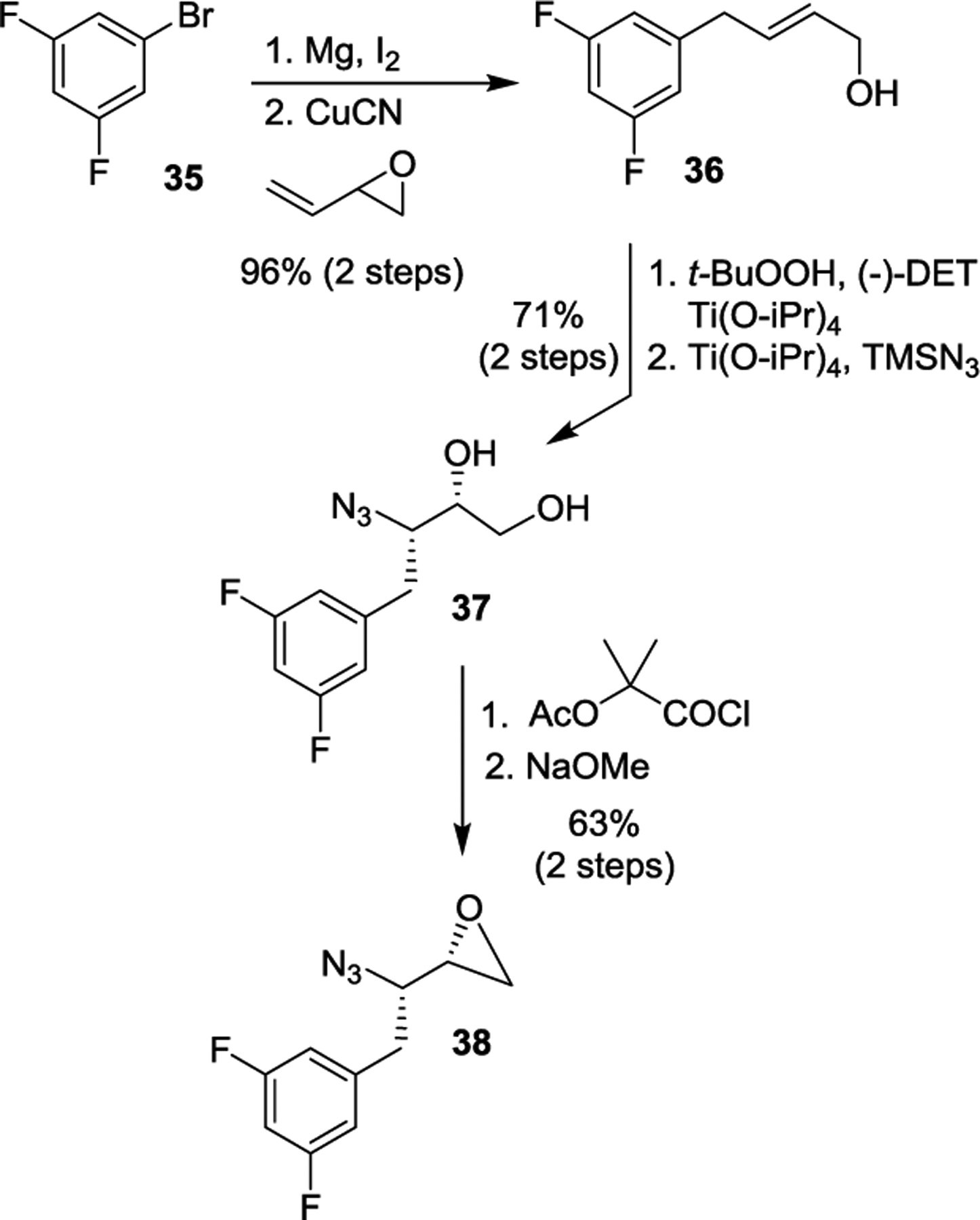
Synthesis of difluoroazidoepoxide 38.
Scheme 6.
