

Abstract
The nucleocapsid (N) protein of SARS-COV-2, a virus responsible for the current COVID-19 pandemic, is considered a potential candidate for the design of new drugs and vaccines. The protein is central to several critical events in virus production, with its highly druggable nature and rich antigenic determinants making it an excellent anti-viral biomolecule. Docking-based virtual screening using the Asinex anti-viral library identified binding of drug molecules at three specific positions: loop 1 region, loop 2 region and β-sheet core pockets, the loop 2 region being the most common binding and stable site for the bulk of the molecules. In parallel, the protein was characterized by vaccine design perspective and harboured three potential B cell-derived T cell epitopes: PINTNSSPD, GVPINTNSS, and DHIGTRNPA. The epitopes are highly antigenic, virulent, non-allergic, non-toxic, bind with good affinity to the highly prevalent DRB*0101 allele and show an average population coverage of 95.04%. A multi-epitope vaccine ensemble which was 83 amino acids long was created. This was highly immunogenic, robust in generating both humoral and cellular immune responses, thermally stable, and had good physicochemical properties that could be easily analyzed in in vivo and in vitro studies. Conformational dynamics of both drug and vaccine ensemble with respect to the receptors are energetically stable, shedding light on favourable conformation and chemical interactions. These facts were validated by subjecting the complexes to relative and absolute binding free energy methods of MMGB/PBSA and WaterSwap. A strong agreement on the system stability was disclosed that supported ligand high affinity potential for the receptors. Collectively, this work sought to provide preliminary experimental data of existing anti-viral drugs as a possible therapy for COVID-19 infections and a new peptide-based vaccine for protection against this pandemic virus.
Keywords: SARS-COV-2, Nucleocapsid (N) protein, COVID-1, Drugs, Vaccine
Graphical abstract
1. Introduction
The ongoing COVID-19 pandemic caused by SARS-COV-2 is a highly infectious disease posing a severe threat to worldwide public health [[1], [2], [3], [4]]. SARS-COV-2 has the incredible potential of human to human spreading, contributing to rapid global dissemination [5,6]. To date, the virus has been reported to cause 24,357,067 cases with 830,150 deaths and 16,890,125 recoveries. Despite tremendous efforts done on mitigating the virus transmission and developing therapeutics in almost every country around the globe, we still have no specific treatment and cure yet [[6], [7], [8], [9]]. This prompted us to devise highly accepted in silico approaches with the ultimate aim to assist in the rapid design of new classes of drugs and vaccines for SARS-COV-2.
SARS-COV-2 is a single-stranded 30 kb long RNA genome virus [6,10,11]. The main region of the genome, named ORF1a/b, covers 2/3 of the length and codes nonstructural proteins (nsp) [12]. The remaining genome encodes four essential structural proteins which include a small envelope (E) protein, a matrix (M) protein, a nucleocapsid (N) protein, and a spike (S) glycoprotein [6,13,14]. The current coronavirus anti-viral regime primarily targets 3c-like (3CL) protease, papain-like (PLP) protease, and the S protein [15,16]. As protease inhibitors may act nonspecifically on the host homologous protease, there is a risk of associated host cell toxicity and can induce severe side effects. The S protein is highly vulnerable to mutations, enabling this protein to acquire a different pattern of host cell receptor binding, which in turn, aid in the protein escape from targeted therapeutics [15]. Considering this, novel strategies are obligatory to curtail infections caused by SARS-COV-2.
The N protein of SARS-COV-2 binds to leader RNA and plays several pivotal roles in RNA transcription and replication, and thus is considered an attractive pharmacological target [[17], [18], [19]]. Its primary function is to produce a ribonucleoprotein (RNP) complex, key to the formation of highly ordered RNA conformation essential for viral RNA replication and transcription and modulating metabolism of infected cells [19]. Additionally, this protein regulates host-pathogen interactions that involve reorganization of actin, the progression of the host cell cycle and apoptosis [20]. From an architectural point of view, the protein is composed of three distinct but highly conserved portions; the C-terminal domain (CTD), the N-terminal domain (NTD), and Ser/Arg (SR) rich linker [17]. CTD functions as a dimerization domain, whereas NTD and SR are responsible for binding to RNA and direct phosphorylation, respectively [6,21]. The crystal structure of the NTD from SARS-COV-2 (PDB ID: 6M3M) has been found to interact with 3′ end of the SARS-COV-2 genome through several key residues mediating infectivity and give specific electrostatic distribution [17]. Such structural information is essential for accelerating drug discovery against this appealing drug target to block SARS-COV-2 production. Furthermore, the N protein has high expression during infection, is highly immunogenic and has the potential to induce protective immune responses targeting SARS-COV-2 [22,23].
Herein, we adopted a comprehensive in silico methodology to identify potent anti-viral leads, protective antigens and diagnostic markers against SARS-COV-2 N protein. Findings of this study may promote the discovery of new anti-viral drugs and vaccination strategies against this high priority virus.
2. Materials and methods
The complete methodology used in the present work is summarized in Fig. 1 .
Fig. 1.

Flow of steps used in the current study.
2.1. SARS-COV-2 N protein preparation
The study commenced with the retrieval of the crystal structure of N protein from the PDB database available with a PDB tag of 6M3M [17]. The structure was determined by X-ray diffraction up to a resolution of 2.70 Å. The 3D structure was then treated in UCSF Chimera [24] for structure editing to optimize receptor energy. During the process, the protein first underwent processing using a steepest descent algorithm keeping the step size to 0.02 Å and number of cycles to 100. Afterwards, the conjugate gradient algorithm was applied on the structure for the default ten steps. Both algorithms tended to clean the structure by improving local interactions, fine-tuning widespread structural errors and moving the structure towards local minima. Next, missing hydrogen atoms were added to the receptor and charges to standard and non-standard residues were assigned by mean of AMBER ff14SB [25] and AM1-BCC [26], respectively. Both initial crystal N protein and post-treated minimized structures were examined using a PDBSum PROCHECK analysis [27] to assist in the selection of starting receptors in a high throughput anti-viral scaffold screening process.
2.2. Ligands preparation
The Asinex anti-viral library delivers a meaningful starting point by arranging chemical entities of potent anti-viral activities and good safety profile for the discovery of new powerful leads. The compounds are also easy to access and purchasable for testing in experimental assays. The anti-viral library was retrieved in SDF format and subsequently filtered in PyRx software [28] to select compounds that fulfil the Lipinski rule of five [29]. As per this rule, only compounds with molecular weight ≤ 500 Da, MlogP ≤4.15, N or O ≤ 10, and NH or OH ≤ 5 are selected. The primary library had 6827 compounds that, after deletion of molecules violating Lipinski rule of five, reduced to 4860. This new list of compounds was minimized for energy minima and converted to .pdbqt format to be ready for docking study.
2.3. Uncovering potential epitopes for SARS-COV-2 N protein
The amino acid sequence of the SARS-COV-2 N protein was scanned for potential B and T cell epitopes capable of evoking strong but protective immunological responses. To accomplish this objective, we employed the Immune Epitope Database (IEDB) [30] where first linear B cell epitopes were predicted using Bepipred Linear Epitope Prediction 2.0 setting cutoff score of >0.5. In parallel, T cell epitopes were also predicted starting with MHC-I alleles using IEDB recommended 2020.04 (NetMHCpanEL 4.0) considering a reference set of alleles (S-Table 1 ). The epitopes were sorted on IC50 score basis, and those with score < 500 nM were selected. The MHC-II alleles prediction was accomplished by selecting IEDB 2.22 method and full HLA reference set (S-Table 1). Each of the predicted epitopes was then evaluated for their potential of evoking immunological response by scanning the epitopes in VaxiJen (cut off antigenic score ≥ 0.4) [31]. Next, the shortlisted immunodominent candidates were checked for allergenicity via AllerTOP version 2.0 [32]. Non-allergens were then toxicity filtered using ToxinPred [33] to opt for non-toxic epitopes. Additionally, the non-homologic and virulent nature of the epitopes was verified by using the epitopes in Blastp search against human (Homosapien: 9606) and VirulentPred [34], respectively. Lastly, IFN-gamma inducing epitopes were filtered via IFNepitope web server [35].
Table 1.
Immunological analysis of SARS-COV-2 N protein.
