

Abstract
Multiple Sclerosis (MS) is a chronic demyelinating disease of the central nervous system (CNS) in which inflammation plays a key pathological role. Recent evidences showed that systemic inflammation induces increasing cell infiltration within meninges and perivascular spaces in the brain parenchyma, triggering resident microglial and astrocytic activation. The anti‐inflammatory enzyme A20, also named TNF associated protein 3 (TNFAIP3), is considered a central gatekeeper in inflammation and peripheral immune system regulation through the inhibition of NF‐kB. The TNFAIP3 locus is genetically associated to MS and its transcripts is downregulated in blood cells in treatment‐naïve MS patients. Recently, several evidences in mouse models have led to hypothesize a function of A20 also in the CNS. Thus, here we aimed to unveil a possible contribution of A20 to the CNS human MS pathology. By immunohistochemistry/immunofluorescence and biomolecular techniques on post‐mortem brain tissue blocks obtained from control cases (CC) and progressive MS cases, we demonstrated that A20 is present in CC brain tissues in both white matter (WM) regions, mainly in few parenchymal astrocytes, and in grey matter (GM) areas, in some neuronal populations. Conversely, in MS brain tissues, we observed increased expression of A20 by perivascular infiltrating macrophages, resident‐activated astrocytes, and microglia in all the active and chronic active WM lesions. A20 was highly expressed also in the majority of active cortical lesions compared to the neighboring areas of normal‐appearing grey matter (NAGM) and control GM, particularly by activated astrocytes. We demonstrated increased A20 expression in the active MS plaques, particularly in macrophages and resident astrocytes, suggesting a key role of this molecule in chronic inflammation.
Keywords: A20/TNFAIP3, active lesions, central nervous system, inflammation, multiple sclerosis, neuropathology
The anti‐inflammatory enzyme A20, codified by the TNFAIP3 gene, is genetically associated to MS and its transcripts is down‐regulated in blood cells of treatment‐naïve MS patients. By immunohistochemistry/immunofluorescence and biomolecular techniques on post‐mortem brain tissues of progressive MS cases, we observed an increased expression of A20 in the active and chronic active white matter lesions and in the active cortical lesions, particularly in activated astrocytes. Our results suggest a key role of A20 also in chronic inflammation of the central nervous system.

Introduction
Multiple Sclerosis (MS) is one of the most common chronic‐inflammatory, immune‐related disease of the central nervous system (CNS), characterized by demyelination, glia alterations, and neuro‐axonal damage (16, 33). In this complex disorder, there is an interplay between inflammatory and neurodegenerative processes, leading to intermittent neurological disturbance followed by progressive accumulation of sensory‐motor dysfunctions and cognitive disability.
Focal inflammatory demyelinating lesions in the white matter (WM), typically called “plaques,” have been historically considered the neuropathological hallmark of MS (33, 34). In the last years, pathological and MRI studies have revealed that lesions are also located and widely extended in the grey matter (GM), especially in the cerebral cortex (9, 11, 17).
Although the etiology of MS remains poorly understood, its complex multifactorial nature involving genetic susceptibility, epigenetic and environmental factors is widely known. Among the genomic regions associated to MS in genome‐wide association studies (GWAS) (3) there is the locus containing the TNF associated protein 3 (TNFAIP3) gene, which encodes for the ubiquitin‐editing enzyme A20.
A20 is an inducible cytoplasmic protein, whose functions make it a central gatekeeper in inflammation and immunity (14). Above all, A20 is considered the most potent negative regulator of the pro‐inflammatory transcription factor NF‐kB. A20 has proved to be necessary for the homeostasis of immune cells such as B cells, T cells, dendritic cells, and macrophages (64). Recently, new functions emerged, including the control of necroptosis and inflammasome activity in immune cells (15).
An A20 dysfunction has been involved in the pathogenesis of several autoimmune and inflammatory disorders, including MS. An expanding list of common coding and noncoding single‐nucleotide polymorphism (SNPs) variants in the TNFAIP3 locus has been associated with an increased susceptibility to inflammatory disorders such as systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), psoriasis, type 1 diabetes, coeliac disease, Crohn's disease, and systemic sclerosis, (6, 10, 32, 38, 48).
A decreased level of A20 transcript has been found in blood cells of patients with MS, RA, or SLE compared to healthy controls (19, 20, 35, 49, 52, 53), and recent evidences describe an altered A20 expression also in immune cells of patients with neurodegenerative disorders such as Parkinson's disease (53). In mice, a specific A20 deletion in immune cells triggers inflammatory phenotypes mimicking those seen in the genetically associated human disorders (12, 24, 26, 31, 42, 60). Consistently, in humans the “Haplo‐insufficiency of A20” (HA20), a monogenic disease characterized by rare familial A20 mutations and a reduced A20 protein expression, leads to generalized inflammation in patients (59).
Besides the role in immune cells, NF‐kB pathway and its inhibitor A20 have a well‐known role in non‐hematogenous organ‐resident cells, particularly in the CNS, in both physiological and pathological conditions. Neurons exhibit a high constitutive activity of NF‐kB, which is involved in learning and memory and in the regulation of neurogenesis, neuritogenesis, and synaptogenesis (43, 44, 50). In contrast, in astrocytes and in microglia the NF‐kB activity is promptly induced by pro‐inflammatory molecules, accompanied by the production of a specific pattern of cytokines and chemokines and reactive nitrogen and oxygen species (58). In fact, it appears that the detrimental action of NF‐kB in CNS disorders takes place mostly in glial cells and not in neurons (2, 27, 29, 37, 41, 57). A chronic activation of NF‐kB pathway in infiltrating and resident glia CNS cells in or near active lesions has been described as an hallmark of the neurodegenerative process occurring in MS and others neurodegenerative disorders (5, 23). The regulatory mechanisms of the NF‐κB activity are not completely understood, but studies in murine models provide evidence that A20 is crucial for NF‐κB pathway regulation also in the CNS. The partial or complete germinal deficiency of A20 in mice causes spontaneous neuroinflammation in puppies with remarkable reactive microgliosis and astrogliosis (22). The selective ablation of A20 in astrocytes, but not in neurons, aggravate the outcome of experimental autoimmune encephalomyelitis (EAE) as a result of increased chemokines production and pro‐inflammatory T lymphocytes recruitment in the spinal cord, due to an increased function of NF‐kB (62). A20 deficiency in microglia determines an increase of the microglial cell number and exacerbates the EAE course, due to the uncontrolled activation of Nlrp3 inflammasome and the subsequent increased IL‐1b secretion and CNS inflammation (45, 61).
Pranski et colleague observed the expression of A20 transcripts and proteins belonging to A20 ubiquitin‐editing complex in several area and cell types of control cases post‐mortem brain tissues, such as neurons in cerebral cortex and glia‐like cells in white matter (55). Furthermore, a recent molecular and neuropathological study of cortical lesions in post‐mortem MS brain tissues has suggested that the overexpression of the A20 gene, together with HSP70, is related to the upregulation of the TNF/TNFR1‐stimulated necroptotic signaling in conditions of increased meningeal inflammation and severe and rapid disease progression (39), which could possibly suggests its role in the inhibition of survival/repair pathways. However, A20 protein expression has never been detected in brain tissues. On this basis, we aimed to perform a neuropathological characterization of A20 protein and gene expression in human healthy and pathological brain tissues, focusing on MS disease.
Materials and Methods
Human post‐mortem snap‐frozen brain tissues
For this study, 38 brain tissue blocks obtained from five control cases (CC) with no neurological diseases and 11 secondary progressive (SP) and six primary progressive (PP) MS cases were used. Demographic and clinical features are reported in Table 1. Post‐mortem brains were cut into blocks (2x2x1cm) and unfixed blocks were immediately frozen by immersing in isopentane, pre‐cooled on a bed of dry ice and stored at −75°C. Human post‐mortem brain tissues were obtained from the United Kingdom (UK) MS Tissue Bank at Imperial College via a UK prospective donor scheme with full ethical approval (08/MRE09/31). Neuropathological confirmation of the diagnosis of MS was carried out according to the International Classification of Diseases of the Nervous System criteria (www.ICDNS.org), and confounding pathologies were excluded, including Alzheimer‐like changes (56).
Table 1.
