

Summary
The Polyomaviridae is a family of ubiquitous dsDNA viruses that establish persistent infection early in life. Screening for human polyomaviruses (HPyVs), which comprise 14 diverse species, relies upon species-specific qPCRs whose validity may be challenged by accelerating genomic exploration of the virosphere. Using this reasoning, we tested 64 published HPyV qPCR assays in silico against the 1781 PyV genome sequences that were divided in targets and nontargets, based on anticipated species specificity of each qPCR. We identified several cases of problematic qPCR performance that were confirmed in vitro and corrected through using degenerate oligos. Furthermore, our study ranked 8 out of 52 tested BKPyV qPCRs as remaining of consistently high quality in the wake of recent PyV discoveries and showed how sensitivity of most other qPCRs could be rescued by annealing temperature adjustment. This study establishes an efficient framework for ensuring confidence in available HPyV qPCRs in the genomic era.
Subject areas: Virology, Omics
Graphical Abstract
Highlights
-
•
In silico testing of 64 HPyV qPCRs versus 1781 (non)targets guides PCR adjustments
-
•
Recognition of 1781 (non)targets by a HPyV qPCR is visualized in 2D/3D oligo Tm maps
-
•
Problematic qPCRs were confirmed in vitro and corrected using degenerate oligos
-
•
In silico analysis finds 8 of 52 BKPyV qPCRs resilient to PyV genomic expansion
Virology; Omics
Introduction
Polyomaviruses are ubiquitous dsDNA viruses that are transmitted early in life and establish asymptomatic persistent infection (Kean et al., 2009; van der Meijden et al., 2013). On average, a healthy individual is infected with nine different human polyomaviruses (HPyVs) during life-time (Gossai et al., 2016; Kamminga et al., 2018). In elderly and immunocompromised patients, HPyVs can cause symptomatic infection, such as nephropathy in kidney transplant patients and progressive multifocal leukoencephalopathy in HIV patients and patients treated with particular immunomodulatory drugs. Natural known diversity of HPyVs and diseases associated with some of the HPyV infections are summarized in Table 1. HPyVs are classified into 14 species, separated by at least 15% residue differences within the large T antigen coding sequence. Thirteen species names include “Human polyomavirus” followed by a number from 1 to 14 in italic (Table 1). For instance, BK polyomavirus (BKPyV) belongs to the species Human polyomavirus 1 within this taxonomic nomenclature framework. One of the species, Human polyomavirus 12, was recently renamed to Sorex araneus polyomavirus 1, because its prototypic virus HPyV12 was found to be almost identical to Shrew polyomavirus 1 in genome sequence analysis (Gedvilaite et al., 2017; Moens et al., 2017).
Table 1.
Polyomavirus species, including human viruses, that were analyzed in this study
| Species | Virus (name acronym) | Main disease associated with infection | Seroprevalence %a | Specimen | Year of virus discovery (reference) |
|---|---|---|---|---|---|
| Human polyomavirus 1 | BK polyomavirus (BKPyV) | Transplant nephropathy; hemorrhagic cystitis | 99 | Urine | 1971 (Gardner et al., 1971) |
| Human polyomavirus 2 | JC polyomavirus (JCPyV) | Progressive multifocal leukoencephalopathy (PML) | 63 | Brain | 1971 (Padgett et al., 1971) |
| Human polyomavirus 3 | Karolinska Institutet polyomavirus (KIPyV) | Respiratory illness | 92 | Nasopharynx | 2007 (Allander et al., 2007) |
| Human polyomavirus 4 | Washington University polyomavirus (WUPyV) | Respiratory illness | 99 | Nasopharynx | 2007 (Gaynor et al., 2007) |
| Human polyomavirus 5 | Merkel cell polyomavirus (MCPyV) | Merkel cell carcinoma | 82 | Skin | 2008 (Feng et al., 2008) |
| Human polyomavirus 6 | Human polyomavirus 6 (HPyV6) | Pruritic and dyskeratotic dermatosis | 84 | Skin | 2010 (Schowalter et al., 2010) |
| Human polyomavirus 7 | Human polyomavirus 7 (HPyV7) | Pruritic and dyskeratotic dermatosis | 72 | Skin | 2010 (Schowalter et al., 2010) |
| Human polyomavirus 8 | Trichodysplasia spinulosa polyomavirus (TSPyV) | Trichodysplasia spinulosa | 80 | Skin | 2010 (van der Meijden et al., 2010) |
| Human polyomavirus 9 | Human polyomavirus 9 (HPyV9) | None | 19 | Serum | 2011 (Sauvage, 2011; Scuda et al., 2011) |
| Human polyomavirus 10 | Malawi polyomavirus (MWPyV) | None | 100 | Stool | 2012 (Buck et al., 2012; Siebrasse et al., 2012) |
| Human polyomavirus 11 | Saint Louis polyomavirus (STLPyV) | None | 65 | Stool | 2012 (Lim et al., 2013) |
| Sorex araneus polyomavirus 1 | Human polyomavirus 12 (HPyV12) | None | 4 | Liver | 2013 (Korup et al., 2013) |
| Human polyomavirus 13 | New Jersey polyomavirus (NJPyV) | Vasculitis, myositis, retinitis | 5 | Muscle | 2014 (Mishra et al., 2014) |
| Human polyomavirus 14 | Lyon IARC polyomavirus (LIPyV) | None | 6 | Skin | 2017 (Gheit et al., 2017) |
As determined previously (Kamminga et al., 2018).
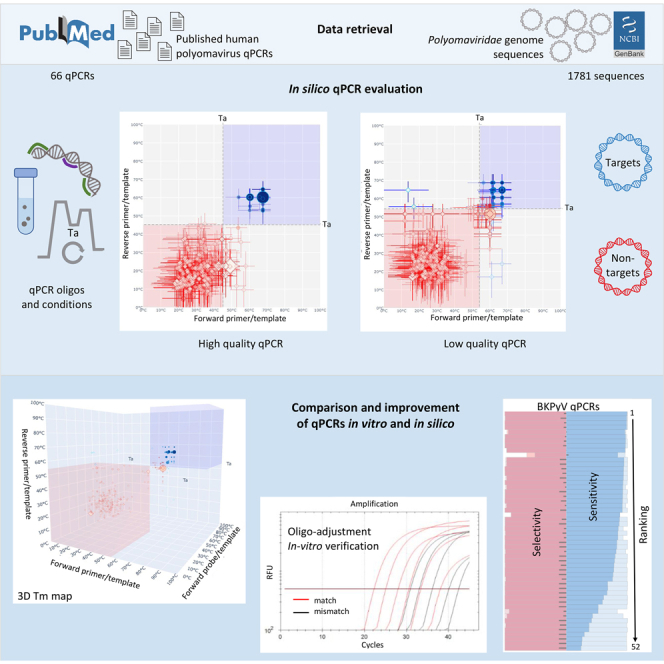
Humans are tested for the presence of HPyVs using “diagnostic” virus-specific PCRs that target viruses of a certain species. These are usually validated and certified at the time of design and thereafter periodically through external quality assessment. Both PCR oligo design and assessment, conducted in silico and in vitro, respectively, are defined by few target viruses. Researchers select target viruses from the available sampling that varies considerably for different polyomaviruses, due to the biased knowledge about natural variation of the respective viruses at the time of the PCR design and availability of the viral genomes and panel samples. Pathogenic strains, of JC polyomavirus (JCPyV) for example, are more likely to be sequenced and thus relatively overrepresented in sequence repositories compared with persistent, avirulent strains. Likewise, closely related variants of the previously identified strains may dominate divergent variants simply because they are readily recognized by currently used PCR primer and probe sets. Ideally, it would be desirable to link updates of diagnostic PCRs to the continuous advancement of our knowledge about natural polyomavirus variation due to expanded genome sequencing (Figure 1), but this may be impractical to do in vitro in a time-wise and cost-effective manner. Use of in silico approaches may present a viable solution to this persisting problem.
Figure 1.

Dynamic of accumulation of complete polyomavirus genome sequences
Shown is annual dynamic of accumulation of the analyzed complete genome sequences of the family Polyomaviridae in GenBank until October 10th 2019 (1781 genomes), according to HAYGENS (https://veb.lumc.nl/HAYGENS/). Genome sequences are dated according to their GenBank entries, which may deviate from the first date of the public sequence release.
For HPyV detection, our laboratory currently uses 14 species-specific qPCRs, of which nine were developed in-house and five adopted from literature, including that against the reassigned HPyV12 (Kamminga et al., 2019). External quality assessment has been performed on a regular basis only for the qPCRs that target the most common polyomaviruses: BKPyV and JCPyV. To address this gap and assess whether these 14 HPyV qPCRs remain as good in the face of expanding genome sequencing as they were at the time of design, here we performed in silico testing of each HPyV qPCR against all currently, publicly available polyomavirus genome sequences, including those of nonhuman origins using a previously described approach (Nijhuis et al., 2018). This analysis was further extended to test a selection of 52 published BKPyV qPCRs. For these purposes, in silico sensitivity and selectivity were calculated for each HPyV qPCR, by analysis of publicly available PyV genome sequences that were divided into target and nontarget groups for each qPCR. We used also in vitro qPCR analysis to test some target genomes that were poorly recognized in silico due to mismatches with PCR oligo(s) or other reasons. Results of the in vitro qPCR and the in silico analysis were in agreement. Furthermore, utilization of target and nontarget datasets facilitated adjustment of annealing temperature (Ta; called thereafter adjusted Ta) in silico in a PCR-specific manner for all analyzed in-house and published HPyV qPCRs.
Results
In silico evaluation of in-house HPyV qPCRs
We started our in silico study by analysis of the in-house qPCRs targeting one of the 14 HPyVs, either in VP1 or LT genes (12 and 2 PCRs, respectively) (Table 2). The in silico workflow used main variables of the PCR assays, including oligos and other components, to calculate PCR sensitivity and selectivity under original and adjusted Ta in the application to the 1,781 complete PyV genome sequences retrieved from GenBank (Figure 2, top panel, Table S2; see STAR Methods). The PyV sequences were assigned to targets and nontargets specific for each PCR, and the recognition of each sequence was assessed numerically and visualized using 2D and 3D Tm maps for each pair of PCR oligos (Figure 2, bottom panel, and Supplemental information). These maps detail temperature-dependent stability of oligo/template complexes and genomic location compatibility of three PCR oligos for product generation, using two measures, T-decision and L-decision, respectively (see STAR Methods for details).
Table 2.