| B cell | T Cell | MHC I | MHC II | MHCpred | Antigenisity | Allergenisity | Toxicity |
|---|---|---|---|---|---|---|---|
| GSRPQGLPNNTAS | GSRPQGLPN | 1.6 | 48 | 93.54 | 0.3955 (Non-antigen) | Non-allergen | |
| RPQGLPNNT | 0.35 | 22 | 43.55 | 0.5758 (Antigenic) | Allergen | ||
| QGLPNNTAS | 0.28 | 7.4 | 79.8 | 0.0743 (Non-antigen) | Allergen | ||
| QHGKEDLKFPRGQGVPINTNSSPDDQIG | PINTNSSPD | 15 | 15 | 5.6 | 0.5204 (Antigenic) | Non-allergen | Non-toxic |
| HGKEDLKFP | 2 | 31 | 20.75 | 1.1473 (Antigenic) | Allergen | ||
| QGVPINTNS | 11 | 15 | 23.12 | 0.3642 (Non-antigenic) | Allergen | ||
| TNSSPDDQI | 2.3 | 17 | 33.19 | 0.4559 (Antigenic) | Allergen | ||
| INTNSSPDD | 2.6 | 22 | 34.99 | 0.4458 (Antigenic) | Allergen | ||
| KFPRGQGVP | 0.04 | 23 | 46.99 | −0.1527 Non Antigenic | Allergen | ||
| KEDLKFPRG | 3.2 | 35 | 47.75 | −0.0113 (Non-antigenic) | Allergen | ||
| GKEDLKFPR | 2 | 35 | 60.39 | 0.7725 (Antigenic) | Allergen | ||
| FPRGQGVPI | 0.04 | 11 | 61.24 | 0.7585 (Antigenic) | Allergen | ||
| GVPINTNSS | 9 | 15 | 62.09 | 0.7585 (Antigenic) | Non-allergen | Non-toxic | |
| DLKFPRGQG | 7.6 | 16 | 69.18 | −0.0028 (Non-antigenic) | Non-allergen | ||
| RIRGGDGKMKDL | RIRGGDGKM | 0.42 | 20 | 58.48 | 0.1803 (Non-antigenic) | Non-allergen | |
| TGPEAGLPYGANK | TGPEAGLPY | 0.56 | 2.7 | 40.83 | −0.0349 (Non-antigenic) | Non-allergen | |
| GALNTPKDHIGTRNPANN | DHIGTRNPA | 7.8 | 12 | 20.23 | 0.4313 (Antigenic) | Non-allergen | Non-toxic |
| IGTRNPANN | 8.2 | 12 | 30.69 | 0.3555 (Non-antigenic) | Allergen | ||
| LNTPKDHIG | 14 | 52 | 83.75 | −1.2393 (Non-antigenic) | Allergen | ||
| ALNTPKDHI | 0.87 | 37 | 86.1 | −0.8423 (Non-antigenic) | Allergen |
2.4. Epitope conservation and population coverage
All entries of the SARS-COV-2 N protein available in the NCBI COVID-19 datahub were retrieved and used to examine predicted IFN epitopes conservation through an online IEDB conservancy analysis tool [36]. It was important to include population coverage of the epitopes in the study as it gave clear directions about the percentage of a specific population likely to respond to the epitopes. This was achieved by employing the IEDB epitope conservation analysis tool [37].
2.5. Construction of vaccine ensemble
The final set of conserved epitopes that provided the greatest population coverage were fused to each other, followed by addition of an adjuvant molecule to the epitope peptide. The 3D structure of the full ensemble was created ab initio using 3Dpro [38]. Loops of the model structure were refined to strengthen structural stability using GalaxyRefine version 2 [39]. Quality assessment of the ensemble 3D model was made employing free available tools: Ramachandran plot of PDBsum [27], ERRAT [40], VERIFY-3D score [41], PROSA Z-score [42]. Then, the structure was minimized to the local energy minima and passed through downward analysis of vaccine design. Different physicochemical parameters of the vaccine were predicted using ProtParam [43]. Solubility and aggregation-prone regions of the vaccine were predicted using Protein-Sol [44] and Aggrescan3D 2.0 [45], respectively.
2.6. MHC clustering analysis and immune simulation of the vaccine ensemble
During the vaccine design process, identification of candidates showing an optimized affinity for a wide range of MHC HLA alleles is fundamental. The MHC clustering analysis was performed using MHCcluster v2.0 [46]. Further, the immune response profile of the vaccine construct was understood using an agent based immune simulator - the C-ImmSim server [47]. The server employed a position specific scoring matrix to spot immune dominant epitopes and machine learning methods to elucidate immune interactions. Most of the simulation parameters were treated as default: adjuvant = 100, number of antigen injection = 1000, random seed chosen = 12,345, and the vaccine was injected with non LPS. The 1000 units of antigen are considered suitable to induce an appropriate immune response to the viral antigen [48]. The simulation steps allowed are 1100 and volume to 110. C-ImmSim server has been successfully applied in several studies to understand host immune system dynamics in response to an antigen [[49], [50], [51], [52], [53]].
2.7. Molecular docking of antiviral ligands and vaccine ensemble
A blind docking approach was applied for docking both anti-viral ligands with the N protein and vaccine ensemble with TLR3 (PDB tag: 1ZIW). In the case of anti-viral ligands, docking was performed with AutoDock4 [54] allowing the central XYZ dimensions search to be restricted to 9.9921 Å on the X-axis, −3.8536 Å on the Y-axis and − 12.7487 Å on the Z-axis. This gives dimensions on the XYZ plane as 31.6231 Å, 46.5257 Å, and 43.2407 Å, respectively, and as net, the whole surface of the receptor molecule was covered to allow ligand molecules to bind freely to the hotspot points of the N protein. Each ligand molecule was docked 10 times to the receptor and the best binding pose was selected by looking for the one with best binding affinity score in kcal/mol (more negative score indicates good binding affinity). The vaccine ensemble docking with innate immune receptor (as a test case here we used TLR3) was assessed in Patchdock [55], the generated complexes were refined with FireDock [56], and the best complex with minimum global energy was considered for visualization and assay [[57], [58], [59], [60]]. Both AutoDock4 and FireDock are best for performing blind docking and generating intermolecular poses that bind best to each other, hence achieving highly stable complexes. Both sets of software are the most citable forms of docking software, widely used and are freely available [56,[61], [62], [63], [64], [65]]. A blind docking approach was used in the present work by providing the complete surface of receptors (N protein in case of drug molecules identification and TLR3 in case of vaccine ensemble docking). This overcomes the limitations of the specific docking that, in majority cases gives false-positive results [66]. Additionally, we proved the docking procedure by docking the co-crystalized ligands to the receptors through the same procedure used in virtual screening of Asinex antiviral library and vaccine ensemble docking to the TLR3 [67]. Both docking protocols results revealed coherent results, thus validating the docking protocol. Further, we employed widely accepted and more accurate molecular dynamics simulation and binding free energies methods to validate the good affinity of the drug molecules and vaccine ensemble [[68], [69], [70]].
2.8. Molecular dynamics simulation assay
Molecular dynamics simulations for both top complexes of anti-viral ligand with N protein and vaccine ensemble with TLR3 were performed using AMBER18 package [71]. The N protein parameters were generated using a ff14SB force field [25] whereas an Amber force field (GAFF) [72] was chosen for anti-viral ligands. To record topology files of both complexes, the leap module [73] was employed. The systems were neutralized by adding an appropriate number of counterions. Next, both systems were submerged in a TIP3 water box, allowing a padding distance of 12 Å. The waterbox with submerged N protein-drug complex and TLR3-vaccine complex are depicted in Fig. 2 . Systems minimization was achieved by running 1500 rounds of steepest descent and conjugate gradient to clean the complexes for unfavorable structural clashes. Heating of systems was done for 100 ps with a gradual increase from 0 K to 300 K, applying pressure of 1 atm. Afterheat, systems were equilibrated for a time period of 100 ps at a constant temperature of 300 K. A production run for each system was completed at time scale of 100 ns. In the process, SHAKE algorithm [74] was applied to constrain all covalently bonded hydrogen atoms of the systems. Periodic boundary conditions were used in the solvation box by the canonical ensemble. Temperature was kept constant at 300 K using a Langevin thermostat [75] and the non-bounded interaction threshold was treated as 8.0 Å. Ewald simulations were utilized for long range interactions. Structural dynamics of the complexes were elucidated through several statistical parameters analyzed through the CPPTRAJ module [76] of the AMBER. Visualization of the snapshots was done by means of UCSF Chimera [24] and VMD software [77].
Fig. 2.

Waterbox presentation of drug (top) and vaccine ensemble complex with respective receptors (colored by yellow cartoon). The drug and vaccine construct is shown by in green sphere.
2.9. Binding free energy estimation
Estimation of binding free energies for biomolecular complexes is a good way of validating intermolecular strength of interactions and shedding light on the dominancy of a particular chemical energy contributing to overall stability. The MMPBSA method of AMBER is an easy and relatively straight forward approach for quantifying binding free energies of ligand(s) docked to a receptor, though this method does not account for entropy contribution [78]. The MMPBSA binding free energy was estimated using equation given below,
where,
ΔG is the net binding free energy for a given system by subtracting the sum of receptor and ligand combine binding energy from the complex. ΔGMM is the gas phase energy change estimated by molecular mechanics and consists of van der Waals and electrostatic energy. ΔGsolv is the solvation free energy change and is the product of polar and non-polar energy. The latter term is calculated via the solvent accessible surface area (SASA).
2.10. Waterswap absolute protein-ligand binding free energies
The Waterswap method implemented in the Sire package permits estimation of absolute protein-ligand binding free energies [79]. The reaction coordinate is constructed during the process which swaps the protein bound ligand to an equivalent volume of water present in the binding pocket. This method uses all available processing cores of a computer node, slow in-process and converges the free energy averages that took at least five days. The average binding free energy is estimated simultaneously through thermodynamic integration (TI), free energy perturbation (FEP) and Bennetts Acceptance Ratio (BAR) methods [70]. Waterswap was run on simulation trajectories of last 10 ns to estimate the absolute binding free energy of the systems.
3. Results and discussion
3.1. SARS-COV-2 N protein and anti-viral library minimization
A primary phase of protein minimization was applied to SARS-COV-2 N protein to lower its overall potential energy. This was necessary to make protein conformation as close as possible to natural biological systems that are dynamic and low in potential energy to ease spontaneous interactions. However, a minimization event may introduce bad contacts in the structure and disturb the conformation of the molecule, which in turn might affect the compound ranking in the docking-based virtual screening process. The minimization process revealed that minimized N protein is a bit better compared to the pre-minimized original N protein structure. Ramachandran plot investigation showed that both original and energy minimized structures have the same percentage of residue distribution in all four quadrants as can be seen in S-Fig. 1. Statistically, both structures secured 89.4% and 10.6% of residues in the most favoured and additionally allowed regions whereas no residue was plotted in generously allowed and disallowed regions. According to the G-factor assay, the minimized protein had a better overall G-factor score of 0.06, reflecting no unusual features in the structure opposed to the original structure (G-factor score of −0.14). Secondly, the PROSA Z-score was calculated which indicated overall good quality and a non-erroneous structure of the minimized N protein. The Z-score for pre-minimized N protein was −5.06 while it was −5.16 for the minimized form. Based on this evidence, we used the minimized N protein structure in the downward framework.