Demographic and clinical data of examined MS patients and CC, provided by the UK MS Tissue Bank. Abbreviations: F = female; M = male; CC = control case; MS = multiple sclerosis; SPMS = secondary progressive multiple sclerosis; PPMS = primary progressive multiple sclerosis.
| Case | Gender | Diagnosis | Disease length (years) | Age of death | Cause of death | Post‐mortem delay (hours) |
|---|---|---|---|---|---|---|
| C08 | F | CC | ‐ | 93 | Broncho‐pneumonia, cerebrovascular accident | 9 |
| C14 | M | CC | ‐ | 64 | Cardiac failure | 18 |
| C15 | M | CC | ‐ | 82 | ‐ | 21 |
| C22 | F | CC | ‐ | 69 | Lung cancer | 33 |
| C25 | M | CC | ‐ | 35 | Carcinoma of tongue | 22 |
| MS104 | M | SPMS | 11 | 53 | Advanced MS, urinary tract infection | 12 |
| MS122 | M | SPMS | 16 | 44 | Broncho‐pneumonia | 16 |
| MS154 | F | SPMS | 10 | 34 | Pneumonia | 12 |
| MS163 | F | SPMS | 6 | 45 | Multi‐organ failure septicemia due to urinary tract infection, MS | 28 |
| MS166 | F | SPMS | 36 | 62 | Broncho‐pneumonia, MS | 7 |
| MS187 | F | SPMS | 30 | 57 | Left ventricular failure, myocardial infarct, ischemic heart disease, sepsis, ms | 13 |
| MS197 | F | SPMS | 27 | 51 | Broncho‐pneumonia, MS | 10 |
| MS200 | F | SPMS | 19 | 44 | Urinary tract infection, sepsis, MS | 20 |
| MS207 | F | SPMS | 25 | 46 | Pneumonia, MS | 10 |
| MS230 | F | SPMS | 20 | 42 | MS | 31 |
| MS245 | M | SPMS | 26 | 64 | Broncho‐pneumonia, MS | 25 |
| MS248 | F | PPMS | 16 | 58 | C.O.P.D., multiple sclerosis | 17 |
| MS273 | M | PPMS | 31 | 61 | Urinary tract infection, septicemia, MS | 24 |
| MS325 | M | PPMS | 2 | 51 | Broncho‐pneumonia | 13 |
| MS383 | M | PPMS | 8 | 42 | Aspiration pneumonia | 17 |
| MS398 | F | PPMS | 28 | 57 | Broncho‐pneumonia, MS | 28 |
| MS473 | F | PPMS | 13 | 39 | Broncho‐pneumonia, MS | 9 |
The project has been approved by the University Bioethics Committee of the University of Turin on 28 February 2015.
Neuropathological characterization of human post‐mortem snap‐frozen brain tissue by immunohistochemistry
Air‐dried 10‐µm‐thick cryosections cut from snap‐frozen tissue blocks were characterized by immunohistochemistry (IHC) with specific antibodies. The complete list of used antibody, the working dilution and the postfixation method are reported in Table 2. Immunostaining for myelin‐oligodendrocyte glycoprotein (MOG) was performed to discriminate presence of white matter (WM) and grey matter (GM) and for the occurrence and extension of demyelinated lesions in WM (WML) and GM (GML). Immunostaining for major histocompatibility complex class II (MHC II) molecules was performed to assess the stage of lesion activity. The polyclonal anti‐A20 antibody was used to detect A20.
Table 2.
List of antibodies used for neuropathological characterization of human post‐mortem snap‐frozen brain tissue by Immunohistochemistry (IHC) and immunofluorescence (IF). Abbreviations: MOG = myelin‐oligodendrocyte glycoprotein; MHC II = major histocompatibility complex class II molecules, GFAP = glial fibrillary acidic protein; NEFH = neurofilament heavy chain; PFA = paraformaldehyde.
| Antigen | Host | Clonality | Clone | Dilution for IHC/IF | Working post‐fixation method | Code | Source | Marker |
|---|---|---|---|---|---|---|---|---|
| MOG | Rabbit | Polyclonal | ‐ | IHC 1:200 | Cold Methanol | 12690‐1‐AP | Proteintech | myelin |
| MHCII | Mouse | Monoclonal | C3/43 | IHC 1:200; IF 1:100 | IHC Ethanol; IF 4% PFA | M0775 | Dako | antigen presenting cells |
| A20 | Rabbit | Polyclonal | ‐ | IHC 1:100; IF 1:50 | 4% PFA | ab74037 | Abcam | A20 |
| CD4 | Mouse | Monoclonal | MT310 | IF 1:50 | 4% PFA | sc‐19641 | Santa Cruz Biotech. | T helper cells |
| CD8 | Mouse | Monoclonal | UCH‐T4 | IF 1:50 | 4% PFA | sc‐1181 | Santa Cruz Biotech. | cytotoxic T cells |
| CD68 | Mouse | Monoclonal | 514H12 | IF 1:50 | 4% PFA | MCA1851T | AbD Serotec | macrophages |
| CD20 | Mouse | Monoclonal | ‐ | IF 1:50 | 4% PFA | M0755 | Dako | B cells |
| GFAP | Mouse | Monoclonal | 5C10 | IF 1:100 | 4% PFA | ab190288 | Abcam | astrocytes |
| NEFH | Mouse | Monoclonal | N52.1.7 | IF 1:100 | 4% PFA | MAB9540 | Abnova | neurons |
Primary antibodies indicated in Table 2 were diluted in PBS containing 0.1% of Triton X‐100 and 10% of normal serum and incubated overnight at 4°C. Next, sections were incubated with the appropriate biotinylated secondary antibodies (Jackson ImmunoResearch Laboratories, West Grove, PA), followed by avidin‐biotin peroxidase complex (ABC; Vector Laboratories Burlingame, CA). The immune reactions were visualized with 3′3‐diaminobenzidine (DAB, Sigma, St. Louis, MO). Hematoxylin was used to counter‐stain nuclei in the slices. All the sections were sealed with DPX (Sigma, St. Louis, MO).
Control blocks contained normal WM and GM areas, while MS blocks included normal‐appearing WM (NAWM), normal‐appearing GM (NAGM), and lesions. Pre‐active (pre‐AL), active (AL), and chronic active (CAL) WM lesions (WML) and active (aGML) or inactive (iGML) GM lesions (GML) were identified based on the anti‐MHC II immunostaining according to Reynolds and colleagues (56).
The quantification of the A20 immunoreactivity in the lesions and in the normal‐appearing areas was performed on IHC images obtained by frozen brain sections deriving from 10 SPMS and 3 PPMS cases. A picture containing lesions and neighboring normal‐appearing matter was obtained for each tissue block by combining several IHC images acquired at 4X magnification with Nikon Eclipse microscope. Total coverage area, number of sections, mean and standard deviation (SD) of the section area were the following: 2.6 × 108 µm2 (8 sections, mean 3.7*107 µm2, SD 4.6 × 107) for the AL; 1.2 × 108 µm2 (12 sections, mean 1.1 × 107 µm2, SD 9.6 × 106) for the CAL; 2.8 × 108 µm2 (20 sections, mean 1.4 × 107 µm2, SD 1.6 × 107) for the matched NAWM; 1.6 × 107 µm2 (27 sections, mean 5.8 × 105 µm2, SD 8.5 × 105) for the aGML; 2.7 × 106 µm2 (9 sections, mean 3.0 × 105 µm2, SD 2.0 × 105) for the iGML; 1.3 × 107 µm2 (36 sections, mean 3.5 × 105 µm2, SD 3.2 × 105) for the NAGM.
Images were acquired and analyzed with Nikon Eclipse microscope. A20 densitometric analysis was manually performed on IHC images using ImageJ. Signal intensity, corresponded to the mean pixel intensity of the area, ranged from 0 (=white or total absent) to 255 (=black or total present). The A20 optic density (OD) in lesions area was normalized for the background by subtracting the OD value of the normal‐appearing neighboring area.
Immunofluorescence
For indirect double immunofluorescence (IF), air‐dried 10‐µm‐thick cryosections cut from snap‐frozen tissue blocks were stained with the A20‐specific polyclonal antibody and the following mouse monoclonal antibodies reported in Table 2: anti‐CD4, anti‐CD8, anti‐CD68, anti‐CD20, anti MHC II, anti‐GFAP, and anti‐NEFH. For antibodies directed against CNS markers (i.e., anti‐MHC II, anti‐GFAP, and anti‐NEFH antibodies) a final incubation with 0.05% Sudan Black B (SBB) (Sigma Aldrich) for 5 minutes a room temperature (RT) was performed to efficiently reduce the auto‐fluorescence signal of the snap‐frozen brain tissues. Staining was visualized using secondary antibodies Alexa Fluor 488 conjugated donkey anti‐mouse IgG and Alexa Fluor 555 conjugated donkey anti‐rabbit IgG (Invitrogen, Eugene, OR). Sections were sealed in ProLong Gold anti‐fade reagent with 4′,6′‐diamidino‐2‐phenylindole (DAPI, Invitrogen, Carlsbad, CA). Images were acquired using Leica TCS SP5 confocal microscope.
