

Abstract
G protein-coupled receptors (GPCRs) are the largest family of transmembrane receptors and are the target of approximately one-third of all Food and Drug Administration (FDA)-approved pharmaceutical drugs. GPCRs interact with many transducers, such as heterotrimeric G proteins, GPCR kinases (GRKs), and β-arrestins. Recent experiments have demonstrated that some ligands can activate distinct effector proteins over others, a phenomenon termed “biased agonism.” These discoveries have raised the potential of developing drugs which preferentially activate therapeutic signaling pathways over those that lead to deleterious side effects. However, to date, only one biased GPCR therapeutic has received FDA approval and many others have either failed to meet their specified primary end points and or demonstrate superiority over currently available treatments. In addition, there is a lack of understanding regarding how biased agonism measured at a GPCR leads to specific downstream physiological responses. Here, we briefly summarize the history and current status of biased agonism at GPCRs and suggest adoption of a “systems pharmacology” approach upon which to develop GPCR-targeted drugs that demonstrate heightened therapeutic efficacy with improved side effect profiles.
Keywords: β-arrestin, biased agonism, drug development, G protein-coupled receptor, systems pharmacology
INTRODUCTION
G protein-coupled receptors (GPCRs) represent the largest family of transmembrane receptors and currently serve as the target for ∼30% of all Food and Drug Administration-approved small molecule drugs (1). GPCRs can signal through many effector proteins; however, the best characterized are the G proteins and β-arrestins. It was recently discovered that some ligands can preferentially activate specific signaling pathways over others, a phenomenon termed “biased agonism” (2). This discovery has spurred many efforts to develop G protein- or β-arrestin-biased drugs that demonstrate heightened therapeutic efficacy with improved side effect profile. However, these efforts have not yet demonstrated enhanced clinical utility (3). With the hindsight of nearly 50 years of scientific advancement in the GPCR field, the binary of G protein versus β-arrestin model of biased receptor signaling is too reductive when considering the pluridimensional signaling observed at GPCRs. Rather than associating a particular signaling output with a single proximal effector protein (G protein or β-arrestin), it may be better to adopt a “systems pharmacology” approach, where the mechanisms underlying a desired or pathological phenotype are explored at multiple levels beyond the receptor. Here, we provide a historical context of GPCR signaling and the discovery of biased agonism. We also discuss the current methods used to study biased agonism at GPCRs and highlight the potential utility of adopting a “systems pharmacology” approach to understand and exploit biased GPCR signaling.
A BRIEF HISTORY OF GPCR SIGNALING
Although the idea of a cell receptor was first conceptualized as early as 1905, the study of receptor regulation and signaling mechanisms was made possible in the early 1970s through the emergence of new tools including photoaffinity labeling, radioligand binding, and affinity chromatography (4, 5). These techniques drastically improved our ability to provide specific and quantitative information about the regulation of receptors and the binding affinity for a wide variety of ligands. Receptor biology research no longer had to rely on inferences made solely through observations of downstream signaling effects. Rather, it allowed for the interpretation of the now commonly used agonist and antagonist binding curves and their incorporation into receptor theory. Eventually, these efforts led to the development of the widely used ternary complex model where receptor binding to an intracellular effector allosterically enhances the binding affinity of the receptor for the ligand, and vice versa (6).
The first proposal of the ternary complex model of G proteins was suggested in 1980 after the successful purification of the heterotrimeric protein in charge of regulating adenylyl cyclase activity, with the classification as a “G protein” reflecting its ability to bind guanine nucleotides (7). In later years, the Lefkowitz laboratory would use chimeric receptors of the α2 and β2 adrenergic receptors to demonstrate that the α2 receptor inhibits adenylyl cyclase through the inhibitory G protein Gαi whereas the β2 adrenergic receptor (β2AR) stimulates adenylyl cyclase through the stimulatory G protein Gαs (8). Subsequently, the purification of the β2AR additionally allowed for the design of oligonucleotide probes used to clone the cDNA and gene of the β2AR, leading to the discovery of canonical features associated with the GPCR family, such as the seven transmembrane domains, sites for regulatory phosphorylation, and sites of consensus sequences for glycosylation at the amino terminus (9).
With the sequencing of the β2AR came the realization that it shared significant homology with rhodopsin, a receptor which has contributed much to our understanding of canonical GPCR signaling (10). Rhodopsin was identified as a protein in the 1930s that underwent signal transduction after exposure to light through cis to trans isomerization of conjugated retinal. During the 1970s into the 1980s, it was found that rhodopsin activated a cyclic nucleotide phosphodiesterase through a GTPase now known as the G protein transducin (10). Around the same time, researchers discovered the existence of rhodopsin kinase, now known as GRK1, which was able to phosphorylate light-activated rhodopsin (11). In addition, reports identified a protein, formerly known as 48 kDa protein or retinal S antigen and now known as visual arrestin, that was involved in the deactivation of rhodopsin (12). As more GPCRs were identified using sequence homology, the focus shifted to understanding the phenomenon of receptor desensitization, which eventually led to the identification of the β-adrenergic receptor kinase (BARK), now known as GPCR kinase 2 (GRK2), one of the seven classified GRKs primarily responsible for phosphorylation of GPCRs (13). β-arrestin, closely related to visual arrestin, was discovered shortly after in 1987 for its ability to “arrest” signaling across the GPCR superfamily (14).
The current paradigm of GPCR signaling involves interaction of the receptor with the heterotrimeric G proteins, the GRKs, and the β-arrestins. Following stimulation, the activated receptor interacts with the G proteins, resulting in a cascade of signaling followed by phosphorylation of the receptor by the GRKs. β-arrestin then binds to the phosphorylated receptor and sterically inhibits further G protein activation, thus resulting in receptor desensitization and a diminished physiological response. It was subsequently shown that β-arrestin can also serve as its own signal transducing system by acting as a scaffold that links the receptor to other intracellular molecules (15). Of these pathways, the most rigorously studied is the ability of β-arrestin to scaffold effectors in the mitogen-activated protein kinase/extracellular signal-related kinase (MAPK/ERK) pathway, and also mediate clathrin-dependent endocytosis, thus proving capable of facilitating receptor desensitization, receptor internalization, and signaling (5).
Further scrutiny of GPCR signaling uncovered the discovery of biased ligands, which refers to the ability of an agonist to selectively activate some pathways, such as those mediated by G proteins or β-arrestins, compared with a reference agonist. Classically, agonists are capable of eliciting maximal signaling responses, partial agonists generate submaximal responses, antagonists do not generate any change in receptor signaling but compete for the receptor orthosteric site, and inverse agonists suppress basal receptor signaling. Importantly, biased agonists do not fit this classical paradigm as they may demonstrate agonist behavior at certain signaling pathways but may be partial agonists or even antagonists at other signaling pathways, i.e., they signal with different efficacies through signaling pathways downstream of a receptor. Recent studies have provided structural evidence that biased agonists stabilize the receptor in particular conformation that differ from the classical “active” and “inactive” states of the receptor, thereby allowing the activation of selective responses (16).
Biased agonists have served as tool compounds to understand specific GPCR signaling pathways that are associated with either β-arrestin or G protein signaling (2). For example, certain GPCRs demonstrate β-arrestin-dependent activation of receptor internalization and activation of MAPK/ERK signaling (15). Conversely, there is evidence for G protein-dependent signaling including the regulation of cyclic adenosine monophosphate (cAMP) levels through adenylyl cyclase, calcium levels through phospholipase C, and the activity of Rho guanine nucleotide exchange factor (17). However, not all β-arrestin- and G protein-mediated signaling pathways are independent of each other. For example, several studies have demonstrated that the MAPK/ERK signaling pathways can be activated by both β-arrestins and G proteins, albeit with differential kinetic and spatial patterns (15). In addition, recent work has observed an agonist-induced physical association between Gαi and β-arrestin, even at GPCRs that do not canonically couple to Gαi (18).
