

Abstract
The paxillin family of proteins, including paxillin, Hic-5, and leupaxin, are focal adhesion adaptor/scaffolding proteins which localize to cell-matrix adhesions and are important in cell adhesion and migration of both normal and cancer cells. Historically, the role of these proteins in regulating the actin cytoskeleton through focal adhesion-mediated signaling has been well documented. However, studies in recent years have revealed additional functions in modulating the microtubule and intermediate filament cytoskeletons to affect diverse processes including cell polarization, vesicle trafficking and mechanosignaling. Expression of paxillin family proteins in stromal cells is also important in regulating tumor cell migration and invasion through non-cell autonomous effects on the extracellular matrix. Both paxillin and Hic-5 can also influence gene expression through a variety of mechanisms, while their own expression is frequently dysregulated in various cancers. Accordingly, these proteins may serve as valuable targets for novel diagnostic and treatment approaches in cancer.
1. Introduction
Cell migration and invasion are finely regulated processes that are critical in many normal physiological processes including during embryonic development, wound repair and immune surveillance (Lauffenburger and Horwitz, 1996; Pollard and Borisy, 2003; Ridley et al., 2003). However, these dynamic cell movements are also crucial in cancer progression and metastasis (Hamidi and Ivaska, 2018). Metastasis is a complex process which includes invasion of primary tumor cells through the neighboring extracellular matrix (ECM) of the tissue stroma, intravasation into the bloodstream, circulatory transport, extravasation, and distant growth of tumor cells in various organs other than the primary site (Chaffer and Weinberg, 2011). Over 90% of cancer mortality is due to metastatic tumor growth, and therefore, understanding the mechanisms by which cells migrate and invade may prove useful in predicting and preventing lethal metastatic disease.
Much of our understanding of the mechanisms underlying cell motility has come from the study of adhesion-based translocation of cells on planar, or two-dimensional (2D) ECM environments (Ridley et al., 2003). However, 2D cell migration rarely occurs in vivo. Instead, most cells, including tumor cells, typically reside within, and migrate through, a complex three-dimensional (3D) ECM, where they may utilize both adhesion-based and adhesion-independent mechanisms of translocation (Friedl and Wolf, 2003; Hamidi and Ivaska, 2018; Harunaga and Yamada, 2011; van Helvert et al., 2018; Yamada and Sixt, 2019). Furthermore, there is now compelling evidence that the organization and composition of the 3D stromal ECM performs critical structural and signaling functions in facilitating tumor cell invasion and migration that involves both cell autonomous and non-cell autonomous signaling (Bonnans et al., 2014; Conklin et al., 2011; Provenzano et al., 2006; Valkenburg et al., 2018).
The paxillin family of cytoskeletal adaptor/scaffold proteins localize to sites of cell-ECM interaction called focal adhesions and have well-established structural and signaling roles in the regulation of the actin cytoskeleton and 2D cell migration (Deakin et al., 2012b; Turner, 2000a). In this chapter, we briefly review these findings and then focus on more recent advances in our understanding of how paxillin proteins contribute to tumor cell migration and invasion in 3D environments, discuss their emerging roles in the regulation of microtubule (MT)- and intermediate filament (IF)-based functions, and how their expression in cancer-associated fibroblasts (CAFs) impacts stroma ECM deposition and mechanosignaling to indirectly influence tumor progression.
2. The paxillin family
Paxillin was first described in 1990 as a 68kDa, tyrosine-phosphorylated cytoskeletal protein that localizes to focal adhesions and interacts with another focal adhesion protein, vinculin (Brown et al., 1996; Turner, 2000a; Turner and Miller, 1994; Turner et al., 1990). Subsequently, two other proteins, Hic-5 (50kDa; also called transforming growth factor beta 1 induced transcript 1 [TGFB1i1] and androgen receptor coactivator 55kDa protein [ARA55]), and leupaxin (45kDa), have been added to the paxillin family, each with similar domain structures, cellular localization, and evolutionarily conserved sequences (Fig. 1, domain maps & interactions) (Fujimoto et al., 1999; Jacob et al., 2016; Lipsky et al., 1998; Shibanuma et al., 1994; Thomas et al., 1999a,b). All three proteins possess an N-terminal domain containing several short, amphipathic, helical leucine- and aspartate-rich LD motifs that participate in multiple protein-protein interactions and thereby facilitate their primary function as molecular adaptor/scaffold proteins (Brown and Turner, 2004; Brown et al., 1998; Tumbarello et al., 2002). The C-termini contain four highly conserved LIM domains, which are double-zinc-finger motifs involved in targeting paxillin proteins to focal adhesions and in binding additional structural and regulatory proteins (Brown et al., 1996; Smith et al., 2013). Signaling is accomplished via multiple phosphorylation sites that are distributed throughout the proteins’ lengths (Bellis et al., 1995; Brown and Turner, 2004; López-Colomé et al., 2017; Turner, 2000a; Turner and Miller, 1994). Paxillin is expressed ubiquitously (Rashid et al., 2017; Turner et al., 1990), while Hic-5 is primarily expressed in smooth muscle tissues including the vasculature (Turner et al., 1991; Yuminamochi et al., 2003). Leupaxin expression is largely restricted to leukocytes (Lipsky et al., 1998), although it has also been observed in certain smooth muscle tissues and various cancers (Deakin et al., 2012b). Due to the lack of robust evidence regarding leupaxin functions in cancer, this review will focus primarily on paxillin and Hic-5.
Fig. 1.
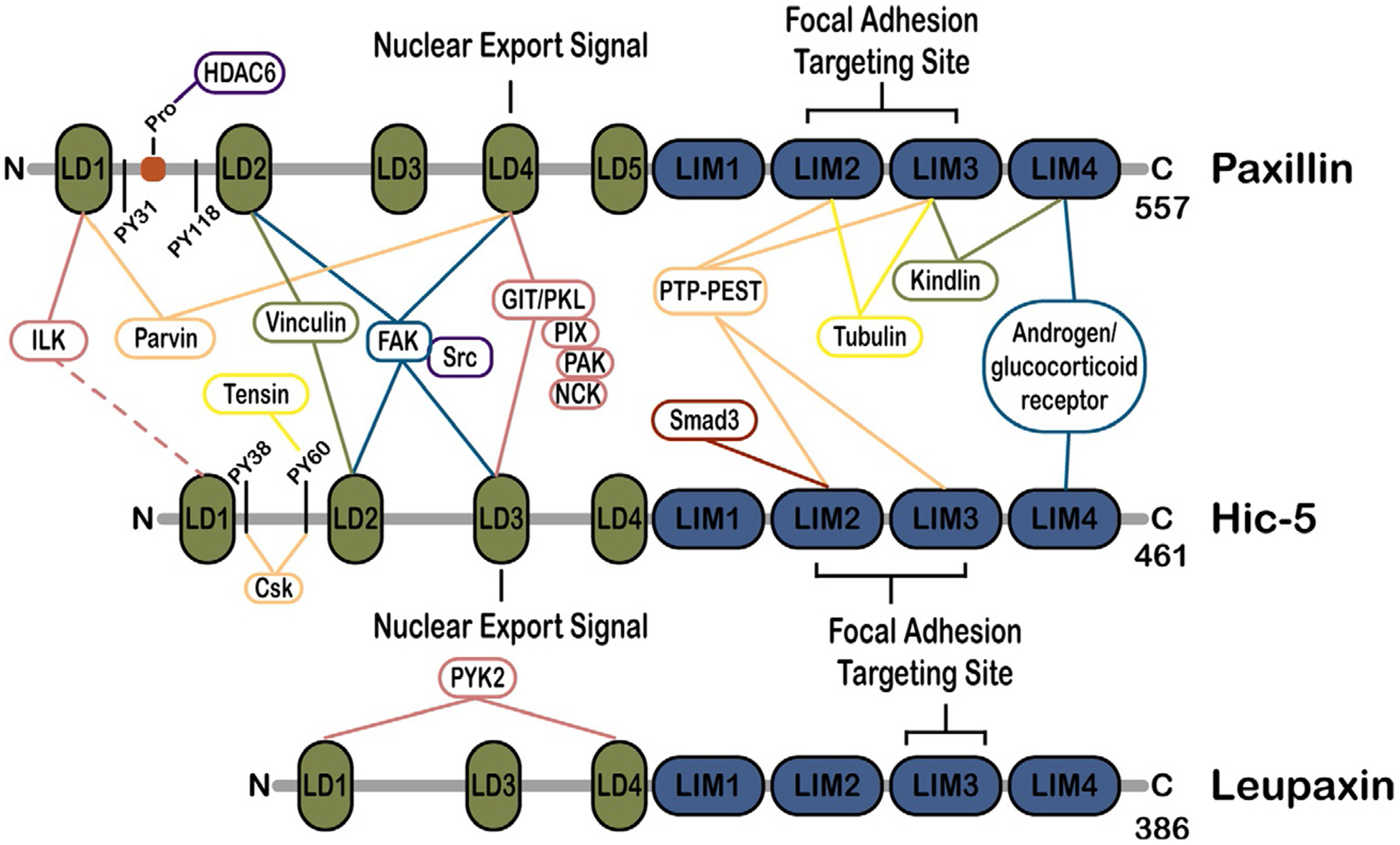
Domain structure of paxillin family members and key binding partners. The three paxillin family members, paxillin, Hic-5 and leupaxin, exhibit extensive homology within the N-terminal leucine aspartate-rich LD motifs, the C-terminal LIM domains and key phosphorylation sites. These domains interact with shared, as well as distinct, binding partners in their role as adapter/scaffold proteins to regulate focal adhesion organization and signaling to control cytoskeletal remodeling and cell migration. A selection of key binding partners is indicated.