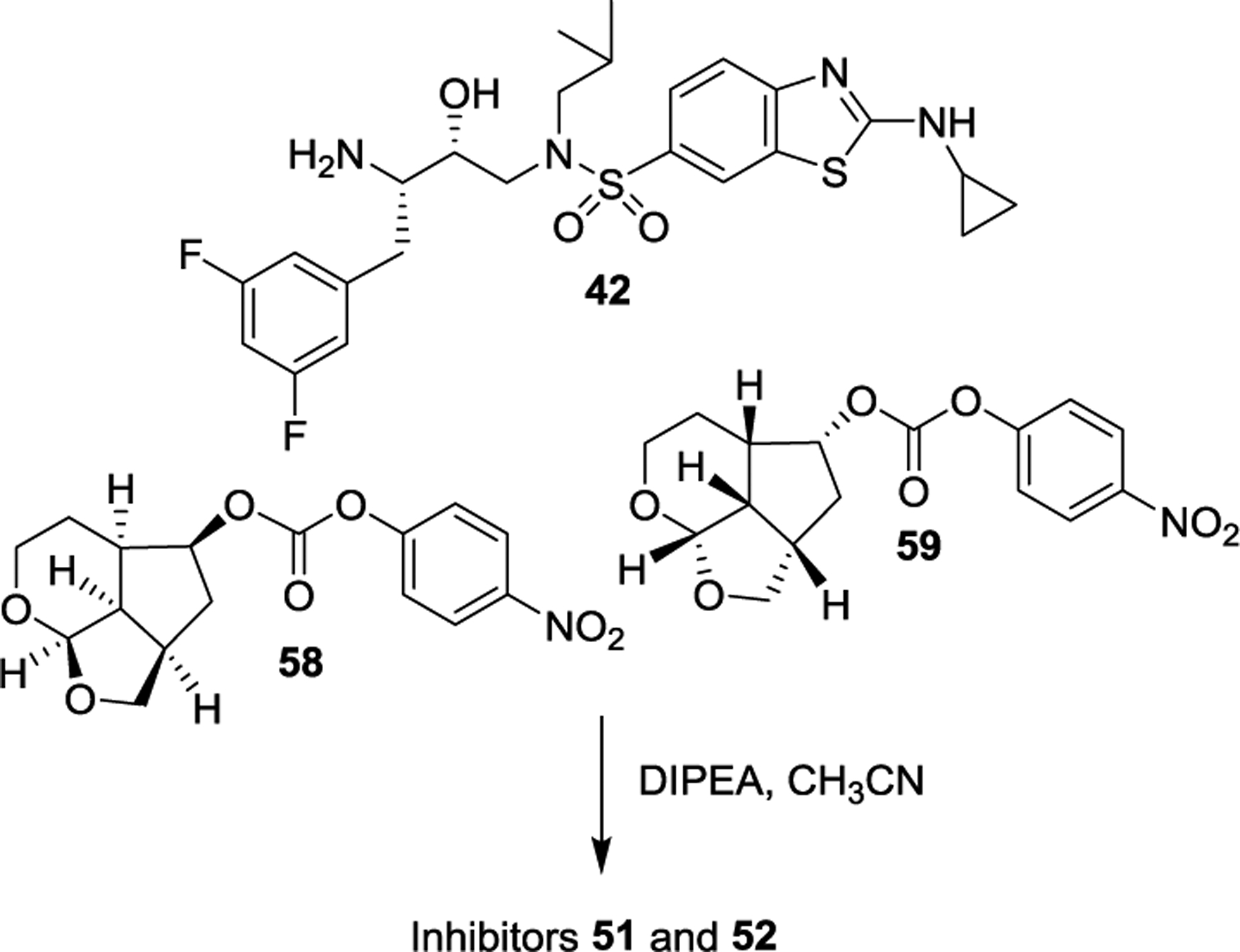
Synthesis of inhibitors 51 and 52.
Acknowledgements
This research was supported by the National Institutes of Health (Grant AI150466, AKG and Grant AI150461, ITW). X-ray data were collected at the Southeast Regional Collaborative Access Team (SER-CAT) beamline 22BM at the Advanced Photon Source, Argonne National Laboratory. Use of the Advanced Photon Source was supported by the US Department of Energy, Basic Energy Sciences, Office of Science, under Contract No. W-31-109-Eng-38. This work was also supported by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health (HM), and in part by grants for the promotion of AIDS research from the Ministry of Health, Welfare and Labor of Japan (HM); grants for the Research Program on HIV/AIDS from the Japan Agency for Medical Research and Development (AMED) under grant numbers JP15fk0410001 and JP18fk0410001 (HM); a grant from the National Center for Global Health and Medicine (NCGM) (HM); and grants from JSPS KAKENHI (HM). The authors would like to thank the Purdue University Center for Cancer Research, which supports the shared NMR and mass spectrometry facilities.
Biographies

Arun K. Ghosh received his BS and MS in Chemistry from the University of Calcutta and Indian Institute of Technology, Kanpur respectively. He obtained his PhD in 1985 at the University of Pittsburgh. He pursued postdoctoral research with Professor E. J. Corey at Harvard University (1985–1988). He was a research fellow at Merck Research Laboratories prior to joining the University of Illinois-Chicago as an assistant Professor in 1994. In 2005, he moved to Purdue University where he is currently the Ian P. Rothwell Distinguished Professor in Chemistry and Medicinal Chemistry. His research interests are in the areas of organic, bioorganic, dru-design, and medicinal chemistry.