CURRENT APPROACHES TO STUDY GPCR-BIASED AGONISM
There are many mechanisms that result in a functionally selective GPCR response. The most commonly studied is “ligand bias,” where a ligand demonstrates biased signaling relative to a reference agonist, commonly the endogenous ligand, at the same receptor (Fig. 1A). For example, G protein-biased agonists of the µ-opioid receptor have distinct physiological effects compared with reference agonists (19–21). In “receptor bias,” a GPCR demonstrates preferential signaling through one transducer over another, largely irrespective of the ligand used to activate the receptor (Fig. 1B), a phenomenon that has been described at atypical chemokine receptors that recruit β-arrestins but do not activate G proteins (22). Methods for quantifying bias based on cellular analyses provide data for one defined set of experimental conditions, whereas in vivo, ligands encounter a range of tissues with different receptor densities, effector expression levels, and agonist sensitivities (23). In “systems bias,” an unbiased ligand and receptor may demonstrate biased signaling between cell types due to differences in the stoichiometry of downstream effectors (24) (Fig. 1C). For example, a β-arrestin-biased D2 dopamine receptor agonist had distinct effects in the cortex and striatum, which was associated with differences in GRK2 expression (25). In “location bias,” the specific subcellular location of a GPCR may demonstrate a signaling profile that is different than that observed at the plasma membrane (Fig. 1D) (26). For example, endosomal signaling by GPCRs has been shown to have distinct effects on transcription (27) and therapeutically targeting neurokinin endosomal signaling has distinct effects on the pain response (28).
Figure 1.
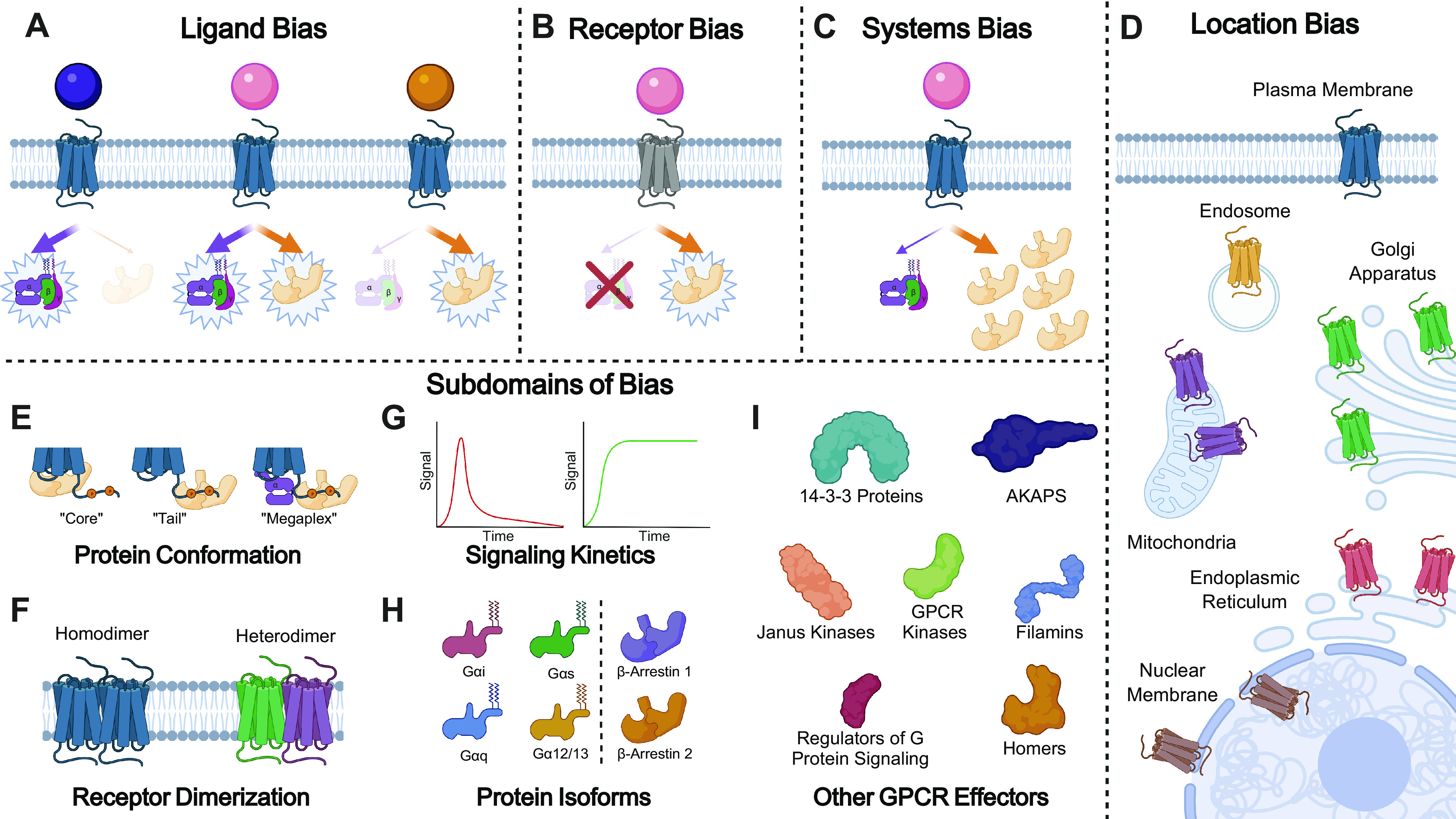
Types of biased agonism observed at GPCRs. A: ligand bias—various ligands can bind to the same receptor but demonstrate differential activation of downstream transducers. The purple ligand (left) demonstrates a G protein-biased ligand, the pink ligand (middle) demonstrates a balanced ligand, and the yellow ligand (right) demonstrates a β-arrestin-biased ligand. B: receptor bias—some GPCRs demonstrate activation of β-arrestins over G proteins, or vice versa, irrespective of the bound ligand. Although the pink balanced ligand can activate both G proteins and β-arrestins at the blue GPCR, it demonstrates preferential activation of β-arrestins at the gray GPCR, as this receptor demonstrates receptor bias. C: systems bias—an unbiased ligand and unbiased receptor can generate a biased signaling response if the cellular system has unequal stoichiometries of signal effectors. D: location bias—GPCRs can exist at many subcellular locations and may demonstrate signaling profiles that are different from that observed at the plasma membrane. E: β-arrestins can adopt many conformations including a state bound to the GPCR core (“core” conformation), GPCR tail (“tail” conformation), or the GPCR tail whereas G proteins are simultaneously engaged with the GPCR core (“megaplex” conformation). These different conformations are related to specific β-arrestin-mediated signaling pathways. F: GPCRs are known to homo- or heterodimerize, as well as oligomerize. These multimeric structures can demonstrate signaling that is different from GPCR monomers. G: different ligands or GPCRs can activate the same effectors at different rates which can translate into different cellular responses. H: there are multiple G protein and β-arrestin isoforms which can simultaneously couple to a single GPCR. In addition to being biased with respect to G proteins and β-arrestins, a GPCR may demonstrate differentially coupling within each effector family. I: there are numerous other identified effectors that can couple to GPCRs, many of which may demonstrate biased behavior. GPCR, G protein-coupled receptor. AKAPS, A-kinase-anchoring protein.