Total RNA extraction, retrotranscription, and gene expression analysis from human brain tissues
For RNA extraction, 50‐μm‐thick cryosections were cut from selected snap‐frozen tissue blocks from CC and MS cases. The area of interest (WM, GM, NAWM, NAGM, and relative lesions) were identified based on immunohistochemistry characterization with anti‐MOG and anti‐MHC II antibodies and manually dissected. The dissection was performed by gently incising the frozen tissue blocks with a needle, before performing the cut of the tissue block. Two or three cryosections were obtained from each tissue block, depending of the size of the area of interest.
Total RNA was extracted from the cryosections using the RNeasy Lipid Tissue Mini Kit (#74804, Qiagen, Hilden, Germany), according to the manufacturer’s instruction. Total RNA was reverse‐transcribed at final concentration of 20 ng/μL using random hexamer primers (High‐Capacity cDNA Reverse Transcription Kit, #4368814, Thermo Fisher Scientific). Gene expression analysis was performed by real‐time PCR using Applied Biosystems’ TaqMan gene expression products (Thermo Fisher Scientific) for the following genes: A20, TNFR1, TNFR2; TNF, IL1B, CXCL10, TGFb, IL10, IL6, NF‐kB, STAT3, TRAFF6, CASP3, CASP8, BCL2, and XIAP. Transcriptional expression was normalized using GA3PDH as a housekeeping gene. Expression levels of the analyzed genes were calculated by the normalized comparative cycle threshold (Ct) method (2−ΔΔCt), using the Universal Human Reference RNA (Stratagene, Santa Clara, California) for calibration.
Statistical analysis
Normality of distributions was assessed by graphical inspection and by the Shapiro–Wilk test. A20 OD was compared between lesioned and neighboring normal‐appearing areas of MS cases by means of the paired Mann–Whitney U test. Normalized OD signals of AL and CAL and of iGML and aGML were compared by the Mann–Whitney U test. The association between A20 levels and clinical and demographical variables was evaluated by regression models. Kruskal–Wallis with Dunn post hoc test was applied to compare gene expression data between CC samples and normal‐appearing or lesioned areas of MS cases. Lesioned and neighboring normal‐appearing areas of MS cases were compared by the paired Mann–Whitney U test. p‐values were adjusted for multiple comparisons using the false discovery rate (fdr) correction. Spearman correlation was evaluated between gene expression data. Statistical analyses were performed using R software 3.5.3 version (www.r‐project.org).
Results
Characterization of A20 protein expression in human post‐mortem control brain tissues
To investigate the A20 protein expression in brain, we performed IHC and double IF using A20‐specific antibodies on brain slices from 5 CC. In both WM (Figure 1A‐D) and GM (Figure 1E‐J) of CC, the IHC revealed the A20 expression in cells morphologically resembling astrocytes (Figure 1C‐C′ and I‐I′) and the double confocal IF confirmed that A20 is basally expressed by scattered GFAP‐positive (GFAP+) astrocytes (Figure 1D, J and Figure S1). In GM, the IHC also showed the A20 expression in the perinuclear area of cells morphologically resembling neurons (Figure 1G‐G′), as confirmed by double confocal IF showing a population of NEFH+ neuronal cells expressing A20 (Figure 1H and Figure S1). We did not detect A20 in MHC II+ microglial cells in either WM or GM (data not shown).
Figure 1.
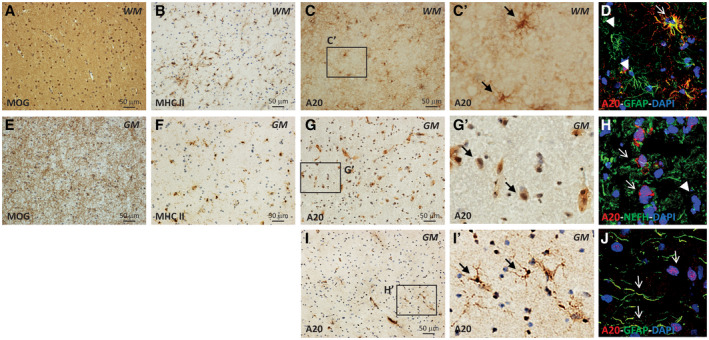
Localization of A20 expression in human post‐mortem CC brain tissues by IHC and IF. WM (A‐D) and GM (E‐J) were identified by anti‐MOG immunostaining (A, E). The presence of microgliosis were identified by anti‐MHC II immunostaining (B, F). In WM, A20 was expressed by cells with astrocyte morphology (C‐C′) expressing GFAP astrocytic marker (D). In GM, A20 was expressed by cells with neuronal morphology with a perinuclear distribution (G‐G′) expressing NEFH neuronal marker (H) and by rare cells with astrocytic morphology (I‐I′) expressing GFAP astrocytic marker (J). The inserts in (C′, G′, and I′) show A20‐positive cells in WM and GM at high magnification. In D, H, and J, the arrows indicate the colocalization of the A20 and the specific cell marker signals, while the arrowheads show the lacking of colocalization. Original confocal microscopy magnification: 40x. [Colour figure can be viewed at wileyonlinelibrary.com]
Characterization of A20 protein expression in NAWM and WML in human post‐mortem MS brain tissues
We further evaluated A20 expression in human post‐mortem brain tissues from progressive MS patients, both in normal‐appearing and demyelinated areas of WM and GM. In NAWM (Figure 2A‐E′) of MS cases, we detected A20 expression on ramified cells resembling astrocytes (Figure 2C‐C′). Double confocal microscopy analysis demonstrated that the A20+ cells were mostly GFAP+ astrocytes, although not all the astrocytes expressed A20 (Figure 2D‐D′ and Figure S2). However, differently from the CC WM, we observed A20 expressed also by few MHC II+ microglia cells (Figure 2E‐E′ and Figure S2). In the demyelinated area (Figure 2F‐M′), including pre‐AL, AL, and CAL, we detected A20 expressed by a huge number of highly ramified resident cells (Figure 2H‐H′ and L‐L′). Particularly in CAL we observed a greater number of ramified cells expressing A20 in the active plaque rim compared to few A20+ cells in the lesion core (Figure 2L‐L′). Double confocal microscopy clarified that in WML A20 is expressed by all the GFAP+ astrocytes (Figure 2I‐I′ and Figure S2) and by numerous MHC II+ microglia cells (Figure 2M‐M′ and Figure S2). In addition, we detected A20 in perivascular infiltrates in or near the active lesions (Figure 3C‐C′). Double confocal microscopy showed that A20 was not expressed by lymphocyte populations including CD4+ T helper (Th), CD8+ cytotoxic T cells, and CD20+ B cells (Figure 3D‐O′). However, it was expressed by almost all the CD68+‐activated microglia/macrophages (Figure 3P‐S′).
Figure 2.
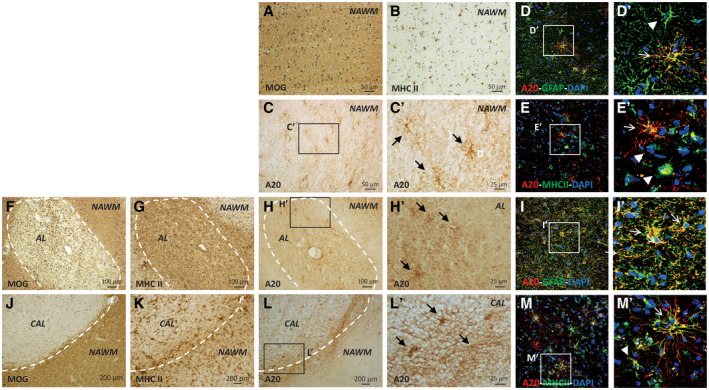
Localization of A20 protein expression in WM of human post‐mortem MS brain tissues by IHC and IF. The presence of NAWM, pre‐active (pre‐AL), active (AL), and chronic active (CAL) lesions was evaluated with MOG and MHC II immunostaining (A, B, F, G, J, K). In NAWM, A20 was expressed by rare scattered ramified cells (C, C') expressing GFAP astrocytic (D, D′) and MHC II microglial markers (E, E′). In the area of active demyelination, including AL and CAL (H, H′, L, L′), A20 was expressed by a huge number of highly ramified cells expressing GFAP astrocytic (I, I′) and MHC II microglia markers (M, M′). The inserts in (C′, D′, E′, H′, I′, L′, and M′) show A20‐positive cells in NAWM and WML at high magnification. The arrows indicate the colocalization between A20 and the cell‐specific markers (MHC II or GFAP); the arrowheads indicate the lack of colocalization. Original confocal microscopy magnification: 40x. [Colour figure can be viewed at wileyonlinelibrary.com]
Figure 3.