The binding partners of, and pathways regulated by, paxillin and its family members have been studied extensively over the past thirty years and have been thoroughly discussed in several excellent reviews (Brown and Turner, 2004; Deakin and Turner, 2008; López-Colomé et al., 2017; Schaller, 2001; Turner, 2000a,b). Briefly, paxillin family members serve as molecular scaffolds/adapters to recruit downstream signaling proteins including kinases, such as focal adhesion kinase (FAK) (Brown et al., 1996; Thomas et al., 1999a,b; Turner and Miller, 1994) and extracellular signal-regulated kinase (ERK) (Ishibe et al., 2004; Ku and Meier, 2000; Subauste et al., 2004), the pseudokinase, integrin-linked kinase (ILK) (Nikolopoulos and Turner, 2001), and phosphatases, such as PTP-PEST (Brown and Turner, 2002; Côté et al., 1999; Jamieson et al., 2005). These phosphorylate or dephosphorylate the multiple serine, threonine, and tyrosine phosphorylation sites in paxillin, thereby regulating its interactions with other proteins (Bellis et al., 1995; Petit et al., 2000). Although they share many of the same binding partners (Fig. 1), there are a number of interactions that appear to be specific to individual paxillin family members. For example, paxillin binds kindlin, tubulin, and Crk (Brown and Turner, 2002; Theodosiou et al., 2016), Hic-5 binds tensin, Csk, and SMADS 3/7 (Goreczny et al., 2018; Shibanuma et al., 2004; Thomas et al., 1999a,b; Wang et al., 2008), and leupaxin binds the FAK-related Pyk2 in leukocytes (Lipsky et al., 1998).
Historically, the effects of paxillin family members on migration are primarily attributed to these phosphorylation changes, direct binding to actin-binding proteins including vinculin, talin and actopaxin/parvin (Humphries et al., 2007; Nikolopoulos and Turner, 2000; Petit et al., 2000; Theodosiou et al., 2016; Webb et al., 2004; Wood et al., 1994), and modulation of the activity of Rho GTPases family members, each of which ultimately impact actin cytoskeleton organization (Deakin and Turner, 2008; Deakin et al., 2012a; Etienne-Manneville and Hall, 2002; Tang et al., 2018). The Rho GTPases are a family of proteins that regulate actin cytoskeletal dynamics and include Cdc42, Rac1, and RhoA, among others (Sit and Manser, 2011). Their functions in 2D cell motility are well-understood (Ridley et al., 2003): Cdc42 is involved in cell polarization and filopodia formation (Faix and Rottner, 2006), Rac1 promotes the formation of broad lamellipodia (Raftopoulou and Hall, 2004), and RhoA promotes the formation of actin stress fibers and is important in the production of contractile forces (Lawson and Burridge, 2014). Both Cdc42 and Rac1 promote the formation of nascent adhesion structures, while RhoA promotes adhesion maturation at the cell’s leading edge and disassembly at the cell rear (Ridley et al., 2003; Spiering and Hodgson, 2011). Through the recruitment of GTPase activating proteins (GAPs) and guanine-nucleotide exchange factors (GEFs) such as β-PIX, DOCK180, and p190 RhoGAP, paxillin family members help maintain the delicate balance between these GTPases that is required for optimal cell migration (Deakin and Turner, 2008; Tsubouchi et al., 2002; Turner et al., 1999; Vallés et al., 2004; Zaidel-Bar et al., 2005). In addition to these canonical regulatory networks, this chapter will emphasize novel ways in which paxillin family members regulate cytoskeletal dynamics and cell motility.
3. Fundamentals of cell migration
Migration and invasion are terms with varying connotations depending on the field in which they are used. In an in vivo, 3D context, migration typically refers to any form of directed cell movement through the interstitial ECM, which is the relatively porous matrix that surrounds cells throughout the body (Kramer et al., 2013). This may require some degree of ECM proteolysis, depending on the ECM environment and the extent of adhesion to other cells and to the matrix (Schaeffer et al., 2014). In contrast, invasion is a form of migration across some sort of tissue barrier, such as the basement membrane that separates epithelia from their surrounding stroma or surround the endothelial cells in blood vessels (Bravo-Cordero et al., 2012; Petrie and Yamada, 2016).
In both physiologic and tumor microenvironments, the ECM is composed of a network of matrix proteins and glycosaminoglycans that can act as both physical barriers to migration and as tracks along which cells can migrate (Mouw et al., 2014). The interstitial matrix consists of molecules including fibronectin, fibrous collagens, elastin, and proteoglycans, which form a 3D lattice (Frantz et al., 2010). In contrast, the basement membrane is composed primarily of laminins and collagen IV, which form a sheet-like layer between epithelial and stromal cells (Frantz et al., 2010). As an epithelial tumor forms within the normal tissue stroma, cancer cells can migrate outward through the adjacent tissue stroma as single cells or as collective groups (De Pascalis and Etienne-Manneville, 2017). Cells that invade collectively maintain their cell-cell adhesions, including cadherin-based adherens junctions, tight junctions, and desmosomes (Friedl and Alexander, 2011). Generally, mesenchymal or mesenchymal-like leader cells will use proteases to degrade and remodel the matrix, therefore forming paths for subsequent cells to follow (Friedl and Wolf, 2003). Through E-cadherin-dependent cell-cell adhesions, they pull follower cells out from the tumor (Padmanaban et al., 2019; Theveneau and Linker, 2017).
Single-cell migration in 3D can occur through two distinct phenotypes and mechanisms described as either amoeboid or mesenchymal (Condeelis and Segall, 2003; Sahai and Marshall, 2003; te Boekhorst and Friedl, 2016). Cells undergoing amoeboid migration are highly deformable and roughly spherical morphologically with leading lamella and few or no cell-matrix adhesions (Friedl and Wolf, 2003; Petrie and Yamada, 2012). Amoeboid migration requires RhoA-mediated cell contractility which drives cells to move via propulsive blebs and by forcing membrane protrusions through small, pre-existing gaps in the matrix (Friedl and Alexander, 2011; Sahai and Marshall, 2003). Therefore, amoeboid cells generally do not need to degrade the ECM in order to translocate (Lämmermann and Sixt, 2009). In contrast, cells undergoing 3D mesenchymal migration are elongated with small Rac-1-driven lamellipodia (Carragher et al., 2006; Lawson and Ridley, 2018; Sanz-Moreno et al., 2008). They form robust adhesions with the ECM and degrade small paths through it using proteolytic enzymes known as matrix metalloproteinases (MMPs), that are both secreted and membrane-bound (te Boekhorst and Friedl, 2016).
Remarkably, some cancer cells can switch frequently between amoeboid and mesenchymal modes of migration in a process known as plasticity, which is important in effectively invading through a variety of microenvironments (Petrie and Yamada, 2016). It has been hypothesized that this migratory plasticity is one of the primary methods by which certain cancer cell types are able to overcome therapies that target MMPs, which are required for effective mesenchymal migration (Friedl and Wolf, 2003). For a cell to transition from mesenchymal to amoeboid migration, it needs to disassemble its adhesions to the matrix and, reciprocally, an amoeboid cell must assemble and stabilize matrix adhesions to begin mesenchymal migration.
Since most migration in vivo occurs in 3D, various tools have been developed over the past several decades to study cell migration in 3D environments and to better understand the mechanisms that regulate these phenotypes (Cukierman et al., 2001). These methods range from simple 3D culture systems, such as in vitro collagen matrices, to highly sophisticated intravital imaging of labeled bioluminescent tumor cells (Condeelis and Weissleder, 2010; Shamir and Ewald, 2014). Many of the in vitro studies described below utilize 3D cell-derived matrices (CDMs), which are generated by culturing fibroblasts at high density for 1–2 weeks (Beacham et al., 2006). The fibroblasts secrete and remodel a matrix that is composed primarily of collagen and fibronectin fibers, similar to the matrix composition found at the interface between a tumor and surrounding normal stroma in vivo (Harunaga and Yamada, 2011). The ways by which members of the paxillin family of adapter proteins influence these various aspects of tumor cell invasion, migration, and stromal matrix remodeling will be discussed below.
4. Roles of paxillin and Hic-5 in regulating migration and invasion
4.1. Actin cytoskeleton-mediated effects
4.1.1. Cell plasticity and 3D migration phenotypes
MDA-MB-231 human breast cancer cells are highly invasive and metastatic in vivo. These cells exhibit plasticity in their 3D migration phenotypes and can invade by both mesenchymal and amoeboid modes, making them a useful tool for studying these processes (Deakin and Turner, 2011; Wolf et al., 2003). Paxillin and Hic-5 are both critical for optimal MDA-MB-231 cell migration and invasion in 3D (Fig. 2, amoeboid/mesenchymal migration) (Deakin and Turner, 2011; Gulvady et al., 2018). Furthermore, both proteins are required for MDA-MB-231 transendothelial migration in vitro and depletion of either protein significantly reduces the number of lung metastases in a mouse model of metastasis (Deakin and Turner, 2011). These data suggest that expression of both paxillin and Hic-5 is necessary for plasticity in vivo, which then enables effective lymphovascular invasion and the establishment of metastatic colonies.