3.2. Virtual screening
High throughput structure-based virtual screening was performed using SARS-COV-2 N protein as a receptor and drug-like molecules from the Asinex anti-viral library as ligands in the process. As a blind docking strategy was employed, the entire surface of the protein was exposed for binding of the drug molecules. The molecules bound to three different binding pockets of loop region 1, β-sheet core, and loop region 2 of the enzyme as shown in Fig. 3 . The majority of the molecules tended to interact with the pockets of the loop 2 region compared to the loop 1 region and β-sheet core pockets. Top 5 complexes were picked based on the binding affinity of the molecules for the pockets and analyzed. The 2D structures of the compounds are provided in S-Fig. 2. The high affinity top 1 compound with binding energy of −8.9 kcal/mol demonstrated binding to the front pocket of a loop 2 region. This pocket is the result of the long loop of β6 connecting to β7, and β2 ending at the N-terminal site through β1. The bulk of the compound structure (2,5,5,7,9-pentamethyl-2,3,4,4a,5,10b-hexahydropyrano[3,2-c]chromene-3-carboxylic acid) aligned horizontally inside the long loop originating from β6 of the β-sheet core, covering 310 helix at the acidic wrist of the protein and ending at the carboxyl site. This chemical moiety formed weak van der Waals, pi-pi stacked, and pi-alkyl interactions with the loop of β6. The 4-isopropylphenol moiety was deep in the cavity, making both hydrogen and hydrophobic contacts with the loops. Compound 3 (binding energy: −8.6 kcal/mol) and 5 (binding energy: −8.2 kcal/mol) followed the same pattern of binding with minimal interaction from strong hydrogen bonding and maximum interaction from weak hydrophobic binding. Compound 2 (binding energy: −8.7 kcal/mol) binding mode was stable at loop region 1 and produced a rich network of hydrogen and van der Waals contacts. The 1-(3-(1,2,4-triazolidin-4-yl)phenyl)ethanone part of the molecule mainly drove the interactions of strong hydrogen bonding with Thr92 and Arg94 β5 of β2 and Asp129 of β6 loop. Compound 4 (binding energy: −8.4 kcal/mol) favoured binding to the back pocket of loop region 2, collectively formed by loops of β2, β5 and β6. The 4-formyl-6-methyl-3-methylene-2,3-dihydroisoxazolo[5,4-b]pyridin-7-ium chemical moiety made hydrogen bonds with Pro74 and Asn76 of β2. The rest of the chemical portion reacted with all three loops through van der Waals bonding. The chemical interactions of the compounds are illustrated in Fig. 4 .
Fig. 3.

Docked pose of different classes of anti-viral inhibitors at different pockets of the SARS-COV-2 N protein. The color order of the top 5 ligands is in the following order: top 1 (tan), top 2 (cyan), top 3 (pink), top 4 (aquamarine), and top 5 (deep pink).(For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Fig. 4.

Chemical network of interaction of the compounds at respective docked sites. From A to E tags represents compound 1 to 5, respectively. The different color of discs can be interpreted as residues of the N protein and can be understand as: dark green discs (hydrogen bonding residues), light green discs (van der Waals residues), pink discs (pi-pi stacked residues), purple discs (pi-sigma residues), and cream discs (alky and pi-alkyl residues).(For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
3.3. SARS-COV-2 N protein epitopes prediction
A complete protein sequence was first analyzed for continuous B cell epitopes that predicted 6 epitopes ranging in length from 7mer to 28mer of score greater than default cut off (0.5). Such B cell epitopes are the recognition and binding sites of the adaptive immunity B cells. Once activated, the B cell transformed into a mature form, differentiate and produce soluble antibodies. Afterward, the antibodies bind to the epitopes and activate humoral adaptive immunity by triggering formation of neutralizing toxins, labeling the pathogen for destruction via T cell immunity. Therefore, B cell epitopes mapping holds a central role in vaccine designing [80]. The predicted B cell epitopes were examined for T cell epitopes. T cell vaccine induces protective cellular immunity and are significant in targeting mutating viruses like SARS-COV-2. Furthermore, T cell vaccines are regarded more effective than conventional B cell epitopes [81,82]. In the present investigation, both B and T cell epitopes were used to design highly efficacious vaccines. Antigens that are displayed on the antigen presenting cells bound to the major histocompatibility complex molecules are recognized by the T cell receptors present on the surface of T cells. T cell epitopes are surface epitopes via two classes: MHC-I and MHC-II, identified by two distinct T cytotoxic and T helper cells. Several T cell epitopes are reported for each B cell epitopes with IC50 values <50 nM depicting high affinity for the reference set of MHC alleles. Next, the T cell epitopes were subjected to MHCphred assay to filter only those epitopes that associated with great affinity with the DRB*0101 allele, a prominent allele in the human population [83]. All T cell epitopes showed great binding with the DRB*0101 allele with an IC50 value of <100 nM. T cell epitopes then analyzed for their ability of provoking the host immune system. This was achieved by passing the T cell epitopes through the VaxiJen server and selecting those with predicted score higher than the default 0.4. Antigenic T cell epitopes were subsequently filtered through allergenic and toxicity check, and only non-allergenic and non-toxic epitopes were picked. The final set of potential epitopes selected for vaccine ensemble design is listed in Table 1 and their exo-membrane topology is shown in Fig. 5 .
Fig. 5.

Surface analysis of final set of T cell epitopes localized at the surface of N protein. A (PINTNSSPD), B (GVPINTNSS) and C (DHIGTRNPA). The amino acids are presented by single letter code.
3.4. Population coverage of T cell epitopes
The MHC molecules are extremely polymeric and thousands of MHC molecules are known that are spread across the world population and in different ethnicities. Thus, peptide-based vaccine design that covers the majority of these alleles could provide a wonderful approach for the design of a broad spectrum and highly effective vaccine that does not show any ethnically biased population coverage. The selected B cell derived T cell epitopes are showing average population coverage of 95.04%. This can be divided up as: Central Africa (94.79%), Central America (53.8%), East Africa (97.08%), East Asia (97.08%), Europe (99.96%), North Africa (99.01%), North America (99.89%), Northeast Asia (97.88%), Oceania (97.88%), South Africa (95.27%), South America (95.15%), South Asia (98.7%), Southeast Asia (97.66%), Southwest Asia (95.79%), West Africa (98.43%), and West Indies (99.69%). The number of combined HLA alleles recognized and cumulative percent of population coverage of the epitopes are given in S-Fig. 3.
3.5. Construction of a chimeric multi-epitope peptide vaccine
Peptide-based vaccines are weakly immunogenic, therefore, the final set of epitopes were fused with the help of rigid AYY linkers to produce a multi-epitope peptide [84,85] (Fig. 6A). AYY linkers aid in keeping the epitopes separated and aid in easy recognition and processing of the epitopes by the host immune system. Also, an adjuvant in the form of β-defensin was added to the N-terminal site of the epitope peptide for further strengthening of the immune provoking ability of the epitope's peptide. β-defensin activates lymphokines production which in turn results in Ig production specific to the antigen along with activation of cellular immunity [86]. Ligation of the adjuvant with the multi-epitope peptide was done through a rigid EAAAK linker. This linker is also rigid and promotes easy immune recognition and processing. The complete construct structure was modelled ab initio as no suitable template was available for homology-based modelling (Fig. 6B). The structure has the majority of its residues (80%) plotted in the most favoured region, 12.90% in the additional allowed region, 5.70% in the generously allowed region, and 1.40% in the disallowed regions (Fig. 6C). From secondary structure point of view, the structure maximum of alpha helix (53.0%), 9.6% of 3–10 helix, and 37.3% of beta turns, and gamma turns (Fig. 6D). The overall vaccine ensemble is antigenic with a score of 0.5060, soluble with a probability score of 0.940 and does not harbour any transmembrane helices; thus it is a good candidate for experimental follow up. The weight of the construct is 8.9 KDa, theoretical PI of 9.36, stability index of 39.9, GRAVY score of −0.602 and aliphatic index of 53.01. The vaccine ensemble is also predicted to be highly soluble with predicted scaled solubility value of 0.775 (threshold, 0.45) and does not contain any aggregation-prone regions. The average aggregation score of the vaccine is −1.11, which is far less than the cutoff score of 0.0.
Fig. 6.

A. Schematic representation of the vaccine, B. 3D structure of the vaccine, C. Ramachandran plot of the vaccine, D. Secondary structure elements of the vaccine, E. Disulfide engineering of the vaccine; both original and mutated strucutures are presented, F. In silico cloning of the vaccine (shown as red) into pET-28a(+) vector. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
3.6. MHC restricted HLA alleles cluster analysis
The vaccine ensemble molecule interacted with a large numbers of HLA alleles in MHC I class compared to MHC II class. This infers the increased potential of the designed vaccine ensemble to be recognized by the vast majority of polymorphic HLA alleles, maximizing many fold epitopes presentation the immune cells leading to providing strong immune protection capabilities. The MHC I and MHC II HLA clusters are tagged as S-Fig. 4 and S-Fig. 5, respectively.
3.7. Loop modelling and refinement of the vaccine ensemble
Loop regions of the vaccine ensemble were modelled/refined to present its closest possible structure for the downward analysis. The structure was refined in consecutive rounds covering regions: Arg14- Ser34, Asn53- Ser57, Gly63- Ser70, Tyr74-Ala83, and Ala47-Ala48. The output structure was further refined for global and local conformations. An improved vaccine ensemble structure was revealed in which the percent of residues in the Rama favoured regions was increased to 96.3% compared to 93.8% in the original model. The clash error score in the original structure was 29.4 and was found only 2.2 in the refined model. The MolProbity score is relatively low (1.241) illustrating the good quality of the refined model. Overall, the galaxy energy of the refined vaccine ensemble is −1744.48 which is significantly less than the initial structure (132.01), a confirmation of good stability.