Characterization of A20 expression in perivascular infiltrates in human post‐mortem MS brain tissue by IHC and IF. In the area of demyelination, A20 was found in perivascular infiltrates (C, C′), not expressing CD4 T helper (D‐G′), CD8+ T cytotoxic (H‐K′), or CD20 B lymphocytes markers (L‐O′). Conversely, A20 was expressed by a high number of CD68+ macrophages infiltrating the active demyelinated lesions in WM (P‐S′). The arrows indicate the colocalization between A20 and CD68 macrophage marker, while the arrowheads indicate the lack of A20 colocalization with cell‐specific markers (CD4, CD8 or CD20). Original confocal microscopy magnification: 40x. [Colour figure can be viewed at wileyonlinelibrary.com]
Characterization of A20 protein expression in NAGM and GML in human post‐mortem MS brain tissues
In the NAGM (Figure 4A‐H), the A20 expression was observed in the perinuclear area of groups of cells morphologically resembling neurons (Figure 4C‐C′) and in rare scattered ramified cells (Figure 4G‐G′). Confocal IF analysis revealed that, similar to CC GM, in NAGM A20 is expressed in the soma of some NEFH+ neurons (Figure 4D and Figure S3) and in sporadic GFAP+ astrocytes (Figure 4H and Figure S3). On the contrary, in aGML (Figure 4I‐L) A20 expression was detected in a network of ramified cells (Figure 4K‐K′), demonstrated to be GFAP+ astrocytes by double confocal IF (Figure 4L and Figure S3). We did not observe a co‐localization of A20 and MHC II+ staining (data not shown), but this could be due to the intense A20 signal of the astrocytes network, thus, we could not exclude that A20 is also expressed by some microglia cells.
Figure 4.
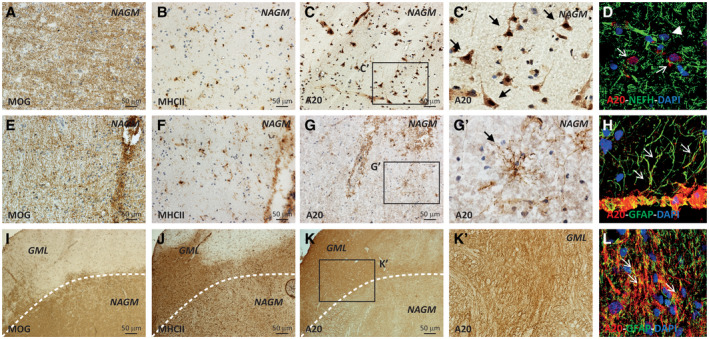
Localization of A20 protein in GM of human post‐mortem MS brain tissues by IHC and IF. The presence of active (aGML) and inactive (iGML) lesions in GM (GML) was evaluated with MOG and MHC II immunostaining (A B, E, F, I, J). In NAGM, A20 was expressed by some populations of cells with neuronal morphology, with somatic distribution (C, C′), expressing the NEFH marker (D). Furthermore, A20 was expressed by rare ramified cells resembling astrocytes (G, G′) expressing the GFAP astrocytic marker (H). Conversely, in the aGML A20 was expressed by a tick mantel of cells with astrocyte morphology (K, K′) expressing the GFAP astrocytic marker (L). The arrows indicate the colocalization between A20 and the neuronal nuclei in the NAGM (D) and the colocalization of A20 with GFAP+ astrocytes in the NAGM (H, L); the arrowheads indicate neurons that do not express A20 (D). Original confocal microscopy magnification: 40x. [Colour figure can be viewed at wileyonlinelibrary.com]
Quantification of A20 protein expression in human post‐mortem MS brain tissues
To quantify the A20 protein expression in both WML and GML, we performed a densitometric analysis on IHC images from 13 MS cases (10 SPMS and 3 PPMS). We excluded major differences in the A20 expression pattern between PPMS and SPMS by graphical inspection. When we examined all the MS cases together, for WML, we measured A20 optic density (OD) in 8 AL, 12 CAL, and 20 neighboring NAWM areas, classified based on MOG and MHC II immunostaining. For AL, we considered the entire area of the lesion, while for CAL only the active rim. A20 OD was significantly higher in both AL and CAL compared to their neighboring NAWM (paired Mann–Whitney U test, P = 0.0078 and P = 0.0005, respectively) (Figure 5A,B). Furthermore, we compared normalized OD signals of AL and CAL, obtained by subtracting the OD value of the neighboring NAWM area to that of the lesion. As a result, we did not observe significant differences (Mann–Whitney U test, P = 0.34) (Figure 5C).
Figure 5.
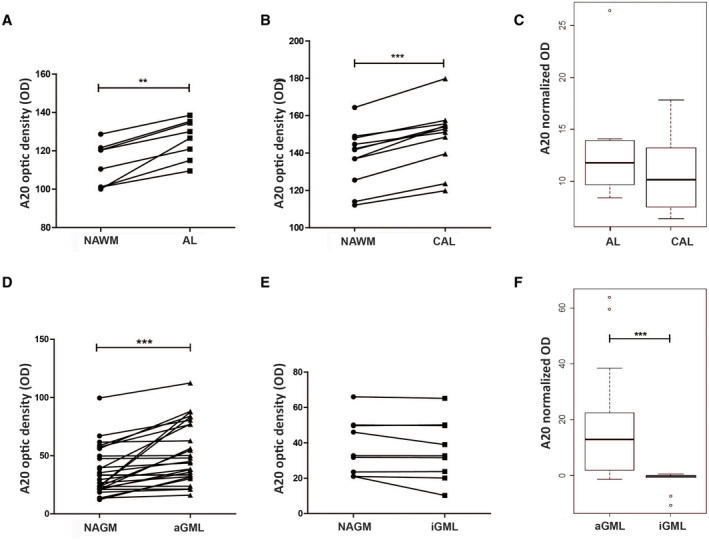
Densitometric analysis of A20 immunoreactivity measured on IHC images of human post‐mortem brain tissues of MS cases. The A20 OD was compared between NAWM and AL (A) or CAL (B) and between NAGM and aGML (D) or iGML (E) (paired Mann–Whitney U‐test, *P < 0.05, **P < 0.001, ***P < 0.0001). Normalized A20 OD signals, calculated by subtracting the OD value of the normal‐appearing area to that of the matched lesion area, were compared between AL and CAL (C) and between aGML and iGML (F) (Mann–Whitney U test, *P < 0.05, **P < 0.001, ***P < 0.0001). [Colour figure can be viewed at wileyonlinelibrary.com]
For GML, we measured A20 OD in 27 aGML, 9 iGML, and 36 neighboring NAGM areas, classified through MOG and MHC II immunostaining. We found higher A20 immunoreactivity in the aGML as compared to the closest NAGM areas (paired Mann–Whitney U test, P < 0.0001) (Figure 5D). In contrast, we did not observed significant differences between A20 immunoreactivity in iGML and neighboring NAGM (paired Mann–Whitney U test, P = 0.10) (Figure 5E). As expected, A20 normalized expression was higher in the aGML compared to iGML (Mann–Whitney U test, P < 0.0001) (Figure 5F).
Also, we did not highlight relevant correlations between A20 immunoreactivity in both WML and aGML and plaque dimension, demographical and clinical features of subjects, such as gender, MS type, disease length, age and cause of death, and post‐mortem delay (data not shown).
Gene expression analysis of A20 and related molecules in human post‐mortem CC and MS brain tissues
We further investigated the snap‐frozen brain tissues of CC and MS cases by quantifying the gene expression level of A20 (Figure 6) and other 13 genes belonging to the A20 pathways and involved in the regulation of inflammation and apoptosis (Figure S4). In particular, we analyzed 4 WM and 4 GM samples from 4 CC and 12 NAWM, 9 WML, 12 NAGM, and 12 GML samples from 10 progressive MS patients. Given the high variability between‐subjects in the gene expression data, we first compared CC samples (WM and GM) with normal‐appearing and lesioned areas of MS cases (Figure 6A,C), then, we restricted the comparison of normal‐appearing and lesioned areas of MS cases to matched samples from the same slice of brain tissue blocks (Figure 6B,D).
Figure 6.