Hiroaki Mitsuya received his MD and PhD from National Kumamoto University School of Medicine (Japan). Following training in oncology/hematology/immunology, he joined National Cancer Institute in Bethesda (USA) in 1982, where he has been Principal Investigator & Chief of Experimental Retrovirology Section since 1991 to present. He also served as Professor of Medicine and Chairman of Departments of Hematology/Rheumatology/Infectious Diseases at Kumamoto University from 1997 to 2016. He now serves as Director, National Center for Global Health & Medicine, Tokyo. His current research focuses on antiviral research for development of therapeutics for HIV/AIDS, hepatitis B, and SARS-CoV-2.

Irene T. Weber received her BS and MS from Cambridge University, England, and obtained her PhD in 1978 from Oxford University, England. She pursued postdoctoral research with Professor Thomas Steitz at Yale University. She was Professor of Microbiology and Immunology at Thomas Jefferson University in Philadelphia from 1991. She moved to Georgia State University, Atlanta, in 2001 where she is a Regents’ Professor of Biology and Chemistry and Georgia Cancer Coalition Distinguished Cancer Scientist. Her research focuses on the structure and activity of enzymes.
Footnotes
Conflicts of interest
There are no conflicts to declare.
Notes and references
- 1.Deeks SG, Archin N, Cannon P, Collins S, Jones RB, de Jong MAWP, Lambotte O, Lamplough R, Ndung’u T, Sugarman J, Tiemessen CT, Vandekerckhove L and Lewin SR, The International AIDS Society (IAS) Global Scientific Strategy working group, Nature Med. 2021, 27, 2085–2098. [DOI] [PubMed] [Google Scholar]
- 2.Frieden TR, Foti KE and Mermin J, N. Engl. J. Med 2015, 373, 2281–2287. [DOI] [PubMed] [Google Scholar]
- 3.UNAIDS/WHO. Global HIV/AIDS Response - Progress Report 2021, November 2021. Available at http://www.unaids.org/en/resources/publications/2011/, Last accessed May 2012.
- 4.de Clercq E, Int. J. Antimicrob. Agents 2009, 33, 307–320. [DOI] [PubMed] [Google Scholar]
- 5.Palella FJ, Delaney KM, Moorman AC, Loveless MO, Fuhrer J, Satten GA, Aschman DJ and Holmberg SD, New Engl. J. Med 1998, 338, 853–860. [DOI] [PubMed] [Google Scholar]
- 6.Sepkowitz KA, N. Engl. J. Med 2001, 344, 1764–1772. [DOI] [PubMed] [Google Scholar]
- 7.Maenza J and Flexner C, Am. Fam. Physician 1998, 57, 2789–2798. [PubMed] [Google Scholar]
- 8.Moreno S, Perno CF, Mallon PW, Behrens G, Corbeau P, Routy J-P and Darcis G, HIV Medicine 2019, 20, 2–12. [DOI] [PubMed] [Google Scholar]
- 9.Deeks SG, Lewin SR and Havlir DV, Lancet 2013, 382, 1525–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Logie C and Gadalla TM, AIDS Care 2009, 21, 742–753. [DOI] [PubMed] [Google Scholar]
- 11.Waters L and Nelson M, Int. J. Clin. Pract 2007, 61, 983–990. [DOI] [PubMed] [Google Scholar]
- 12.Menendez-Arias L, Antiviral Res. 2010, 85, 210–231. [DOI] [PubMed] [Google Scholar]
- 13.HIV-associated neurocognitive disorder, by Smail RC and Brew BJ, Chapter 7, pages 75–97. In Handbook of Clinical Neurology, Vol. 152 (3rd series), The Neurology of HIV Infection, Brew BJ, Ed; 2018. Elsevier; [DOI] [PubMed] [Google Scholar]
- 14.Currier JS, Top. Antivir. Med 2018, 25, 133–137. [PMC free article] [PubMed] [Google Scholar]
- 15.Vitoria M, Rangaraj A, Ford N and Doherty M, Curr. Opin. HIV and AIDS 2019, 14, 143–149. [DOI] [PubMed] [Google Scholar]
- 16.Ghosh AK, J. Med. Chem 2009, 52, 2163–2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ghosh AK, J. Org. Chem 2010, 75, 7967–7989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ghosh AK and Chapsal BD, Design of the anti-HIV protease inhibitor darunavir. In Introduction to Biological and Small Molecule Drug Research and Development; Ganellin CR, Jefferis R, Roberts S, Eds.; Elsevier: Amsterdam, 2013; pp 355–385. [Google Scholar]
- 19.Ghosh AK, Osswald HL and Prato G, J. Med. Chem 2016, 59, 5172–5208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ghosh AK, Dawson ZL and Mitsuya H, Bioorg. Med. Chem 2007, 15, 7576–7580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koh Y, Nakata H, Maeda K, Ogata H, Bilcer G, Devasamudram T, Kincaid JF, Boross P, Wang Y-F, Tie Y, Volarath P, Gaddis L, Harrison RW, Weber IT, Ghosh AK and Mitsuya H, Antimicrob. Agents Chemother 2003, 47, 3123–3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de Meyer S, Azijn H, Surleraux D, Jochmans D, Tahri A, Pauwels R, Wigerinck P and de Bethune M-P, Antimicrob. Agents Chemother 2005, 49, 2314–2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Deeks ED, Drugs 2014, 74, 99–125. [DOI] [PubMed] [Google Scholar]
- 24.On June 23, 2006, FDA approves darunavir for previously treated HIV-infected patients, and in December 13, 2008, for all HIV/AIDS patients including pediatrics who did not receive antiretroviral therapies previously. Available at:http://www.accessdata.fda.gov/drugsatfda_docs/label/2008/021976s009lbl.pdf (accessed May 2012).