In addition, granularity of biased agonism exists that allows for even greater signaling specificity beyond the canonical G protein versus β-arrestin paradigm. We refer to this granularity in biased agonism as “subdomains” of bias. For example, β-arrestin is known to adopt many different conformations (tail, core, and megaplex) which are directly associated with specific functions, such as receptor internalization or inhibition of G protein signaling (29) (Fig. 1E). GPCRs are now appreciated to hetero- or homodimerize, and even oligomerize, with other receptors or with accessory proteins, which impact their signaling (30) (Fig. 1F). The biased agonism observed by a GPCR may also vary based on the specific kinetic profile of the observed signaling (31) (Fig. 1G). In addition, a GPCR can simultaneously couple to many different isoforms of both G protein and β-arrestins, and these isoforms can demonstrate divergent signaling (32) (Fig. 1H). Finally, there are many identified signaling effectors that couple to GPCRs beyond G proteins and β-arrestins, which also may demonstrate biased signaling (Fig. 1I).
Our understanding of biased agonism has been aided by technological breakthroughs, such as those in receptor pharmacology, structural biology, and biotechnology, enabling more rapid, thorough, and targeted detailing of GPCR structural properties and signaling activity (Table 1). In particular is the adoption of resonance energy transfer (RET)-based assays with fluorescence (FRET) and bioluminescence (BRET) biosensors. Using these technologies, multiple aspects of GPCR signaling including heterotrimeric G protein dissociation (35), G protein nucleotide status (36), β-arrestin recruitment and trafficking (33), β-arrestin conformation (40), second messenger accumulation (54), and receptor dimerization (39) can be interrogated. We do not discuss the methods used to investigate the structural mechanisms underlying biased agonism as they are beyond the scope of this review.
Table 1.
Examples of recent technologies to assess biased signaling at GPCRs.
| Signaling Activities | Assay | Example | Ref. |
|---|---|---|---|
| Transducer recruitment and trafficking | BRET/FRET | BRET between β-arrestin-RlucII and GFP-location marker to monitor β-arrestin trafficking to subcellular locations | (33) |
| Enzyme complementation | Split nanoluciferase assay to assess β-arrestin recruitment and intracellular trafficking | (34) | |
| Transducer activation | G protein dissociation | TRUPATH BRET assay to assess Gα dissociation from Gβɣ | (35) |
| G protein nucleotide status | BERKY BRET biosensors detecting GTP-bound Gα | (36) | |
| Receptor internalization and trafficking | BRET/FRET | Assessing GPCR-RlucII trafficking to GFP-tagged markers for different cellular locations (plasma membrane, endosome, etc.) | (33) |
| Microscopy | Visualizing the redistribution of fluorescently labeled receptors | (37) | |
| Protein-protein interactions | Proximity labeling | APEX proximity labeling for identification of interacting partners at the AT1R and the β2AR using biased ligands | (38) |
| Time-resolved RET | BRET/FRET for GPCR oligomerization | (39) | |
| Tripartite protein interactions | Gαi:β-arrestin:ERK complex formation at the vasopressin receptor 2 | (18) | |
| Conformation | Intramolecular RET | FlAsH intramolecular BRET biosensors to assess β-arrestin conformation at six different locations | (40) |
| Spectroscopy | NMR and DEER to assess AT1R structure using biased ligands | (16, 41) | |
| Atomic structure determination | Crystallography, cryo-electron microscopy | (42,43) | |
| In silico methods | V-SYNTHES—structure-based screening of 11 billion compounds to identify targets of the cannabinoid receptor | (44) | |
| Molecular dynamics simulation of β-arrestin at vasopressin receptor 2 C-terminal mutants | (45) | ||
| Post-translational modification | Phosphorylation | Mass spectrometry-based analysis of GPCR C-terminus at CXCR4, mass spectrometry of global phosphoproteome | (46–48) |
| Ubiquitination | BRET-based real-time monitoring of β-arrestin ubiquitination | (49) | |
| Receptor-ligand interactions | Fluorescence polarization | HTS of novel small molecule binders of NTS1 receptor | (50) |
| BRET/FRET | FRET between fluorescent ligand and fluorescently tagged receptor | (51) | |
| Surface plasmon resonance | Measure direct binding between an immobilized receptor and its ligands for high throughput drug screening | (52,53) | |
| Signaling | Second messenger accumulation | RET biosensor to quantify intracellular levels of cAMP, DAG, IP3, pERK | (46, 54–56) |
| Fluorescent dyes for intracellular calcium influx | (57) | ||
| Transcription | Reporter assays (luciferase, β-galactosidase), RNA-seq | (57,58) |
AT1R, angiotensin II type 1 receptor; β2AR, β2 adrenergic receptor; BRET, bioluminescence resonance energy transfer; cAMP, cyclic adenosine monophosphate; CXCR4, chemokine receptor 4; DAG, diacylglycerol; DEER, double electron-electron spectroscopy; ERK, extracellular signal-related kinase; FlAsH, fluorescein arsenical hairpin; FRET, fluorescence resonance energy transfer; GFP, green fluorescent protein; GPCR, G protein-coupled receptor; HTS, high throughput screen; IP3, inositol triphosphate; NTS1, neurotensin receptor 1; NMR, nuclear magnetic resonance spectroscopy; pERK, phosphorylated extracellular signal-related kinase; RlucII, Renilla luciferase II; RNA-seq, RNA sequencing.
Many assessments of biased agonism relied on the detection of an amplified signal, where the efficacy of GPCR signaling through a particular pathway can be easily misinterpreted (59). Many of these RET-based assays are stoichiometric in nature, allowing for accurate assessments and direct comparison of relative coupling with G proteins and β-arrestins. Because RET-based technologies can interrogate many of the aforementioned subdomains of biased agonism, they have become widely adapted as tools to assess GPCR functional selectivity. Using these technologies, one can mathematically model, either quantitatively or qualitatively, the extent of biased agonism using bias plots or by calculating bias factors (59). A limitation to these RET-based assays is that many of them require fusion to a fluorescent or bioluminescent probe that may impact the normal function of the protein being studied. However, novel methods have been developed to use RET methods in a manner that allows for interrogation of GPCR transducers, such as G protein nucleotide status, without the need to modify the protein of interest (36).
It is next important to determine whether the observed biased pharmacological response corresponds to a functionally selective cellular response. Quantitative imaging technologies, such as high content imaging (HCS) (60), label-free optical tools including dynamic mass redistribution (DMR) (61) and surface plasmon resonance (SPR) (62), and electric biosensor technologies, such as cellular impedance spectroscopy (63) have provided a more comprehensive tool set for determining phenotypes associated with GPCR activity. Such methods can report on cellular processes such as cell proliferation, migration, cell death, cell-cell communication, and adhesion (64). These whole cell phenotypical profiling approaches can delineate collective cellular responses and ultimate biological outputs of GPCR signaling activity. Importantly, these assays can be used in conjunction with tools that manipulate GPCR signaling to determine the contribution of G proteins and β-arrestins to the cellular response. For example, one can study the function of a biased agonist in the context of pharmacological inhibitors of specific G protein isoforms, or by using cell lines devoid of G proteins or β-arrestins (15).