Fig. 2.
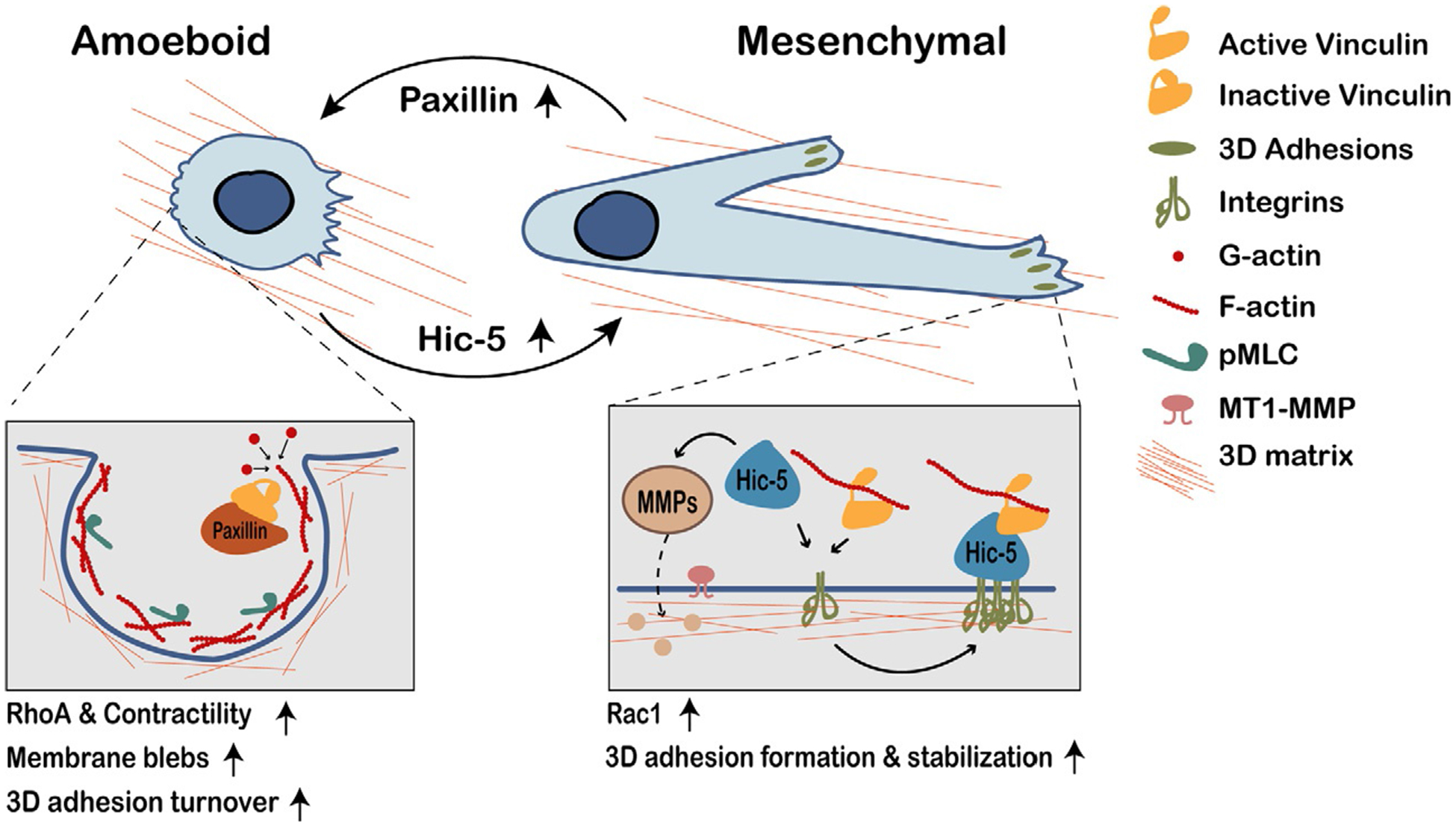
Paxillin and Hic-5 play opposing roles in regulating tumor cell 3D migration phenotype. Tumor cells with high ratios of paxillin:Hic-5 expression (low Hic-5 levels, since paxillin expression is fairly consistent among tumor cells) more commonly exhibit amoeboid migration, which requires RhoA-mediated contractility. These cells have limited, or no 3D adhesions; any adhesions that form tend to disassemble rapidly. Paxillin may sequester inactive vinculin to promote adhesion disassembly. Amoeboid cells migrate by squeezing through existing spaces in the ECM via F-actin-rich plasma membrane blebs. Conversely, tumor cells with low paxillin:Hic-5 ratios (high Hic-5 levels) can exhibit plasticity and are more likely to exhibit mesenchymal migration, which is Rac1-mediated. These cells form relatively robust, stable 3D adhesions, and use MMPs to degrade the ECM. In these cells, Hic-5 binds active vinculin to promote 3D adhesion formation and stabilization. It is unclear precisely how paxillin versus Hic-5 “activity” is regulated to promote dynamic migration plasticity.
Interestingly, while paxillin and Hic-5 co-ordinately regulate single cell 3D migration in vitro, they have dramatically opposing roles in dictating the migration phenotype utilized, which suggests that their relative endogenous expression levels, and/or their respective signaling activity, could play an important role in determining how tumor cells migrate in vivo (Deakin and Turner, 2011; Gulvady et al., 2018). Specifically, when MDA-MB-231 cells are seeded in a 3D matrix, acute knockdown of paxillin or overexpression of Hic-5 results in a hypermesenchymal phenotype, characterized by an exaggerated, elongated cell shape and robust, stable focal adhesions (Deakin and Turner, 2011). These robust adhesions take more time to assemble and disassemble, resulting in slower cell motility. Conversely, Hic-5 knockdown or paxillin overexpression results in cells exhibiting an amoeboid phenotype (Deakin and Turner, 2011; Gulvady et al., 2018). These stark differences in phenotypes are not recapitulated when the cells are plated on a 2D ECM, likely due in part to the fact that amoeboid movement is not observed in 2D cell culture. Instead, cells are well-spread with robust focal adhesions regardless of Hic-5 or paxillin expression levels, thus further emphasizing the importance of cellular context (Deakin and Turner, 2011; Gulvady et al., 2018).
Since paxillin and Hic-5 have highly homologous protein structures, including conserved LD motifs, LIM domains, and key phosphorylation sites, it is remarkable that they play such opposing roles in regulating 3D migration phenotypes (Deakin and Turner, 2011). Many factors could contribute to these differing roles, including differences in spatiotemporal localization, binding affinities for effectors, proteolysis, rates of autophagy of focal adhesions, regulation by kinases, and regulation of Rho GTPases (Cortesio et al., 2011; Deakin et al., 2012a; Nishiya et al., 2001; Sharifi et al., 2016, 2017). For example, tyrosine-phosphorylated paxillin binds the adapter protein Crk to promote migration in bladder carcinoma cells, while Hic-5 is unable to interact with Crk (Petit et al., 2000; Thomas et al., 1999a,b), but selectively interacts with the Src-inhibitory kinase, Csk (Thomas et al., 1999a,b). Importantly, the Rho GTPases play a key role in modulating 3D migration phenotypes: amoeboid migration requires elevated RhoA activity and mesenchymal migration requires Rac1 activity (Friedl et al., 2014). Numerous studies describe roles for both paxillin and Hic-5 in regulating Rho GTPase activity and their differing affinities for binding partners could affect the balance of this signaling. For example, it is known that paxillin and Hic-5 have different affinities for the GTPase activating protein GIT1 (Nishiya et al., 2002). Studies in 2D have shown that paxillin binds GIT1/2 through its LD4 motif, and the associated GIT-PIX-PAK-NCK protein complex regulates local Rac1 activity (Loo et al., 2004; Nayal et al., 2006; Turner et al., 1999; West et al., 2001). Other factors that may contribute to these opposing roles will be discussed in the following paragraphs.