Overview of lab-developed, in-house used HPyV qPCRs
| HPyV target (species Human polyomavirus #) | GenBank ID | Target genea | Expected product length (bp) | Forward primer sequence (5′–3′) | Probe sequence (polarity) (5′–3′ for forward and 3′–5′ for reverse) | Reverse primer sequence (3′–5′) | Year of design | References |
|---|---|---|---|---|---|---|---|---|
| BKPyV (1) | NC_001538 | VP1 | 90 | GAAAAGGAGAGTGTCCAGGG | FAM-CCAAAAAGCCAAAGGAACCC (F) | GAACTTCTACTCCTCCTTTTATTAGT | 2003 | van der Meijden et al. (2014) |
| JCPyV (2) | NC_001699 | LT | 129 | GTCTCCCCATACCAACATTAGCTT | YAK-TCTTTCCACTGCACAATCCTCTCATGAATG (F) | GGTTTAGGCCAGTTGCTGACTT | 2006 | Pal et al., (2006) |
| KIPyV (3) | NC_009238 | VP1 | 148 | AAGTTCCCCGGGTACAAACTC | TXR-GGTAGAAGTACTAGCCGCAGTACCACTGT (F) | CCATCCTGAGCAGCTGTTGTA | 2016 | Kamminga et al. (2019) |
| WUPyV (4) | NC_009539 | VP1 | 74 | AACCAGGAAGGTCACCAAGAAG | TXR-CAACCCACAAGAGTGCAAAGCCTTCC (F) | CTACCCCTCCTTTTCTGACTTGTTT | 2011 | Rao et al. (2011) |
| MCPyV (5) | NC_010277 | LT | 149 | CCACAGCCAGAGCTCTTCCT | CY5-TCCCAGGCTTCAGACTCCCAb (F) | TGGTGGTCTCCTCTCTGCTACTG | 2009 | Goh et al. (2009) |
| HPyV6 (6) | NC_014406 | VP1 | 150 | GTAGGGTATGCTGGTAAC | YAK-CTCTCCTCTGTCTGAAGTGAACTCTAA (R) | CAGGAATTGTCTAAACATCATATC | 2012 | Purdie et al. (2018) |
| HPyV7 (7) | NC_014407 | VP1 | 116 | GTGCTGATATGGTTGGAA | TXR-AGCCTGTACTGTTCTCTGGTTACT (R) | TCTGCAGTGGACTCTAAA | 2012 | Purdie et al. (2018) |
| TSPyV (8) | NC_014361 | VP1 | 104 | GAGTCTAAGGACAACTATGG | Q705-CTTGTCCTGGTCACTGCTGTT (R) | CTAGCTGTACTGTAGGTTG | 2012 | van der Meijden et al. (2016) |
| HPyV9 (9) | NC_015150 | VP1 | 109 | CCTGTAAGCTCTCTCCTTA | FAM-CTTGTTCTCTGGTCTTATGCCTCA (F) | CCTGATAAATTCTGACTTCTTC | 2012 | van der Meijden et al. (2014) |
| MWPyV (10) | NC_018102 | VP1 | 86 | GACACCACAATGACAGTTGAG | CY5-CCAAGGATGGGCAATGATGTAAAAACA (F) | GGATCACTGTAGCCATACCAT | 2016 | Kamminga et al. (2019) |
| STLPyV (11) | NC_020106 | VP1 | 101 | TTGAAAATGGCTCCAAAAAGAAAATCT | CY5-AGATGCACCTCACAGACATGTCCAATGGA (F) | TGGCACGGATCATATTCACATCT | 2016 | Kamminga et al. (2019) |
| HPyV12c | NC_020890 | VP1 | 139 | AAGGGCTGTAAGAAATCC | FAM-CCAGTATCTGCTCTCCTAACCAGT (F) | CTCCAAACCCTCATATACC | 2015 | Kamminga et al. (2019) |
| NJPyV (13) | NC_024118 | VP1 | 135 | CCCACCAAGTAAAGTAAC | YAK-AAGTGTCCTATACCTACTCCAGTGC (F) | CAGAGTTCAATTTCAGTAGTA | 2015 | Kamminga et al. (2019) |
| LIPyV (14) | NC_034253 | VP1 | 83 | TGACAGGTGACAATTCCCAGG | Q705-AGAGGAAGTACGCGTCTATGATGGCAGAG (F) | CCTTGGCAGATCTAACCCTCC | 2017 | Kamminga et al. (2019) |
Abbreviations: LT: Large T; VP1: Viral Protein 1; F: Forward; R: Reverse.
Probe modified compared with original article.
HPyV12 was formerly in species Human polyomavirus 12 but has been reassigned to species Sorex araneus polyomavirus 1 (https://talk.ictvonline.org/taxonomy/p/taxonomy-history?taxnode_id=201904426).
Figure 2.

Schematic workflow of in silico PCR testing and example of results visualization
Presented are main stages of in silico analysis of a publicly available HPyV qPCR using genome sequences of polyomaviruses (top two panels), as well as an example of results visualization (bottom panel). This pipeline is also applied to analysis of in-house HPyV qPCRs, which provided PCR variables. All calculations are performed for each genome sequence and PCR oligos set and are detailed in the STAR Methods section. Results of in silico evaluation of the qPCR in respect to T-decision ranges and L-decision binary of qPCR oligos annealed to target (blue) and nontarget (red) templates are presented using a Tm map for each pair of qPCR oligos, three in total. Each Tm map is divided into four nonoverlapping orthogonal zones delimited by two internal boundaries set at temperature (T) corresponding to the Ta of the presented qPCR and distinguished by three background colors. Light blue zone: T-ranges favorable for annealing of both oligos to template and facilitating qPCR; light red zone: T-ranges unfavorable for both oligos to facilitate qPCR; two light gray zones: a T-range is unfavorable for one of two oligos to facilitate qPCR. When the coordinates of the calculated qPCR product conforms to the sequence location boundaries delimited by the corresponding L-decision = 1, Tm values of a pair of oligos annealed to the respective sequence are labeled with a circle, otherwise they are a diamond. Two labels may overlap, partly or fully, on the map, and size of circle or diamond label is proportional to the number of labels, when they fully overlap. Position of each label on the map corresponds to Tm of the oligo/template complex for two oligos under which the T-decision is equal to 0.5. Each label occupies middle position in two bars, vertical (bottom-to-top) and horizontal (left-to-right), which delimit T-ranges for the respective oligo/template complexes corresponding to the T-decision [0.95–0.05] ranges (see STAR Methods). The opacity of each label corresponds to its T-decision value within the respective target or nontarget color gradient. Interactive versions of 2D and 3D Tm maps for the data presented in this study are available on the resource website. The user can zoom into any label to learn from a pop-up about Tm, template GenBank ID, and other characteristics of oligo/template complexes. When a label represents several fully overlapping oligo/template complexes, the pop-up informs about the number of sequences involved in the overlap and details characteristics of a single sequence only. Note that an overlap may involve sequences from the same or different groups, namely targets and nontargets. The user may explore three 2D Tm maps of a PCR simultaneously using a 3D Tm map that can facilitate understanding the basis of sensitivity and selectivity.
Selectivity was consistently high for all 14 analyzed qPCRs (between 0.98 and 1), regardless of the use of standard or adjusted Ta, suggesting a high specificity of all original qPCRs in respect to the sequenced nontarget HPyV genomes (Table 3). In contrast, the in silico sensitivity under the standard Ta was quite variable (between 0.19 and 0.97), indicating target-dependence of some qPCRs. Ta adjustment considerably improved qPCR sensitivity, up to the range of 0.92–1. No link between the calculated qPCR sensitivity and the presence of mismatches, before and after Ta adjustment, was observed. Below we describe in silico analysis of three HPyV qPCRs (directed against Trichodysplasia spinulosa polyomavirus (TSPyV), JCPyV, and BKPyV) in detail, and use in vitro testing to verify the sensitivity estimation for several targets and extending it also to oligos with corrected mismatches.
Table 3.
In silico evaluation of in-house HPyV qPCRs, ranked in descending order according to sensitivity under standard Ta
| qPCR and its HPyV target | Total number of target genome sequences | Oligo/target mismatches (number of target templates and oligos involved)a |
Sensitivity2 for |
Selectivityb for |
Adjusted Ta, °C | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Number of target sequences with oligo mismatches (% of all targets) | 5′primer/target mismatches | Probe/target mismatches | 3′primer/target mismatches | Standard Tac | Adjusted Ta | Standard Tac | Adjusted Ta | |||
| STLPyV | 7 | 0 | 0 | 0 | 0 | 0.97 | 1.00 | 1.00 | 1.00 | 46.8 |
| WUPyV | 147 | 25 (17%) | 0 | 25 | 3 | 0.97 | 1.00 | 1.00 | 1.00 | 46.0 |
| KIPyV | 12 | 0 | 0 | 0 | 0 | 0.95 | 1.00 | 1.00 | 1.00 | 46.7 |
| MCPyV | 63 | 0 | 0 | 0 | 0 | 0.95 | 1.00 | 1.00 | 1.00 | 45.9 |
| JCPyV | 690 | 96 (14%) | 81 | 10 | 9 | 0.94 | 1.00 | 1.00 | 1.00 | 46.7 |
| LIPyV | 2 | 1 (50%) | 0 | 1 | 1 | 0.80 | 1.00 | 1.00 | 1.00 | 45.1 |
| BKPyV | 522 | 120 (23%) | 1 | 107 | 12 | 0.76 | 0.99 | 1.00 | 0.98 | 50.8 |
| MWPyV | 21 | 1 (5%) | 1 | 0 | 0 | 0.76 | 1.00 | 1.00 | 1.00 | 45.2 |
| HPyV9 | 4 | 0 | 0 | 0 | 0 | 0.39 | 1.00 | 1.00 | 1.00 | 45.0 |
| HPyV6 | 17 | 1 (6%) | 1 | 0 | 0 | 0.37 | 0.99 | 1.00 | 1.00 | 45.0 |
| HPyV7 | 10 | 1 (10%) | 1 | 0 | 0 | 0.32 | 0.99 | 1.00 | 1.00 | 45.0 |
| TSPyV-deg | 23 | 0 | 0 | 0 | 0 | 0.26 | 0.99 | 1.00 | 1.00 | 45.0 |
| TSPyV | 23 | 7 (30%) | 7 | 0 | 0 | 0.23 | 0.96 | 1.00 | 1.00 | 45.0 |
| NJPyV | 1 | 0 | 0 | 0 | 0 | 0.20 | 0.99 | 1.00 | 1.00 | 45.0 |
| HPyV12 | 5 | 3 (60%) | 2 | 3 | 0 | 0.19 | 0.92 | 1.00 | 1.00 | 45.0 |
| Total | 1,524 | 94 | 146 | 25 | ||||||
Some target sequences may have mismatches with more than one type of the oligo, so the sum of the number of target sequences with mismatches to oligos may exceed the number of affected sequences.
Sensitivity and selectivity were calculated by overall detection of the respective target and nontarget templates by an in-house qPCR.
Standard Ta: 60°C.