- 25.Curran A and Pascuet ER, Enferm. Infecc. Microbiol. Clin 2008, 10, 14–22. [DOI] [PubMed] [Google Scholar]
- 26.Mills AM, Nelson M, Jayaweera D, Ruxrungtham K, Cassetti I, Girard P-M, Workman C, Dierynck I, Sekar V, Abeele CV and Lavreys L, AIDS 2009, 23, 1679–1688. [DOI] [PubMed] [Google Scholar]
- 27.Ghosh AK, Sridhar PR, Kumaragurubaran N, Koh Y, Weber IT and Mitsuya H, ChemMedChem 2006, 1, 939–950. [DOI] [PubMed] [Google Scholar]
- 28.Ghosh AK, Chapsal BD and Mitsuya H, Darunavir, a New PI with Dual Mechanism: from a Novel Drug Design Concept to New Hope against Drug-Resistant HIV. In Aspartic Acid Proteases as Therapeutic Targets; Ghosh AK, Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weiheim, Germany, 2010; Vol. 45, pp 205–243. [Google Scholar]
- 29.Ghosh AK, Chapsal BD, Weber IT and Mitsuya H, Acc. Chem. Res 2008, 41, 78–86. [DOI] [PubMed] [Google Scholar]
- 30.Tie Y, Boross PI, Wang YF, Gaddis L, Hussain AK, Leshchenko S, Ghosh AK, Louis JM, Harrison RW and Weber IT, J. Mol. Biol 2004, 338, 341–352. [DOI] [PubMed] [Google Scholar]
- 31.Kovalevsky AY, Liu F, Leshchenko S, Ghosh AK, Louis JM, Harrison RW and Weber IT, J. Mol. Biol 2006, 363, 161–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yoshimura K, Kato R, Kavlick MF, Nguyen A, Maroun V, Maeda K, Hussain KA, Ghosh AK, Gulnik SV, Erickson JW and Mitsuya H, J. Virol 2002, 76, 1349–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ghosh AK and Gemma S, Development of Direct Thrombin Inhibitor, Dabigatran Etexilate, as an Anticoagulant Drug. In Structure-Based Design of Drugs and Other Bioactive Molecules: Tools and Strategies; Wiley-VCH:Weinheim, Germany, 2014; pp 337–354. [Google Scholar]
- 34.Ghosh AK, Anderson DD, Weber IT and Mitsuya H, Angew. Chem., Int. Ed 2012, 51, 1778–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hong L, Zhang X, Hartsuck JA and Tang J, Protein Sci. 2000, 9, 1898–1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Laco GS, Schalk-Hihi C, Lubkowski J, Morris G, Zdanov A, Olson A, Elder JH, Wlodawer A and Gustchina A, Biochemistry 1997, 36, 10696–10708. [DOI] [PubMed] [Google Scholar]
- 37.Ghosh AK and Chapsal B, Second-Generation Approved HIV-1 Protease Inhibitors for the treatment of HIV/AIDS. In Aspartic Acid Proteases as Therapeutic Targets; Ghosh AK, Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weiheim, Germany, 2010; Vol. 45, pp 169–204. [Google Scholar]
- 38.Newman DJ and Cragg GM, J. Nat. Prod 2012, 75, 311–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li JW-H and Vederas JC, Science 2009, 325, 161–165. [DOI] [PubMed] [Google Scholar]
- 40.Clardy J and Walsh C, Nature 2004, 432, 829–837. [DOI] [PubMed] [Google Scholar]
- 41.Nakanishi K, Bioorg. Med. Chem 2005, 13, 4987–5000. [DOI] [PubMed] [Google Scholar]
- 42.Quinoa E, Kakou Y and Crews P, J. Org. Chem 1988, 53, 3642–3644. [Google Scholar]
- 43.Corley DG, Herb R, Moore RE, Scheuer PJ and Paul VJ, J. Org. Chem 1988, 53, 3644–3646. [Google Scholar]
- 44.Zanno PR, Miura I, Nakanishi K and Elder DL, J. Am. Chem. Soc 1975, 97, 1975–1977. [DOI] [PubMed] [Google Scholar]
- 45.Wang J, Soisson SM, Young K, Shoop W, Kodali S, Galgoci A, Painter R, Parthasarathy G, Tang YS, Cummings R, Ha S, Dorso K, Motyl M, Jayasuriya H, Ondeyka J, Herath K, Zhang C, Hernandez L, Allocco J, Basilio Á, Tormo JR, Genilloud O, Vicente F, Pelaez F, Colwell L, Lee SH, Michael B, Felcetto T, Gill C, Silver LL, Hermes JD, Bartizal K, Barrett J, Schmatz D, Becker JW, Cully D and Singh SB, Nature 2006, 441, 358–361. [DOI] [PubMed] [Google Scholar]