3.8. Immune responses simulation of the vaccine ensemble
An in silico immune response model of the vaccine ensemble was generated that illustrated primary, secondary and tertiary immune responses are credible in clearing the pathogen (Fig. 7 ). The IgM + IgG is critical in showing an immune protection response to the antigen whereas IgG2 is the least productive. Similarly, significant interleukin and cytokine reactions are witnessed, IFN-gamma being the most key immune protective factor (Fig. 7). Formation of different isotypes of the B cell immunity to the antigen demonstrates a fundamental role of humoral immunity against the pathogen and subsequent creation of antigen memory. Increased production of cytotoxic and helper T cell populations with corresponding memory formation further affirm the role of T cell immunity complementing B cell immunity in protecting the host from the pathogen. A high population of dendritic cells, as well as that of macrophages, makes it evident that the set of epitopes used in the construction of multi-epitope vaccine ensembles is attractive for activation of all components of the host immune system aiding in combating the pathogen.
Fig. 7.

Host immune system simulation in response to the vaccine ensemble antigen; production of different immunoglobulin response (top) and cytokines and interleukins with Simpson index (bottom).
3.9. Vaccine ensemble docked conformation with TLR3
The vaccine ensemble was docked with TLR3, an immune receptor from innate immunity that prompts viral recognition and induces type I interferon production. Blinded protein-peptide docking was performed to predict the predominant orientation of the vaccine ensemble with respect to the TLR3 receptor. In total, 100 conformations of the vaccine ensemble were generated, followed by refinement of each complex to opt for the highly stable conformation. Different energy values for the top 10 refined solutions of the docked vaccine ensemble and TLR3 complexes are presented in Table 2 . Solution 9 was ranked as the top solution due to global energy of −7.93 kJ/mol (attractive van der Waals energy: −33.10 kJ/mol, repulsive van der Waals energy: 14.31 kJ/mol, atomic contact energy: 15.51 kJ/mol, and hydrogen bonding energy: −3.66 kJ/mol). Visual interpretation of solution 9 unveils deep central docking of the vaccine ensemble at the TLR3 cavity. The vaccine generates a network of hydrophobic and hydrophilic chemical interactions with TLR3 residues. Ser387, Glu460, Tyr462, Tyr465, and Asn662 are hydrogen bonding contributing residues from the TLR3 with Ser56, Thr68, Asp59, Tyr62, and Arg12 of the vaccine, respectively. Van der Waals interactions from the TLR3 that played a significant role in binding and anchoring of the vaccine are: Lys330, Glu363, Tyr383, Ile411, Thr415, Lys416, Val435, Phe459, Arg484, Arg488, Ile534, His565, Ile566, Phe657, and Asn659. From the vaccine, residues Val13, Arg14, Lys50, Asn55, Ser57, Pro58, Ile66, Ala73, Tyr74, Ile77, and Asn81 participated in van der Waals interactions. The docked conformation of the vaccine ensemble with TLR3 can be seen in Fig. 8 .
Table 2.
Top 10 refined TLR3-vaccine solutions ranked on basis of global energy in KJ/mol.
| Rank | Solution Number | Global Energy | Attractive Van der Waals Energy | Repulsive Van der Waals Energy | Atomic Contact Energy | Hydrogen bonding Energy |
|---|---|---|---|---|---|---|
| 1 | 9 | −7.93 | −33.10 | 14.31 | 15.51 | −3.66 |
| 2 | 7 | −2.54 | −31.03 | 22.05 | 14.68 | −6.93 |
| 3 | 3 | 0.83 | −7.43 | 1.96 | 2.17 | −1.19 |
| 4 | 2 | 1.88 | −14.95 | 2.48 | 14.70 | −5.13 |
| 5 | 6 | 6.71 | −2.19 | 0.00 | 3.38 | 0.00 |
| 6 | 10 | 17.93 | −7.56 | 13.93 | 9.32 | −2.48 |
| 7 | 5 | 32.79 | −43.26 | 90.77 | 14.74 | −3.88 |
| 8 | 8 | 45.32 | −18.23 | 6.96 | 15.53 | −1.49 |
| 9 | 4 | 123.17 | −11.32 | 142.99 | 4.43 | −2.45 |
| 10 | 1 | 200.68 | −14.32 | 256.72 | 5.08 | −0.91 |
Fig. 8.
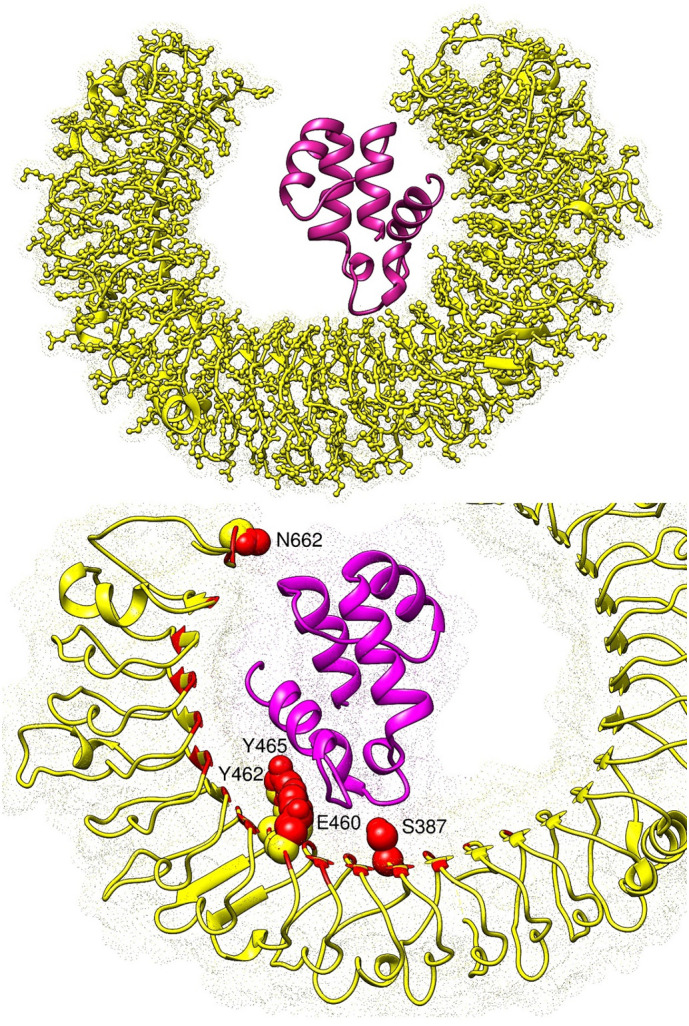
The top figure presents docked conformation of the vaccine ensemble (shown in dark maroon) at the active pocket of TLR3 (yellow cartoon surface as dot). The bottom figure illustrates closed view of the vaccine interacting with TLR3 residues (shown in red bubbles) via hydrogen bonding. The rest of red highlighted regions in sticks are those involved in van der Waals interactions with the TLR3 residues.
3.10. Disulfide engineering and codon optimization of the vaccine ensemble
Disulfide engineering is a directed approach of introducing disulfide bonds in a protein, and is a logical approach of emulating molecular interactions stabiilty required in many industrial and biomedical applications [87]. The chimeric vaccine sequence was subjected to Design 2.0 server which highlighted 5 pairs of residues with high bond energy ranging from 1.26 kcal/mo to 6.75 kcal/mol. The pairs of residues are: Ile2-Ala61, Arg17-Cys40, Val20-Cys41, Cys21-Lys26, and Pro51-Pro62 were mutated to enhanced vacine stability (S-Table 2). The original and mutated vaccine structures are shown in Fig. 6E. Furthermore, the vaccine sequence was reverse translated to improve the codon usage of the sequence as per Escherichia coli expression system to get maximum expression of the vaccine during wet lab experimentation. The codon adaptation index of the vaccine is 1 which is an ideal value for enhance expression. Lastly, the vaccine sequence was cloned into pET28a(+) expression vector (Fig. 6F).