Hic-5 knockdown in MDA-MB-231 cells promotes an amoeboid phenotype through increased activity of RhoA/ROCK and non-muscle myosin II (Deakin and Turner, 2011), which are critical for amoeboid motility due to their contribution to membrane blebbing, retraction, and contractility (Sahai and Marshall, 2003; Yoshida and Soldati, 2006). However, excessive RhoA and myosin activity, without balanced Rac1 activity, may result in dysregulation of blebbing and contractile dynamics that result in the decreases in migration velocity and invasion observed when MDA-MB-231 cells lacking Hic-5 are plated on 3D CDMs (Deakin and Turner, 2011; Ridley, 2015). It is important to note that Hic-5’s effect on contractility has not always been consistent in the literature and varies between cell types. For example, it inhibits actomyosin contractility in smooth muscle and osteoblast-like cells by relocating from adhesion sites to actin stress fibers (Guignandon et al., 2006; Kim-Kaneyama et al., 2005), but also stimulates RhoA activity, stress fiber formation, and cell migration upon transforming growth factor beta (TGF-β)-induced epithelial-mesenchymal transition (Tumbarello and Turner, 2007). Therefore, Hic-5’s role in modulating RhoA activity is likely cell- and context-dependent. It is also important to note that, despite paxillin’s structural homology with Hic-5 and its role as one of the first proteins recruited to focal adhesions, paxillin is not sufficient to nucleate and stabilize 3D adhesions in MDA-MB-231 cells in the absence of Hic-5 (Deakin and Turner, 2011). Cells with an amoeboid morphology that cannot form adhesions and switch their 3D migration phenotype when the microenvironment favors mesenchymal motility are less able to effectively invade (Petrie and Yamada, 2012).
Conversely, the hypermesenchymal phenotype, caused by paxillin depletion in MDA-MB-231 cells, is associated with a significant increase Rac1 activity, which may impair migration persistence by promoting increased lateral protrusions that do not contribute to directed migration (Deakin and Turner, 2011). Paxillin knockdown also inhibits non-muscle myosin II activity, thereby impairing the actomyosin contractility crucial for amoeboid migration; contractility is important for releasing cell adhesions at the cell rear, so diminished contractility may promote a hypermesenchymal phenotype, partially through this failure of adhesion disassembly (Parsons et al., 2010). In a 2D ECM environment, paxillin has been shown to regulate only adhesion disassembly rates (Webb et al., 2004), but in 3D, paxillin depletion decreases both adhesion assembly and disassembly rates and increases the frequency of both short-lived, dynamic adhesions and stable, elongated, long-lasting adhesions (Deakin and Turner, 2011). This difference may be due in part to the fact that cell-matrix adhesions in 3D exhibit different functions, composition, and localization than those classically observed in 2D (Cukierman et al., 2001). With more stable, long-lasting adhesions and slower adhesion disassembly, cell motility is slowed because cells remain tethered more tightly to the matrix (Liu et al., 2015). These changes in adhesion dynamics are potentially due to significantly diminished levels of FAK activity following paxillin depletion (as measured by FAK Y397 phosphorylation) (Deakin and Turner, 2011), since FAK promotes both adhesion maturation and disassembly in 2D (Dumbauld et al., 2010; Webb et al., 2004). Interestingly, FAK Y397 phosphorylation is absent in the 3D adhesions formed by fibroblasts in CDMs (Cukierman et al., 2001) and FAK phosphorylation levels in epithelial cells are dictated by the compliance of the surrounding stromal matrix (Provenzano and Keely, 2011).
Consistent with the experimental manipulation of paxillin and Hic-5 levels described above, the endogenous Hic-5 expression level in tumor cells is indeed a reliable indicator of the cancer cells’ migratory phenotype in 3D CDMs, and could therefore be used to predict how cells migrate in tumors in vivo (Gulvady et al., 2018). For example, in a panel of cancer cell lines derived from melanoma, fibrosarcoma, breast and pancreatic cancers, increased ratios of Hic-5-to-paxillin expression correlated with a more mesenchymal phenotype, as well as increased migration velocity, in vitro invasiveness, and plasticity (Gulvady et al., 2018). Additionally, ectopic overexpression of Hic-5 in the cells with low endogenous levels (e.g., A375MEA-3 and A375P melanoma cells) increased the plasticity and the percentage of cells exhibiting a mesenchymal phenotype and stimulated increased migration through 3D matrices. Interestingly, paxillin expression was relatively high and constant in the same panel of cells and thus was not a good indicator of their 3D phenotype (Gulvady et al., 2018). Nevertheless, this may indicate the absolute requirement for paxillin in tumor cells, possibly for functions unrelated to adhesion and motility. Therefore, Hic-5 expression is a primary determinant of cell morphology and migratory phenotype. Accordingly, Hic-5 levels could potentially be used to predict the predominant mode of motility in patient tumor samples and thereby guide treatment choices to best reduce cell invasion and metastasis. For example, cells from tumors with low Hic-5 expression may favor amoeboid migration and therefore be more resistant to MMP-targeting drugs such as andecaliximab, which has been used in clinical trials for treatment of several cancers (Winer et al., 2018).
Hic-5’s role in promoting focal adhesion formation and a 3D mesenchymal phenotype depends on vinculin, another focal adhesion and actin-binding protein and direct binding partner of both Hic-5 and paxillin (Deakin et al., 2012a; Turner et al., 1990) (Fig. 2). Vinculin is well known for its role in mechanosensing/mechanotransduction and in 2D adhesion maturation and importantly, vinculin-null fibroblasts also exhibit a rounded, amoeboid morphology in 3D, which suggests that it may cooperate with Hic-5 to enable mesenchymal migration (Dumbauld et al., 2013; Thievessen et al., 2015). Indeed, experiments with vinculin mutants showed that ectopic expression of vinculin A50I, an “inactive” mutant, in MDA-MB-231 cells phenocopies Hic-5 knockdown, while expression of vinculin T12, a constitutively active vinculin mutant, promotes a mesenchymal phenotype (Gulvady et al., 2018). Importantly, co-expression of vinculin A50I with Hic-5 in cells with low endogenous Hic-5 expression (A375MEA-3 melanoma cells) blocks Hic-5 rescue of a mesenchymal phenotype (Gulvady et al., 2018). These data imply that both Hic-5 and active vinculin are required for driving a mesenchymal phenotype in 3D. Importantly, although Hic-5 does not interact with vinculin A50I at the plasma membrane, paxillin is known to do so and could therefore play a role in keeping vinculin, as well as talin, inactive at the membrane (Atherton et al., 2020; Deakin et al., 2012a). This could partially explain paxillin’s ability to promote adhesion disassembly and an amoeboid morphology and to antagonize the activity of Hic-5 (Fig. 2).
As previously described, Rac1 activity is important for 3D mesenchymal migration and Hic-5’s role in mesenchymal migration may be partially mediated through Rac1 (Gulvady et al., 2018; Pankov et al., 2005; Petrie et al., 2012; Sanz-Moreno et al., 2008). Treatment of A375MEA-3 cells overexpressing Hic-5 with a Rac1 inhibitor prevents Hic-5-mediated induction of a mesenchymal phenotype, further connecting these proteins in mesenchymal migration (Gulvady et al., 2018). Additionally, Rac1 and RhoA activity both influence the binding preference of vinculin for Hic-5 versus paxillin in either 2D or 3D ECMs, suggesting that Rho GTPase activity and competition between Hic-5 and paxillin for vinculin binding may be a major mechanism regulating tumor cell migration and plasticity (Deakin et al., 2012a).
4.1.2. Invadopodia and matrix degradation
Cells undergoing mesenchymal migration degrade the ECM by several methods. Expression of membrane-bound membrane-type 1 MMP (MT1-MMP/MMP14) and of secreted MMP1, MMP9, MMP10, MMP11, and MMP13 is often upregulated in almost every cancer type and plays a key role in degrading the ECM, basement membranes, and vascular basal lamina to promote invasion and metastasis (Gobin et al., 2019). Hic-5 has been implicated in regulation of several of these proteins, which will be discussed in later sections. Briefly, Hic-5 is known to promote MT1-MMP localization to the membrane in both endothelial cells and fibroblasts (Dave et al., 2016; Petropoulos et al., 2016) and to promote MMP-9 expression in several cancer lines (Mori et al., 2019).
Cancer cells also utilize specialized adhesion structures known as invadopodia, which are actin-rich membrane protrusions that exhibit abundant MMP localization and activity, including MT1-MMP (Eddy et al., 2017; Linder, 2009; Murphy and Courtneidge, 2011). Although invadopodia have primarily been studied in vitro, intravital imaging and careful immunohistochemical staining provide evidence of their existence and importance for directed ECM degradation in vivo, where they have been observed in close proximity to areas of basement membrane degradation and at sites of tumor cell extravasation through the endothelium (Leong et al., 2014; Lohmer et al., 2014). Broadly speaking, individual invadopodia consist of a core of F-actin and actin regulatory and binding proteins, which are often surrounded by a ring of adhesion-associated proteins, including integrins, and various Rho GTPase family members (Eddy et al., 2017). The tyrosine kinase Src is particularly important in invadopodia formation (Chen et al., 1985), while cortactin, TKS4, and TKS5 play key roles in modulating their maturation and activity (Buschman et al., 2009; Oser et al., 2009; Sharma et al., 2013). In 2D, invadopodia (and related structures such as podosomes) are visualized as small, actin-rich puncta that colocalize with areas of matrix degradation. However, invadopodia can also self-assemble in 2D culture to form large superstructures known as rosettes which can degrade much larger areas of matrix (Linder, 2007). Both paxillin and Hic-5 localize to invadopodia and regulate their dynamics (Badowski et al., 2008; Gulvady et al., 2019; Petropoulos et al., 2016; Pignatelli et al., 2012b).