Once a biased ligand is associated with a desirable cellular or in vivo response, one can identify GPCR therapeutics using screening-based approaches on a desired set of pharmacological properties. There are also efforts to use in silico methods to develop GPCR therapeutics based on existing ligand-bound GPCR structures (65). For example, the development of the FDA-approved antagonist Maraviroc, used in the treatment of human immunodeficiency virus (HIV), began with high throughput screening (HTS) using a CC-chemokine receptor 5 radioligand-binding assay, which identified a weak agonist that had no cellular antiviral activity, but served as a starting point for compound modification and synthesis based on structure-activity relationship studies (66). However, attempts to translate such pharmacological findings for biased agonists to therapeutics is difficult, particularly due to problems with drug efficacy, toxicity, or unexpected biology (67). Cell culture conditions, in which many signaling studies are conducted, are often not analogous to biological conditions. For example, studies in Chinese hamster ovary (CHO) cells have shown that when the mouse β3-adrenoceptor is expressed at high concentrations, cAMP-mediated signaling predominates, but when the same receptor is expressed at low concentrations, MAPK/ERK pathways become the most significant signaling pathway (68). Due to the context-based specificity of biased signaling, addressing such discrepancies is critical for successful biased drug development.
UTILIZING SYSTEMS PHARMACOLOGY TO UNDERSTAND AND EXPLOIT GPCR-BIASED AGONISM
The discovery of biased agonism at GPCRs raised the possibility of developing functionally selective pharmaceutical drugs which activate therapeutic signaling pathways while simultaneously avoiding deleterious effects (2). Currently, the general framework in developing or discovering biased GPCR therapeutics is to associate biased signaling of compounds at the level of the GPCR with a desired or pathological physiological output in vivo or in cell culture. Frequently, the observed phenotype is associated with signaling through the proximal transducers with little insight into the signaling pathways that are directly responsible for the observed phenotype (Fig. 2A). This approach is best highlighted at the µ-opioid receptor, where previous work suggested that signaling via β-arrestins leads to constipation, respiratory, and tolerance whereas signaling through G protein leads to antinociception (69, 70). However, more recent studies have challenged those findings, and it is unclear if this paradigm is as simple as previously described (71). This general approach relies on associating differential activation of a few signaling events proximal to the receptor, but the overall effect of an agonist in vivo is driven by the activity of multiple pathways whose activity is collectively modulated by the ligand.
Figure 2.
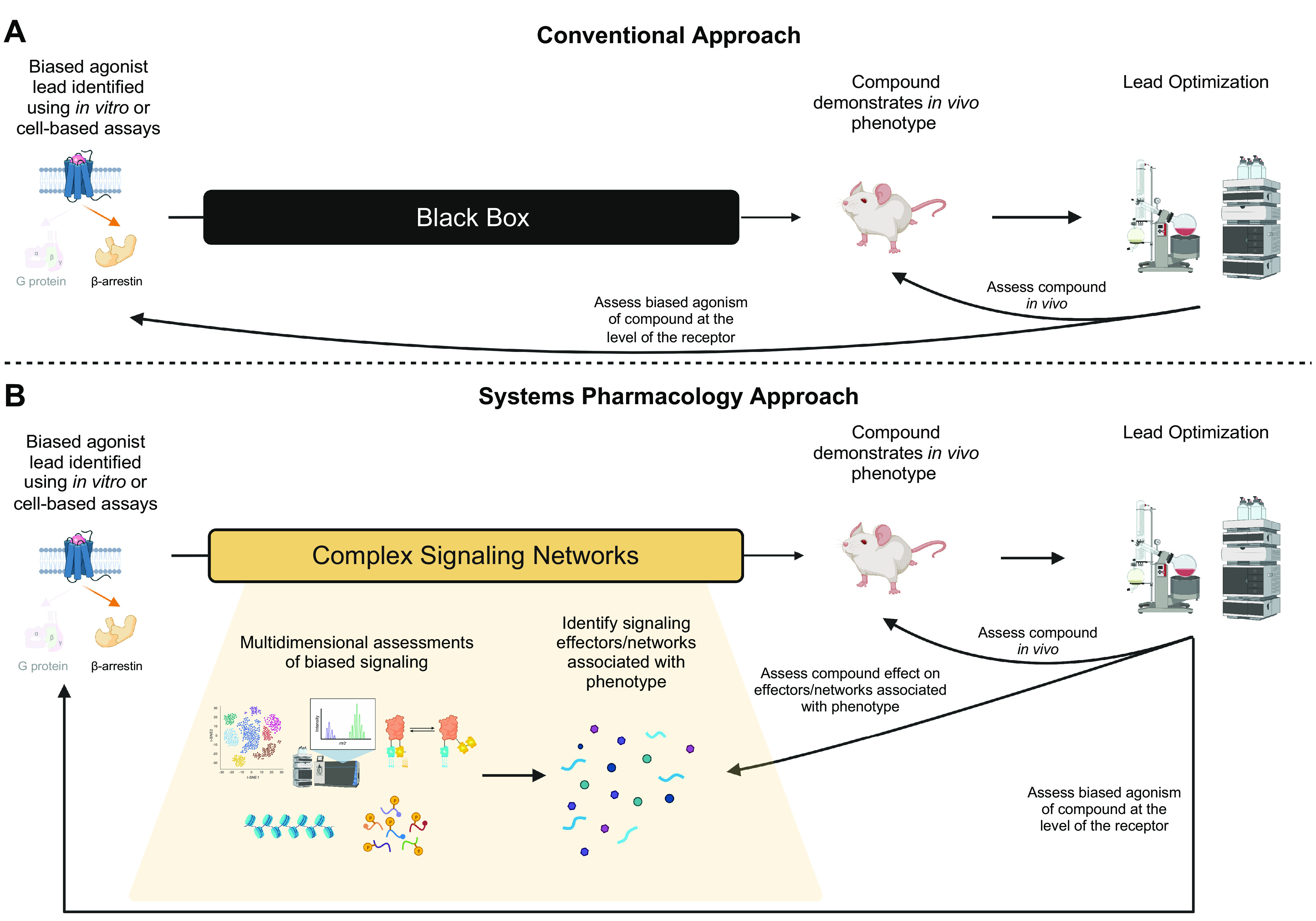
A revised approach to the development of biased GPCR therapeutics. A: conventional approach to develop biased GPCR ligands: an endogenous or synthetic ligand demonstrates biased agonism at a GPCR, and this specific signaling is associated with a desired cellular or in vivo response. The signaling pathways responsible for the in vivo phenotype are largely considered as a black box. These compounds are then tested for the ability to generate the desired cellular or in vivo response. Medicinal chemistry or high throughput screening (HTS) is used to refine the ligand or identify other biased agonists which can then be tested at earlier stages in this approach. B: a systems pharmacology approach to develop biased GPCR ligands: a similar process is repeated as above; however, after the association between biased GPCR signaling and a desired cellular or in vivo phenotype is determined, multiple analyses are carried out to better define the signaling pathways that underlie the desired response. For example, methods include RNA-seq, mass spectrometry-based proximity labeling, proteomics, or phosphoproteomics, ChIP-seq, and RET-based biosensors. Identification of signaling pathways that associate with the desired phenotype can then serve as direct drug targets, or markers upon which to screen drug compounds. Similarly, the identified compounds using medicinal chemistry or HTS are then tested for their ability to generate the desired cellular or in vivo response, after which the process can be repeated and refined. ChIP-seq; chromatin immunoprecipitation sequencing; GPCR, G protein-coupled receptor; RET, resonance energy transfer.