3.11. Evaluation of conformational stability
The conformational stability of the SARS-COV-2 N protein in complex with the top drug molecule and vaccine ensemble was deciphered by running 50-ns of MD simulations. Structural stability of the systems was monitored first by calculating carbon alpha distance of superimposed 50,000 snapshots of the MD simulation (Fig. 9 (top left)). For the drug complex, a small number of minor structure fluctuations were noticed in the protein suggesting the structure has endured less conformational changes during the course of simulation. The average rmsd estimated for the drug complex is 1.38 Å with maximum observed is 2.53 Å. The variations were investigated by visualizing snapshots at regular intervals (0-ns, 10-ns, 20-ns, 30-ns, 40-ns, and 50-ns) and superimposed in UCSF Chimera [88] (Fig. 10A). The 3D alignment revealed an rmsd of 1.3 Å that clearly showed the drug complex stability. In the process, the compound was seen in different conformations where 4-isopropylphenol region of the molecule is showing stability at the initial docked site but the 2,5,5,7,9-pentamethyl-2,3,4,4a,5,10b-hexahydropyrano[3,2-c]chromene-3-carboxylic acid is stretching along the length allowing the molecule entry deep in the cavity with formation of extra van der Waals contacts as depicted by the binding free energy estimation in section. Further, these movements are the result of flexible loop regions of the N protein that propel the molecule to alter its conformation by adjusting the molecule along the channel of the receptor. This depicts binding free energy methods, with the compound enjoying binding at the cavity site and is an attempt at shwing stable binding as simulations progress. In the same way, rmsd analysis of the vaccine ensemble with TLR3 was carried out (Fig. 9 (top left)). The average rmsd of the TLR3-vaccine ensemble is 3.34 Å. Variations in the rmsd are mainly because of the ligand structure move in an attempt to get a highly stable and immune cell recognizable pose, resulting in force applied on the flexible regions of the TLR3. This was affirmed by taking regular snapshots at interval of 10-ns and superimposition in UCSF Chimera that revealed an rmsd of 1.124 Å (Fig. 10B). The receptor rmsd was followed by ligand rmsd to affirm the ligand binding stability with the receptors (Fig. 9 (top right)). An average rmsd of 1.30 Å and maximum of 2.41 Å were reported for the drug compound. Again minor ups and downs in ligand rmsd were spotted, reflecting conformational moves as stated earlier, though these adjustments are in favour of the increased system stability. The vaccine ensemble rmsd is very low (0.15 Å) demonstrating very stable behavior of the vaccine with TLR3. Next, compactness of the N protein structure was tested using the radius of gyration tool (Fig. 9 (bottom left)). The average gyration value of the N protein in the presence of the drug along the simulation is 14.56 Å and maximum value observed is 14.87 Å. This clearly shows that the secondary elements are highly compact in the N protein 3D structure and show stability in the drug molecule presence. The same compact complex system was noted for the TLR3 and vaccine ensemble with a mean radius of gyration value of 33.72 Å. The number of hydrogen bonds formed between the drug/vaccine with the receptors is shown in Fig. 9 (bottom right). On average, the drug molecule is in contact with the N protein docked site via 1 hydrogen bond through the simulation period whereas the vaccine produced a robust network of hydrogen bonds with TLR3 that, on average, is higher in each simulation frame.
Fig. 9.

Different MD based analysis: protein rmsd (top left), ligand rmsd (top right), protein ROG (bottom left) and hydrogen bonds plotting (bottom right).
Fig. 10.

A. Binding conformation adjustments of drug molecule (shown in different color sticks) at the binding pocket of SARS-COV2 N protein (shown by gray surface), B. Binding conformation adjustments of vaccine (shown in different color cartoons) at the binding pocket of TLR3. The colouring pattern can be interpreted as: 0-ns (tan), 10-ns (cyan), 20-ns (orchid), 30-ns (light green), 40-ns (salmon), and 50-ns (gray).(For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
3.12. Binding free energy calculation
The MMPBSA is now commonly applied on biological systems to model molecular recognition and is a central focus in molecular simulations [89]. This method uses reasonable computational cost and is in routine use to estimate binding free energies of small ligand molecules bound to a large biomolecule receptor [89]. The MMGBSA analysis, which is complementary to the MMPBSA, revealed binding with considerable affinity to the N protein as depicted by the net total energy of −30.4504 kcal/mol. This net energy can be split into components as: complex (−10,452.14 kcal/mol), receptor (−10,414.58 kcal/mol), and ligand (−7.10 kcal/mol). The gas phase energy and solvation energy contribution to the net energy of the system are significant; the latter dominates by polar energy as the favourable (−15.27 kcal/mol) opposed to minor support from non-polar energy (−4.27 kcal/mol). The gas phase energy is the attribute of van der Waals energy (−37.49 kcal/mol) in contrast to the non-favourable part from electrostatic energy (26.60 kcal/mol). The detailed N protein and drug molecule MMGBSA binding energies of the complex, receptor, ligand and the net energies are provided in S-Table 3 . In the MMPBSA method, the system net total binding energy is −23.59 kcal/mol (delta gas phase (10.89 kcal/mol) + delta solvation energy (−12.70 kcal/mol)). Together, both gas and solvation contributions play a balanced role in the binding affinity of the compound for the receptor. As in MMGBSA, the solvation energy is the output of a good polar solvation role (−9.43 kcal/mol) compared to less non-polar energy (−3.27 kcal/mol) in MMPBSA. The gas phase energy van der Waals component as reported in the MMGBSA is the prime factor in the compound interaction (37.49 kcal/mol) with the protein whereas, due to the lack of formation of ionic moieties that can give the electrostatic nature of interactions, the columbic interaction is insignificant (26.60 kcal/mol). The complete data of the MMPBSA analysis for N protein and drug molecule are presented in S-Table 4. Likewise, the TLR3-vaccine ensemble was highly stable with MMPBSA net binding energy of −47.96 kcal/mol. The stabilizing factor was revealed to be the gas phase electrostatic energy (−1359.83 kcal/mol) in the system lower energy state along with −67.79 kcal/mol of van der Waals energy. The polar solvation energy is the non-favourable contributor (1390.67 kcal/mol) to the net solvation energy (1379.67 kcal/mol), in contrast to little favourable energy from non-polar energy (−10.99 kcal/mol). The full data of MMGBSA and MMPBSA for TLR3 and vaccine ensemble are given as S-Table 5 and S-Table 6, respectively.
Table 3.
Hotspot residues with binding free energy values.
| Residue/Ligand | MMGBSA | MMPBSA | Residue/Ligand | MMGBSA | MMPBSA |
|---|---|---|---|---|---|
| Drug molecule | −13.3381 | −10.6936 | Vaccine Ensemble | −22.57 | −24.57 |
| Trp5 | −3.52555 | −2.19901 | Ser387 | −4.45 | −5.67 |
| Thr29 | −1.88196 | −1.01 | Glu460 | −3.48 | −5.98 |
| Asn30 | −3.23374 | −1.68084 | Tyr462 | −2.78 | −3.14 |
| Ile94 | −1.02521 | −1.12 | Tyr465 | −1.24 | −1.78 |
| Ile105 | −3.05031 | −2.58066 | Asn662 | −3.65 | −4.14 |
3.13. Energy contribution of key receptor residues and ligands
To underline residues that are essential to ligand binding and are favourable in complex stability, per residue decomposition was performed. Common residues of MMGBSA and MMPBSA with negative average binding energy were categorized as essential amino acids and were vital in interactions with the ligand (Table 3). For instance, residues such Trp5, Thr29, Asn30, Ile94, and Ile105 were demonstrated as hot spot residues due to their profound contribution in complex stabilization. For the TLR3-vaccine, Ser387, Glu460, Tyr462, Tyr465, and Asn662 are hotspots in anchoring the vaccine ensemble at the docked position.
3.14. WaterSwap absolute binding free energy
To estimate the absolute binding free energy of the system, WaterSwap was used. Unlike MMPBSA, this uses an explicit water model thus does not suffers from the shortcomings of the usually applied MMPBSA method. The binding free energy is calculated by means of three highly sophisticated approaches including TI, FEP and BAR. The resultant value is considered good when the mentioned techniques are converged with difference of ≤1 kcal/mol. The drug complex produced a TI value of −22.314 kcal/mol, FEP of −22.145 kcal/mol, and BAR of −23.87 kcal/mol. Similarly, the vaccine ensemble in complex TLR3 was reported to have TI, FEP and BAR values of −47.45 kcal/mol, −48.74 kcal/mol, and − 48.91 kcal/mol, respectively.
4. Concluding remarks
Efficient development of new vaccines and biologically useful drug molecules usually take years of research efforts and is a multibillion dollar gamble. The use of available pharmaceutically active and safe anti-viral agents in silico and in wet lab experiments as a substitute is a swift approach for uncovering medication which may efficiently deal with deadly and evolving viral infections. The conventional drug discovery pipeline takes a decade for safe anti-viral therapy development [90,91]. For diseases that are highly contagious like COVID-19, we don't have enough time and require to speed up the process by screening available drugs and repurposing them against this new disease threat in a process called drug repurposing. In an NIH-funded study published in the journal “Nature”, scientists screened a library of 12,000 existing drugs against SARS-CoV-2 using laboratory-grown human cell lines and non-human primates. They found 21 drugs that showed potential to thwart SARS-CoV-2, 13 out of which can be safely given to people. Most of these drugs have tested clinically against autoimmune diseases, HIV, osteoporosis and other complications [92]. Recently, an international team led by Sumit Chanda at Sanford Burnham Prebys Medical Discovery Institute, together with Yuen Kwok-Yung's team at the University of Hong Kong, employed a high throughput method to rapidly screen 1987 compounds from the ReFRAME library [93] that shortlisted 100 drugs that reliably hinder the virus growth by at least 40%. Further, 21 drugs were filtered based on dose-response relationship. Out of these 21 drugs, one drug was remdesivir, which is the FDA approved drug originally developed against Ebola virus, as a possible treatment option against COVID-19 [94]. Additionally, studies on the most potent drugs in the list reduced the viral load by 65% to 85%. Most potent in the list were apilimod (a drug in clinical trials to treat rheumatoid arthritis, and Crohn's disease) and clofazimine (a 70 year old FDA drug for the treatment of leprosy) (https://directorsblog.nih.gov/2020/08/04/exploring-drug-repurposing-for-covid-19-treatment/). These findings suggest the use of existing and experimental drugs has the potential to treat COVID-19. Likewise, immune-informatics tools can be employed to recognize immunological active sites in the viral genome for the purpose of developing epitope-based vaccine candidates. Such vaccines have several advantages over whole organism-based vaccines as they are safe and easy to produce [[95], [96], [97]]. Peptide-based vaccines significantly limit reactogenic and allergenic complications, and triggers stimulation of B cells and T cells or both simultaneously [96]. There are low batch to batch differences in peptide-based vaccine production and can be easily standardized [98]. The peptide structure is well known and structure-function can be easily correlated in contrast to a traditional vaccine. Furthermore, the peptide can be easily formulated to conjugated structures and multi-epitope vaccines [98]. Despite the many benefits of using peptide vaccines, they possess lower immunogenic ability which can be overcome by fusing adjuvants in peptide vaccine formulations [96]. Such peptide-based vaccines are currently being considered and hold substantial promise to prevent human viruses like hepatitis C virus and HIV [96,97]. In the work reported herein, an integrated study of computational drugs leads to identification and peptide-based vaccine ensemble design against SARS-COV-2 N protein is performed. The scientific and medical hunt for COVID-19 drugs and vaccines is in full bloom to stop the pandemic and subsequent waves of virus spread. This can be witnessed by an exponential number of COVID-19 publications in journals or in preprint archives (around 22, 000 in PubMed and 5000 preprint in BioRxiv/MedRxiv) covering SARS-COV-2 drugs and vaccines directly or indirectly [99]. Making the process of data sharing and joint global push is much needed for prioritizing drug and vaccine candidates, clinical trial streamlining. Coordinating on a regulatory process might be a prompt way of dealing with this deadly and ghastly virus. Finally, we acknowledge several limitations of this work due to lack of experimental support but nevertheless these results are promising and can save time and resources for scientific personnel involved directly in experimental therapeutics design. In this way, the translational distance between preclinical and clinical products might be reduced considerably and will aid in paving a path for rapid practical development of drugs and the much anticipated vaccine.