- 46.Singh SB and Barrett JF, Biochem. Pharmacol 2006, 71, 1006–1015. [DOI] [PubMed] [Google Scholar]
- 47.Towle MJ, Salvato KA, Budrow J, Wels BF, Kuznetsov G, Aalfs KK, Welsh S, Zheng W, Seletsky BM, Palme MH, Habgood GJ, Singer LA, DiPietro LV, Wang Y, Chen JJ, Quincy DA, Davis A, Yoshimatsu K, Kishi Y, Yu MJ and Littlefield BA, Cancer Res. 2001, 61, 1013–1021. [PubMed] [Google Scholar]
- 48.Singh SB, Ondeyka JG, Herath KB, Zhang C, Jayasuriya H, Zink DL, Parthasarathy G, Becker JW, Wang J and Soisson SM, Bioorg. Med. Chem. Lett 2009, 19, 4756–4759. [DOI] [PubMed] [Google Scholar]
- 49.Martens E and Demain AL, J. Antibiot 2011, 64, 705–710. [DOI] [PubMed] [Google Scholar]
- 50.Prota AE, Bargsten K, Northcote PT, Marsh M, Altmann K-H, Miller JH, Díaz JF and Steinmetz MO, Angew. Chem. Int. Ed. Engl 2014, 53, 1621–1625. [DOI] [PubMed] [Google Scholar]
- 51.Rudolf JD, Dong L-B and Shen B, Biochem. Pharmacol 2017, 133, 139–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Craik DJ, Fairlie DP, Liras S and Price D, Chem. Biol. Drug Design 2012, 81, 136–147. [DOI] [PubMed] [Google Scholar]
- 53.Otvos L Jr., Wade JD, Front. Chem 2014, 2, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.O’Brien WA III, AIDS Read. 2006, 16, 38–44. [PubMed] [Google Scholar]
- 55.Roberts NA, Martin JA, Kinchington D, Broadhurst AV, Craig JC, Duncan IB, Galpin SA, Handa BK, Kay J, Kröhn A, Lambert RW, Merrett JH, Mills JS, Parkes KE, Redshaw S, Ritchie AJ, Taylor DL, Thomas GJ and Machin PJ, Science 1990, 248, 358–361. [DOI] [PubMed] [Google Scholar]
- 56.Ghosh AK, Anderson DD and Mitsuya H, The FDA approved HIV-1 protease inhibitors for treatment of HIV/AIDS. In Burger’s Medicinal Chemistry, Drug Discovery and Development, 7th ed.; Abraham DJ, Rotella DP, Eds.; 2010; Vol. 7, pp 1–72. [Google Scholar]
- 57.Lv Z, Chu Y and Wang Y, HIV AIDS (Auckl.) 2015, 7, 95–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ghosh AK, Thompson WJ, Lee HY, McKee SP, Munson PM, Duong TT, Darke PL, Zugay JA, Emini EA, Schleif WA, Huff JR and Anderson PS, J. Med. Chem 1993, 36, 924–927. [DOI] [PubMed] [Google Scholar]
- 59.Ghosh AK, Lee HY, Thompson WJ, Culberson C, Holloway MK, McKee SP, Munson PM, Duong TT, Smith AM, Darke PL, Zugay JA, Emini EA, Schleif WA, Huff JR and Anderson PS, J. Med. Chem 1994, 37, 1177–1188. [DOI] [PubMed] [Google Scholar]
- 60.Ghosh AK, Thompson WJ, Munson PM, Liu W and Huff JR, Bioorg. Med. Chem. Lett 1995, 5, 83–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ghosh AK, Kincaid JF, Walters DE, Chen Y, Chaudhuri NC, Thompson WJ, Culberson C, Fitzgerald PMD, Lee HY, McKee SP, Munson PM, Duong TT, Darke PL, Zugay JA, Schleif WA, Axel MG, Lin J and Huff JR, J. Med. Chem 1996, 39, 3278–3290. [DOI] [PubMed] [Google Scholar]
- 62.Ghosh AK, Kincaid JF, Cho W, Walters DE, Krishnan K, Hussain KA, Koo Y, Cho H, Rudall C, Holland L and Buthod J, Bioorg. Med. Chem. Lett 1998, 8, 687–690. [DOI] [PubMed] [Google Scholar]
- 63.Ghosh AK, Pretzer E, Cho H, Hussain KA and Duzgunes N, Antivir. Res 2002, 54, 29–36. [DOI] [PubMed] [Google Scholar]
- 64.Erickson JW, Anonymous; Gulnik SV, Mitsuya H and Ghosh AK, Fitness Assay and Associated Methods. US: 07470506, 2008. [Google Scholar]
- 65.Gulnik SV, Suvorov LI, Liu BS, Yu B, Anderson B, Mitsuya H and Erickson JW, Biochemistry 1995, 34, 9282–9287. [DOI] [PubMed] [Google Scholar]