A20 gene expression analysis in human post‐mortem brain tissues of MS and CC. The A20 expression was compared between WM of CC and NAWM and WML of MS cases (A), and between GM of CC and NAGM and aGML of MS cases (C) (Kruskal–Wallis with Dunn post hoc test, *P < 0.05, **P < 0.001, ***P < 0.0001). In addition, the A20 expression was compared between matched samples of normal‐appearing and lesioned areas of MS cases (B, D) (paired Mann–Whitney U test, *P < 0.05, **P < 0.001, ***P < 0.0001). Relative gene expression was calculated by the normalized comparative cycle threshold (Ct) method 2−ΔΔC. [Colour figure can be viewed at wileyonlinelibrary.com]
The A20 expression level was significantly higher in WML compared to both WM of CC and matched NAWM (WM vs. WML: Kruskal–Wallis with Dunn post hoc test, P = 0.048; NAWM vs. WML: paired Mann–Whitney U test, P = 0.008) (Figure 6A,B). A20 was more expressed also by GML compared to matched NAGM, although the difference was less pronounced than in the WM (paired Mann–Whitney U test, P = 0.02) (Figure 6D). On the contrary, we did not observe significant differences between NAGM or aGML and GM of CC (Kruskal–Wallis test, P = 0.27) (Figure 6C).
None of the genes belonging to the A20 pathways and involved in the regulation of inflammation and apoptosis showed significant differences among GM tissues (Figure S4). However, most of them showed differences between WM tissues. TNF receptors 1 (TNFR1) and 2 (TNFR2), whose stimulation by TNFα induces an A20 upregulation, were higher expressed in the WML compared to the matched NAWM (paired Mann–Whitney U test, P = 0.02 and P = 0.02, respectively) (Figure S4A‐B). The pro‐inflammatory cytokines TNFα and IL‐1β, both able to upregulate A20 expression through the binding to their receptors, showed higher levels in the WML compared to the neighboring matched NAWM (paired Mann–Whitney U test, P = 0.02 and P = 0.02, respectively) (Figure S4C‐D). However, we did not observe any difference in the expression of the pro‐inflammatory cytokine IL6 (Figure S4E). The anti‐inflammatory/regulatory cytokines IL10 and TGFβ were highly expressed in the WML compared to the matched NAWM (paired Mann–Whitney U test, P = 0.04 and P = 0.008, respectively) and to WM of CC, although for TGFβ this difference was not significant after adjusting for multiple comparisons (Kruskal–Wallis with Dunn post hoc test, P = 0.03 and P = 0.11, respectively) (Figure S4F‐G). Regarding the apoptotic pathways, we observed higher expression levels of the caspases CASP3 and CASP8 in the WML compared to the neighboring NAWM (paired Mann–Whitney U test, P = 0.02 and P = 0.008, respectively) and, for CASP8 only, also compared to WM of CC (Kruskal–Wallis with Dunn post hoc test, P = 0.04) (Figure S4H‐I). The anti‐apoptotic molecule XIAP, but not BCL2, showed an increased expression in WML compared to matched NAWM (paired Mann–Whitney U test, P = 0.02) (Figure S4J‐K). Finally, the pro‐inflammatory transcription factors NF‐kB and STAT3 were highly expressed in WML compared to the matched NAWM (paired Mann–Whitney U test, P = 0.02 and P = 0.03, respectively) (Figure S4L‐M). As shown in the correlation plots, A20 expression was significantly correlated with the expression level of most of the genes in its pathway, in both WML and GML (Spearman correlation coefficient, P < 0.05) (Figure S4N‐O). Notably, in WML the strongest correlation was observed between A20 and TNF‐related molecules including TNF, TNFR1, and TNFR2, the caspases CASP3 and CASP8, the chemokines IL10 and TGFβ, the transcription factors STAT3 and NF‐kB, which induce the expression of A20 itself (Spearman correlation coefficient ≥ 0.7, P < 0.001). TNFR1, IL10, and CASP8 showed the higher correlation with A20 also in the GML (Spearman correlation coefficient ≥ 0.7, P < 0.0001).
Discussion
The key role of A20 in immune system regulation and in restricting the inflammatory processes in periphery (38, 40), as well as its involvement in inflammatory and autoimmune diseases such as MS, is widely known (10, 14, 19, 20, 52, 53, 63). However, recent data suggested an involvement of A20 also in the pathophysiology of CNS, especially in mouse model of MS (1, 22, 36, 45, 51, 61, 62). In humans, the distribution of some components of the A20 ubiquitin‐editing complex has been described in post‐mortem brain tissues by Pranski and coauthors, but the study of A20 has been limited by the difficulty to find an anti‐A20 primary antibody working on this type of tissues (55). Undoubtedly, close to the identification a suitable antibody working on snap‐frozen human brain samples, the high quality of the tissues obtained by the UK tissue bank, probably due to the standardized procedure of preservation and freezing adopted, eased to detect the protein.
Here, we aimed to perform, for the first time to our knowledge, a neuropathological characterization of the A20 expression in post‐mortem brain tissues obtained from CC and MS patients, with both PP and SP form of the disease. We observed that A20 protein and transcript are expressed in brain tissues of healthy donors, both in the GM, by several neurons, and in the WM, by few parenchymal astrocytes. These data are in accordance with those of Pranski and colleagues (55), which described the presence of components of the A20 ubiquitin‐editing complex prevalently in neurons, in which NF‐kB pathway is constitutively activated and involved in cell homeostasis and survival, and more rarely in glia cells. Unlike neurons, in glia cells as well as in the immune cells, A20 expression is minimal at the steady state. Conversely, A20 rapidly increases its expression upon NF‐kB activation following stimulation with different factors including inflammatory cytokines and microbial derived products. In this way, A20 controls and restricts the NF‐kB activity by a negative feedback loop mechanism (28).
In MS brain tissues, we observed a similar pattern of A20 expression in PP and SPMS, suggesting that they share, at least partially, some common inflammatory and/or regulatory mechanisms of responses to pathological conditions. In addition, the distribution of the A20 protein in CNS resident cells of no‐lesioned area was similar to that identified in the CC. Particularly, in the NAGM A20 was expressed by several neurons in the soma, while in the NAWM it was expressed by some highly ramified astrocytes and by few microglia cells.
On the contrary, A20 showed a very different pattern of expression in the demyelinated active lesions in both WM and GM. A20 protein was highly expressed in all the WML examined. In particular, in the CAL A20 was mostly expressed by a very dense network of reactive astrocytes in the active rim of the lesions, while in the AL it was mainly expressed by infiltrating macrophages along the whole area of injury, but not by B and T lymphocytes. In AL, as in the CAL, A20 was also expressed by astrocytes characterized by massive enlargement of the cell soma and reduced proves density, a hypertrophic morphology typical of astrogliosis reaction. We also observed A20 expression in some population of microglia, but the number of A20‐positive microglia cells could be underestimated due to the massive presence of A20‐positive reactive astrocytes in the same areas. Corroborating these data, Voet and colleagues described an overexpression of the A20 transcript in WML compared to NAWM control tissue of human post‐mortem MS brain tissues (61).
Similarly to WML, in the majority of aGML a tick mantle of reactive astrocytes expressed A20 at high levels. We did not observed the A20 expression in other cell types, but, as for the WML, we cannot exclude it could have been hidden by the strong and compact A20 immunofluorescence signal in astrocytes.
To this regard, neurons present in cortical lesions were previously demonstrated to overexpress A20 in combination with TNFR1. This profile of expression was associated to a gradient of neuronal loss from the CSF/pial surface toward the inner cortical layers ( 39 ).
In addition, we were not able to detect A20 in oligodendrocytes, due to the lack of validated protocols for oligodendrocytes markers working after PFA fixation in frozen brain tissues (39).
Finally, we described the upregulation of the gene expression of several A20‐related molecules in the WML compared to NAWM, including upstream receptors inducing A20 expression such as TNFR1 and TNFR2, pro‐inflammatory cytokines such as IL‐1b and TNF‐a, the transcription factor NF‐kB and STAT3, and the pro‐apoptotic‐molecules CASP3 and CASP8. We also observed the upregulation of regulatory cytokines such us TGFβ and IL10 and anti‐apoptotic enzymes such us XIAP, suggesting that negative feedback processes might possibly occur in some active lesioned areas. However, in the GML, we did not observe significant modulation of these molecules.