The following are the supplementary data related to this article.Supplementary File: S-Fig. 1. Superimposition of minimized SARS-COV-2 N protein structure (blue) over pre-minimized SARS-COV-2 N protein structure (red).
S-Fig. 2 2D structures of the top 5 compounds shortlisted after virtual screening.
S-Fig. 3. Population coverage by the selected set of epitopes.
S-Fig. 4. Cluster analysis of MHC I HLA alleles. Red color presents stronger interaction whereas yellow color stands for weaker interactions.
S-Fig. 5. Cluster analysis of MHC II HLA alleles. Red color presents stronger interaction whereas yellow color stands for weaker interactions.
S-Table 1. List of reference alleles used for prediction of epitopes.
S-Table 2. Pair of residues selected for disulfide engineering.
S-Table 3. MMGBSA binding free energies for N protein and drug molecule complex.
S-Table 4. MMPBSA binding free energies for N protein and drug molecule complex.
S-Table 5. MMGBSA binding free energies for TLR3 and vaccine complex.
S-Table 6. MMPBSA binding free energies for TLR3 and vaccine complex.
Authors contributions
Sajjad Ahmad: Conceptulatization, Data Curation, Formal Analysis, Software, Writing-Review & Editing. Yasir Waheed: Conceptualization, Funding acquisition, Supervision, Writing-review & editing. Saba Ismail: Data Curation, Formal Analysis, Methodology, Software. Sumra Wajid Abbasi: Data Curation, Formal Analysis, Methodology, Software. Muzammil Hasan Najmi: Investigation, Writing-review & editing.
Declaration of Competing Interest
The authors in this study have no conflict of interest.
Acknowledgment
Dr. Yasir Waheed is highly grateful to the Foundation University, Islamabad, for granting financial assistance to accomplish this work.
References
- 1.Zhu N., Zhang D., Wang W., Li X., Yang B., Song J., Zhao X., Huang B., Shi W., Lu R., Niu P., Zhan F., Ma X., Wang D., Xu W., Wu G., Gao G.F., Tan W. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 2020;0 doi: 10.1056/NEJMoa2001017. null. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guan W., Ni Z., Hu Y., Liang W., Ou C., He J., Liu L., Shan H., Lei C., Hui D.S.C., et al. Clinical characteristics of coronavirus disease 2019 in China. N. Engl. J. Med. 2020;382(18):1708–1720. doi: 10.1056/NEJMoa2002032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cao B., Wang Y., Wen D., Liu W., Wang J., Fan G., Ruan L., Song B., Cai Y., Wei M., et al. A trial of lopinavir--ritonavir in adults hospitalized with severe Covid-19. N. Engl. J. Med. 2020;382(19):1787–1799. doi: 10.1056/NEJMoa2001282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ahmed T., Noman M., Almatroudi A., Shahid M., Khurshid M., Tariq F., Qamar M.T. ul, Yu R., Li B. 2020. A Novel Coronavirus 2019 Linked with Pneumonia in China: Current Status and Future Prospects. [Google Scholar]
- 5.Chan J.F.-W., Yuan S., Kok K.-H., K.K.-W. To, Chu H., Yang J., Xing F., Liu J., Yip C.C.-Y., Poon R.W.-S., et al. A familial cluster of pneumonia associated with the 2019 Novel coronavirus indicating person-to-person transmission: a study of a family cluster. Lancet. 2020;395:514–523. doi: 10.1016/S0140-6736(20)30154-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gil C., Ginex T., Maestro I., Nozal V., Barrado-Gil L., Cuesta-Geijo M.A., Urquiza J., Ramírez D., Alonso C., Campillo N.E., et al. COVID-19: drug targets and potential treatments. J. Med. Chem. 2020 doi: 10.1021/acs.jmedchem.0c00606. [DOI] [PubMed] [Google Scholar]
- 7.Wang L., Wang Y., Ye D., Liu Q. A review of the 2019 novel coronavirus (COVID-19) based on current evidence. Int. J. Antimicrob. Agents. 2020;105948 doi: 10.1016/j.ijantimicag.2020.105948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li H., Liu S.-M., Yu X.-H., Tang S.-L., Tang C.-K. Coronavirus disease 2019 (COVID-19): current status and future perspective. Int. J. Antimicrob. Agents. 2020;105951 doi: 10.1016/j.ijantimicag.2020.105951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chakraborty I., Maity P. COVID-19 outbreak: migration, effects on society, global environment and prevention. Sci. Total Environ. 2020;138882 doi: 10.1016/j.scitotenv.2020.138882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stadler K., Masignani V., Eickmann M., Becker S., Abrignani S., Klenk H.-D., Rappuoli R. SARS—beginning to understand a new virus. Nat. Rev. Microbiol. 2003;1:209–218. doi: 10.1038/nrmicro775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu F., Zhao S., Yu B., Chen Y.-M., Wang W., Song Z.-G., Hu Y., Tao Z.-W., Tian J.-H., Pei Y.-Y., et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579:265–269. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chan J.F.-W., Kok K.-H., Zhu Z., Chu H., K.K.-W. To, Yuan S., Yuen K.-Y. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg. Microbes Infect. 2020;9:221–236. doi: 10.1080/22221751.2020.1719902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li G., De Clercq E. 2020. Therapeutic Options for the 2019 Novel Coronavirus (2019-nCoV) [DOI] [PubMed] [Google Scholar]
- 14.Morse J.S., Lalonde T., Xu S., Liu W.R. Learning from the Past: Possible Urgent Prevention and Treatment Options for Severe Acute Respiratory Infections Caused by 2019-nCoV. Chembiochem. 2020;21(5):730–738. doi: 10.1002/cbic.202000047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ramajayam R., Tan K.-P., Liang P.-H. 5th ed. Vol. 39. Biochem Soc Trans; 2011. Recent Development of 3C and 3CL Protease Inhibitors for Anti-Coronavirus and Anti-Picornavirus Drug Discovery; pp. 1371–1375. [DOI] [PubMed] [Google Scholar]
- 16.Wrapp D., Wang N., Corbett K.S., Goldsmith J.A., Hsieh C.-L., Abiona O., Graham B.S., McLellan J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science (80) 2020;367:1260–1263. doi: 10.1126/science.abb2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kang S., Yang M., Hong Z., Zhang L., Huang Z., Chen X., He S., Zhou Z., Zhou Z., Chen Q., et al. Crystal structure of SARS-CoV-2 nucleocapsid protein RNA binding domain reveals potential unique drug targeting sites. Acta Pharm. Sin. B. 2020;10(7):1228–1238. doi: 10.1016/j.apsb.2020.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nelson G.W., Stohlman S.A., Tahara S.M. High affinity interaction between nucleocapsid protein and leader/intergenic sequence of mouse hepatitis virus RNA. J. Gen. Virol. 2000;81:181–188. doi: 10.1099/0022-1317-81-1-181. [DOI] [PubMed] [Google Scholar]
- 19.Cong Y., Ulasli M., Schepers H., Mauthe M., V’kovski P., Kriegenburg F., Thiel V., de Haan C.A.M., Reggiori F. Nucleocapsid protein recruitment to replication-transcription complexes plays a crucial role in coronaviral life cycle. J. Virol. 2020;94 doi: 10.1128/JVI.01925-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Du L., Zhao G., Lin Y., Chan C., He Y., Jiang S., Wu C., Jin D.-Y., Yuen K.-Y., Zhou Y., et al. Priming with rAAV encoding RBD of SARS-CoV S protein and boosting with RBD-specific peptides for T cell epitopes elevated humoral and cellular immune responses against SARS-CoV infection. Vaccine. 2008;26:1644–1651. doi: 10.1016/j.vaccine.2008.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chang C., Sue S.-C., Yu T., Hsieh C.-M., Tsai C.-K., Chiang Y.-C., Lee S., Hsiao H., Wu W.-J., Chang W.-L., et al. Modular organization of SARS coronavirus nucleocapsid protein. J. Biomed. Sci. 2006;13:59–72. doi: 10.1007/s11373-005-9035-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ahmed S.F., Quadeer A.A., McKay M.R. Preliminary identification of potential vaccine targets for the COVID-19 coronavirus (SARS-CoV-2) based on SARS-CoV immunological studies. Viruses. 2020;12:254. doi: 10.3390/v12030254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu S.-J., Leng C.-H., Lien S., Chi H.-Y., Huang C.-Y., Lin C.-L., Lian W.-C., Chen C.-J., Hsieh S.-L., Chong P. Immunological characterizations of the nucleocapsid protein based SARS vaccine candidates. Vaccine. 2006;24:3100–3108. doi: 10.1016/j.vaccine.2006.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. UCSF chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 25.Case D.A., Babin V., Berryman J.T., Betz R.M., Cai Q., Cerutti D.S., Cheatham T.E., III, Darden T.A., Duke R.E., Gohlke H., et al. Vol. 14. Amber; 2014. The FF14SB force field; pp. 29–31. [Google Scholar]
- 26.Kerrigan J.E. 2009. AMBER 10.0 Introductory Tutorial. [Google Scholar]
- 27.Laskowski R.A. PDBsum: summaries and analyses of PDB structures. Nucleic Acids Res. 2001;29:221–222. doi: 10.1093/nar/29.1.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dallakyan S., Olson A.J. Chem. Biol. Springer; 2015. Small-molecule library screening by docking with PyRx; pp. 243–250. [DOI] [PubMed] [Google Scholar]