Paxillin has been shown to regulate invadopodia dynamics in Rous sarcoma virus (RSV)-transformed baby hamster kidney (BHK) cells and osteoclasts (Badowski et al., 2008). Tyrosine-phosphorylated paxillin is enriched at the inner rim of rosettes, and overexpression of a non-phosphorylatable paxillin mutant (Y31,118F) impaired disassembly of invadopodia actin cores (Badowski et al., 2008). This phenotype can be recapitulated using inhibitors of either calpain or ERK. Paxillin tyrosine phosphorylation activates Erk, which then activates calpain, a protease that promotes both adhesion and invadopodium disassembly through proteolysis (Boateng and Huttenlocher, 2012; Calle et al., 2006; Cortesio et al., 2011).
Although Hic-5 is not expressed in most epithelial cells, it promotes the formation of invadopodia following its upregulation during TGF-β-induced epithelial-mesenchymal transition (EMT) or ectopic overexpression in MCF10A normal mammary epithelial cells (Fig. 3A, invadopodia) (Pignatelli et al., 2012b). This is particularly relevant considering Hic-5’s previously discussed role in promoting 3D mesenchymal migration, where cells must degrade the basement membrane in order to invade through the tumor stroma. Hic-5 tyrosine phosphorylation at Y38 and Y60 by Src is critical in this system, as overexpression of the Y38/Y60F non-phosphorylatable mutant or treatment with a Src inhibitor both prevent formation of invadopodia. Downstream signaling from Hic-5 requires RhoC activity for efficient invadopodia formation and p38 MAPK signaling (through Rac1) for matrix degradation, cell migration, and invasion (Fig. 3B) (Pignatelli et al., 2012b).
Fig. 3.
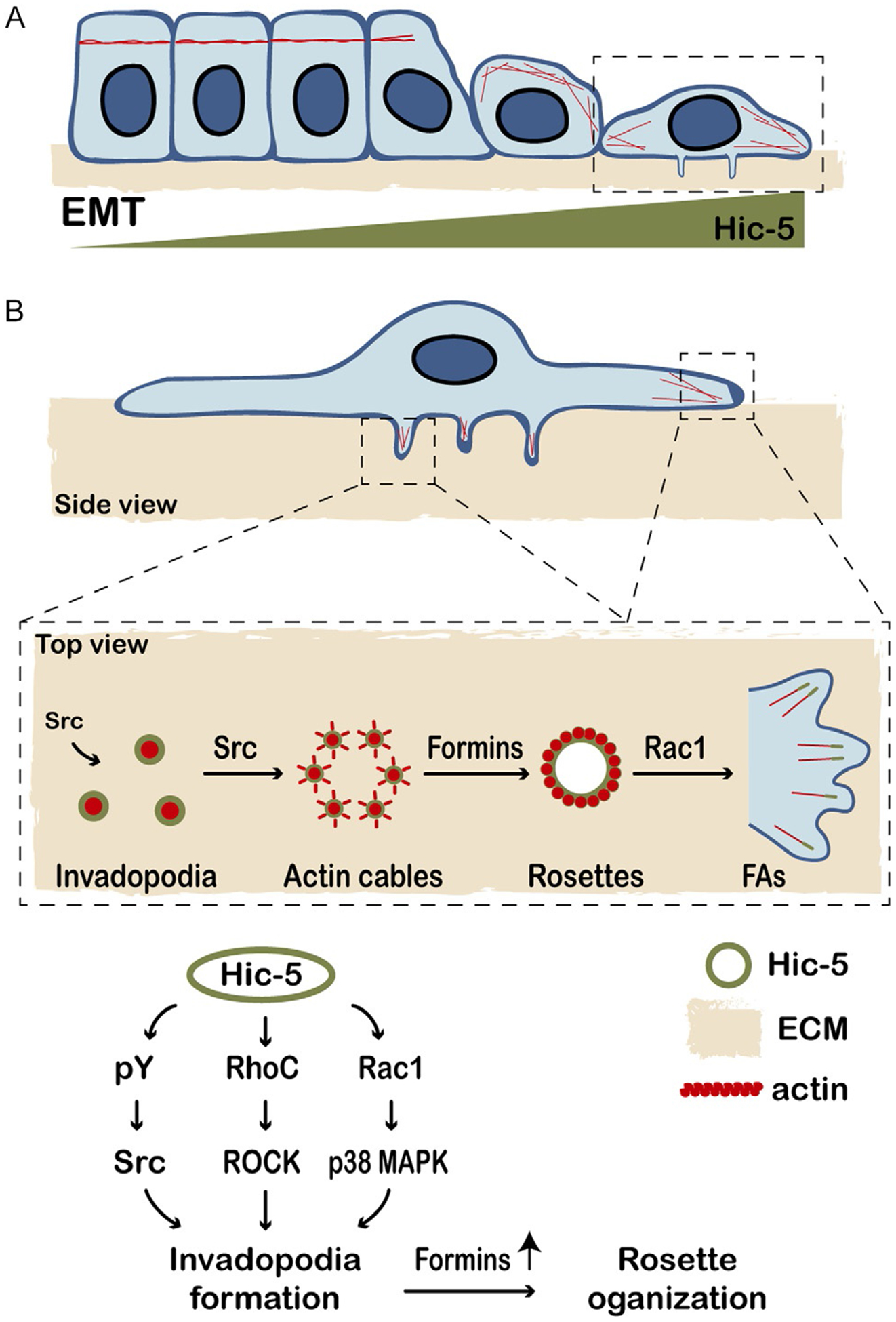
Hic-5 contributes to the formation and dynamics of invadopodia and higher order rosette structures. (A) Hic-5 is upregulated during TGF-β-induced epithelial-mesenchymal transition (EMT) to promote RhoA/ROCK-mediated actin stress fiber ormation. (B) Hic-5 upregulation in normal mammary epithelial cells also promotes the formation of matrix-degrading invadopodia, via Src, Rac1 and RhoC, leading to increased cell invasion. Both paxillin and Hic-5 localize to, and regulate, the dynamics of individual invadopodia. Hic-5 also promotes the coalescence of numerous invadopodia into higher order rosettes in Src-transformed cells, involving formin-regulated actin assembly. Rapid rosette disassembly is Hic-5-dependent and is frequently associated with bursts of Rac1-driven lamellipodial extensions and the formation of new focal adhesions.
Hic-5 is also necessary for invadopodia rosette formation in NIH/3T3 Src-transformed fibroblasts (Gulvady et al., 2019). In this system, Hic-5 localization to invadopodia requires its LIM domains and complex rosette assembly requires its LD2 and LD3 motifs and phosphorylation at Y38/Y60. The expression of a Hic-5 mutant lacking its LD2 and LD3 domains (Hic-5 ΔLD2,3) or a non-phosphorylatable mutant (Hic-5 Y38,60F) does not prevent invadopodia formation but does alter their dynamics and impairs their organization into rosette structures. Furthermore, FAK kinase activity, Rac1 GTPase activity, and activity of the actin nucleating/polymerizing formin proteins are all required for this Hic-5 dependent invadopodia coalescence into rosettes (Gulvady et al., 2019). Pharmacologic inhibition of any of these proteins mimics the Hic-5 ΔLD2,3 phenotype. However, constitutively active FAK does not rescue rosette formation in cells expressing Hic-5 ΔLD2,3, implying that FAK acts upstream of Hic-5 to promote invadopodia coalescence into rosettes. Therefore, it is possible that FAK/Src-mediated phosphorylation of Hic-5 regulates its interaction with, or activation of, a Rac1 GEF, thereby promoting Rac1 activity that then induces formin-mediated actin assembly to interconnect invadopodia (Gulvady et al., 2019; Panzer et al., 2016) (Fig. 3).
4.2. Crosstalk with microtubules
To migrate effectively and persistently, cells must establish front-rear polarization and efficient anterograde vesicle trafficking to enable directed delivery of factors required for migration to the leading edge of the cell and for recycling of focal adhesion-associated integrins (Mellman and Nelson, 2008; Paul et al., 2015; Petrie et al., 2009; Wilson et al., 2018). The microtubule (MT) network is a major mediator of cell polarization through its role in coordinating cell-ECM adhesions, in positioning of the Golgi apparatus, and in facilitating vesicle trafficking (Garcin and Straube, 2019).
Microtubules are 25-nm wide tubes composed of protofilaments of tubulin subunits (Garcin and Straube, 2019). These subunits can be post-translationally modified by acetylation, phosphorylation, and tyrosination, among others, and these modifications affects MT dynamics (Janke and Bulinski, 2011). MT acetylation is commonly associated with longer-lived and more stable microtubules (Janke and Montagnac, 2017). Importantly, acetylated MTs are enriched at the Golgi apparatus and toward the leading edge in cells undergoing both 2D and 3D cell migration, suggesting that this post-translational modification may be important in establishing a polarized phenotype (Doyle et al., 2009; Ryan et al., 2012). One important regulator of MT acetylation and stability is the histone deacetylase 6 (HDAC6), which also deacetylates α-tubulin (Asthana et al., 2013). In fact, HDAC6 inhibitors have been successful in reducing tumor cell migration and invasion in pre-clinical studies and are currently being used in a number of clinical trials (Bian et al., 2018; Li et al., 2018).