Evaluation and refinement of the TSPyV qPCR
Analysis of the Tm oligo/template maps for the TSPyV qPCR identified 7 out of the 23 (30.4%) analyzed TSPyV genome sequences for which T-decision [0.95-0.05] ranges of at least one oligo/template were not within the favorable Tm zone for the confident genome detection under the standard Ta (Figures 3A and 3B). Those seven relatively poorly recognized genomes are prototyped by KM007161.1 and were sequenced after the original TSPyV qPCR was designed. They have a recurrent mismatch in the forward primer annealing site (GAGTCTAAGGA[C→G]AACTATGG), which correlated with a Tm drop for the oligo/template complex of this primer from 60.9°C to 51.0°C (Figures 3A and 3B and resource website: https://veb.lumc.nl/MANUSCRIPTS/Polyomaviridae2021.cgi).
Figure 3.

In silico and in vitro testing and refinement of a TSPyV qPCR
(A and B) Results of in silico testing of the in-house TSPyV qPCR under standard Ta for qPCR oligos annealed to 23 target (blue) and 1758 nontarget (red) templates are presented using two oligo/template Tm maps: forward versus reverse primer pair (A), forward primer versus reverse probe pair (B). A Tm shift for oligo/template complex of seven TSPyV genomes, typified by KM007161.1, after including a degenerated base into the forward primer (Table S3) is shown with the bold arrow. Overall design of the Tm maps is explained in the legend of Figure 2.
(C) In vitro dilution series of two TSPyV genomes with either full-match (NC_014361) (qPCR efficiency = 97.6%, R2 = 0.996, slope = −3.382) or a mismatch (KM007161.1) to the forward oligo (qPCR efficiency = 97.1%, R2 = 0.995, slope = −3.394). A relative poor recognition of the mismatch genome is evident.
(D) The same as in (C) except for using forward primer with a degenerate base. Note similar recognition of the two genomes.
The predicted detrimental effect of this mismatch on the qPCR sensitivity was analyzed in vitro. It caused an 8 Cq-increase and therefore a drop in analytical sensitivity of the qPCR toward the respective sequences compared with the original TSPyV sequence (Figure 3C). Inclusion of the degenerate base in the forward primer (GAGTCTAAGGASAACTATGG) increased the Tm of its complex with respective targets to 59.3°C in silico (resource website), and, accordingly, almost entirely rescued the TSPyV qPCR analytical sensitivity toward KM007161.1 at the standard Ta (Figure 3D). These results revealed excellent agreement between the in vitro and in silico results.
According to in silico evaluation shown in Table 3, the PCR sensitivity could be rescued by lowering the Ta as well. This Ta adjustment would allow efficient recognition of all TSPyV genomes with the original PCR set of oligos with a predicted sensitivity of 0.96.
Testing and refinement of the JCPyV qPCR
In contrast to TSPyV, none of the analyzed 690 genome sequences of JCPyV were found in a problematic region of three Tm maps under standard Ta, although T-decision [0.95–0.05] ranges of 89 JCPyV genomes partially overlapped with at least one such region (Figure 4 and resource website). The latter genome sequences all contained a mismatch within annealing region to a respective primer or probe (Table 3). Because the T-decision [0.95–0.05] range of most of these oligo/target complexes were predominantly above the qPCR standard Ta and they accounted only for 13% of targets, these mismatches decreased average sensitivity of this qPCR only to 0.94. Compared with other JCPyV, a mismatch in the forward primer annealing region (GTCTCCCCAT[A→G]CCAACATTAGCTT) affected 76 JCPyV genome sequences (prototyped by AF015535.1) out of 89 (11% of total) was associated with comparable decreases in Tm in silico (4°C decrease from 68.4°C to 64.4°C, Figures 4A–4C) and in vitro (0.6 Cq increase from 24.2 to 24.8 at 105 copies/reaction, Figure 4D). This effect was even smaller for the remaining 13 JCPyV genome sequences in silico, which also showed that sensitivity of the JCPyV qPCR could be increased from 0.94 to 1.0 by adjustment of Ta from 60.0°C to 46.7°C (Table 3).
Figure 4.

In silico and in vitro testing of a JCPyV qPCR
(A–C) Results of in silico evaluation of the in-house JCPyV qPCR under standard Ta for qPCR oligos annealed to 690 target (blue) and 1,091 nontarget (red) templates are presented using three oligo/template Tm maps: forward versus reverse primer oligo pair (A); forward primer versus forward probe oligo pair (B); and reverse primer versus forward probe oligo pair (C). Overall design of the Tm maps is explained in the legend of Figure 2. Fully interactive versions of these maps and a 3D melting temperature map are available on the resource website.
(D) The influence of a common mismatch present in 72/690 JCPyV genomes was tested by comparing the performance of the qPCR on the regular control plasmid without mismatch (Mismatch−, efficiency = 98.7%, R2 = 0.998, slope = −3.353) and the plasmid containing the mismatch (Mismatch+, efficiency = 104.4%, R2 = 0.994, slope = −3.222) (D) (e.g. AF015535.1, forward primer annealing region: GTCTCCCCAT[A→G]CCAACATTAGCTT). A small difference in Cq values is seen when the mismatch is present.
Testing and refinement of the in-house BKPyV qPCR
The most complex results were obtained for the in-house BKPyV qPCR analyzed against 522 target and 1,259 nontarget genome sequences, as evident from the Tm plots under the standard Ta (Figures 5A–5C; interactive 2D and 3D Tm plots are shown on the resource website). A relatively small overlap with a nonfavorable Tm map zone was observed for the T-decision [0.95–0.05] ranges of oligo/template complexes for all 522 BKPyV genome sequences. Furthermore, the T-decision [0.95–0.05] ranges for oligo/template complexes with two BKPyV genome sequences (MF627830.1 and AY628231.1) were predominantly outside the favorable zone in two of three oligo/target Tm 2D maps. As a result, average sensitivity of the in-house BKPyV qPCR was 0.76 under the standard Ta.
Figure 5.

In silico and in vitro testing of a BKPyV qPCR
(A–C) Results of in silico testing of the in-house BKPyV qPCR under standard Ta for qPCR oligos annealed to 522 target (blue) and 1,259 nontarget (red) sequences are presented using three quadrant oligo/template Tm maps as detailed in legend to Figure 2: forward versus reverse primer oligo pair (A); forward primer versus forward probe oligo pair (B); and reverse primer versus forward probe oligo pair (C). Selected genome sequences discussed in the text are indicated with arrows accompanied by their GenBank numbers.
(D) In vitro evaluation of impact of a common single nucleotide mismatch in the probe-to-target annealing region on the qPCR performance against GenBank ID AB211375.1. An increase of about 1 Cq for the target with the mismatch (efficiency = 94%, R2 = 0.993, slope = −3.475) relative to the matching target was observed (efficiency = 94.7%, R2 = 0.999, slope = −3.456).
One hundred twenty genome sequences (23% of the analyzed BKPyVs) include mismatches between oligos and corresponding template annealing sites, primarily in the probe target region (107 of 120 genomes; Table 3). The most prevalent mismatch (CCAAAAAGCCAAAGGA[A→C]CCC), found in the probe annealing site of 105 genome sequences and prototyped by AB211375.1, caused the estimated probe/template Tm to decrease from 66.14 to 64.28°C (Figures 5B and 5C and resource website). This common mismatch resulted in recurrent decrease of approximately 1 Cq in vitro (Figure 5D). This decrease was reverted by linking a minor groove binder (MGB) to the probe (Figure S1), which accordingly increased the Tm of the probe/template complex by 15°C (resource website).
Two other mismatches in the probe annealing site caused a drop in predicted Tm of oligo/target complexes for deviant genome sequences, compared with the “wild-type” genome (prototyped by NC_001538.1), from 66.14°C to 56.96°C for MF627830.1 (CCAAAAAGCCAAAGGAA[C→T]CC) and to 60.38°C for AY628231.1 (CCAAAAAGCCA[A→G]AGGAACCC) (Figures 5B and 5C and resource website). Because the genome sequences with these mismatches represented only 0.4% of the sequenced BKPyV genomes, their detection was not tested in vitro.
In silico evaluation of BKPyV qPCRs described in literature
To extend utility of our in silico qPCR evaluation strategy, we applied it to a substantial subset of BKPyV qPCRs described in literature. A database of 52 BKPyV-specific qPCRs taken from 32 papers (Bárcena-Panero et al., 2012; Bergallo et al., 2018; Bressollette-Bodin et al., 2005; Dadhania et al., 2008; Delbue et al., 2015; Dumoulin and Hirsch, 2011; Funahashi et al., 2010; Gard et al., 2015; Greer et al., 2015; Gustafsson et al., 2013; Hammarin et al., 2011; Hasan et al., 2016; Hoffman et al., 2008; Kamminga et al., 2019; Keith et al., 2018; Ledesma et al., 2012; Marchetti et al., 2007; Marinelli et al., 2007; Mitui et al., 2013; Muldrew and Lovett, 2013; Pal et al., 2006; Pang et al., 2007; Pietilä et al., 2015; Priftakis et al., 2003; Ryschkewitsch et al., 2004; Şahiner et al., 2014; Sarmento et al., 2019; Signorini et al., 2014; Si-Mohamed et al., 2006; Stolt et al., 2005; Thomas et al., 2007; Yamamoto et al., 2015) was created by searching PubMed using a text-mining approach detailed in the STAR methods section. Each selected BKPyV qPCR is listed in Table S1 with its reference, original and adjusted Ta values, and degeneracy of its oligos. Figure S2 shows the location of each PCR oligo annealing site on a reference BKPyV genome, as well as the sensitivity and selectivity calculated using the Ta value described in the original paper.
Under the original Ta, both sensitivity and selectivity were better than 0.95 for at least ten BKPyV qPCRs (Figure 6). Overall, sensitivity varied considerably among the published PCRs, being <0.9 and as low as in the range of 0.2–0.5 for 30 and 8 BKPyV qPCRs, respectively. This low sensitivity was primarily due to overlap of T-decision [0.95–0.05] ranges for some oligo/template complexes for target genomes with nonfavorable zones of Tm map, indicative of suboptimal Ta. Adjusting the individual BKPyV qPCR Ta-values according to the BCR criterion (average of sensitivity and selectivity, see STAR Methods) substantially increased the sensitivity of almost all affected BKPyV qPCRs, with sensitivity of none of qPCR being below 0.8 after the Ta adjustment. In contrast to sensitivity, only two BKPyV qPCRs displayed selectivity below 0.9 (Delbue et al., 2015; Yamamoto et al., 2015) after the Ta adjustment, which was caused by a high similarity between BKPyV and JCPyV at sites complementary to the PCR oligos in a fraction of JCPyV genomes (resource website) (Delbue et al., 2015; Tremolada et al., 2010; Watzinger et al., 2004; Yamamoto et al., 2015). This complication was not resolved by the Ta adjustment, which also had minor effect on already high selectivity of other BKPyV qPCR.
Figure 6.