- 29.Lipinski C.A. Lead- and drug-like compounds: the rule-of-five revolution. Drug Discov. Today Technol. 2004;1:337–341. doi: 10.1016/j.ddtec.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 30.Vita R., Mahajan S., Overton J.A., Dhanda S.K., Martini S., Cantrell J.R., Wheeler D.K., Sette A., Peters B. The immune epitope database (IEDB): 2018 update. Nucleic Acids Res. 2018;47:D339–D343. doi: 10.1093/nar/gky1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Doytchinova I.A., Flower D.R. VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 2007;8:4. doi: 10.1186/1471-2105-8-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dimitrov I., Flower D.R., Doytchinova I. BMC Bioinformatics. 2013. AllerTOP-a server for in silico prediction of allergens; p. S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gupta S., Kapoor P., Chaudhary K., Gautam A., Kumar R., Raghava G.P.S., Consortium O.S.D.D., et al. In silico approach for predicting toxicity of peptides and proteins. PLoS One. 2013;8:e73957. doi: 10.1371/journal.pone.0073957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Garg A., Gupta D. VirulentPred: a SVM based prediction method for virulent proteins in bacterial pathogens. BMC Bioinform. 2008;9:62. doi: 10.1186/1471-2105-9-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dhanda S.K., Vir P., Raghava G.P.S. Designing of interferon-gamma inducing MHC class-II binders. Biol. Direct. 2013;8:30. doi: 10.1186/1745-6150-8-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bui H.-H., Sidney J., Li W., Fusseder N., Sette A. Development of an epitope conservancy analysis tool to facilitate the design of epitope-based diagnostics and vaccines. BMC Bioinform. 2007;8:361. doi: 10.1186/1471-2105-8-361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bui H.-H., Sidney J., Dinh K., Southwood S., Newman M.J., Sette A. Predicting population coverage of T-cell epitope-based diagnostics and vaccines. BMC Bioinform. 2006;7:1–5. doi: 10.1186/1471-2105-7-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cheng J., Randall A.Z., Sweredoski M.J., Baldi P. SCRATCH: a protein structure and structural feature prediction server. Nucleic Acids Res. 2005;33:W72–W76. doi: 10.1093/nar/gki396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Heo L., Park H., Seok C. GalaxyRefine: protein structure refinement driven by side-chain repacking. Nucleic Acids Res. 2013;41:W384–W388. doi: 10.1093/nar/gkt458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Colovos C., Yeates T.O. ERRAT: an empirical atom-based method for validating protein structures. Protein Sci. 1993;2:1511–1519. doi: 10.1002/pro.5560020916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Eisenberg D., Lüthy R., Bowie J.U. Methods Enzymol. Elsevier; 1997. VERIFY3D: assessment of protein models with three-dimensional profiles; pp. 396–404. [DOI] [PubMed] [Google Scholar]
- 42.Wiederstein M., Sippl M.J. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007;35:W407–W410. doi: 10.1093/nar/gkm290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.ProtParam E. 2017. ExPASy-ProtParam Tool. [Google Scholar]
- 44.Hebditch M., Carballo-Amador M.A., Charonis S., Curtis R., Warwicker J. Protein--sol: a web tool for predicting protein solubility from sequence. Bioinformatics. 2017;33:3098–3100. doi: 10.1093/bioinformatics/btx345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zambrano R., Jamroz M., Szczasiuk A., Pujols J., Kmiecik S., Ventura S. AGGRESCAN3D (A3D): server for prediction of aggregation properties of protein structures. Nucleic Acids Res. 2015;43:W306–W313. doi: 10.1093/nar/gkv359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thomsen M., Lundegaard C., Buus S., Lund O., Nielsen M. MHCcluster, a method for functional clustering of MHC molecules. Immunogenetics. 2013;65:655–665. doi: 10.1007/s00251-013-0714-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rapin N., Lund O., Castiglione F. 2012. C-Immsim 10.1 Server. [Google Scholar]
- 48.Qamar M. ul, Shokat Z., Muneer I., Ashfaq U.A., Javed H., Anwar F., Bari A., Zahid B., Saari N. Multiepitope-based subunit vaccine design and evaluation against respiratory syncytial virus using reverse vaccinology approach. Vaccines. 2020;8:288. doi: 10.3390/vaccines8020288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shey R.A., Ghogomu S.M., Esoh K.K., Nebangwa N.D., Shintouo C.M., Nongley N.F., Asa B.F., Ngale F.N., Vanhamme L., Souopgui J. In-silico design of a multi-epitope vaccine candidate against onchocerciasis and related filarial diseases. Sci. Rep. 2019;9:4409. doi: 10.1038/s41598-019-40833-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dong R., Chu Z., Yu F., Zha Y. Contriving multi-epitope subunit of vaccine for COVID-19: immunoinformatics approaches. Front. Immunol. 2020;11:1784. doi: 10.3389/fimmu.2020.01784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kumar N., Sood D., Chandra R. Design and optimization of a subunit vaccine targeting COVID-19 molecular shreds using an immunoinformatics framework. RSC Adv. 2020;10:35856–35872. doi: 10.1039/d0ra06849g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Abdelmoneim A.H., Mustafa M.I., Abdelmageed M.I., Murshed N.S., Dawoud E. dk, Ahmed E.M., Eldein R.M. Kamal, Elfadol N.M., Sati A.O.M., Makhawi A.M. Immunoinformatics design of multiepitopes peptide-based universal cancer vaccine using matrix metalloproteinase-9 protein as a target. Immunol. Med. 2020:1–18. doi: 10.1080/25785826.2020.1794165. [DOI] [PubMed] [Google Scholar]
- 53.Chaudhuri D., Datta J., Majumder S., Giri K. In silico designing of peptide based vaccine for hepatitis viruses using reverse vaccinology approach. Infect. Genet. Evol. 2020;104388 doi: 10.1016/j.meegid.2020.104388. [DOI] [PubMed] [Google Scholar]
- 54.Trott O., Olson A.J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schneidman-Duhovny D., Inbar Y., Nussinov R., Wolfson H.J. PatchDock and SymmDock: servers for rigid and symmetric docking. Nucleic Acids Res. 2005;33:W363–W367. doi: 10.1093/nar/gki481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Andrusier N., Nussinov R., Wolfson H.J. FireDock: fast interaction refinement in molecular docking. Proteins Struct. Funct. Bioinforma. 2007;69:139–159. doi: 10.1002/prot.21495. [DOI] [PubMed] [Google Scholar]
- 57.Nezafat N., Karimi Z., Eslami M., Mohkam M., Zandian S., Ghasemi Y. Designing an efficient multi-epitope peptide vaccine against Vibrio cholerae via combined immunoinformatics and protein interaction based approaches. Comput. Biol. Chem. 2016;62:82–95. doi: 10.1016/j.compbiolchem.2016.04.006. [DOI] [PubMed] [Google Scholar]
- 58.Jaiswal A.K., Tiwari S., Jamal S.B., Barh D., Azevedo V., Soares S.C. An in silico identification of common putative vaccine candidates against treponema pallidum: a reverse vaccinology and subtractive genomics based approach. Int. J. Mol. Sci. 2017;18 doi: 10.3390/ijms18020402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chauhan V., Rungta T., Goyal K., Singh M.P. Designing a multi-epitope based vaccine to combat Kaposi Sarcoma utilizing immunoinformatics approach. Sci. Rep. 2019;9:2517. doi: 10.1038/s41598-019-39299-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Enayatkhani M., Hasaniazad M., Faezi S., Guklani H., Davoodian P., Ahmadi N., Einakian M.A., Karmostaji A., Ahmadi K. Reverse vaccinology approach to design a novel multi-epitope vaccine candidate against COVID-19: an in silico study. J. Biomol. Struct. Dyn. 2020:1–19. doi: 10.1080/07391102.2020.1756411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vieira T.F., Sousa S.F. Comparing AutoDock and Vina in ligand/decoy discrimination for virtual screening. Appl. Sci. 2019;9:4538. [Google Scholar]
- 62.Sousa S.F., Fernandes P.A., Ramos M.J. Protein--ligand docking: current status and future challenges. Proteins Struct. Funct. Bioinforma. 2006;65:15–26. doi: 10.1002/prot.21082. [DOI] [PubMed] [Google Scholar]
- 63.Wang Z., Sun H., Yao X., Li D., Xu L., Li Y., Tian S., Hou T. Comprehensive evaluation of ten docking programs on a diverse set of protein--ligand complexes: the prediction accuracy of sampling power and scoring power. Phys. Chem. Chem. Phys. 2016;18:12964–12975. doi: 10.1039/c6cp01555g. [DOI] [PubMed] [Google Scholar]
- 64.Azam S.S., Abbasi S.W. Molecular docking studies for the identification of novel melatoninergic inhibitors for acetylserotonin-O-methyltransferase using different docking routines. Theor. Biol. Med. Model. 2013;10:63. doi: 10.1186/1742-4682-10-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kangueane P., Nilofer C. Protein-Protein Domain-Domain Interact. Springer; 2018. Protein-protein docking: Methods and tools; pp. 161–168. [Google Scholar]