It has long been known that paxillin is involved in establishing and maintaining mesenchymal cell front-rear polarity through its LD4 motif. Expression of a paxillin mutant lacking LD4 impairs cells’ ability to orient their Golgi apparatus toward the leading edge and to migrate in a directed manner, but promotes the formation of random, non-directed protrusions, likely as a result of dysregulated Rac1 activity (West et al., 2001). However, recent work has more clearly established another mechanism for paxillin’s crucial role in cell polarity.
Paxillin regulates MT acetylation by binding to and inhibiting HDAC6 (Fig. 4A, MT acetylation, polarity, & trafficking) (Deakin and Turner, 2014). Depletion of paxillin results in a significant decrease in MT acetylation but does not affect overall tubulin expression or distribution. The change in acetylation also reduces cell migration and importantly, can be reversed by treatment with the HDAC6-specific inhibitor tubacin or by HDAC6 knockdown (Deakin and Turner, 2014). Together with data demonstrating HDAC6 enrichment adjacent to focal adhesions and paxillin/HDAC6 binding in cells, these observations suggest that paxillin inhibition of HDAC6 functions to permit normal MT acetylation, thereby promoting cell motility, and is potentially linked to MT-mediated focal adhesion turnover (Efimov et al., 2008; Kaverina et al., 1999). However, since HDAC6 inhibitors can reduce tumor invasion in vivo as mentioned above, it is likely that a delicate balance of HDAC6 activity and microtubule acetylation is required for optimal cell migration and invasion.
Fig. 4.
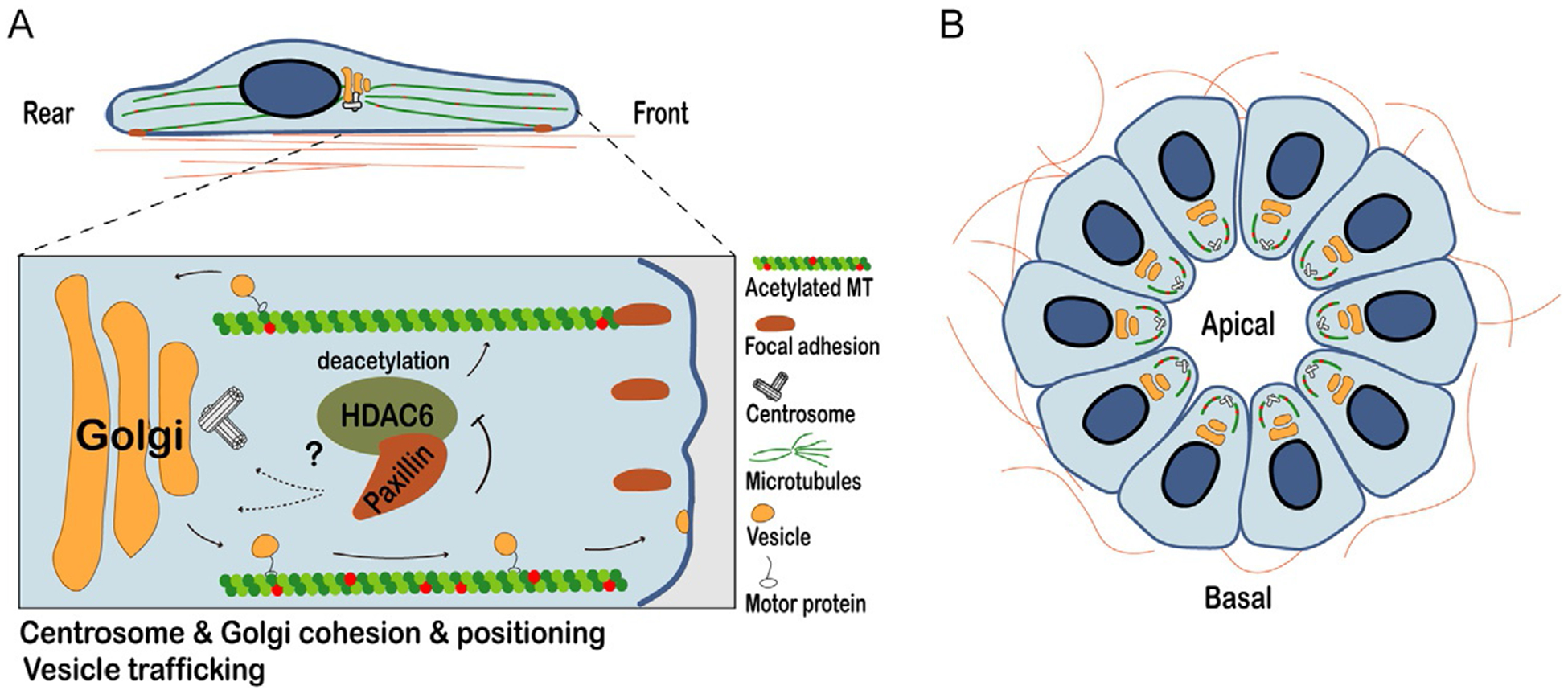
Paxillin-dependent changes in microtubule acetylation and their effect on cell polarity, vesicle trafficking, and Golgi/centrosome cohesion. (A) Paxillin promotes front-rear polarity in mesenchymal cells through its regulation of microtubule (MT) acetylation. Paxillin binding to, and inhibition of the deacetylase HDAC6 promotes MT acetylation. Paxillin-dependent MT acetylation is also required for Golgi cohesion and positioning, as well as vesicle trafficking of pro-migratory factors, such as integrins from the endoplasmic reticulum via the Golgi apparatus to the plasma membrane. Microtubule targeting to focal adhesions promotes their turnover in a paxillin-dependent manner. Although primarily a focal adhesion protein, paxillin also localizes to the centrosome and is required for its cohesion and polarization during migration. (B) Paxillin also plays a key role in maintaining apical-basal polarity and lumen formation in normal epithelia through regulation of HDAC6 to control microtubule acetylation. Apically enriched acetylated microtubules facilitate basal-apical trafficking, cell shape and the integrity of cell-cell junctions.
Paxillin depletion and resulting MT deacetylation also affect the integrity and positioning of the Golgi apparatus and centrosomes (Deakin and Turner, 2014; Dubois et al., 2017). Paxillin knockdown causes fragmentation of the Golgi and prevents its efficient, polarized orientation toward the leading edge in 1D, 2D, and 3D systems (Dubois et al., 2017). This is likely due to MT-dependent effects, since MTs are known to tether Golgi stacks together and support their cohesion (Lippincott-Schwartz et al., 2000). Interestingly, paxillin is also required for centrosome polarization and cohesion—the maintenance of close proximity between centrioles in non-dividing cells (Fig. 4A) (Dubois et al., 2017). A small amount of paxillin localizes to the centrosome and paxillin depletion leads to significant centriole separation (Dubois et al., 2017). Centrioles are typically closely associated in normal cells during interphase (Gönczy, 2015). However, they are often aberrantly duplicated and separated in cancer cells and it is believed that these changes contribute to genomic instability due to chromosome segregation errors during mitosis (Pease and Tirnauer, 2011). Centriole cohesion also depends on MT integrity, suggesting that paxillin-mediated MT acetylation changes may be responsible for the separation phenotype previously observed following paxillin knockdown in MDA-MB-231 cells (Burakov et al., 2003; Dubois et al., 2017). Once again, these centrosome- and Golgi-related phenotypes can be rescued in paxillin-depleted cells by tubacin treatment or HDAC6 RNAi, confirming that they are mediated through paxillin-dependent effects on MT acetylation (Deakin and Turner, 2014; Dubois et al., 2017). However, the precise mechanism connecting paxillin, MT acetylation, and Golgi/centrosome cohesion and orientation has yet to be clearly elucidated.
Importantly, the Golgi apparatus and centrosomes are both microtubule-organizing centers (MTOCs) in the cell (Wu and Akhmanova, 2017). Therefore, paxillin is required for the cohesion and effective polarization of each of the primary MTOCs in both normal and cancer cells. This mechanism is independent of paxillin localization to focal adhesions, since the observed phenotypes are evident even when cells were plated on poly-l-lysine-coated dishes, which prevents focal adhesion formation (Dubois et al., 2017). Thus, reduced paxillin expression in certain cancers could result in loss of cell polarization to impair effective cell migration/invasion, while a loss of centriole cohesion could promote carcinogenesis due to genomic instability.
Pharmacologic inhibition of FAK, an important signaling partner of paxillin, also prevents paxillin localization to the centrosome and mimics many of the phenotypes which occurred following paxillin depletion, suggesting that these proteins likely work together in regulating MT acetylation (Dubois et al., 2017). FAK stimulates MT acetylation through RhoA activation, so activated FAK could potentially modulate paxillin’s regulation of HDAC6 activity (Palazzo et al., 2004).