The high levels of A20 expression that we observed in the macrophages infiltrating the WML is line with the well‐known regulatory role of the enzyme in the immune system and in particular in the circulating myeloid cells, where it controls cell homeostasis and activation to prevent an abnormal pro‐inflammatory response (15, 40). Macrophages are the most predominant immune cell type in inflammatory demyelinating MS lesions. It is well established that foamy macrophages primarily promote MS disease progression by internalizing myelin debris, presenting brain‐derived autoantigens, and secreting pro‐inflammatory mediators (13, 25). However, early studies show that they also promote axonal sprouting and remyelination (21, 30). This dual role can be explained by the fact that macrophages comprise several different phenotypical and functional subpopulations (46, 47), thus, further experiments are needed to clarify the role of the enzyme in these cells.
Glia cells as well display different phenotypes and functions. On the one hand, the microglia promotes neuroinflammatory and neurodegenerative events in MS by releasing inflammatory mediators. On the other hand, it supports the CNS repair through the production of neurotrophic factors and the clearance of myelin debris (4). Also astrocytes are early and active players in the lesion pathology (7, 54) and could exert both detrimental and beneficial effects (54). In active lesions, astrocytes assume a hypertrophic morphology and may sustain damage through the production of pro‐inflammatory cytokines and reactive oxygen species (7). In a later stage of lesion development, astrocytes may dampen inflammation through the secretion of BDNF and the expression of TLRs promoting neuroprotection and lesion repair (8, 18).
Contrary to the vast literature on A20 in blood peripheral cells, less is known on its role in glia cells. The completely germinal deficiency of A20 in mice causes spontaneous neuroinflammation in puppies, with remarkable reactive microgliosis and astrogliosis (22). Consistently, the selective ablation of A20 in astrocytes resulted in a more severe clinical course of the EAE, as a result of increased chemokines production and enhanced recruitment of pro‐inflammatory T lymphocytes in the spinal cord (62). The specific A20 deletion in microglia increases microglial cell number, affects microglial regulation of neuronal synaptic functions, and also exacerbates MS‐like disease (45, 61).
According to the literature, here, we demonstrated the overexpression of A20 protein and transcript in glia cells, predominantly astrocytes, in both WML and GML of MS patients. In particular, we found reactive astrocytes expressing A20 in the active margin of demyelinating lesion extending into neighboring NAWM, suggesting possible contribution to lesion development. It would be of interest to investigate which type of astrocytes expresses the enzyme through double staining for A20 and astrocytic markers as well as in vitro or in vivo experiments on mouse models of demyelination.
Conclusion
Overall, our results support the hypothesis of a specific role of A20 in activated astrocytes and microglia, close to the well know role in the myeloid cells. However, they do not allow to determine whether it is beneficial or a detrimental, since glia cells display spatial‐ and temporal‐dependent identities in inflamed CNS. A20 could be involved in an attempt to regulate inflammation, or could actively induce demyelination and cell death sustaining the chronic‐inflammatory state, according to the inflammatory stimuli. Functional studies are necessary to shed light on this debate.
The knowledge of the role of the A20 pathway in these cells and in the ongoing inflammation during the progressive stage of MS could be crucial to design and address new therapeutic approaches.
Conflict of Interest
The authors declare no conflict of interest.
Author Contributions
Study concept and design: Simona Perga, Francesca Montarolo, Antonio Bertolotto, and Roberta Magliozzi. Analysis and interpretation of data: Simona Perga, Serena Martire, and Francesca Montarolo. Drafting of the manuscript: Simona Perga. Critical revision of the manuscript: Simona Perga, Serena Martire, Francesca Montarolo, and Roberta Magliozzi. Statistical analysis: Serena Martire. Obtained funding: Simona Perga and Antonio Bertolotto. Study supervision: Antonio Bertolotto and Roberta Magliozzi.
Supporting information
Figure S1. Identification of cell types expressing A20 in human post‐mortem CC brain tissues by IHC and IF. A20 was expressed by rare astrocytes in WM and GM, as validated by double confocal immunofluorescence and confocal microscopy with GFAP astrocytic marker (A‐D′ and I‐L′). Additionally, in GM, A20 was also expressed by some population of NEFH+ neurons, with a perinuclear distribution, as shown by the colocalization of A20 with the DAPI nuclear staining (E‐H′). The inserts in (D′, H′, and L′) show A20‐positive cells in WM and GM at high magnification. Original magnification: 40X. The arrows indicate the colocalization between A20 and the cell‐specific markers; the arrowheads indicate cells that do not express A20.
{kind=link}
Figure S2. Identification of cell type expressing A20 in human post‐mortem MS brain tissues by IHC and IF. In NAWM, A20 was expressed by a subgroup of GFAP+ astrocyte (A‐D′) and MHC II+ microglia cells (E‐H′), while in the WML, A20 was expressed by all the GFAP+ astrocytes (I‐L′) and by a higher number of MHC II+ cells (M‐P′), as validated by double immunofluorescence and confocal microscopy. The inserts in (D′, H′, L′, and P′) show A20‐positive cells in NAWM and WML at high magnification. Original magnification: 40X. The arrows indicate the colocalization between A20 and the cell‐specific markers; the arrowheads indicate the lack of colocalization.
{kind=link}
Figure S3. Identification of cell type expressing A20 in human post‐mortem MS brain tissues by IHC and IF. In NAGM, A20 was expressed in the perinuclear area of some population of neurons, identified with anti‐NEFH neuronal marker (A‐D′), and by some GFAP+ astrocytes (I‐L′). In the area with aGML, A20 is expressed by neurons (E‐H′) and by a fit network of astrocytes, identified with GFAP‐specific marker (M‐P′) similarly to NAGM). The inserts in (D′, H′, L′, and P′) show A20‐positive cells in NAGM and aGML at high magnification. Original magnification: 40X. The arrows indicate the colocalization between A20 and the cell‐specific markers; the arrowheads indicate cells that do not express A20.
{kind=link}
Figure S4. Gene expression analysis of molecules in the A20 pathway: TNF receptors (A‐B), pro‐ and anti‐inflammatory cytokines (C‐G), molecules involved in the apoptosis regulation (H‐K) and pro‐inflammatory transcription factors (L‐M). The expression levels were compared between WM of CC and NAWM and WML of MS cases, and between GM of CC and NAGM and aGML of MS cases (Kruskal–Wallis with Dunn post hoc test, *P < 0.05, **P < 0.001, ***P < 0.0001). In addition, the A20 expression was compared between matched samples of normal‐appearing and lesioned areas of MS cases (paired Mann–Whitney U test, *P < 0.05, **P < 0.001, ***P < 0.0001). Relative gene expression was calculated by the normalized comparative cycle threshold (Ct) method 2−ΔΔC. Correlation plot of genes in WM (N) and GM (O) (Spearman correlation coefficient, P < 0.05).
{kind=link}
Fig S4E‐G
{kind=link}
Fig S4H‐K
{kind=link}
Fig S4L‐M
{kind=link}
Fig S4N‐O
{kind=link}
Acknowledgments
This study was funded by the Fondazione Italiana Sclerosi Multipla (grant 2014/R/14 to S.P.) and the Italian Ministry of Health (Bando Giovani Ricercatori 2010—Grant number GR‐2010‐2315964 to S.P.). The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication. Brain post‐mortem tissue samples and the associated clinical and genetic data were supplied by the United Kingdom Multiple Sclerosis Tissue Bank at Imperial College of London (https://www.imperial.ac.uk/medicine/multiple‐sclerosis‐and‐parkinsons‐tissue‐bank).
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the article and its supplementary materials.