- 66.Zhang W., Bell E.W., Yin M., Zhang Y. EDock: blind protein--ligand docking by replica-exchange Monte Carlo simulation. Aust. J. Chem. 2020;12:1–17. doi: 10.1186/s13321-020-00440-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shivanika C., Kumar D., Ragunathan V., Tiwari P., Sumitha A., et al. Molecular docking, validation, dynamics simulations, and pharmacokinetic prediction of natural compounds against the SARS-CoV-2 main-protease. J. Biomol. Struct. Dyn. 2020:1. doi: 10.1080/07391102.2020.1815584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Santos L.H.S., Ferreira R.S., Caffarena E.R. Docking Screens Drug Discov. Springer; 2019. Integrating molecular docking and molecular dynamics simulations; pp. 13–34. [DOI] [PubMed] [Google Scholar]
- 69.Genheden S., Ryde U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discovery. 2015;10:449–461. doi: 10.1517/17460441.2015.1032936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Woods C.J., Malaisree M., Michel J., Long B., McIntosh-Smith S., Mulholland A.J. Rapid decomposition and visualisation of protein-ligand binding free energies by residue and by water. Faraday Discuss. 2014;169:477–499. doi: 10.1039/c3fd00125c. [DOI] [PubMed] [Google Scholar]
- 71.Case D., Ben-Shalom I., Brozell S., Cerutti D., Cheatham T., III, Cruzeiro V., Darden T., Duke R., Ghoreishi D., Gilson M., et al. Univ. California; San Fr.: 2018. AMBER 18. (n.d.) [Google Scholar]
- 72.Wang J., Wolf R.M., Caldwell J.W., Kollman P.A., Case D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 73.Schafmeister C., Ross W.S., Romanovski V. Univ; Calif: 1995. The Leap Module of AMBER. [Google Scholar]
- 74.Kräutler V., Van Gunsteren W.F., Hünenberger P.H. A fast SHAKE algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations. J. Comput. Chem. 2001;22:501–508. [Google Scholar]
- 75.Izaguirre J.A., Catarello D.P., Wozniak J.M., Skeel R.D. Langevin stabilization of molecular dynamics. J. Chem. Phys. 2001;114:2090–2098. [Google Scholar]
- 76.Roe D.R., Cheatham T.E., III PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013;9:3084–3095. doi: 10.1021/ct400341p. [DOI] [PubMed] [Google Scholar]
- 77.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 78.Miller B.R., McGee T.D., Swails J.M., Homeyer N., Gohlke H., Roitberg A.E. MMPBSA.py: an efficient program for end-state free energy calculations. J. Chem. Theory Comput. 2012;8:3314–3321. doi: 10.1021/ct300418h. [DOI] [PubMed] [Google Scholar]
- 79.Woods C.J., Malaisree M., Hannongbua S., Mulholland A.J. A water-swap reaction coordinate for the calculation of absolute protein-ligand binding free energies. J. Chem. Phys. 2011;134 doi: 10.1063/1.3519057. [DOI] [PubMed] [Google Scholar]
- 80.Sanchez-Trincado J.L., Gomez-Perosanz M., Reche P.A. Fundamentals and methods for T-and B-cell epitope prediction. J Immunol Res. 2017;2017 doi: 10.1155/2017/2680160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Saadi M., Karkhah A., Nouri H.R. Development of a multi-epitope peptide vaccine inducing robust T cell responses against brucellosis using immunoinformatics based approaches. Infect. Genet. Evol. 2017;51:227–234. doi: 10.1016/j.meegid.2017.04.009. [DOI] [PubMed] [Google Scholar]
- 82.Tian Y., da Silva Antunes R., Sidney J., Arlehamn C.S.L., Grifoni A., Dhanda S.K., Paul S., Peters B., Weiskopf D., Sette A. A review on T cell epitopes identified using prediction and cell-mediated immune models for Mycobacterium tuberculosis and Bordetella pertussis. Front. Immunol. 2018;9 doi: 10.3389/fimmu.2018.02778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Naz A., Awan F.M., Obaid A., Muhammad S.A., Paracha R.Z., Ahmad J., Ali A. Identification of putative vaccine candidates against helicobacter pylori exploiting exoproteome and secretome: a reverse vaccinology based approach. Infect. Genet. Evol. 2015;32:280–291. doi: 10.1016/j.meegid.2015.03.027. [DOI] [PubMed] [Google Scholar]
- 84.Ul Qamar M.T., Saleem S., Ashfaq U.A., Bari A., Anwar F., Alqahtani S. Epitope-based peptide vaccine design and target site depiction against Middle East respiratory syndrome coronavirus: an immune-informatics study. J. Transl. Med. 2019;17:362. doi: 10.1186/s12967-019-2116-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhang L. Multi-epitope vaccines: a promising strategy against tumors and viral infections. Cell. Mol. Immunol. 2018;15:182. doi: 10.1038/cmi.2017.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Diamond G., Beckloff N., Weinberg A., Kisich K.O. The roles of antimicrobial peptides in innate host defense. Curr. Pharm. Des. 2009;15:2377–2392. doi: 10.2174/138161209788682325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dombkowski A.A., Sultana K.Z., Craig D.B. Protein disulfide engineering. FEBS Lett. 2014;588:206–212. doi: 10.1016/j.febslet.2013.11.024. [DOI] [PubMed] [Google Scholar]
- 88.Kaliappan S., Bombay I.I.T. 2016. UCSF Chimera-Superimposing and Morphing. [Google Scholar]
- 89.Hou T., Wang J., Li Y., Wang W. 2011. Assessing the Performance of the MM_PBSA and MM_GBSA Methods. 1. The Accuracy. pdf; pp. 69–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Qamar M.T. ul, Alqahtani S.M., Alamri M.A., Chen L.-L. Structural basis of SARS-CoV-2 3CLpro and anti-COVID-19 drug discovery from medicinal plants. J. Pharm. Anal. 2020;10(4):313–319. doi: 10.1016/j.jpha.2020.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Qamar M.T. ul, Kiran S., Ashfaq U.A., Javed M.R., Anwar F., Ali M.A., Gilani A.H. ul. Discovery of novel dengue NS2B/NS3 protease inhibitors using pharmacophore modeling and molecular docking based virtual screening of the zinc database. Int. J. Pharmacol. 2016;12:621–632. [Google Scholar]
- 92.Riva L., Yuan S., Yin X., Martin-Sancho L., Matsunaga N., Pache L., Burgstaller-Muehlbacher S., De Jesus P.D., Teriete P., Hull M.V., et al. Discovery of SARS-CoV-2 antiviral drugs through large-scale compound repurposing. Nature. 2020:1–11. doi: 10.1038/s41586-020-2577-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Janes J., Young M.E., Chen E., Rogers N.H., Burgstaller-Muehlbacher S., Hughes L.D., Love M.S., Hull M.V., Kuhen K.L., Woods A.K., et al. The ReFRAME library as a comprehensive drug repurposing library and its application to the treatment of cryptosporidiosis. Proc. Natl. Acad. Sci. 2018;115:10750–10755. doi: 10.1073/pnas.1810137115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Poduri R., Joshi G., Jagadeesh G. Drugs targeting various stages of the SARS-CoV-2 life cycle: exploring promising drugs for the treatment of Covid-19. Cell. Signal. 2020;74:109721. doi: 10.1016/j.cellsig.2020.109721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Skwarczynski M., Toth I. Peptide-based synthetic vaccines. Chem. Sci. 2016;7:842–854. doi: 10.1039/c5sc03892h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Li W., Joshi M., Singhania S., Ramsey K., Murthy A. Peptide vaccine: progress and challenges. Vaccines. 2014;2:515–536. doi: 10.3390/vaccines2030515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Malonis R.J., Lai J.R., Vergnolle O. Peptide-based vaccines: current progress and future challenges. Chem. Rev. 2019;120:3210–3229. doi: 10.1021/acs.chemrev.9b00472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Topuzoğullari M., Acar T., Arayici P.P., Uçar B., Uğurel E., Abamor E. Şefik, Arasoğlu T., BALIK D.I., Derman S. An insight into the epitope-based peptide vaccine design strategy and studies against COVID-19. Turk. J. Biol. 2020;44:215–227. doi: 10.3906/biy-2006-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Funk C.D., Laferrière C., Ardakani A. A snapshot of the global race for vaccines targeting SARS-CoV-2 and the COVID-19 pandemic. Front. Pharmacol. 2020;11:937. doi: 10.3389/fphar.2020.00937. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following are the supplementary data related to this article.Supplementary File: S-Fig. 1. Superimposition of minimized SARS-COV-2 N protein structure (blue) over pre-minimized SARS-COV-2 N protein structure (red).
S-Fig. 2 2D structures of the top 5 compounds shortlisted after virtual screening.
S-Fig. 3. Population coverage by the selected set of epitopes.
S-Fig. 4. Cluster analysis of MHC I HLA alleles. Red color presents stronger interaction whereas yellow color stands for weaker interactions.
S-Fig. 5. Cluster analysis of MHC II HLA alleles. Red color presents stronger interaction whereas yellow color stands for weaker interactions.
S-Table 1. List of reference alleles used for prediction of epitopes.
S-Table 2. Pair of residues selected for disulfide engineering.
S-Table 3. MMGBSA binding free energies for N protein and drug molecule complex.
S-Table 4. MMPBSA binding free energies for N protein and drug molecule complex.
S-Table 5. MMGBSA binding free energies for TLR3 and vaccine complex.
S-Table 6. MMPBSA binding free energies for TLR3 and vaccine complex.