- 66.Yoshimura K, Kato R, Kavlick MF, Nguyen A, Maroun V, Maeda K, Hussain KA, Ghosh AK, Gulnik SV, Erickson JW and Mitsuya HA, J. Virol 2002, 76, 1349–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ghosh AK, Kulkarni S, Anderson DD, Hong L, Baldridge A, Wang Y-F, Chumanevich AA, Kovalevsky AY, Tojo Y, Amano M, Koh Y, Tang J, Weber IT and Mitsuya H, J. Med. Chem 2009, 52, 7689–7705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lefebvre E and Schiffer CA, AIDS Rev. 2008, 10, 131–142. [PMC free article] [PubMed] [Google Scholar]
- 69.Koh Y, Aoki M, Danish ML, Aoki-Ogata H, Amano M, Das D, Shafer RW, Ghosh AK and Mitsuya H, J. Virol 2011, 85, 10079–10089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hayashi H, Takamune N, Nirasawa T, Aoki M, Morishita Y, Das D, Koh Y, Ghosh AK, Misumi S and Mitsuya H, Proc. Natl. Acad. Sci. U. S. A 2014, 111, 12234–12239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yedidi RS, Garimella H, Aoki M, Aoki-Ogata H, Desai DV, Chang SB, Davis DA, Fyvie WS, Kaufman JD, Smith DDW, Das D, Wingfield PT, Maeda K, Ghosh AK and Mitsuya H, Antimicrob. Agents Chemother 2014, 58, 3679–3688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gustchina A, Sansom C, Prevost M, Richelle J, Wodak SY, Wlodawer A and Weber IT, Protein Eng. 1994, 7, 309–316. [DOI] [PubMed] [Google Scholar]
- 73.Wlodawer A, Miller M, Jaskólski M, Sathyanarayana BK, Baldwin E, Weber IT, Selk LM, Clawson L, Schneider J and Kent SBH, Science 1989, 245, 616–621. [DOI] [PubMed] [Google Scholar]
- 74.Ghosh AK, Chapsal BD, Baldridge A, Steffey MP, Walters DE, Koh Y, Amano M and Mitsuya H, J. Med. Chem 2011, 54, 622–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ghosh AK, Yashchuk S, Mizuno A, Chakraborty N, Agniswamy J, Wang YF, Aoki M, Gomez PM, Amano M, Weber IT and Mitsuya H, ChemMedChem 2015, 10, 107–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Salcedo Gomez PM, Amano M, Yashchuk S, Mizuno A, Das D, Ghosh AK and Mitsuya H, Antimicrob. Agents Chemother 2013, 57, 6110–6121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ghosh AK, Xu CX, Rao KV, Baldridge A, Agniswamy J, Wang YF, Weber IT, Aoki M, Miguel SG, Amano M and Mitsuya H, ChemMedChem 2010, 5, 1850–1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Amano M, Tojo Y, Salcedo-Gomez PM, Campbell JR, Das D, Aoki M, Xu CX, Rao KV, Ghosh AK and Mitsuya H, Antimicrob. Agents Chemother 2013, 57, 2036–2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ghosh AK, Sridhar PR, Leshchenko S, Hussain AK, Li J, Kovalevsky AY, Walters DE, Wedekind JE, Grum-Tokars V, Das D, Koh Y, Maeda K, Gatanaga H, Weber IT and Mitsuya H, J. Med. Chem 2006, 49, 5252–5261. [DOI] [PubMed] [Google Scholar]
- 80.Koh Y, Das D, Leschenko S, Nakata H, Ogata-Aoki H, Amano M, Nakayama M, Ghosh AK and Mitsuya H, Antimicrob. Agents Chemother 2009, 53, 997–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ghosh AK, Gemma S, Takayama J, Baldridge A, Leshchenko-Yashchuk S, Miller HB, Wang YF, Kovalevsky AY, Koh Y, Weber IT and Mitsuya H, Org. Biomol. Chem 2008, 6, 3703–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Delino NS, Aoki M, Hayashi H, Hattori S-I, Chang SB, Takamatsu Y, Martyr CD, Das D, Ghosh AK and Mitsuya H, Antimicrob. Agents Chemother 2018, 62, e02060–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ghosh AK, Rao KV, Nyalapatla PR, Osswald HL, Martyr CD, Aoki M, Hayashi H, Agniswamy J, Wang YF, Bulut H, Das D, Weber IT and Mitsuya H, J. Med. Chem 2017, 60, 4267–4278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Amano M, Salcedo-Gomez PM, Yedidi RS, Delino NS, Nakata H, Rao KV, Ghosh AK and Mitsuya H, Sci. Rep 2017, 71, 12235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Agniswamy J, Kneller DW, Ghosh AK and Weber IT, Biochem. Biophysical. Res. Comm 2021, 566, 30–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ali R and Siddiqui N, J. Chem 2013, 2013, 345198. [Google Scholar]