Sensitivity and selectivity for BKPyV qPCRs under original and adjusted Ta
For each published BKPyV qPCR specified at the right, selectivity and sensitivity are depicted schematically with contrasting colors under original and adjusted Ta along with difference between these Ta. Impact of Ta adjustment is shown as increase (SN+, SL+) or decrease (SN−, SL−) of the corresponding original sensitivity and selectivity values (SN and SL). PCRs are listed in the descending order according to the sensitivity under adjusted Ta. Ta difference = (adjusted Ta−original Ta) is shown with gray bars.
As a rule, the Ta adjustment was associated with a temperature decrease, most often in a range from 5°C to 10°C (Figure 6). For two PCRs, Ta adjustment was minor (<1°C) that did not affect either sensitivity or selectivity, which were already good under standard Ta: >0.95 for Keith et al., 2018 and >0.87 for Delbue et al., 2015. For other two PCRs (Signorini et al., 2014; Yamamoto et al., 2015), adjustment of Ta led to improved selectivity as the result of decreased false-positive detection of the closely related nontargets, although it was accompanied by a decrease in PCR sensitivity.
Discussion
In this report we demonstrated how fast accumulating genome sequences of the family Polyomaviridae could be utilized for testing species-specific HPyV qPCRs in silico in an efficient manner. This analysis identified qPCRs, which can detect and discriminate all known HPyVs, as well as those PCRs that require upgrade. We showed how this improvement could be achieved using either degenerate nucleotides in oligos or through adjustment of Ta in a procedure assisted by the use of nontarget PyVs. Below we briefly discuss its premise, main findings, including apparent agreement between in silico and in vitro results, as well as limitations and challenges of the approach that may be addressed in future research.
Design of conventional PCR involves calculation of key variables, including PCR Ta. It is informed by target sequences and concerns oligos number, size and template location, as well as Tm of oligo/template complexes. This computation-based foundation of the PCR analysis enables running a PCR also in silico, as we previously demonstrated in an analysis of human astroviruses (Nijhuis et al., 2018) and used here for HPyVs. Compared with its in vitro counterpart, qPCR analysis in silico offers scalability in a cost- and time-effective manner that affords HPyV qPCR testing against 1781 PyV genome sequences in this study rather than against a few PyV genomes, as it is common otherwise. Analyzing so many genome sequences is also associated with an increasing risk of sequencing mistakes affecting the evaluation. Based on these considerations we excluded several sequences of low quality from the analysis. Otherwise, we considered this complication of low impact and unbiased in respect to the evaluated qPCRs, some of which had notably performed well on the entire dataset. In future, a procedure for quality control of genome sequences could be incorporated in the pipeline. Because PyV discovery and characterization are fast pacing and firmly sequence based, accumulation of target sequences can inform periodic HPyV qPCR testing in silico for keeping it up to date. Given that species demarcation of polyomaviruses is genomic based (King et al., 2018; Moens et al., 2017), future updates of target and nontarget groups could be unlinked from GenBank records, used in this study. As we demonstrated for the analyzed 66 HPyV qPCRs, including the qPCR modified in this study, the obtained in silico results could be integrated on a web-page (resource website). It facilitates inspection of all results in respect to separate oligos, templates, and species in either a tabular format or using interactive Tm maps under choice of Ta.
To measure quality of target sequence recognition by species-specific HPyV qPCR, we calculated sensitivity that was averaged over all known target genome sequences of the species. Composition of target genome sequence datasets reflects sampling and full genome sequencing of the known natural diversity of the respective species, which both may be biased. Accordingly, the obtained sensitivity values could be skewed when they were below the maximum possible value of 1. This bias could be partially addressed by sequence weighting and including partial genome sequences, where it is feasible, in the analysis. We note in this respect that validation of HPyV qPCR in vitro also depends on limited choice of known HPyVs for testing.
Besides sensitivity, we also calculated selectivity of species-specific HPyV qPCR in a similar manner. It measured discrimination of nontarget polyomaviruses by the respective qPCR and may serve as a proxy for its expected false-positive rate. Nontarget sequences included all known PyVs of different host origins, including HPyVs that did not belong to the PCR targets. New HPyVs continue to be discovered and often they cluster phylogenetically with PyVs of nonhuman origins, which supports the family wide choice of the nontarget datasets in our study. This broad host range of nontarget PyVs sequences combined with bias of PyV genome sequencing affects estimation of selectivity, when its values fall below the maximum possible value of 1, as it was discussed for the sensitivity calculation above. This estimation may be adjusted by limiting nontarget sequences only to those that are most closely related to target HPyV species; this choice might be especially warranted if the respective species co-circulate, as observed for BKPyV and JCPyV.
Sensitivity and selectivity of qPCR depends on choice of Ta, which is commonly selected within the 45–65°C range, as seen, e.g., in the dataset of 52 published BKPyV qPCRs analyzed in silico. The selected Ta must be below the respective Tm of the involved oligo/template complexes of targets and above those of nontargets. By combining sensitivity and selectivity of qPCR into a single BCR characteristic, we were able to propose Ta adjustment that led to improved overall quality for most of qPCRs analyzed. This result illustrated benefits of the computational framework for HPyV detection that was informed by the current state of genome sequencing. It may be especially valuable in respect to nontarget sequences, which are relatively undercharacterized compared with target sequences upon conventional in vitro evaluation. Besides nontarget PyV sequences, DNA of other origins, if present in considerable excess to target sequences, may be a factor affecting qPCR by depleting PCR oligos through nonspecific annealing. This concern, which is pertinent for relatively low Ta could lead to false-negative results and may be addressed in future in vitro testing.
Our study was prompted by the need to evaluate 14 in-house qPCRs, which we designed or adopted from literature over prior years. We learned that five qPCRs had in silico sensitivity 0.9 or higher, three in the range of 0.5–0.9, and six below 0.5, due to uneven recognition of some newly sequenced genomes in most of the cases. For in vitro characterization, we chose three qPCRs with different sensitivities in the aforementioned ranges, 0.94, 0.76, and 0.23, respectively. The most pronounced drop of sensitivity to 0.23 was in TSPyV qPCR due to a primer mismatch with a relatively large fraction of target genomes that were sequenced after the original qPCR was designed. Incorporation of a degenerate base in the forward primer restored the TSPyV qPCR capacity to recognize the full-known diversity spectrum of this HPyV, as was shown both in silico and in vitro. Such agreement was also observed with respect to two other qPCRs, JCPyV and BKPyV, although we decided against modifying oligos of those qPCRs after considering other factors affecting scale of sensitivity gain. For BKPyV qPCR, this decision was informed by low frequency of the poorly recognized targets, which accounts to only 0.4% of BKPyV genome sequences in GenBank. Should this number be revised upward significantly in the future, the BKPyV qPCR design could be revisited or another BKPyV qPCR be adopted from literature (see below). Drop of the qPCR sensitivity for poorly compared with properly recognized targets was defined by Tm decrease of target/oligo complexes in silico and accompanied by Cq increase in vitro. To establish the exact relationship between the changes of Tm and Cq, which may depend on many factors, additional analysis is required.
In silico analysis also provided a unified platform for comparison of numerous BKPyV qPCRs that were designed by different labs in different years against different genomic loci and tested under different conditions using different panels of viruses. The need for this type of comparison was raised in literature but not met (Blackard et al., 2020; Solis et al., 2015). Our study identified several BKPyV qPCRs that proved to be resilient in the face of the continuing expansion of BKPyV genome sequencing and may be the best choice to go forward. We also provide suggestions toward how analytical sensitivity for other published BKPyV qPCRs could be improved by adjustment of Ta. We believe that combined with characterization of the in-house HPyV qPCRs, these results facilitate efficient use of the accumulated PyV genome sequences for improving confidence in the detection of HPyVs by the available qPCRs.
Limitations of the study
We used a nearest-neighbor model (McTigue et al., 2004; SantaLucia, 1998) for Tm calculation of the oligo/template complexes that also relied on several thermodynamics parameters. We have not evaluated how accuracy and availability of the parameter values might have affected the obtained computational results.
The presented in silico evaluation of qPCRs relies on quality of genome sequencing which may vary for the analyzed sequences and produce genomic variations of technical origins. The erroneously sequenced individual nucleotides may not be confidently distinguished from the natural variation that is underrepresented in public databases. They might have inflated the known actual diversity of both target and nontarget sequences that was analyzed in our study. If a computational procedure for quality control of genome sequences is developed, it could be incorporated in the pipeline. Accurate separation of analyzed sequences into target and nontarget groups is the key for calculation of qPCR quality indicators and Ta adjustment. We have ensured the proper partitioning of polyomaviruses into the two groups by manual inspection of sequence annotations and obtained results. However, this approach is poorly scalable. Future updates of target and nontarget groups could be made firmly algorithm based using virus classification techniques, e.g. DEmARC (Lauber and Gorbalenya, 2012; Moens et al., 2017).