Rather than acting discretely through G proteins or through β-arrestins, GPCRs signal through multiple transducers/effectors through a complex and interconnected signaling network. There are many potential explanations underlying this signaling including direct interactions between G proteins and β-arrestins (18), convergence of initially divergent signaling pathways, differential kinetic and spatial patterns of activation of the same pathways, multiple drug targets by a specific ligand, or the integration of both GPCR and non-GPCR signaling. Associating proximal cellular signaling assays of G protein and β-arrestin signaling with specific phenotypes may be too reductive when trying to develop biased pharmaceuticals which promote the desired physiological effects. For those reasons, we advocate for a “systems pharmacology” approach when developing biased therapeutics (Fig. 2B). Systems pharmacology has been defined as “the study of a biological process in which all of the relevant components are studied together in parallel to discover integrative mechanisms” (72). In practice, after a particular phenotype and proximal GPCR signaling profile are associated, a more rigorous approach is taken to dissect the complex signaling that underlies this association. Given the widespread availability and reasonable cost of current technologies, one can assess many aspects of cellular biology and biochemistry. Mass spectrometry can identify differential expression of proteins, posttranslational modifications such as phosphorylation, and even GPCR effectors using proximity-based assays (46,47, 73). RNA sequencing (RNA-seq), single-cell RNA-seq, chromatin immunoprecipitation sequencing (ChIP-seq), and assay for transposase-accessible chromatin using sequencing (ATAC-seq) allow for high throughput assessment of transcription, promoter function, and DNA accessibility (74). Various fluorescent and bioluminescent biosensors exist to assess kinase activity and second messengers such as cAMP and calcium (57). Use of multidimensional assays and the associated computational tools to analyze such large data sets may uncover key and novel GPCR signaling pathways that are better correlated to or even directly associated with the phenotype being analyzed. Then, drugs can be screened or developed that demonstrate a signaling profile that appropriately regulate the effector(s) identified in addition to or in place of G proteins and β-arrestins. This systems pharmacology approach has already been applied to understand the mechanisms underlying the V2R-mediated regulation of the aquaporin-2 channel in water balance disorders (74). Similarly, a systems pharmacology approach was used to understand the mechanisms underlying retinal degeneration and identify novel combination therapies that target multiple GPCRs to potentially treat this disease (75, 76). Importantly, because laboratories can now generate large -omic data sets that interrogate cellular signaling across multiple dimensions, there has been a revolution in data science to analyze “multi-omic” data sets which integrate multiple data sets to study complex biological processes (77). An ideal systems pharmacology approach will require a concerted effort that lies at the intersection of biochemistry, molecular biology, structural biology, pharmacology, and data science to completely understand how a receptor-ligand interaction is translated into a biological phenotype. Adopting a systems pharmacology approach to biased agonism at GPCRs may unveil novel effectors or complex signaling networks that are better associated with cellular and animal physiology and increase the likelihood of developing a successful biased GPCR therapeutic.
CONCLUSIONS
Biased agonism at GPCRs presents an exciting opportunity to develop targeted therapeutics with heightened clinical efficacy and an improved side effect profile. Currently, biased agonism is generally described as the relative ability of a GPCR to differentially activate proximal transducers, such as G proteins or β-arrestins. Efforts to develop biased agonists as drugs have met with limited success up to this point, possibly because attributing the complex GPCR signaling and phenotypes to proximal transducers is too reductive. Rather, we believe that a systems pharmacology approach that investigates both proximal and distal aspects of GPCR signaling may unveil mechanisms underlying various pathologies and identify effectors and networks that can be targeted directly or through a single or multiple GPCRs. Although G proteins and β-arrestins are undoubtedly critical signaling effectors that regulate both normal and abnormal physiology, they are simply the tip of the iceberg that underlie biased GPCR signaling. The complexity of GPCR signaling networks presents an exciting opportunity to redefine how we as a research community define biased GPCR signaling and how we search for and design novel GPCR therapeutics.
GRANTS
This work was supported by the National Institute of General Medical Sciences Grants T32GM00717 (to D.S.E) and R01GM122798 (to S.R); the Duke University Medical Scientist Training Program (to D.S.E); and the American Heart Association Predoctoral Fellowship No. 20PRE35120592 (to D.S.E).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
This article is part of the special collection “Advances in GPCRs: Structure, Mechanisms, Disease, and Pharmacology.” Wei Kong, MD, PhD, and Jinpeng Sun, PhD, served as Guest Editors of this collection.
AUTHOR CONTRIBUTIONS
D.S.E. prepared figures; D.S.E., U.P., J.G., and C.H. drafted manuscript; D.S.E., U.P., J.G., C.H., and S.R. edited and revised manuscript; D.S.E., U.P., J.G., C.H., and S.R. approved final version of manuscript.
ACKNOWLEDGMENTS
Graphical figures were created using BioRender.com.
REFERENCES
- 1.Sriram K, Insel PA. G protein-coupled receptors as targets for approved drugs: how many targets and how many drugs? Mol Pharmacol 93: 251–258, 2018. doi: 10.1124/mol.117.111062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Smith JS, Lefkowitz RJ, Rajagopal S. Biased signalling: from simple switches to allosteric microprocessors. Nat Rev Drug Discov 17: 243–260, 2018. doi: 10.1038/nrd.2017.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Michel MC, Charlton SJ. Biased agonism in drug discovery-is it too soon to choose a path? Mol Pharmacol 93: 259–265, 2018. doi: 10.1124/mol.117.110890. [DOI] [PubMed] [Google Scholar]
- 4.Barwich A-S, Bschir K. The manipulability of what? The history of G-protein coupled receptors. Biol Philos 32: 1317–1339, 2017. doi: 10.1007/s10539-017-9608-9. [DOI] [Google Scholar]
- 5.Lefkowitz RJ. A brief history of G-protein coupled receptors (Nobel Lecture). Angew Chem Int Ed Engl 52: 6366–6378, 2013. doi: 10.1002/anie.201301924. [DOI] [PubMed] [Google Scholar]