References
- 1. Abbasi A, Forsberg K, Bischof F, Garratt AN, Déglon N (2015) The role of the ubiquitin‐editing enzyme A20 in diseases of the central nervous system and other pathological processes. Front Mol Neurosci 8:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Baiguera C, Alghisi M, Pinna A, Bellucci A, De Luca MA, Frau L et al (2012) Late‐onset Parkinsonism in NFκB/c‐Rel‐deficient mice. Brain 135:2750–2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Beecham AH, Patsopoulos NA, Xifara DK, Davis MF, Kemppinen A, Cotsapas C et al (2013) Analysis of immune‐related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat Genet 45:1353–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bogie JFJ, Stinissen P, Hendriks JJA (2014) Macrophage subsets and microglia in multiple sclerosis. Acta Neuropathol 128:191–213. [DOI] [PubMed] [Google Scholar]
- 5. Bonetti B, Stegagno C, Cannella B, Rizzuto N, Moretto G, Raine CS (1999) Activation of NF‐kappaB and c‐jun transcription factors in multiple sclerosis lesions. Implications for oligodendrocyte pathology. Am J Pathol 155:1433–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bowes J, Lawrence R, Eyre S, Panoutsopoulou K, Orozco G, Elliott KS et al (2010) Rare variation at the TNFAIP3 locus and susceptibility to rheumatoid arthritis. Hum Genet 128:627–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Brosnan CF, Raine CS (2013) The astrocyte in multiple sclerosis revisited. Glia 61:453–465. [DOI] [PubMed] [Google Scholar]
- 8. Bsibsi M, Persoon‐Deen C, Verwer RWH, Meeuwsen S, Ravid R, Van Noort JM (2006) Toll‐like receptor 3 on adult human astrocytes triggers production of neuroprotective mediators. Glia 53:688–695. [DOI] [PubMed] [Google Scholar]
- 9. Calabrese M, Filippi M, Gallo P (2010) Cortical lesions in multiple sclerosis. Nat Rev Neurol 6:438–444. [DOI] [PubMed] [Google Scholar]
- 10. Catrysse L, Vereecke L, Beyaert R, van Loo G (2014) A20 in inflammation and autoimmunity. Trends Immunol 35:22–31. [DOI] [PubMed] [Google Scholar]
- 11. Chard D, Miller D (2009) Grey matter pathology in clinically early multiple sclerosis: Evidence from magnetic resonance imaging. J Neurol Sci 282:5–11. [DOI] [PubMed] [Google Scholar]
- 12. Chu Y, Vahl JC, Kumar D, Heger K, Bertossi A, Wójtowicz E et al (2011) B cells lacking the tumor suppressor TNFAIP3/A20 display impaired differentiation and hyperactivation and cause inflammation and autoimmunity in aged mice. Blood 117:2227–2236. [DOI] [PubMed] [Google Scholar]
- 13. Chuang T‐Y, Guo Y, Seki SM, Rosen AM, Johanson DM, Mandell JW et al (2016) LRP1 expression in microglia is protective during CNS autoimmunity. Acta Neuropathol Commun 4:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Coornaert B, Carpentier I, Beyaert R (2009) A20: central gatekeeper in inflammation and immunity. J Bio Chem 284:8217–8221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Das T, Chen Z, Hendriks RW, Kool M (2018) A20/tumor necrosis factor α‐induced protein 3 in immune cells controls development of autoinflammation and autoimmunity: lessons from mouse models. Front Immunol 9:104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dutta R, Trapp BD (2006) Pathology and definition of multiple sclerosis. Rev Prat 56:1293–1298. [PubMed] [Google Scholar]
- 17. Filippi M, Rocca MA (2010) MRI and cognition in multiple sclerosis. Neurol Sci 31(Suppl. 2):S231–S234. [DOI] [PubMed] [Google Scholar]
- 18. Fulmer CG, VonDran MW, Stillman AA, Huang Y, Hempstead BL, Dreyfus CF (2014) Astrocyte‐Derived BDNF supports myelin protein synthesis after cuprizone‐induced demyelination. J Neurosci 34:8186–8196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gilli F, Lindberg RLP, Valentino P, Marnetto F, Malucchi S, Sala A et al (2010) Learning from nature: Pregnancy changes the expression of inflammation‐related genes in patients with multiple sclerosis. PLoS One 5:e8962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gilli F, Navone ND, Perga S, Marnetto F, Caldano M, Capobianco M et al (2011) Loss of braking signals during inflammation. Arch Neurol 68:1–10. [DOI] [PubMed] [Google Scholar]
- 21. Grajchen E, Hendriks JJA, Bogie JFJ (2018) The physiology of foamy phagocytes in multiple sclerosis. Acta Neuropathol Commun 6:124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Guedes RP, Csizmadia E, Moll HP, Ma A, Ferran C, da Silva CG (2014) A20 deficiency causes spontaneous neuroinflammation in mice. J Neuroinflammation 11:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gveric D, Kaltschmidt C, Cuzner ML, Newcombe J (1998) Transcription factor NF‐kappaB and inhibitor I kappaBalpha are localized in macrophages in active multiple sclerosis lesions. J Neuropathol Exp Neurol 57:168–178. [DOI] [PubMed] [Google Scholar]
- 24. Hammer GE, Turer EE, Taylor KE, Fang CJ, Advincula R, Oshima S et al (2011) Expression of A20 by dendritic cells preserves immune homeostasis and prevents colitis and spondyloarthritis. Nat Immunol 12:1184–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hendriks JJA, Teunissen CE, de Vries HE, Dijkstra CD (2005) Macrophages and neurodegeneration. Brain Res Brain Res Rev 48:185–195. [DOI] [PubMed] [Google Scholar]
- 26. Hövelmeyer N, Reissig S, Thi Xuan N, Adams‐Quack P, Lukas D, Nikolaev A et al (2011) A20 deficiency in B cells enhances B‐cell proliferation and results in the development of autoantibodies. Eur J Immunol 41:595–601. [DOI] [PubMed] [Google Scholar]
- 27. Hsiao HY, Chen YC, Chen HM, Tu PH, Chern Y (2013) A critical role of astrocyte‐mediated nuclear factor‐κB‐dependent inflammation in huntington’s disease. Hum Mol Genet 22:1826–1842. [DOI] [PubMed] [Google Scholar]
- 28. Kaltschmidt B, Kaltschmidt C (2009) NF‐ kB in the nervous system. Cold Spring Harb Perspect Biol 1:a001271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kaltschmidt B, Sparna T, Kaltschmidt C (1999) Activation of NF‐kappa B by reactive oxygen intermediates in the nervous system. Antioxid Redox Signal 1:129–144. [DOI] [PubMed] [Google Scholar]
- 30. Kigerl KA, Gensel JC, Ankeny DP, Alexander JK, Donnelly DJ, Popovich PG (2009) Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J Neurosci 29:13435–13444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kool M, van Loo G, Waelput W, De Prijck S, Muskens F, Sze M et al (2011) The ubiquitin‐editing protein a20 prevents dendritic cell activation, recognition of apoptotic cells, and systemic autoimmunity. Immunity 35:82–96. [DOI] [PubMed] [Google Scholar]
- 32. Koumakis E, Giraud M, Dieudé P, Cohignac V, Cuomo G, Airò P et al (2012) Brief report: Candidate gene study in systemic sclerosis identifies a rare and functional variant of the TNFAIP3 locus as a risk factor for polyautoimmunity. Arthritis Rheum 64:2746–2752. [DOI] [PubMed] [Google Scholar]
- 33. Lassmann H, Brück W, Lucchinetti CF (2007) The immunopathology of multiple sclerosis: An overview. Brain Pathol 17:210–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lassmann H, van Horssen J, Mahad D (2012) Progressive multiple sclerosis: pathology and pathogenesis. Nat Rev Neurol 8:647–656. [DOI] [PubMed] [Google Scholar]
- 35. Li D, Wang L, Fan Y, Song L, Guo C, Zhu F et al (2012) Down‐regulation of A20 mRNA expression in peripheral blood mononuclear cells from patients with systemic lupus erythematosus. J Clin Immunol 32:1287–1291. [DOI] [PubMed] [Google Scholar]
- 36. Liu X, He F, Pang R, Zhao D, Qiu W, Shan K et al (2014) Interleukin‐17 (IL‐17)‐induced microRNA 873 (miR‐873) contributes to the pathogenesis of experimental autoimmune encephalomyelitis by targeting A20 ubiquitin‐editing Enzyme. J Biol Chem 289:28971–28986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. van Loo G, De Lorenzi R, Schmidt H, Huth M, Mildner A, Schmidt‐Supprian M et al (2006) Inhibition of transcription factor NF‐κB in the central nervous system ameliorates autoimmune encephalomyelitis in mice. Nat Immunol 7:954–961. [DOI] [PubMed] [Google Scholar]
- 38. Ma A, Malynn BA (2012) A20: linking a complex regulator of ubiquitylation to immunity and human disease. Nat Rev Immunol 12:774–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Magliozzi R, Howell OW, Durrenberger P, Aricò E, James R, Cruciani C et al (2019) Meningeal inflammation changes the balance of TNF signalling in cortical grey matter in multiple sclerosis. J Neuroinflammation 16:259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Majumdar I, Paul J (2014) The deubiquitinase A20 in immunopathology of autoimmune diseases. Autoimmunity 47:307–319. [DOI] [PubMed] [Google Scholar]