Paxillin’s regulation of MT acetylation also indirectly impacts anterograde vesicle trafficking (Fig. 4A) (Dubois et al., 2017). As mentioned previously, effective transport of pro-migratory components to the leading edge is crucial for optimal directed migration, and this transport often occurs along MTs (Naslavsky and Caplan, 2018). In paxillin-depleted cells, trafficking of GFP-tagged vesicular stomatitis virus G (VSVG)—a marker of anterograde trafficking—from the endoplasmic reticulum to the Golgi and from the Golgi to the plasma membrane are both impaired (Dubois et al., 2017). The mechanism by which this occurs remains to be elucidated, but it is possible that paxillin-dependent changes in MT acetylation affect the binding and/or activity of the molecular motors that traffic cargo along MTs or the activity of Rab GTPases, which function in vesicle trafficking and integrin recycling and therefore, focal adhesion dynamics (Garcin and Straube, 2019; Paul et al., 2015). Regardless of the mechanism, this result suggests an interesting focal adhesion-independent mechanism by which paxillin regulates cell migration.
Using a conditional paxillin knockout mouse, it has been shown that paxillin is also critical for the establishment of apical-basal polarity in normal mouse mammary glands in vivo and the formation of 3D polarized acini in a mammary epithelial cell in vitro model system, including the apical enrichment of acetylated MTs (Fig. 4B) (Xu et al., 2019). This newly described role for paxillin in normal epithelial cells could become dysregulated during carcinogenesis, as cancer cells often undergo an EMT and lose their apical-basal polarity prior to invasion (EMT will be discussed in a subsequent section). Indeed, 3D acini of mammary epithelial cells derived from the normal mammary gland of the paxillin knockout mice closely resemble the disorganized morphology and defects in cell-cell junction organization seen in tumor organoids generated using tumor fragments from conditional paxillin knockout MMTV-PyMT breast cancer mice (our unpublished results). Remarkably, pharmacologic inhibition of HDAC6 with tubacin rescued the defects in acini polarization (Xu et al., 2019), suggesting that paxillin regulation of MT acetylation not only affects front-rear polarization in single cells, but can also affect the polarization of multicellular units, potentially through its effects on intracellular trafficking and crosstalk with the actin cytoskeleton (Booth et al., 2014).
4.3. Crosstalk with intermediate filaments
Intermediate filaments (IFs) generally play an important role in determining cell morphology and response to mechanical stress due to their characteristic structural integrity (Leduc and Etienne-Manneville, 2015). IFs are built from several proteins and protein families, including cytokeratins, lamins, neurofilaments, desmin, and vimentin, and their expression varies widely by cell type and between normal and cancerous cells (Lowery et al., 2015). In fact, several IFs have been used clinically as markers for disease grade and tumor invasiveness for decades (Osborn and Weber, 1983). For example, vimentin expression is typically limited to mesenchymal cells, so its expression in epithelial cells may indicate a more malignant phenotype (Korsching et al., 2005). Furthermore, cytokeratins are used as markers for tumors of epithelial origin, and individual cytokeratins may be enriched in certain cell populations. For example, cells leading strands of collectively migrating cells in breast cancer are known to express cytokeratin 14 (Diepenbruck and Christofori, 2016). Different IFs have differing effects on cell migration (Leduc and Etienne-Manneville, 2015).
Vimentin is a type III IF expressed primarily in fibroblasts and cells that have undergone EMT (Lowery et al., 2015). Recent studies have shown a role for vimentin in promoting directional persistence of migrating cells, and vimentin filaments occasionally terminate at focal adhesions, suggesting some degree of crosstalk between the actin and IF cytoskeletons at these sites (Battaglia et al., 2018), a process that appears to be dependent on Hic-5 (Vohnoutka et al., 2019). Additionally, vimentin expression in fibroblasts protects against nuclear rupture and DNA damage as cells migrate, by forming a cage around the nucleus (Patteson et al., 2019). A similar mechanism could be important for protecting cancer cells migrating through tight spaces in the ECM, especially since DNA damage is an important part of carcinogenesis through its impact on genomic stability (Hanahan and Weinberg, 2011). Further evidence of focal adhesion/vimentin crosstalk and the role of Hic-5 will be discussed below.
4.4. Epithelial-mesenchymal transition
As briefly described above, epithelial-mesenchymal transition (EMT) is a key process in both normal development and in cancer cell invasion and metastasis, although its role in cancer is highly controversial. (Diepenbruck and Christofori, 2016; Ye and Weinberg, 2015). This controversy stems largely from the inability to monitor the transient and reversible EMT that occurs in vivo and evidence that EMT may not be required for metastasis in certain cancers (Fischer et al., 2015). Furthermore, immunohistochemical staining of tumors often show cells migrating collectively, which does not require a complete EMT; in fact, cells which have undergone a total EMT cannot invade collectively (Clark and Vignjevic, 2015). However, new tools have improved our understanding of EMT and its contribution to invasion in certain contexts, and a whole range of intermediate EMT states have been described (Pastushenko et al., 2018).
The process of EMT involves the disassembly of cell-cell junctions, the down-regulation of cell-cell junction components such as E-cadherin, and reorganization of the actin cytoskeleton to promote cell-matrix adhesion and increased migration (Brabletz et al., 2018). EMT also results in a switch from epithelial markers (including several cytokeratins) to mesenchymal markers (including vimentin) (Ye and Weinberg, 2015). Mesenchymal cells are much more motile than epithelial cells in both 2D and 3D culture systems (Aiello et al., 2018; Brabletz et al., 2018). TGF-β, a versatile cytokine which can, depending on the context, act as both an oncogene and a tumor suppressor, is a potent inducer of EMT and stimulates upregulation of Hic-5 expression which, as noted, is typically very low in epithelial cells (Fig. 3A) (Tumbarello and Turner, 2007). As mentioned above, Hic-5 is also known as transforming growth factor beta 1 induced transcript 1 (TGFB1i1) and participates in the non-canonical arm of TGF-β signaling, which primarily impacts RhoGTPase signaling and the actin cytoskeleton (Varney et al., 2016).
Following TGF-β stimulation, Hic-5 facilitates EMT through a downstream pathway involving RhoA and ROCK (Tumbarello and Turner, 2007). Hic-5 RNAi after TGF-β stimulation results in reduced cell migration and the retention of epithelial morphology. Additionally, Hic-5 RNAi prevents TGF-β-induced RhoA activation, while ectopic Hic-5 overexpression promotes ROCK kinase activity and the formation of ROCK-dependent stress fibers (Tumbarello and Turner, 2007). Interestingly, inhibiting RhoA/ROCK activity by expressing dominant negative RhoA or by using a ROCK inhibitor prevents an increase in Hic-5 expression following TGF-β-induced EMT, suggesting that Hic-5 and RhoA/ROCK work in a positive feedback loop to promote EMT and maintain a mesenchymal phenotype. As discussed earlier, Hic-5 upregulation in response to TGF-β also promotes the formation of invadopodia that are important for the resulting mesenchymal invasion (Pignatelli et al., 2012a) (Fig. 3B).
5. Stroma-mediated effects on tumor invasion
Tumor cells are surrounded by an ECM which is deposited by resident fibroblasts and, along with local immune cells and the vasculature, makes up the tumor stroma (Quail and Joyce, 2013). While the idea that the microenvironment can affect cancer progression has been around since at least the proposition of the “seed-and-soil” hypothesis of metastasis, recent work has greatly increased out understanding of this field (Bissell et al., 2002; Humphrey et al., 2014; Kalluri, 2016; Langley and Fidler, 2011; Paget, 1889). In fact, multiple gene expression studies of the tumor stroma provide a more accurate prediction of patient outcomes than expression studies of the tumor epithelial cells themselves in several cancers, including colorectal and breast cancer (Calon et al., 2015; Finak et al., 2008; Todd et al., 2016). With these facts in mind, it is important to consider the roles of paxillin family members in stromal cells and how their expression in these cells can indirectly affect tumor cell migration and invasion.
5.1. Extracellular matrix deposition
During wound healing, resident tissue fibroblasts differentiate to become myofibroblasts, which participate in wound contraction and fibrotic changes (Bochaton-Piallat et al., 2016; Dabiri et al., 2008). In a similar manner, resident tumor fibroblasts are activated by ECM rigidity- and signaling-mediated changes to become smooth muscle actin-positive, highly contractile cancer-associated fibroblasts (CAFs), which generate and remodel the cancer ECM and play a significant role in tumor progression (Kalluri, 2016). Crosstalk between CAFs and tumor cells can produce a feed-forward loop resulting in increased tumor cell invasion and metastasis through changes in the rigidity and molecular composition of the ECM and secretion of cytokines. For instance, more rigid and aligned matrices rich in fibrillar collagens and fibronectin generally promote cell invasion and, clinically, increasing tumor rigidity is associated with cancer progression, metastasis, and worse prognoses in breast and colorectal cancer, among others (Emon et al., 2018; Levental et al., 2009; Wei and Yang, 2016). Increased ECM rigidity can result from a number of factors, including increased expression of the collagen cross-linking protein lysyl oxidase (Omoto et al., 2018; Wei et al., 2017) or increased deposition of ECM fibers such as collagen and hyaluronan (Gkretsi and Stylianopoulos, 2018). The relative abundance of ECM proteins is also important for tumor stroma crosstalk. For example, increased stromal expression levels of fibronectin and tenascin C correlate with decreased overall patient survival in breast cancer (Ioachim et al., 2002). Increased collagen density and alignment, quantified by a “tumor-associated collagen signature” score, are also associated with negative patient outcomes (Provenzano et al., 2008).