Due to biased genome sequencing toward viruses of high public interest, the obtained sensitivity values could be skewed upward or downward, when they were below the maximum possible value of 1. Incorporating sequence weighting and including partial genome sequences, where it is feasible, may be worth considering in the future. The employed partitioning of all polyomaviruses into either targets or nontargets, based on qPCR design, has merits. However, in certain circumstances, when two or more virus species are most closely related and co-circulate, as observed for BKPyV and JCPyV, the composition of the nontarget group could be limited to the co-circulating virus, which was not explored in our study. Besides nontarget PyV sequences, DNA of other origins, if present in considerable excess to target sequences, may be a factor affecting qPCR by depleting PCR oligos through nonspecific annealing. These effects were outside the presented analysis. Our study concerned 66 HPyV qPCRs mostly involved BKPyV and selected from literature. Many other qPCRs, especially those designed for JCPyV, were left outside this study due to its scale limitations; they may include high-quality qPCRs as well.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| Qiagen HotStarTaq Master Mix kit | Qiagen, Venlo, the Netherlands | Qiagen Cat. No 203446 |
| gBlocks | IDT, San Jose, CA, USA | http://www.idtdna.com/gBlocks |
| Critical commercial assays | ||
| Qubit dsDNA HS Assay | Thermo Fisher Scientific, Waltham, MA, USA | Thermofisher Cat. No Q32851 |
| QuikChange kit | Agilent Technologies, Santa Clara, CA, USA | Agilent Technologies Cat. No 200523 |
| NucleoSpin Plasmid EasyPure, | Macherey-Nagel, Düren, Germany | Macherey-Nagel Cat. No. 740727.50 |
| TOPO TA Cloning Kit | Invitrogen, Waltham, Massachusetts, USA | Invitrogen Cat. No. K4575J10 |
| Oligonucleotides | ||
| Please see Table 1 for a list of oligonucleotides and their respective sources used in vitro | This paper | |
| Please see Table S3 for an overview of all oligo sequences and PCR cycling conditions used in the in silico comparison of BKPyV PCRs | This paper | |
| Recombinant DNA | ||
| pBR322 plasmid with BKPyV full genome insert | Kamminga et al. (2019) | V01108, Addgene #25466 |
| pBR322 plasmid with JCPyV full genome insert | Kamminga et al. (2019) | NC_001699, Addgene #25626 |
| pUC19 plasmid with TSPyV full genome insert | Kamminga et al. (2019) | NC_014361, Addgene #70033 |
| pGEX5x3 plasmid with KIPyV VP1 insert | Kamminga et al., 2019 | NC_009238 |
| pGEX5x3 plasmid with WUPyV VP1 insert | Kamminga et al. (2019) | NC_009539 |
| pZERO-2 plasmid with MCPyV full genome insert | Kamminga et al. (2019) | KF266963 |
| pFunnyFarm plasmid with HPyV6 full genome insert | Kamminga et al. (2019) | HM011560 |
| pFunnyFarm plasmid with HPyV7 full genome insert | Kamminga et al. (2019) | HM011566 |
| pGEX5x3 plasmid with HPyV9 VP1 insert | Kamminga et al. (2019) | NC_015150 |
| pGEX5x3 plasmid with MWPyV VP1 insert | Kamminga et al. (2019) | NC_018102 |
| pGEX5x3 plasmid with STLPyV VP1 insert | Kamminga et al. (2019) | NC_020106 |
| pGEX5x3 plasmid with HPyV12 VP1 insert | Kamminga et al. (2019) | NC_020890 |
| pGEX5x3 plasmid with NJPyV VP1 insert | Kamminga et al. (2019) | NC_024118 |
| pGEX5x3 plasmid with LIPyV VP1 insert | Kamminga et al. (2019) | NC_034253 |
| Software and algorithms | ||
| Python package Plotly | Plotly Technologies Inc., 2015 | https://plotly.com/ |
| R 3.6.1 software and its readxl, readr, and Tidyverse packages | R Core Team, 2020; Wickham et al. (2018, 2019); Wickham and Bryan (2019) | https://cran.r-project.org/ |
| Biopython 1.73 | Cock et al. (2009) | https://biopython.org/ |
| Bio-Rad CFX Manager version 3.1 | Bio-Rad Laboratories, Hercules, CA, USA | Bio-rad software # #1845000 |
| Other | ||
| Bio-Rad CFX96 Real-Time PCR Detection System | Bio-Rad Laboratories, Hercules, CA, USA | Bio-rad Cat. No. 184-5096 |
| Resource website for in silico evaluation of the 66 qPCRs using 1781 genome sequences of the Polyomaviridae | This paper | https://veb.lumc.nl/MANUSCRIPTS/Polyomaviridae2021.cgi |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Sergio Kamminga (s.kamminga@lumc.nl)
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Plasmids were acquired from Addgene for BKPyV (Addgene #25466), JCPyV (#25626), TSPyV (#70033), HPyV6 (#24727), HPyV7 (#24728) and MCPyV (#24729) in DH5α competent cells. For the other polyomaviruses, synthetic DNA sequences of VP1 (gBlocks; IDT, San Jose, CA, USA), were inserted into pGEX5x3 plasmid in XL-1 blue competent cells using a TOPO TA cloning kit (Invitrogen, Waltham, Massachusetts, USA). Cultures were incubated overnight and plasmids were extracted with NucleoSpin Plasmid EasyPure kit (Macherey-Nagel, Düren, Germany). Plasmid concentrations were measured with Qubit dsDNA HS Assay (Thermo Fisher Scientific, Waltham, MA, USA) and diluted to a stock concentration of 1 pg/μL.
Method details
In-house HPyV qPCRs
For this study, we selected 14 HPyV-specific qPCRs used in our laboratory to detect BKPyV, JCPyV, KIPyV, WUPyV, MCPyV, HPyV6, HPyV7, TSPyV, HPyV9, MWPyV, STLPyV, HPyV12, NJPyV, and LIPyV respectively (Kamminga et al., 2019). Table 2 lists these qPCRs along with information on the targeted viral gene, amplicon length, oligo sequences, and reference to its first description in literature (Goh et al., 2009; Kamminga et al., 2019; Pal et al., 2006; Purdie et al., 2018; Rao et al., 2016; van der Meijden et al., 2014, 2016). All HPyV qPCRs had their primer and probe oligos perfectly matched to the respective HPyV genome sequence regions at the time when they were designed. Primers are specified as “forward” or “reverse” by authors of qPCR. In contrast, polarity may be not consistently defined for probe. To address this gap, we defined probe as either “forward” or “reverse” according to the respective qualifier of a primer which belongs to the same strand sequence as the probe.
Literature-compiled BKPyV qPCRs
In August 2019, we compiled a dataset of BKPyV qPCRs reported in literature with the help of text-mining. Biopython 1.73 was used to search through PubMed’s Entrez Database (Cock et al., 2009) (Figure 2, top panel). The case-insensitive search terms used were "polyoma AND qPCR AND human". The result (n = 453 articles) was then parsed using R 3.6.1 software and its readxl, readr, and Tidyverse packages (R Core Team, 2020; Wickham et al., 2019, 2018; Wickham and Bryan, 2019). Subsequently, abstracts of these articles were scanned for the presence of the following terms: "bk" and at least one of "qpcr", "real-time pcr", "taqman". The following papers were excluded: a) with no oligo sequences specified; b) using exact copies of previously published qPCRs; c) with no Taqman probe specified; d) not publicly accessible. The extracted articles with these terms (n = 96) were further screened individually for inclusion in this study. Finally, we considered 52 Taqman-based qPCRs (the most common qPCR method) targeting viruses belonging to the species Human polyomavirus 1, including BKPyV, and collected information about primer and probe oligo sequence, concentration, polarity, dye linkage and inclusion of modified bases (The polarity of primers and probe was derived as specified in the In-house HPyV qPCRs section). Concentration of Mg2+ and other variables of the qPCR buffer, cycling conditions and the reaction volume were also documented whenever available. When neither of the latter was reported, default values of our qPCR test-system (see below) were used in subsequent analyses. Each qPCR was assigned with a unique name that included the last name of the first author of the paper, year of the publication, qPCR virus target name and, if necessary, an additional identifier when multiple qPCRs were described in a single paper.
Sequence database of polyomavirus genomes: target and nontarget groups
In August 2019, complete polyomavirus genome sequences were downloaded from the National Center for Biotechnology Information (NCBI) GenBank and RefSeq into the VirAliS platform (Gorbalenya et al., 2010), using the HAYGENS tool (version 2.4, results available at the http://veb.lumc.nl/HAYGENS) (Figure 1). In brief, HAYGENS combined results of homology search in GenBank for polyomavirus genomes using HMMER tool version 3.2.1 (Eddy, 2011) and a hidden Markov model profile of the alignment of 1199 PyV Large T-antigens (LT) (Lauber et al., 2015) with queries of the analyzed GenBank entries on the presence of the term “polyomavirus. Using lengths of 85 polyomavirus genomes from RefSeq and the location of LT in these genomes as entries for training, HAYGENS separated full and partial genome sequences of other entries. With this approach, 1784 PyV genomes (1522 of human and 262 nonhuman origin) were recognized as (nearly) complete and their sequences were used in this study. Sequences of three MCPyV genomes were subsequently excluded due to low sequencing quality (MG241581; MG241582; MG241579), leading to a total of 1781 sequences included for analysis (Table S2). For evaluation of each qPCR, the PyV genome sequences were divided into PCR-specific target and nontarget groups, respectively, based on their description and taxonomy annotation in GenBank entries. In total, 15 groups of genome sequences were formed: 14 species-specific HPyVs and a group combining all nonhuman PyVs (Table S2). Three genome sequences of Sorex Araneus polyomavirus were treated as part of HPyV12 for this study, although they as well as the prototype human virus have been re-assigned later to species Sorex araneus polyomavirus 1. For evaluation of each HPyV qPCR, the target group includes HPyV genome sequences belonging to the respective polyomavirus species; all other polyomavirus genomes, of either human or nonhuman origins, were considered nontarget for this qPCR. Sequences of GenBank PyV entry and its complement were analyzed in our study in silico (see below). Every evaluated qPCR was designed against its target HPyV strand coding for T antigen; this strand sequence is predominant among PyV entries in GenBank. Accordingly, the forward and reverse orientations of primers and probe are defined against this strand in all qPCRs. However, some PyV entries, including notably several MCPyV entries, are represented by the complementary sequence in the GenBank. These complementary sequences were converted into the predominant strand form to facilitate the use of common genomic coordinates for the respective group of targets during in silico analysis.
Quantification and statistical analysis: In silico evaluation of polyomavirus qPCR and results visualization
General aspects
The in silico evaluations performed here estimate the ability of HPyV qPCR to discriminate targeted from nontargeted HPyV genome sequences, using a modified computational procedure that we applied previously to astroviruses (Nijhuis et al., 2018). Briefly, this procedure analyzed oligo/template complexes for a given set of oligos at all possible genomic sites under specified concentrations (SantaLucia, 1998) to locate sites with maximal melting temperatures (Tm) of the oligo/template annealing complexes. It followed a nearest-neighbor approach, with Tm being the temperature at which the oligo and template molecules are equally probable to be separated or annealed (McTigue et al., 2004). For degenerate oligos with degeneracy d, Tm value for each oligo/template combination was calculated assuming that concentration of each unique oligo is 1/d of the total oligos concentration, and Tm of the reaction was at the maximum value of Tm for the considered oligo/template combination (Table S3, “Compounds”, “Degeneracy”). For oligos conjugated with a minor groove binder, Tm value was increased by 15°C (Table S3, “Oligos”, shown with [MGB]), due to the average value of such increase estimated previously (Afonina et al., 1997; Kutyavin et al., 2000). The same concentration of target DNA (1 nM) was used in each qPCR during the in silico evaluation (Table S3, “Compounds”). As detailed below, the in silico qPCR evaluation involves calculation of several characteristics, including T- and L-decisions, for each genomic sequence to assess sensitivity and selectivity of qPCR.
Temperature (T)-decision
Each PCR specifies annealing temperature Ta that is selected within a range of 45°C–60°C, and commonly set to 60°C (standard Ta, see Table S3, “Annealing, Ta/sec”). It must be substantially below the respective Tm, that ensures target recognition (true positives), but high enough to avoid recognition of nontargets (false positives). To facilitate comparison of Ta-related results for oligo/template complexes of all analyzed sequences by a PCR, we introduced a continuous T-decision function that changes from 0 to 1; it equals to 0.5, when Tm of oligo/template complex matches PCR Ta. Practically, we used T-ranges corresponding to the T-decision [0.95–0.05] range to evaluate formation of the respective oligo/template complexes under a given Ta (Figure 2). T-decisions were calculated for each pair of DNA template, target and nontarget, and PCR oligonucleotide, two primers and probe, which then were used to calculate a T-decision for a DNA template as a product of its T-decisions for all individual PCR oligos (template T-decision).