- 87.Seth S Antiinflamm. Antiallergy Agents Med. Chem 2015, 14, 98–112. [DOI] [PubMed] [Google Scholar]
- 88.Park BK, Kitteringham NR and O’Neill PM, Annu. Rev. Pharmacol. Toxicol 2001, 41, 443–470. [DOI] [PubMed] [Google Scholar]
- 89.Smart BE, J. Fluor. Chem 2001, 109, 3–11. [Google Scholar]
- 90.Ghosh AK, Rao KV, Nyalapatla PR, Kovela S, Brindisi M, Osswald HL, Sekhara Reddy B, Agniswamy J, Wang YF, Aoki M, Hattori SI, Weber IT and Mitsuya H, ChemMedChem 2018, 13, 803–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Aoki M, Hayashi H, Rao KV, Das D, Higashi-Kuwata N, Bulut H, Aoki-Ogata H, Takamatsu Y, Yedidi RS, Davis DA, Hattori SI, Nishida N, Hasegawa K, Takamune N, Nyalapatla PR, Osswald HL, Jono H, Saito H, Yarchoan R, Misumi S, Ghosh AK and Mitsuya H, eLife 2017, 6, e28020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hattori S-I, Hayashi H, Bulut H, Rao KV, Nyalapatla PR, Hasegawa K, Aoki M, Ghosh AK and Mitsuya H, Antimicrob. Agents Chemother 2019, 63, e02635–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bulut H, Hattori S.-i., Aoki-Ogata H, Hayashi H, Das D, Aoki M, Davis DA, Rao KV, Nyalapatla PR, Ghosh AK and Mitsuya H, Sci. Rep 2020, 10, 10664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Koh Y, Amano M, Towata T, Danish M, Leshchenko-Yashchuk S, Das D, Nakayama M, Tojo Y, Ghosh AK and Mitsuya H, J. Virol 2010, 84, 11961–11969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Aoki M, Hayashi H, Yedidi RS, Martyr CD, Takamatsu Y, Aoki-Ogata H, Nakamura T, Nakata H, Das D, Yamagata Y, Ghosh AK and Mitsuya H, J. Virol 2016, 90, 2180–2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ghosh AK, Chapsal BD, Baldridge A, Ide K, Koh Y and Mitsuya H, Org. Lett 2008, 10, 5135–5138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ghosh AK, Chapsal BD, Parham GL, Steffey M, Agniswamy J, Wang Y-F, Amano M, Weber IT and Mitsuya H, J. Med. Chem 2011, 54, 5890–5901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Katsuki T and Sharpless KB, J. Am. Chem. Soc 1980, 102, 5974–5976. [Google Scholar]
- 99.Gao Y, Hanson RM, Klunder JM, Ko SY, Masamue H and Sharpless KB, J. Am. Chem. Soc 1987, 109, 5765–5780. [Google Scholar]
- 100.Ghosh AK, Schiltz GE, Rusere LN, Osswald HL, Walters DE, Amano M and Mitsuya H, Org. Biomol. Chem 2014, 12, 6842–6854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Mukherjee S and Corey EJ, Org. Lett 2010, 12, 1024–1027. [DOI] [PubMed] [Google Scholar]
- 102.Reddy KM, Bhimireddy E, Thirupathi B, Breitler S, Yu S and Corey EJ, J. Am. Chem. Soc 2016, 138, 2443–2453. [DOI] [PubMed] [Google Scholar]
- 103.Lee J-E, Kwon J and Yun J, Chem. Commun 2008, 6, 733–734. [DOI] [PubMed] [Google Scholar]
- 104.Nicolaou KC, Adsool VA and Hale CRH, Org. Lett 2010, 12, 1552–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Caron M, Carlier K and Sharpless KB, J. Org. Chem 1988, 53, 5185–5187. [Google Scholar]
- 106.Ghosh AK, McKee SP, Lee HY and Thompson WT, J. Chem. Soc. Chem. Commun 1992, 1992, 273–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ghosh AK, Cardenas EL and Brindisi M, Tetrahedron Lett. 2017, 58, 4062–4065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ghosh AK, Nyalapatla PR, Kovela S, Rao KV, Brindisi M, Osswald HL, Amano M, Aoki M, Agniswamy J, Wang Y-F, Weber IT and Mitsuya H, J. Med. Chem 2018, 61, 4561–4577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Koh Y, Amano M, Towata T, Danish M, Leshchenko-Yashchuk S, Das D, Nakayama M, Tojo Y, Ghosh AK and Mitsuya H, J. Virol 2010, 84, 11961–11969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Aoki M, Danish ML, Aoki-Ogata H, Amano M, Ide K, Das D, Koh Y and Mitsuya H, J. Virol 2012, 86, 13384–13396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Dulcere JP, Crandal J, Faure R, Santelli M, Agati V and Mihoubi MN, J. Org. Chem 1993, 58, 5702–5708. [Google Scholar]