- 6.De Lean A, Stadel JM, Lefkowitz RJ. A ternary complex model explains the agonist-specific binding properties of the adenylate cyclase-coupled beta-adrenergic receptor. J Biol Chem 255: 7108–7117, 1980. doi: 10.1016/S0021-9258(20)79672-9. [DOI] [PubMed] [Google Scholar]
- 7.Northup JK, Sternweis PC, Smigel MD, Schleifer LS, Ross EM, Gilman AG. Purification of the regulatory component of adenylate cyclase. Proc Natl Acad Sci USA 77: 6516–6520, 1980. doi: 10.1073/pnas.77.11.6516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kobilka BK, Kobilka TS, Daniel K, Regan JW, Caron MG, Lefkowitz RJ. Chimeric α2-,β2-adrenergic receptors: delineation of domains involved in effector coupling and ligand binding specificity. Science 240: 1310–1316, 1988. doi: 10.1126/science.2836950. [DOI] [PubMed] [Google Scholar]
- 9.Dixon RA, Kobilka BK, Strader DJ, Benovic JL, Dohlman HG, Frielle T, Bolanowski MA, Bennett CD, Rands E, Diehl RE, Mumford RA, Slater EE, Sigal IS, Caron MG, Lefkowitz RJ, Strader CD. Cloning of the gene and cDNA for mammalian β-adrenergic receptor and homology with rhodopsin. Nature 321: 75–79, 1986. doi: 10.1038/321075a0. [DOI] [PubMed] [Google Scholar]
- 10.Costanzi S, Siegel J, Tikhonova IG, Jacobson KA. Rhodopsin and the others: a historical perspective on structural studies of G protein-coupled receptors. Curr Pharm Des 15: 3994–4002, 2009. doi: 10.2174/138161209789824795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Benovic JL. Historical perspective of the g protein-coupled receptor kinase family. Cells 10: 555, 2021. doi: 10.3390/cells10030555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Caron MG, Barak LS. A brief history of the β-arrestins. Methods Mol Biol 1957: 3–8, 2019. doi: 10.1007/978-1-4939-9158-7_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Benovic JL, Strasser RH, Caron MG, Lefkowitz RJ. β-adrenergic receptor kinase: identification of a novel protein kinase that phosphorylates the agonist-occupied form of the receptor. Proc Natl Acad Sci USA 83: 2797–2801, 1986. doi: 10.1073/pnas.83.9.2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kühn H, Wilden U. Deactivation of photoactivated rhodopsin by rhodopsin-kinase and arrestin. J Recept Res 7: 283–298, 1987. doi: 10.3109/10799898709054990. [DOI] [PubMed] [Google Scholar]
- 15.Luttrell LM, Wang J, Plouffe B, Smith JS, Yamani L, Kaur S, Jean-Charles PY, Gauthier C, Lee MH, Pani B, Kim J, Ahn S, Rajagopal S, Reiter E, Bouvier M, Shenoy SK, Laporte SA, Rockman HA, Lefkowitz RJ. Manifold roles of β-arrestins in GPCR signaling elucidated with siRNA and CRISPR/Cas9. Sci Signal 11: eaat7650, 2018. doi: 10.1126/scisignal.aat7650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wingler LM, Elgeti M, Hilger D, Latorraca NR, Lerch MT, Staus DP, Dror RO, Kobilka BK, Hubbell WL, Lefkowitz RJ. Angiotensin analogs with divergent bias stabilize distinct receptor conformations. Cell 176: 468–478.e11, 2019. doi: 10.1016/j.cell.2018.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wootten D, Christopoulos A, Marti-Solano M, Babu MM, Sexton PM. Mechanisms of signalling and biased agonism in G protein-coupled receptors. Nat Rev Mol Cell Biol 19: 638–653, 2018. doi: 10.1038/s41580-018-0049-3. [DOI] [PubMed] [Google Scholar]
- 18.Smith JS, Pack TF, Inoue A, Lee C, Zheng K, Choi I, Eiger DS, Warman A, Xiong X, Ma Z, Viswanathan G, Levitan IM, Rochelle LK, Staus DP, Snyder JC, Kahsai AW, Caron MG, Rajagopal S. Noncanonical scaffolding of Gαi and β-arrestin by G protein-coupled receptors. Science 371: eaay1833, 2021. doi: 10.1126/science.aay1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Soergel DG, Subach RA, Burnham N, Lark MW, James IE, Sadler BM, Skobieranda F, Violin JD, Webster LR. Biased agonism of the μ-opioid receptor by TRV130 increases analgesia and reduces on-target adverse effects versus morphine: A randomized, double-blind, placebo-controlled, crossover study in healthy volunteers. Pain 155: 1829–1835, 2014. doi: 10.1016/j.pain.2014.06.011. [DOI] [PubMed] [Google Scholar]
- 20.Viscusi ER, Webster L, Kuss M, Daniels S, Bolognese JA, Zuckerman S, Soergel DG, Subach RA, Cook E, Skobieranda F. A randomized, phase 2 study investigating TRV130, a biased ligand of the μ-opioid receptor, for the intravenous treatment of acute pain. Pain 157: 264–272, 2016. doi: 10.1097/j.pain.0000000000000363. [DOI] [PubMed] [Google Scholar]
- 21.Manglik A, Lin H, Aryal DK, McCorvy JD, Dengler D, Corder G, Levit A, Kling RC, Bernat V, Hübner H, Huang XP, Sassano MF, Giguère PM, Löber S, Da D, Scherrer G, Kobilka BK, Gmeiner P, Roth BL, Shoichet BK. Structure-based discovery of opioid analgesics with reduced side effects. Nature 537: 185–190, 2016. doi: 10.1038/nature19112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rajagopal S, Kim J, Ahn S, Craig S, Lam CM, Gerard NP, Gerard C, Lefkowitz RJ. β-arrestin- but not G protein-mediated signaling by the “decoy” receptor CXCR7. Proc Natl Acad Sci USA 107: 628–632, 2010. doi: 10.1073/pnas.0912852107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kenakin T, Christopoulos A. Signalling bias in new drug discovery: detection, quantification and therapeutic impact. Nat Rev Drug Discov 12: 205–216, 2013. doi: 10.1038/nrd3954. [DOI] [PubMed] [Google Scholar]
- 24.Onfroy L, Galandrin S, Pontier SM, Seguelas MH, N'Guyen D, Sénard JM, Galés C. G protein stoichiometry dictates biased agonism through distinct receptor-G protein partitioning. Sci Rep 7: 7885, 2017. doi: 10.1038/s41598-017-07392-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Urs NM, Gee SM, Pack TF, McCorvy JD, Evron T, Snyder JC, Yang X, Rodriguiz RM, Borrelli E, Wetsel WC, Jin J, Roth BL, O'Donnell P, Caron MG. Distinct cortical and striatal actions of a β-arrestin-biased dopamine D2 receptor ligand reveal unique antipsychotic-like properties. Proc Natl Acad Sci USA 113: E8178–E8186, 2016. doi: 10.1073/pnas.1614347113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mohammad Nezhady MA, Rivera JC, Chemtob S. Location bias as emerging paradigm in GPCR biology and drug discovery. iScience 23: 101643, 2020. doi: 10.1016/j.isci.2020.101643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tsvetanova NG, von Zastrow M. Spatial encoding of cyclic AMP signaling specificity by GPCR endocytosis. Nat Chem Biol 10: 1061–1065, 2014. doi: 10.1038/nchembio.1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jensen DD, Lieu T, Halls ML, Veldhuis NA, Imlach WL, Mai QN, Poole DP, Quach T, Aurelio L, Conner J, Herenbrink CK, Barlow N, Simpson JS, Scanlon MJ, Graham B, McCluskey A, Robinson PJ, Escriou V, Nassini R, Materazzi S, Geppetti P, Hicks GA, Christie MJ, Porter CJH, Canals M, Bunnett NW. Neurokinin 1 receptor signaling in endosomes mediates sustained nociception and is a viable therapeutic target for prolonged pain relief. Sci Transl Med 9: eaal3447, 2017. doi: 10.1126/scitranslmed.aal3447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thomsen ARB, Plouffe B, Cahill TJ 3rd, Shukla AK, Tarrasch JT, Dosey AM, Kahsai AW, Strachan RT, Pani B, Mahoney JP, Huang L, Breton B, Heydenreich FM, Sunahara RK, Skiniotis G, Bouvier M, Lefkowitz RJ. GPCR-G Protein-β-arrestin super-complex mediates sustained G protein signaling. Cell 166: 907–919, 2016. doi: 10.1016/j.cell.2016.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rocheville M, Lange DC, Kumar U, Patel SC, Patel RC, Patel YC. Receptors for dopamine and somatostatin: formation of hetero-oligomers with enhanced functional activity. Science 288: 154–157, 2000. doi: 10.1126/science.288.5463.154. [DOI] [PubMed] [Google Scholar]