Length (L)-decision
Template recognition by PCR depends also on proper spacing of its annealing sites for PCR primer and probe oligos. The latter must conform to product length maximum size and the lack of overlap between annealing sites for certain oligo pairs, collectively forming “length” constraints of qPCR. The maximum amplicon length was set either at 400 nucleotides or at 200 nucleotides (according to the expected maximum length of the PCR product, see Table S3, “Oligo location limitations” and “Product”) and minimum distances between 5′- and -3′ ends of the corresponding oligos were set to 0 for oligos with the same polarity. Due to evolutionary considerations, these constraints are most likely fulfilled for targets although not necessarily for nontargets, if those diverged considerably at the expected cognate sites of annealing. For these sequences, maximal Tm may be observed at alternative sites, either compatible or not with the product length constraints of the qPCR. We called the respective binary outcome of comparison of the calculated product lengths with permitted size ranges “L-decision”; it equals either 1 or 0 when length constraint was satisfied or not, respectively. L-decision was calculated for each pair of oligos, including forward and reverse primers and a single probe, resulting in three L-decisions for a template, target or nontarget. Also, template-wide L-decision was calculated as a product of (three) individual oligo-based L-decisions (template L-decision).
Template cumulative decision
Finally, we calculated decision function for detection of individual templates and template groups (species) that combined T- and L-decisions. The former was calculated using a product of T- and L-decisions for the respective template. Detection of a group of target or nontarget templates was further estimated by summarizing template cumulative decision values for all genome sequences in the group and dividing the obtained value by the number of viruses in the group.
Sensitivity and selectivity of qPCR in silico
To characterize quality of template recognition by a PCR, we used sensitivity and selectivity. The latter term is a counterpart to specificity, common in the field, and was defined as the extent to which the method can be used to determine particular analytes in mixtures or matrices without interferences from other components of similar behavior, as per International Union of Pure and Applied Chemistry (IUPAC) recommendation (Vessman et al., 2001).
Using both T- and L-decision values for a PyV template complexed with all oligos used by a PCR, an overall template detection (p) by this PCR was calculated; ranging between 0 and 1. The calculated value p is interpreted as true positive (TP), and 1-p as false negative (FN) for target sequences; and for nontarget sequence, p is interpreted as false positive (FP) and 1-p as true negative (TN). Accordingly, cumulative TP + FN of target sequences is always equal to the total number of targets and cumulative FP + TN of nontarget sequences is equal to the total number of nontargets.
Finally, in silico sensitivity and selectivity of qPCR in respect to separate target sequences or an entire species were calculated using respective TP, FN, FP, and TN values under standard Ta, original Ta supplied in publications, or adjusted Ta (Table S3, “Sensitivity/Selectivity” and “Ta adjusted and original”). The latter corresponds to the temperature that maximized the average of the respective sensitivity and selectivity (balanced classification rate, BCR).
Visualization of results
The results of a given HPyV qPCR evaluation were visualized using Tm maps generated with the original software and Python/Perl package Plotly (Plotly Technologies Inc., 2015) (Figure 2). Tm was calculated for complexes of oligos (primers or probes) and DNA templates, target and nontarget, and plotted using either 2D Tm maps for each pair of oligos (forward primer vs. reverse primer, forward primer vs. probe, probe vs. reverse primer; probe polarity specified), or a single 3D Tm map for integral presentation of the results with all three oligos involved. These Tm plots for each of 66 qPCRs analyzed in this study are available at the https://veb.lumc.nl/MANUSCRIPTS/Polyomaviridae2021.cgi.
In vitro qPCR quality assessment
HPyV qPCRs were evaluated in vitro using Bio-Rad CFX Manager version 3.1. Cycling conditions were 95°C for 15 min, followed by 45 cycles of 95°C for 30 s, 60°C for 30 s and 72°C for 30 s. Baseline threshold values were determined separately for each target and fluorescence drift correction was applied. For qPCR optimization, plasmids containing the relevant HPyV full genome or the Viral Protein 1 (VP1)-coding sequence were used. To obtain HPyV plasmid 10-fold dilution series (10.000–1 copy/reaction), total DNA concentration of each target was measured in a Qubit 4 Fluorometer using a Qubit dsDNA HS Assay (Thermo Fisher Scientific, Waltham, MA, USA) following manufacturer’s instructions. Plasmids containing a mismatch in the annealing region were created by site-directed mutagenesis, using the QuikChange kit (Agilent Technologies, Santa Clara, CA, USA) according to manufacturer’s instructions.
Additional resources
Resource website, related to Figures 2, 3, 4, 5, and 6: Detailed description of each PCR and interactive Tm maps (66 qPCRs in total, including modifications to BKPyV and TSPyV qPCR described above), accessible via: https://veb.lumc.nl/MANUSCRIPTS/Polyomaviridae2021.cgi.
Acknowledgments
This work was supported and funded in part by Sanquin Blood Supply Foundation, a not-for-profit organization, the EU Horizon 2000 EVAg project (https://www.european-virus-archive.com/), and Molecular Diagnostics of Infectious Diseases Working Group, Dutch Association for Medical Microbiology (WDMI/NVMM, https://www.nvmm.nl/vereniging/commissies-en-werkgroepen/wmdi/). IAS and AEG thank Dmitry Samborskiy for help with administrating the VirAliS software.
Author contributions
Conceptualization: AEG and IAS; conceived and designed the research: SK, IAS, AEG, HZ, and MCWF; collected the data: IAS, SK, MT, EM, and CB; contributed analysis tools: IAS, SK, MT, and AEG; analyzed results: SK, IAS, MT, and AEG; wrote the manuscript: SK, IAS, AEG, and MCWF.
Declaration of interests
The authors declare no competing interests.
Published: January 21, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.103613.
Contributor Information
Sergio Kamminga, Email: s.kamminga@lumc.nl.
Igor A. Sidorov, Email: i.sidorov@lumc.nl.
Mariet C.W. Feltkamp, Email: m.c.w.feltkamp@lumc.nl.
Alexander E. Gorbalenya, Email: a.e.gorbalenya@lumc.nl.
Supplemental information
Data and code availability
Data: The data are fully presented in the main manuscript and Supplemental information.
Code: The code used to generate in silico results may not be extracted from a multipurpose software package developed in our group; it will be described separately. To assist the reader in this respect, we set-up a dedicated web-page, which provides access to the code functionalities relevant to this study https://veb.lumc.nl/MANUSCRIPTS/Polyomaviridae2021.cgi.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Afonina I., Zivarts M., Kutyavin I., Lukhtanov E., Gamper H., Meyer R.B. Efficient priming of PCR with short oligonucleotides conjugated to a minor groove binder. Nucleic Acids Res. 1997;25:2657–2660. doi: 10.1093/nar/25.13.2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allander T., Andreasson K., Gupta S., Bjerkner A., Bogdanovic G., Persson M.A.A., Dalianis T., Ramqvist T., Andersson B. Identification of a third human polyomavirus. J. Virol. 2007;81:4130–4136. doi: 10.1128/JVI.00028-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bárcena-Panero A., Echevarría J.E., Romero-Gómez M.P., Royuela E., Castellanos A., González I., Fedele G. Development and validation with clinical samples of internally controlled multiplex real-time PCR for diagnosis of BKV and JCV infection in associated pathologies. Comp. Immunol. Microbiol. Infect. Dis. 2012;35:173–179. doi: 10.1016/j.cimid.2011.12.010. [DOI] [PubMed] [Google Scholar]
- Bergallo M., Daprà V., Fava P., Ponti R., Calvi C., Montanari P., Novelli M., Quaglino P., Galliano I., Fierro M.T. DNA from human polyomaviruses, MWPyV, HPyV6, HPyV7, HPyV9 and HPyV12 in cutaneous T-cell lymphomas. Anticancer Res. 2018;38:4111–4114. doi: 10.21873/anticanres.12701. [DOI] [PubMed] [Google Scholar]
- Blackard J.T., Davies S.M., Laskin B.L. BK polyomavirus diversity—why viral variation matters. Rev. Med. Virol. 2020;30:e2102. doi: 10.1002/rmv.2102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bressollette-Bodin C., Coste-Burel M., Hourmant M., Sebille V., Andre-Garnier E., Imbert-Marcille B.M. A prospective longitudinal study of BK virus infection in 104 renal transplant recipients. Am. J. Transpl. 2005;5:1926–1933. doi: 10.1111/j.1600-6143.2005.00934.x. [DOI] [PubMed] [Google Scholar]
- Buck C.B., Phan G.Q., Raiji M.T., Murphy P.M., McDermott D.H., McBride A.A. Complete genome sequence of a tenth human polyomavirus. J. Virol. 2012;86:10887. doi: 10.1128/JVI.01690-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cock P.J.A., Antao T., Chang J.T., Chapman B.A., Cox C.J., Dalke A., Friedberg I., Hamelryck T., Kauff F., Wilczynski B., de Hoon M.J.L. Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics. 2009;25:1422–1423. doi: 10.1093/bioinformatics/btp163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dadhania D., Snopkowski C., Ding R., Muthukumar T., Chang C., Aull M., Lee J., Sharma V.K., Kapur S., Suthanthiran M. Epidemiology of BK virus in renal allograft recipients: independent risk factors for BK virus replication. Transplantation. 2008;86:521–528. doi: 10.1097/TP.0b013e31817c6447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delbue S., Elia F., Carloni C., Pecchenini V., Franciotta D., Gastaldi M., Colombo E., Signorini L., Carluccio S., Bellizzi A., et al. JC virus urinary excretion and seroprevalence in natalizumab-treated multiple sclerosis patients. J. Neurovirol. 2015;21:645–652. doi: 10.1007/s13365-014-0268-0. [DOI] [PubMed] [Google Scholar]