- 112.Shambayati S, Crewel WE and Schreiber SL, S. L. Tetrahedron Lett. 1990, 31, 5289–5292. [Google Scholar]
- 113.Brummond KM and Kent JL, Tetrahedron 2000, 56, 3263–3283. [Google Scholar]
- 114.Ghosh AK and Chen Y, Tetrahedron Lett. 1995, 36, 505–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Bianchi D, Cesti P and Battistel E, J. Org. Chem 1988, 53, 5531–34. [Google Scholar]
- 116.Ghosh AK, Fyvie WS, Brindisi M, Steffey M, Agniswamy J, Wang Y-F, Aoki M, Amano M, Weber IT and Mitsuya H, ChemMedChem 2017, 12, 1942–1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Yedidi RS, Maeda K, Fyvie WS, Steffey M, Davis DA, Palmer I, Aoki M, Kaufman JD, Stahl SJ, Garimella H, Das D, Wingfield PT, Ghosh AK and Mitsuya H, Antimicrob. Agents Chemother 2013, 57, 4920–4927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ghosh AK, Xia Z, Kovela S, Robinson WL, Johnson ME, Kneller DW, Wang Y-F, Aoki M, Takamatsu Y, Weber IT and Mitsuya H, ChemMedChem 2019, 14, 1863–1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Windsor IW, Palte MJ, Lukesh III JC, Gold B, Forest KT and Raines RT, J. Am. Chem. Soc 2018, 140, 14015–14018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Rusere LN, Lockbaum GJ, Lee S-K, Henes M, Kosovrasti K, Spielvogel E, Nalivaika EA, Swanstrom R, Yilmaz NK, Schiffer CA and Ali A, J. Med. Chem 2019, 62, 8062–8079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Rusere LN, Lockbaum GJ, Henes M, Lee S-K, Spielvogel E, Rao DN, Kosovrasti K, Nalivaika EA, Swanstrom R, Yilmaz NK, Schiffer CA and Ali JA. Med. Chem 2020, 63, 8296–8319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Chen X, Li L, Kempf DJ, Sham H, Wideburg NE, Saldivar A, Vasavanonda S, Marsh KC, McDonald E and Norbeck DW, Bioorg. Med. Chem. Lett 1996, 6, 2847–2852. [Google Scholar]
- 123.Bai X, Yang Z, Zhu M, Dong B, Zhou L, Zhang G, Wang J and Wang Y, Eur. J. Med. Chem 2017, 137, 30–44. [DOI] [PubMed] [Google Scholar]
- 124.Yang Z-H, Bai X-G, Zhou L, Wang J-X, Liu H-T and Wang Y-C, Bioorg. Med. Chem. Lett 2015, 25, 1880–1883. [DOI] [PubMed] [Google Scholar]
- 125.Bungard CJ, Williams PD, Schulz J, Wiscount CM, Holloway MK, Loughran HM, Manikowski JJ, Su H-P, Bennett DJ, Chang L, Chu X-J, Crespo A, Dwyer MP, Keertikar K, Morriello GJ, Stamford AW, Waddell ST, Zhong B, Hu B, Ji T, Diamond TL, Bahnck-Teets C, Carroll SS, Fay JF, Min X, Morris W, Ballard JE, Miller MD and McCauley JA, ACS Med. Chem. Lett 2017, 8, 1292–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Bungard CJ, Williams PD, Ballard JE, Bennett DJ, Beaulieu C, Bahnck-Teets C, Carroll SS, Chang RK, Dubost DC, Fay JF, Diamond TL, Greshock TJ, Hao L, Holloway MK, Felock PJ, Gesell JJ, Su H-P, Manikowski JJ, McKay DJ, Miller M, Min X, Molinaro C, Moradei OM, Nantermet PG, Nadeau C, Sanchez RI, Satyanarayana T, Shipe WD, Singh SK, Truong VL, Vijayasaradhi S, Wiscount CM, Vacca JP, Crane SN, and McCauley JA, ACS Med. Chem. Lett 2016, 7, 702–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Coburn CA, Holloway MK, Vacca JP, Preparation of phenylsulfonylpyrrolidinylaminoethyl diphenylalaninamides as HIV protease inhibitors. WO2010138338AI, 2010. [Google Scholar]