- 41. Marcora E, Kennedy MB (2010) The Huntington’s disease mutation impairs Huntingtin’s role in the transport of NF‐κB from the synapse to the nucleus. Hum Mol Genet 19:4373–4384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Matmati M, Jacques P, Maelfait J, Verheugen E, Kool M, Sze M et al (2011) A20 (TNFAIP3) deficiency in myeloid cells triggers erosive polyarthritis resembling rheumatoid arthritis. Nat Genet 43:908–912. [DOI] [PubMed] [Google Scholar]
- 43. Methot L, Hermann R, Tang Y, Lo R, Al‐Jehani H, Jhas S et al (2013) Interaction and antagonistic roles of NF‐ B and Hes6 in the regulation of cortical neurogenesis. Mol Cell Biol 15:2797–2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mihalas AB, Araki Y, Huganir RL, Meffert MK (2013) Opposing action of nuclear factor κB and Polo‐like kinases determines a homeostatic end point for excitatory synaptic adaptation. J Neurosci 33:16490–16501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Montarolo F, Perga S, Tessarolo C, Spadaro M, Martire S, Bertolotto A (2020) TNFAIP3 deficiency affects monocytes, monocytes‐derived cells and microglia in mice. Int J Mol Sci 21:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mosser DM (2003) The many faces of macrophage activation. J Leukoc Biol 73:209–212. [DOI] [PubMed] [Google Scholar]
- 47. Mosser DM, Edwards JP (2008) Exploring the full spectrum of macrophage activation. Nat Rev Immunol 8:958–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Musone S, Taylor K, Nititham J, Chu C, Poon A, Liao W et al (2011) Sequencing of TNFAIP3 and association of variants with multiple autoimmune diseases. Genes Immun 1264:176–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Navone ND, Perga S, Martire S, Berchialla P, Malucchi S, Bertolotto A (2014) Monocytes and CD4+ T cells contribution to the under‐expression of NR4A2 and TNFAIP3 genes in patients with multiple sclerosis. J Neuroimmunol. 272:99–102. [DOI] [PubMed] [Google Scholar]
- 50. O’Mahony A, Raber J, Montano M, Foehr E, Han V, Lu S et al (2006) NF‐kappaB/Rel regulates inhibitory and excitatory neuronal function and synaptic plasticity. Mol Cell Biol 26:7283–7298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Peluffo H, Gonzalez P, Acarin L, Arís A, Beyaert R, Villaverde A et al (2013) Overexpression of the nuclear factor kappaB inhibitor A20 is neurotoxic after an excitotoxic injury to the immature rat brain. Neurol Res 35:308–319. [DOI] [PubMed] [Google Scholar]
- 52. Perga S, Martire S, Montarolo F, Giordani I, Spadaro M, Bono G et al (2018) The footprints of poly‐autoimmunity: evidence for common biological factors involved in multiple sclerosis and Hashimoto’s thyroiditis. Front Immunol 9:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Perga S, Martire S, Montarolo F, Navone ND, Calvo A, Fuda G et al (2017) A20 in multiple sclerosis and Parkinson’s disease: clue to a common dysregulation of anti‐inflammatory pathways? Neurotox Res 32:1–7. [DOI] [PubMed] [Google Scholar]
- 54. Ponath G, Park C, Pitt D (2018) The role of astrocytes in multiple sclerosis. Front Immunol 9:217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Pranski EL, Van Sanford CD, Dalal NV, Orr AL, Karmali D, Cooper DS et al (2012) Comparative distribution of protein components of the A20 ubiquitin‐editing complex in normal human brain. Neurosci Lett 520:104–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Reynolds R, Roncaroli F, Nicholas R, Radotra B, Gveric D, Howell O (2011) The neuropathological basis of clinical progression in multiple sclerosis. Acta Neuropathol 122:155–170. [DOI] [PubMed] [Google Scholar]
- 57. Sarnico I, Lanzillotta A, Benarese M, Alghisi M, Baiguera C, Battistin L et al (2009) NF‐KappaB dimers in the regulation of neuronal survival. Int Rev Neurobiol 85:351–362. [DOI] [PubMed] [Google Scholar]
- 58. Shabab T, Khanabdali R, Moghadamtousi SZ, Kadir HA, Mohan G (2017) Neuroinflammation pathways: a general review. Int J Neurosci 127:624–633. [DOI] [PubMed] [Google Scholar]
- 59. Takagi M, Ogata S, Ueno H, Yoshida K, Yeh T, Hoshino A et al (2017) Haploinsufficiency of TNFAIP3 (A20) by germline mutation is involved in autoimmune lymphoproliferative syndrome. J Allergy Clin Immunol 139:1914–1922. [DOI] [PubMed] [Google Scholar]
- 60. Tavares RM, Turer EE, Liu CL, Advincula R, Scapini P, Rhee L et al (2010) The ubiquitin modifying enzyme A20 restricts B cell survival and prevents autoimmunity. Immunity 33:181–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Voet S, Mc Guire C, Hagemeyer N, Martens A, Schroeder A, Wieghofer P et al (2018) A20 critically controls microglia activation and inhibits inflammasome‐dependent neuroinflammation. Nat Commun 9:2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wang X, Deckert M, Xuan NT, Nishanth G, Just S, Waisman A et al (2013) Astrocytic A20 ameliorates experimental autoimmune encephalomyelitis by inhibiting NF‐κB‐ and STAT1‐dependent chemokine production in astrocytes. Acta Neuropathol 126:711–724. [DOI] [PubMed] [Google Scholar]
- 63. Wertz I, Dixit V (2014) A20–a bipartite ubiquitin editing enzyme with immunoregulatory potential. Adv Exp Med Biol 809:1–12. [DOI] [PubMed] [Google Scholar]
- 64. Zhang M, Peng LL, Wang Y, Wang JS, Liu J, Liu MM et al (2016) Roles of A20 in autoimmune diseases. Immunol Res 64:337–344. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Identification of cell types expressing A20 in human post‐mortem CC brain tissues by IHC and IF. A20 was expressed by rare astrocytes in WM and GM, as validated by double confocal immunofluorescence and confocal microscopy with GFAP astrocytic marker (A‐D′ and I‐L′). Additionally, in GM, A20 was also expressed by some population of NEFH+ neurons, with a perinuclear distribution, as shown by the colocalization of A20 with the DAPI nuclear staining (E‐H′). The inserts in (D′, H′, and L′) show A20‐positive cells in WM and GM at high magnification. Original magnification: 40X. The arrows indicate the colocalization between A20 and the cell‐specific markers; the arrowheads indicate cells that do not express A20.
Figure S2. Identification of cell type expressing A20 in human post‐mortem MS brain tissues by IHC and IF. In NAWM, A20 was expressed by a subgroup of GFAP+ astrocyte (A‐D′) and MHC II+ microglia cells (E‐H′), while in the WML, A20 was expressed by all the GFAP+ astrocytes (I‐L′) and by a higher number of MHC II+ cells (M‐P′), as validated by double immunofluorescence and confocal microscopy. The inserts in (D′, H′, L′, and P′) show A20‐positive cells in NAWM and WML at high magnification. Original magnification: 40X. The arrows indicate the colocalization between A20 and the cell‐specific markers; the arrowheads indicate the lack of colocalization.
Figure S3. Identification of cell type expressing A20 in human post‐mortem MS brain tissues by IHC and IF. In NAGM, A20 was expressed in the perinuclear area of some population of neurons, identified with anti‐NEFH neuronal marker (A‐D′), and by some GFAP+ astrocytes (I‐L′). In the area with aGML, A20 is expressed by neurons (E‐H′) and by a fit network of astrocytes, identified with GFAP‐specific marker (M‐P′) similarly to NAGM). The inserts in (D′, H′, L′, and P′) show A20‐positive cells in NAGM and aGML at high magnification. Original magnification: 40X. The arrows indicate the colocalization between A20 and the cell‐specific markers; the arrowheads indicate cells that do not express A20.
Figure S4. Gene expression analysis of molecules in the A20 pathway: TNF receptors (A‐B), pro‐ and anti‐inflammatory cytokines (C‐G), molecules involved in the apoptosis regulation (H‐K) and pro‐inflammatory transcription factors (L‐M). The expression levels were compared between WM of CC and NAWM and WML of MS cases, and between GM of CC and NAGM and aGML of MS cases (Kruskal–Wallis with Dunn post hoc test, *P < 0.05, **P < 0.001, ***P < 0.0001). In addition, the A20 expression was compared between matched samples of normal‐appearing and lesioned areas of MS cases (paired Mann–Whitney U test, *P < 0.05, **P < 0.001, ***P < 0.0001). Relative gene expression was calculated by the normalized comparative cycle threshold (Ct) method 2−ΔΔC. Correlation plot of genes in WM (N) and GM (O) (Spearman correlation coefficient, P < 0.05).
Fig S4E‐G
Fig S4H‐K
Fig S4L‐M
Fig S4N‐O
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the article and its supplementary materials.