- Dumoulin A., Hirsch H.H. Reevaluating and optimizing polyomavirus BK and JC real-time PCR assays to detect rare sequence polymorphisms. J. Clin. Microbiol. 2011;49:1382–1388. doi: 10.1128/JCM.02008-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy S.R. Accelerated Profile HMM Searches. PLoS Comput Biol. 2011;10 doi: 10.1371/journal.pcbi.1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng H., Shuda M., Chang Y., Moore P.S. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science. 2008;319:1096–1100. doi: 10.1126/science.1152586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funahashi Y., Iwata S., Ito Y., Kojima S., Yoshikawa T., Hattori R., Gotoh M., Nishiyama Y., Kimura H. Multiplex real-time PCR assay for simultaneous quantification of BK polyomavirus, JC polyomavirus, and adenovirus DNA. J. Clin. Microbiol. 2010;48:825–830. doi: 10.1128/JCM.01699-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gard L., Niesters H.G.M., Riezebos-Brilman A. A real time genotyping PCR assay for polyomavirus BK. J. Virol. Methods. 2015;221:51–56. doi: 10.1016/j.jviromet.2015.04.024. [DOI] [PubMed] [Google Scholar]
- Gardner S.D., Field A.M., Coleman D.V., Hulme B. New human papovavirus (BK) isolated from urine after renal transplantation. Lancet. 1971;1:1253–1257. doi: 10.1016/S0140-6736(71)91776-4. [DOI] [PubMed] [Google Scholar]
- Gaynor A.M., Nissen M.D., Whiley D.M., Mackay I.M., Lambert S.B., Wu G., Brennan D.C., Storch G.A., Sloots T.P., Wang D. Identification of a novel polyomavirus from patients with acute respiratory tract infections. PLoS Pathog. 2007;3:e64. doi: 10.1371/journal.ppat.0030064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gedvilaite A., Tryland M., Ulrich R.G., Schneider J., Kurmauskaite V., Moens U., Preugschas H., Calvignac-Spencer S., Ehlers B. Novel polyomaviruses in shrews (Soricidae) with close similarity to human polyomavirus 12. J. Gen. Virol. 2017;98:3060–3067. doi: 10.1099/jgv.0.000948. [DOI] [PubMed] [Google Scholar]
- Gheit T., Dutta S., Oliver J., Robitaille A., Hampras S., Combes J.-D., McKay-Chopin S., Le Calvez-Kelm F., Fenske N., Cherpelis B., et al. Isolation and characterization of a novel putative human polyomavirus. Virology. 2017;506:45–54. doi: 10.1016/j.virol.2017.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goh S., Lindau C., Tiveljung-Lindell A., Allander T. Merkel cell polyomavirus in respiratory tract secretions. Emerg. Infect. Dis. 2009;15:489–491. doi: 10.3201/eid1503.081206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbalenya A.E., Lieutaud P., Harris M.R., Coutard B., Canard B., Kleywegt G.J., Kravchenko A.A., Samborskiy D.V., Sidorov I.A., Leontovich A.M., Jones T.A. Practical application of bioinformatics by the multidisciplinary VIZIER consortium. Antiviral Res. 2010;87:95–110. doi: 10.1016/j.antiviral.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gossai A., Waterboer T., Nelson H.H., Michel A., Willhauck-Fleckenstein M., Farzan S.F., Hoen A.G., Christensen B.C., Kelsey K.T., Marsit C.J., et al. Seroepidemiology of human polyomaviruses in a US population. Am. J. Epidemiol. 2016;183:61–69. doi: 10.1093/aje/kwv155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer A.E., Forman M.S., Valsamakis A. Comparison of BKV quantification using a single automated nucleic acid extraction platform and 3 real-time PCR assays. Diagn. Microbiol. Infect. Dis. 2015;82:297–302. doi: 10.1016/j.diagmicrobio.2015.04.006. [DOI] [PubMed] [Google Scholar]
- Gustafsson B., Priftakis P., Rubin J., Giraud G., Ramqvist T., Dalianis T. Human polyomaviruses were not detected in cerebrospinal fluid of patients with neurological complications after hematopoietic stem cell transplantation. Future Virol. 2013;8:809–814. doi: 10.2217/fvl.13.55. [DOI] [PubMed] [Google Scholar]
- Hammarin A.-L., Öqvist B., Wahlgren J., Falk K.I. Systematic screening of BK virus by real-time PCR prevents BK virus associated nephropathy in renal transplant recipients. J. Med. Virol. 2011;83:1959–1965. doi: 10.1002/jmv.22196. [DOI] [PubMed] [Google Scholar]
- Hasan M.R., Tan R., Al-Rawahi G., Thomas E., Tilley P. Comparative evaluation of laboratory developed real-time PCR assays and RealStar® BKV PCR Kit for quantitative detection of BK polyomavirus. J. Virol. Methods. 2016;234:80–86. doi: 10.1016/j.jviromet.2016.04.009. [DOI] [PubMed] [Google Scholar]
- Hoffman N.G., Cook L., Atienza E.E., Limaye A.P., Jerome K.R. Marked variability of BK virus load measurement using quantitative real-time PCR among commonly used assays. J. Clin. Microbiol. 2008;46:2671–2680. doi: 10.1128/JCM.00258-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamminga S., Meijden E., Feltkamp M.C.W., Zaaijer H.L. Seroprevalence of fourteen human polyomaviruses determined in blood donors. PLoS One. 2018;13:e0206273. doi: 10.1371/journal.pone.0206273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamminga S., van der Meijden E., de Brouwer C., Feltkamp M., Zaaijer H. Prevalence of DNA of fourteen human polyomaviruses determined in blood donors. Transfusion. 2019;59:3689–3697. doi: 10.1111/trf.15557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kean J.M., Rao S., Wang M., Garcea R.L. Seroepidemiology of human polyomaviruses. PLoS Pathog. 2009;5:e1000363. doi: 10.1371/journal.ppat.1000363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keith K.A., Hartline C.B., Bowlin T.L., Prichard M.N. A standardized approach to the evaluation of antivirals against DNA viruses: polyomaviruses and lymphotropic herpesviruses. Antiviral Res. 2018;159:122–129. doi: 10.1016/j.antiviral.2018.09.016. [DOI] [PubMed] [Google Scholar]
- King A.M.Q., Lefkowitz E.J., Mushegian A.R., Adams M.J., Dutilh B.E., Gorbalenya A.E., Harrach B., Harrison R.L., Junglen S., Knowles N.J., et al. Changes to taxonomy and the International code of virus classification and nomenclature ratified by the International Committee on taxonomy of viruses (2018) Arch. Virol. 2018;163:2601–2631. doi: 10.1007/s00705-018-3847-1. [DOI] [PubMed] [Google Scholar]
- Korup S., Rietscher J., Calvignac-Spencer S., Trusch F., Hofmann J., Moens U., Sauer I., Voigt S., Schmuck R., Ehlers B. Identification of a novel human polyomavirus in organs of the gastrointestinal tract. PLoS One. 2013;8:e58021. doi: 10.1371/journal.pone.0058021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutyavin I.V., Afonina I.A., Mills A., Gorn V.V., Lukhtanov E.A., Belousov E.S., Singer M.J., Walburger D.K., Lokhov S.G., Gall A.A., et al. 3′-Minor groove binder-DNA probes increase sequence specificity at PCR extension temperatures. Nucleic Acids Res. 2000;28:655–661. doi: 10.1093/nar/28.2.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauber C., Gorbalenya A.E. Partitioning the genetic diversity of a virus family: approach and evaluation through a case study of picornaviruses. J. Virol. 2012;86:3890–3904. doi: 10.1128/JVI.07173-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauber C., Kazem S., Kravchenko A.A., Feltkamp M.C.W., Gorbalenya A.E. Interspecific adaptation by binary choice at de novo polyomavirus T antigen site through accelerated codon-constrained Val-Ala toggling within an intrinsically disordered region. Nucleic Acids Res. 2015;43:4800–4813. doi: 10.1093/nar/gkv378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledesma J., Muñoz P., Garcia de Viedma D., Cabrero I., Loeches B., Montilla P., Gijon P., Rodriguez-Sanchez B., Bouza E., BKV Study Group BK virus infection in human immunodeficiency virus-infected patients. Eur. J. Clin. Microbiol. Infect. Dis. 2012;31:1531–1535. doi: 10.1007/s10096-011-1474-9. [DOI] [PubMed] [Google Scholar]
- Lim E.S., Reyes A., Antonio M., Saha D., Ikumapayi U.N., Adeyemi M., Stine O.C., Skelton R., Brennan D.C., Mkakosya R.S., et al. Discovery of STL polyomavirus, a polyomavirus of ancestral recombinant origin that encodes a unique T antigen by alternative splicing. Virology. 2013;436:295–303. doi: 10.1016/j.virol.2012.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchetti S., Graffeo R., Siddu A., Santangelo R., Ciotti M., Picardi A., Favalli C., Fadda G., Cattani P. BK virus DNA detection by real-time polymerase chain reaction in clinical specimens. New Microbiol. 2007;30:119–126. [PubMed] [Google Scholar]
- Marinelli K., Bagnarelli P., Gaffi G., Trappolini S., Leoni P., Paggi A.M., Della Vittoria A., Scalise G., Varaldo P.E., Menzo S. PCR real time assays for the early detection of BKV-DNA in immunocompromised patients. New Microbiol. 2007;30:275–278. [PubMed] [Google Scholar]
- McTigue P.M., Peterson R.J., Kahn J.D. Sequence-dependent thermodynamic parameters for locked nucleic acid (LNA)-DNA duplex formation. Biochemistry. 2004;43:5388–5405. doi: 10.1021/bi035976d. [DOI] [PubMed] [Google Scholar]
- Mishra N., Pereira M., Rhodes R.H., An P., Pipas J.M., Jain K., Kapoor A., Briese T., Faust P.L., Lipkin W.I. Identification of a novel polyomavirus in a pancreatic transplant recipient with retinal blindness and vasculitic myopathy. J. Infect. Dis. 2014;210:1595–1599. doi: 10.1093/infdis/jiu250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitui M., Leos N.K., Lacey D., Doern C., Rogers B.B., Park J.Y. Development and validation of a quantitative real time PCR assay for BK virus. Mol. Cell. Probes. 2013;27:230–236. doi: 10.1016/j.mcp.2013.08.001. [DOI] [PubMed] [Google Scholar]
- Moens U., Calvignac-Spencer S., Lauber C., Ramqvist T., Feltkamp M.C.W., Daugherty M.D., Verschoor E.J., Ehlers B. ICTV Report Consortium. ICTV Virus Taxonomy Profile: Polyomaviridae. J Gen Virol. 2017;98:1159–11160. doi: 10.1099/jgv.0.000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moens U., Krumbholz A., Ehlers B., Zell R., Johne R., Calvignac-Spencer S., Lauber C. Biology, evolution, and medical importance of polyomaviruses: an update. Infect. Genet. Evol. 2017;54:18–38. doi: 10.1016/j.meegid.2017.06.011. [DOI] [PubMed] [Google Scholar]
- Muldrew K.L., Lovett J.L. An in-house assay for BK polyomavirus quantification using the Abbott m2000 RealTime system. J. Med. Microbiol. 2013;62:1714–1720. doi: 10.1099/jmm.0.058388-0. [DOI] [PubMed] [Google Scholar]