- 31.Klein Herenbrink C, Sykes DA, Donthamsetti P, Canals M, Coudrat T, Shonberg J, Scammells PJ, Capuano B, Sexton PM, Charlton SJ, Javitch JA, Christopoulos A, Lane JR. The role of kinetic context in apparent biased agonism at GPCRs. Nat Commun 7: 10842, 2016. doi: 10.1038/ncomms10842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ahn S, Wei H, Garrison TR, Lefkowitz RJ. Reciprocal regulation of angiotensin receptor-activated extracellular signal-regulated kinases by β-arrestins 1 and 2. J Biol Chem 279: 7807–7811, 2004. doi: 10.1074/jbc.C300443200. [DOI] [PubMed] [Google Scholar]
- 33.Namkung Y, Le Gouill C, Lukashova V, Kobayashi H, Hogue M, Khoury E, Song M, Bouvier M, Laporte SA. Monitoring G protein-coupled receptor and β-arrestin trafficking in live cells using enhanced bystander BRET. Nat Commun 7: 12178, 2016. doi: 10.1038/ncomms12178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hauge Pedersen M, Pham J, Mancebo H, Inoue A, Asher WB, Javitch JA. A novel luminescence-based β-arrestin recruitment assay for unmodified receptors. J Biol Chem 296: 100503, 2021. doi: 10.1016/j.jbc.2021.100503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Olsen RHJ, DiBerto JF, English JG, Glaudin AM, Krumm BE, Slocum ST, Che T, Gavin AC, McCorvy JD, Roth BL, Strachan RT. TRUPATH, an open-source biosensor platform for interrogating the GPCR transducerome. Nat Chem Biol 16: 841–849, 2020. doi: 10.1038/s41589-020-0535-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maziarz M, Park JC, Leyme A, Marivin A, Garcia-Lopez A, Patel PP, Garcia-Marcos M. Revealing the activity of trimeric G-proteins in live cells with a versatile biosensor design. Cell 182: 770–785.e16, 2020. doi: 10.1016/j.cell.2020.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jullié D, Valbret Z, Stoeber M. Optical tools to study the subcellular organization of GPCR neuromodulation. J Neurosci Methods 366: 109408, 2022. doi: 10.1016/j.jneumeth.2021.109408. [DOI] [PubMed] [Google Scholar]
- 38.Paek J, Kalocsay M, Staus DP, Wingler L, Pascolutti R, Paulo JA, Gygi SP, Kruse AC. Multidimensional tracking of GPCR signaling via peroxidase-catalyzed proximity labeling. Cell 169: 338–349.e11, 2017. doi: 10.1016/j.cell.2017.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cottet M, Faklaris O, Maurel D, Scholler P, Doumazane E, Trinquet E, Pin JP, Durroux T. BRET and Time-resolved FRET strategy to study GPCR oligomerization: from cell lines toward native tissues. Front Endocrinol (Lausanne) 3: 92, 2012. doi: 10.3389/fendo.2012.00092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee MH, Appleton KM, Strungs EG, Kwon JY, Morinelli TA, Peterson YK, Laporte SA, Luttrell LM. The conformational signature of β-arrestin2 predicts its trafficking and signalling functions. Nature 531: 665–668, 2016. doi: 10.1038/nature17154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shimada I, Ueda T, Kofuku Y, Eddy MT, Wüthrich K. GPCR drug discovery: integrating solution NMR data with crystal and cryo-EM structures. Nat Rev Drug Discov 18: 59–82, 2019. doi: 10.1038/nrd.2018.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Safdari HA, Pandey S, Shukla AK, Dutta S. Illuminating GPCR signaling by cryo-EM. Trends Cell Biol 28: 591–594, 2018. doi: 10.1016/j.tcb.2018.06.002. [DOI] [PubMed] [Google Scholar]
- 43.Congreve M, de Graaf C, Swain NA, Tate CG. Impact of GPCR structures on drug discovery. Cell 181: 81–91, 2020. doi: 10.1016/j.cell.2020.03.003. [DOI] [PubMed] [Google Scholar]
- 44.Sadybekov AA, Sadybekov AV, Liu Y, Iliopoulos-Tsoutsouvas C, Huang XP, Pickett J, Houser B, Patel N, Tran NK, Tong F, Zvonok N, Jain MK, Savych O, Radchenko DS, Nikas SP, Petasis NA, Moroz YS, Roth BL, Makriyannis A, Katritch V. Synthon-based ligand discovery in virtual libraries of over 11 billion compounds. Nature 601: 452–459, 2022. doi: 10.1038/s41586-021-04220-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dwivedi-Agnihotri H, Chaturvedi M, Baidya M, Stepniewski TM, Pandey S, Maharana J, Srivastava A, Caengprasath N, Hanyaloglu AC, Selent J, Shukla AK. Distinct phosphorylation sites in a prototypical GPCR differently orchestrate β-arrestin interaction, trafficking, and signaling. Sci Adv 6: eabb8368, 2020.doi: 10.1126/sciadv.abb8368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tsvetanova NG, Trester-Zedlitz M, Newton BW, Peng GE, Johnson JR, Jimenez-Morales D, Kurland AP, Krogan NJ, von Zastrow M. Endosomal cAMP production broadly impacts the cellular phosphoproteome. J Biol Chem 297: 100907, 2021. doi: 10.1016/j.jbc.2021.100907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lone AM, Giansanti P, Jorgensen MJ, Gjerga E, Dugourd A, Scholten A, Saez-Rodriguez J, Heck AJR, Tasken K. Systems approach reveals distinct and shared signaling networks of the four PGE2 receptors in T cells. Sci Signal 14: eabc8579, 2021.doi: 10.1126/scisignal.abc8579. [DOI] [PubMed] [Google Scholar]
- 48.Busillo JM, Armando S, Sengupta R, Meucci O, Bouvier M, Benovic JL. Site-specific phosphorylation of CXCR4 is dynamically regulated by multiple kinases and results in differential modulation of CXCR4 signaling. J Biol Chem 285: 7805–7817, 2010. doi: 10.1074/jbc.M109.091173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Perroy J, Pontier S, Charest PG, Aubry M, Bouvier M. Real-time monitoring of ubiquitination in living cells by BRET. Nat Methods 1: 203–208, 2004. doi: 10.1038/nmeth722. [DOI] [PubMed] [Google Scholar]
- 50.Heine P, Witt G, Gilardi A, Gribbon P, Kummer L, Plückthun A. High-throughput fluorescence polarization assay to identify ligands using purified G protein-coupled receptor. SLAS Discov 24: 915–927, 2019. doi: 10.1177/2472555219837344. [DOI] [PubMed] [Google Scholar]