- Nijhuis R.H.T., Sidorov I.A., Chung P.K., Wessels E., Gulyaeva A.A., de Vries J.J., Claas E.C.J., Gorbalenya A.E. PCR assays for detection of human astroviruses: in silico evaluation and design, and in vitro application to samples collected from patients in The Netherlands. J. Clin. Virol. 2018;108:83–89. doi: 10.1016/j.jcv.2018.09.007. [DOI] [PubMed] [Google Scholar]
- Padgett B.L., Zurhein G.M., Walker D.L., Eckroade R.J., Dessel B.H. Cultivation of papova-like virus from human brain with progressive multifocal leucoencephalopathy. Lancet. 1971;1:1257–1260. doi: 10.1016/S0140-6736(71)91777-6. [DOI] [PubMed] [Google Scholar]
- Pal A., Sirota L., Maudru T., Peden K., Lewis A.M., Jr. Real-time, quantitative PCR assays for the detection of virus-specific DNA in samples with mixed populations of polyomaviruses. J. Virol. Methods. 2006;135:32–42. doi: 10.1016/j.jviromet.2006.01.018. [DOI] [PubMed] [Google Scholar]
- Pang X.L., Doucette K., LeBlanc B., Cockfield S.M., Preiksaitis J.K. Monitoring of polyomavirus BK virus viruria and viremia in renal allograft recipients by use of a quantitative real-time PCR assay: one-year prospective study. J. Clin. Microbiol. 2007;45:3568–3573. doi: 10.1128/JCM.00655-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietilä T., Nummi M., Auvinen P., Mannonen L., Auvinen E. Expression of BKV and JCV encoded microRNA in human cerebrospinal fluid, plasma and urine. J. Clin. Virol. 2015;65:1–5. doi: 10.1016/j.jcv.2015.01.019. [DOI] [PubMed] [Google Scholar]
- Plotly Technologies Inc . Plotly Technologies Inc; 2015. Collaborative Data Science. [Google Scholar]
- Priftakis P., Bogdanovic G., Kokhaei P., Mellstedt H., Dalianis T. BK virus (BKV) quantification in urine samples of bone marrow transplanted patients is helpful for diagnosis of hemorrhagic cystitis, although wide individual variations exist. J. Clin. Virol. 2003;26:71–77. doi: 10.1016/s1386-6532(02)00040-9. [DOI] [PubMed] [Google Scholar]
- Purdie K.J., Proby C.M., Rizvi H., Griffin H., Doorbar J., Sommerlad M., Feltkamp M.C., Meijden E.V., Inman G.J., South A.P., et al. The role of human papillomaviruses and polyomaviruses in BRAF-inhibitor induced cutaneous squamous cell carcinoma and benign squamoproliferative lesions. Front. Microbiol. 2018;9:1806. doi: 10.3389/fmicb.2018.01806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team . R Foundation for Statistical Computing; Vienna, Austria: 2020. R: a language and environment for statistical computing. https://www.R-project.org/ [Google Scholar]
- Rao S., Garcea R.L., Robinson C.C., Simões E.A.F. WU and KI polyomavirus infections in pediatric hematology/oncology patients with acute respiratory tract illness. J. Clin. Virol. 2011;52:28–32. doi: 10.1016/j.jcv.2011.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao S., Lucero M.G., Nohynek H., Tallo V., Lupisan S.P., Garcea R.L., Simões E.A.F. Wu and KI polyomavirus infections in Filipino children with lower respiratory tract disease. J. Clin. Virol. 2016;82:112–118. doi: 10.1016/j.jcv.2016.07.013. [DOI] [PubMed] [Google Scholar]
- Ryschkewitsch C., Jensen P., Hou J., Fahle G., Fischer S., Major E.O. Comparison of PCR-southern hybridization and quantitative real-time PCR for the detection of JC and BK viral nucleotide sequences in urine and cerebrospinal fluid. J. Virol. Methods. 2004;121:217–221. doi: 10.1016/j.jviromet.2004.06.021. [DOI] [PubMed] [Google Scholar]
- Şahiner F., Gümral R., Yildizoğlu Ü., Babayiğit M.A., Durmaz A., Yiğit N., Saraçli M.A., Kubar A. Coexistence of Epstein–Barr virus and Parvovirus B19 in tonsillar tissue samples: quantitative measurement by real-time PCR. Int. J. Pediatr. Otorhinolaryngol. 2014;78:1288–1293. doi: 10.1016/j.ijporl.2014.05.012. [DOI] [PubMed] [Google Scholar]
- SantaLucia J. A unified view of polymer, dumbbell, and oligonucleotide DNA nearest-neighbor thermodynamics. Proc. Natl. Acad. Sci. U S A. 1998;95:1460–1465. doi: 10.1073/pnas.95.4.1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarmento D.J.S., Palmieri M., Galvão G.S., Tozetto-Mendoza T.R., Canto C.M., Pierrotti L.C., David-Neto E., Agena F., Gallottini M., Pannuti C.S., et al. BK virus salivary shedding and viremia in renal transplant recipients. J. Appl. Oral Sci. 2019;27:e20180435. doi: 10.1590/1678-7757-2018-0435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauvage V. Human polyomavirus related to African green monkey lymphotropic polyomavirus. Emerg. Infect. Dis. 2011;17:1364–1370. doi: 10.3201/eid1708.110278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schowalter R.M., Pastrana D.V., Pumphrey K.A., Moyer A.L., Buck C.B. Merkel cell polyomavirus and two previously unknown polyomaviruses are chronically shed from human skin. Cell Host Microbe. 2010;7:509–515. doi: 10.1016/j.chom.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scuda N., Hofmann J., Calvignac-Spencer S., Ruprecht K., Liman P., Kühn J., Hengel H., Ehlers B. A novel human polyomavirus closely related to the African green monkey-derived lymphotropic polyomavirus. J. Virol. 2011;85:4586–4590. doi: 10.1128/JVI.02602-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siebrasse E.A., Reyes A., Lim E.S., Zhao G., Mkakosya R.S., Manary M.J., Gordon J.I., Wang D. Identification of MW polyomavirus, a novel polyomavirus in human stool. J. Virol. 2012;86:10321–10326. doi: 10.1128/JVI.01210-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Signorini L., Belingheri M., Ambrogi F., Pagani E., Binda S., Ticozzi R., Ferraresso M., Ghio L., Giacon B., Ferrante P., Delbue S. High frequency of Merkel cell polyomavirus DNA in the urine of kidney transplant recipients and healthy controls. J. Clin. Virol. 2014;61:565–570. doi: 10.1016/j.jcv.2014.10.012. [DOI] [PubMed] [Google Scholar]
- Si-Mohamed A., Goff J.L., Désiré N., Maylin S., Glotz D., Bélec L. Detection and quantitation of BK virus DNA by real-time polymerase chain reaction in the LT-ag gene in adult renal transplant recipients. J. Virol. Methods. 2006;131:21–27. doi: 10.1016/j.jviromet.2005.06.025. [DOI] [PubMed] [Google Scholar]
- Solis M., Meddeb M., Sueur C., Domingo-Calap P., Soulier E., Chabaud A., Perrin P., Moulin B., Bahram S., Stoll-Keller F., et al. French BKV Study Group Sequence variation in amplification target genes and standards influences interlaboratory comparison of BK virus DNA load measurement. J. Clin. Microbiol. 2015;53:3842–3852. doi: 10.1128/JCM.02145-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolt A., Kjellin M., Sasnauskas K., Luostarinen T., Koskela P., Lehtinen M., Dillner J. Maternal human polyomavirus infection and risk of neuroblastoma in the child. Int. J. Cancer. 2005;113:393–396. doi: 10.1002/ijc.20573. [DOI] [PubMed] [Google Scholar]
- Thomas L.D., Vilchez R.A., White Z.S., Zanwar P., Milstone A.P., Butel J.S., Dummer S. A prospective longitudinal study of polyomavirus shedding in lung-transplant recipients. J. Infect. Dis. 2007;195:442–449. doi: 10.1086/510625. [DOI] [PubMed] [Google Scholar]
- Tremolada S., Delbue S., Castagnoli L., Allegrini S., Miglio U., Boldorini R., Elia F., Gordon J., Ferrante P. Mutations in the external loops of BK virus VP1 and urine viral load in renal transplant recipients. J. Cell Physiol. 2010;222:195–199. doi: 10.1002/jcp.21937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Meijden E., Bialasiewicz S., Rockett R.J., Tozer S.J., Sloots T.P., Feltkamp M.C.W. Different serologic behavior of MCPyV, TSPyV, HPyV6, HPyV7 and HPyV9 polyomaviruses found on the skin. PLoS One. 2013;8:e81078. doi: 10.1371/journal.pone.0081078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Meijden E., Horváth B., Nijland M., Vries K., Rácz E., Diercks G.F., de Weerd A.E., Groningen M.C.C., van der blij-de Brouwer C.S., van der Zon A.J., et al. Primary polyomavirus infection, not reactivation, as the cause of trichodysplasia spinulosa in immunocompromised patients. J. Infect. Dis. 2016;215:1080–1084. doi: 10.1093/infdis/jiw403. [DOI] [PubMed] [Google Scholar]
- van der Meijden E., Janssens R.W.A., Lauber C., Bavinck J.N.B., Gorbalenya A.E., Feltkamp M.C.W. Discovery of a new human polyomavirus associated with Trichodysplasia spinulosa in an immunocompromized patient. PLoS Pathog. 2010;6:e1001024. doi: 10.1371/journal.ppat.1001024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Meijden E., Wunderink H.F., van der Blij-de Brouwer C.S., Zaaijer H.L., Rotmans J.I., Bavinck J.N.B., Feltkamp M.C.W. Human polyomavirus 9 infection in kidney transplant patients. Emerg. Infect. Dis. 2014;20:991–999. doi: 10.3201/eid2006.140055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vessman J., Stefan R.I., Staden J.F.V., Danzer K., Lindner W., Burns D.T., Fajgelj A., Müller H. Selectivity in analytical chemistry: (IUPAC Recommendations 2001) 2001. [DOI]
- Watzinger F., Suda M., Preuner S., Baumgartinger R., Ebner K., Baskova L., Niesters H.G.M., Lawitschka A., Lion T. Real-time quantitative PCR assays for detection and monitoring of pathogenic human viruses in immunosuppressed pediatric patients. J. Clin. Microbiol. 2004;42:5189–5198. doi: 10.1128/JCM.42.11.5189-5198.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H., Averick M., Bryan J., Chang W., McGowan L., François R., Grolemund G., Hayes A., Henry L., Hester J., et al. Welcome to the Tidyverse. J. Open Source Softw. 2019;4:1686. doi: 10.21105/joss.01686. [DOI] [Google Scholar]
- Wickham H., Bryan J. readxl: Read Excel Files. R package version 1.3.1. 2019 https://CRAN.R-project.org/package=readxl. [Google Scholar]
- Wickham H., Hester J., Francois R. readr: Read Rectangular Text Data. R package version 1.3.1. 2018 https://CRAN.R-project.org/package=readr. [Google Scholar]
- Yamamoto Y., Morooka M., Ihira M., Yoshikawa T. The kinetics of urinary shedding of BK virus in children with renal disease. Microbiol. Immunol. 2015;59:37–42. doi: 10.1111/1348-0421.12212. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data: The data are fully presented in the main manuscript and Supplemental information.
Code: The code used to generate in silico results may not be extracted from a multipurpose software package developed in our group; it will be described separately. To assist the reader in this respect, we set-up a dedicated web-page, which provides access to the code functionalities relevant to this study https://veb.lumc.nl/MANUSCRIPTS/Polyomaviridae2021.cgi.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.