- 51.Ciruela F, Jacobson KA, Fernández-Dueñas V. Portraying G protein-coupled receptors with fluorescent ligands. ACS Chem Biol 9: 1918–1928, 2014. doi: 10.1021/cb5004042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Capelli D, Parravicini C, Pochetti G, Montanari R, Temporini C, Rabuffetti M, Trincavelli ML, Daniele S, Fumagalli M, Saporiti S, Bonfanti E, Abbracchio MP, Eberini I, Ceruti S, Calleri E, Capaldi S. Surface plasmon resonance as a tool for ligand binding investigation of engineered GPR17 receptor, a G protein coupled receptor involved in myelination. Front Chem 7: 910, 2019. doi: 10.3389/fchem.2019.00910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Huber S, Casagrande F, Hug MN, Wang L, Heine P, Kummer L, Plückthun A, Hennig M. SPR-based fragment screening with neurotensin receptor 1 generates novel small molecule ligands. PLoS One 12: e0175842, 2017. doi: 10.1371/journal.pone.0175842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jiang LI, Collins J, Davis R, Lin KM, DeCamp D, Roach T, Hsueh R, Rebres RA, Ross EM, Taussig R, Fraser I, Sternweis PC. Use of a cAMP BRET sensor to characterize a novel regulation of cAMP by the sphingosine 1-phosphate/G13 pathway. J Biol Chem 282: 10576–10584, 2007. doi: 10.1074/jbc.M609695200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tewson PH, Martinka S, Shaner NC, Hughes TE, Quinn AM. New DAG and cAMP sensors optimized for live-cell assays in automated laboratories. J Biomol Screen 21: 298–305, 2016. doi: 10.1177/1087057115618608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Harvey CD, Ehrhardt AG, Cellurale C, Zhong H, Yasuda R, Davis RJ, Svoboda K. A genetically encoded fluorescent sensor of ERK activity. Proc Natl Acad Sci USA 105: 19264–19269, 2008. doi: 10.1073/pnas.0804598105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yasi EA, Kruyer NS, Peralta-Yahya P. Advances in G protein-coupled receptor high-throughput screening. Curr Opin Biotechnol 64: 210–217, 2020. doi: 10.1016/j.copbio.2020.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen L, Jung HJ, Datta A, Park E, Poll BG, Kikuchi H, Leo KT, Mehta Y, Lewis S, Khundmiri SJ, Khan S, Chou CL, Raghuram V, Yang CR, Knepper MA. Systems biology of the vasopressin V2 receptor: new tools for discovery of molecular actions of a GPCR. Annu Rev Pharmacol Toxicol 62: 595–616, 2022. doi: 10.1146/annurev-pharmtox-052120-011012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gundry J, Glenn R, Alagesan P, Rajagopal S. A practical guide to approaching biased agonism at G protein coupled receptors. Front Neurosci 11: 17, 2017. doi: 10.3389/fnins.2017.00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rausch O. High content cellular screening. Curr Opin Chem Biol 10: 316–320, 2006. doi: 10.1016/j.cbpa.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 61.Deng H, Sun H, Fang Y. Label-free cell phenotypic assessment of the biased agonism and efficacy of agonists at the endogenous muscarinic M3 receptors. J Pharmacol Toxicol Methods 68: 323–333, 2013. doi: 10.1016/j.vascn.2013.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Maynard JA, Lindquist NC, Sutherland JN, Lesuffleur A, Warrington AE, Rodriguez M, Oh SH. Surface plasmon resonance for high-throughput ligand screening of membrane-bound proteins. Biotechnol J 4: 1542–1558, 2009. doi: 10.1002/biot.200900195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Peters MF, Scott CW. Evaluating cellular impedance assays for detection of GPCR pleiotropic signaling and functional selectivity. J Biomol Screen 14: 246–255, 2009. doi: 10.1177/1087057108330115. [DOI] [PubMed] [Google Scholar]
- 64.Scott CW, Peters MF. Label-free whole-cell assays: expanding the scope of GPCR screening. Drug Discov Today 15: 704–716, 2010. doi: 10.1016/j.drudis.2010.06.008. [DOI] [PubMed] [Google Scholar]
- 65.Zhang R, Xie X. Tools for GPCR drug discovery. Acta Pharmacol Sin 33: 372–384, 2012. doi: 10.1038/aps.2011.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dorr P, Westby M, Dobbs S, Griffin P, Irvine B, Macartney M, Mori J, Rickett G, Smith-Burchnell C, Napier C, Webster R, Armour D, Price D, Stammen B, Wood A, Perros M. Maraviroc (UK-427,857), a potent, orally bioavailable, and selective small-molecule inhibitor of chemokine receptor CCR5 with broad-spectrum anti-human immunodeficiency virus type 1 activity. Antimicrob Agents Chemother 49: 4721–4732, 2005. doi: 10.1128/AAC.49.11.4721-4732.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bowes J, Brown AJ, Hamon J, Jarolimek W, Sridhar A, Waldron G, Whitebread S. Reducing safety-related drug attrition: the use of in vitro pharmacological profiling. Nat Rev Drug Discov 11: 909–922, 2012. doi: 10.1038/nrd3845. [DOI] [PubMed] [Google Scholar]
- 68.Sato M, Horinouchi T, Hutchinson DS, Evans BA, Summers RJ. Ligand-directed signaling at the β3-adrenoceptor produced by 3-(2-Ethylphenoxy)-1-[(1,S)-1,2,3,4-tetrahydronapth-1-ylamino]-2S-2-propanol oxalate (SR59230A) relative to receptor agonists. Mol Pharmacol 72: 1359–1368, 2007. doi: 10.1124/mol.107.035337. [DOI] [PubMed] [Google Scholar]
- 69.Bohn LM, Lefkowitz RJ, Gainetdinov RR, Peppel K, Caron MG, Lin FT. Enhanced morphine analgesia in mice lacking β-arrestin 2. Science 286: 2495–2498, 1999. doi: 10.1126/science.286.5449.2495. [DOI] [PubMed] [Google Scholar]
- 70.Bohn LM, Gainetdinov RR, Lin FT, Lefkowitz RJ, Caron MG. μ-opioid receptor desensitization by β-arrestin-2 determines morphine tolerance but not dependence. Nature 408: 720–723, 2000. doi: 10.1038/35047086. [DOI] [PubMed] [Google Scholar]
- 71.Kliewer A, Gillis A, Hill R, Schmiedel F, Bailey C, Kelly E, Henderson G, Christie MJ, Schulz S. Morphine-induced respiratory depression is independent of β-arrestin2 signalling. Br J Pharmacol 177: 2923–2931, 2020. doi: 10.1111/bph.15004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Knepper MA. Systems biology of diuretic resistance. J Clin Invest 125: 1793–1795, 2015. doi: 10.1172/JCI81505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lobingier BT, Hüttenhain R, Eichel K, Miller KB, Ting AY, von Zastrow M, Krogan NJ. An approach to spatiotemporally resolve protein interaction networks in living cells. Cell 169: 350–360.e12, 2017. doi: 10.1016/j.cell.2017.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chen L, Jung HJ, Datta A, Park E, Poll BG, Kikuchi H, Leo KT, Mehta Y, Lewis S, Khundmiri SJ, Khan S, Chou CL, Raghuram V, Yang CR, Knepper MA. Systems biology of the vasopressin V2 receptor: new tools for discovery of molecular actions of a GPCR. Annu Rev Pharmacol Toxicol 62: 595–616, 2021. doi: 10.1146/annurev-pharmtox-052120-011012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chen Y, Palczewska G, Mustafi D, Golczak M, Dong Z, Sawada O, Maeda T, Maeda A, Palczewski K. Systems pharmacology identifies drug targets for Stargardt disease-associated retinal degeneration. J Clin Invest 123: 5119–5134, 2013. doi: 10.1172/JCI69076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chen Y, Palczewska G, Masuho I, Gao S, Jin H, Dong Z, Gieser L, Brooks MJ, Kiser PD, Kern TS, Martemyanov KA, Swaroop A, Palczewski K. Synergistically acting agonists and antagonists of G protein-coupled receptors prevent photoreceptor cell degeneration. Sci Signal 9: ra74, 2016. doi: 10.1126/scisignal.aag0245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Subramanian I, Verma S, Kumar S, Jere A, Anamika K. Multi-omics data integration, interpretation, and its application. Bioinform Biol Insights 14: 1177932219899051, 2020. doi: 10.1177/1177932219899051. [DOI] [PMC free article] [PubMed] [Google Scholar]