Information about ECM rigidity and molecular composition is at least partially transmitted through focal adhesions in both tumor cells and CAFs. Multiple focal adhesion proteins can act as mechanosensors, including paxillin and Hic-5, as well as vinculin, talin, and integrins (Jansen et al., 2015). Tumor cells can then respond to these changes through complex signaling pathways involving activation of proteins such as Rho GTPases, MAPKs (mitogen-activated protein kinases), YAP/TAZ (Yes-associated protein/transcriptional coactivator with PDZ-binding motif), FAK, and MRTF (myocardin-related transcription factor) (Maller et al., 2013; Provenzano and Keely, 2011). Together, these pathways induce changes in cytoskeletal organization and gene expression that will be discussed in more detail below.
Hic-5 expression regulates myofibroblast function in non-cancerous wound healing and is also important in regulating CAF function in numerous cancers and pre-cancerous lesions (Fig. 5, CAFs & ECM deposition) (Dabiri et al., 2008; Du et al., 2019; Goreczny et al., 2017; Omoto et al., 2018; Varney et al., 2016). For example, Hic-5 deficiency in pancreatic fibroblasts (known as pancreatic stellate cells or PSCs) protects from cerulein-induced pancreatitis in mice (Chen et al., 2020). In humans, chronic pancreatitis predisposes patients to pancreatic cancer, and the PSCs of patients with pancreatitis exhibit increased Hic-5 expression. If pancreatic cancer develops, Hic-5 expression in PSCs then promotes proliferation, survival, and invasion of pancreatic cancer cells in vitro when these cells are cultured together. Additionally, Hic-5 depletion in PSCs results in decreased MMP-9 expression in the fibroblasts (Qian et al., 2020). Furthermore, higher Hic-5 expression correlates with decreased post-operative survival. These data suggest that Hic-5-mediated fibrosis may play an important role in tissue stiffening and other pre-cancerous changes that may ultimately lead to cancer.
Fig. 5.
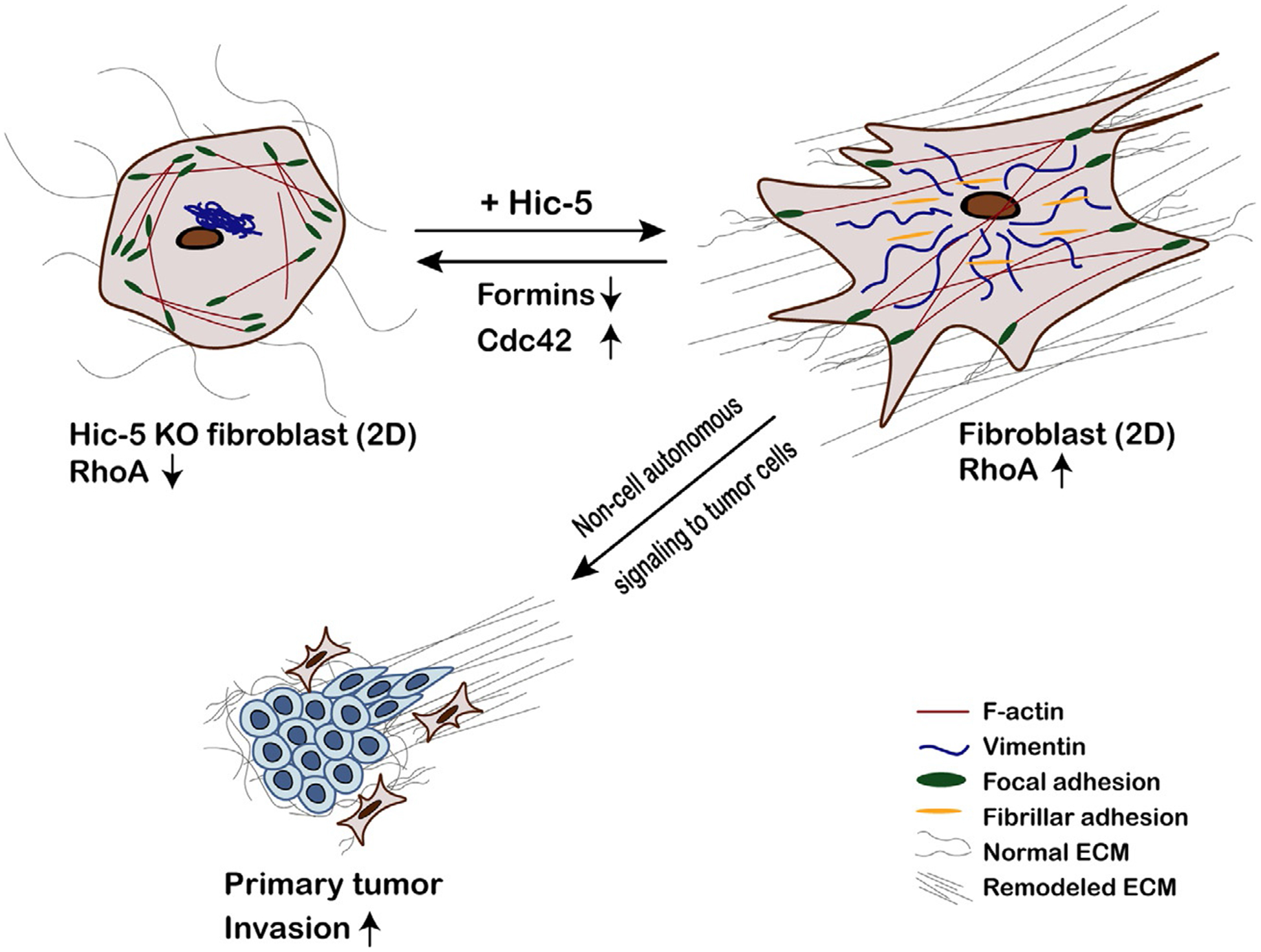
Hic-5 expression in cancer-associated fibroblasts is required for effective extracellular matrix deposition and organization. Hic-5 expression promotes increased RhoA-mediated contractility in cancer-associated fibroblasts (CAFs), leading to a more “activated” myofibroblast-like phenotype. Hic-5 knockout CAFs are deficient in extracellular matrix (ECM) deposition and remodeling in 2D and 3D when compared to those expressing Hic-5; this is at least partially mediated by diminished fibronectin fiber assembly at fibrillar adhesions involving Src-regulated Hic-5 interactions with tensin. Furthermore, Hic-5 knockout CAFs exhibit a central F-actin “hole” and a collapsed vimentin intermediate filament cytoskeleton. Both of these phenotypes can be mimicked in Hic-5-expressing CAFs by pharmacologic formin inhibition or rescued in Hic-5 knockout CAFs by pharmacologic inhibition of the RhoGTPase Cdc42. Hic-5-dependent deposition and remodeling of the stromal ECM in vivo promotes non-cell autonomous signaling in the tumor cells to stimulate their invasion and dissemination.
A recent study using patient-derived CAFs shows that Hic-5 expression is important in colorectal cancer (Omoto et al., 2018). Relative to normal fibroblasts from tumor-adjacent healthy tissue, colorectal CAFs have significantly higher Hic-5 expression. Hic-5 expression was induced in the normal fibroblasts by treatment with culture media that had been conditioned by CAFs or with cytokines including TGF-β, IL-1 β, and SDF-1/CXCL12. Colon cancer cell growth was shown to be inhibited in the presence of CAFs with low Hic-5 levels in in vitro assays, compared with cells cultured with wild-type CAFs (Omoto et al., 2018). Most interestingly, Hic-5 knockout mice did not develop any tumors in an azoxymethane-induced colorectal cancer model, in contrast to a >50% incidence in wild-type mice (Omoto et al., 2018). This was suggested to be partially due to Hic-5 stimulation of lysyl oxidase and collagen I expression. Lysyl oxidase is a secreted protein responsible for cross-linking collagen in vivo, and its expression correlates with increasing tumor stiffness, a negative prognostic marker in many cancers (Levental et al., 2009). Therefore, Hic-5 expression in human CAFs promotes a pro-tumorigenic stroma in colorectal cancer. Similar results were observed in oesophageal cancer CAFs (Du et al., 2019).
In the MMTV-PyMT mouse model of breast cancer, Hic-5 is surprisingly not expressed in the invasive tumor cells but its expression in the CAFs promotes tumor growth, decreases tumor latency, and enhances lung metastasis (Goreczny et al., 2017). Immunostaining of tumors from Hic-5 heterozygous and knockout mice show that collagen and fibronectin staining in the tumor stroma is decreased in the absence of Hic-5. This may be partially due to Hic-5’s role in promoting fibrillar adhesion formation and maturation through its phosphorylation-dependent interaction with tensin-1 (Fig. 5) (Goreczny et al., 2018). These adhesions are important for assembly of fibronectin fibrils, which also affects collagen deposition and organization in vivo. Isolated CAFs from Hic-5 knockout mouse tumors are also less contractile than their Hic-5-expressing counterparts as a result of significantly reduced RhoA activity. In addition, the Hic-5 knockout CAFs produce thinner 3D CDMs with poorly aligned fibronectin and collagen fibers in vitro (Goreczny et al., 2017). This is a potentially clinically relevant phenotype since, as mentioned above, increasing density and alignment of tumor ECMs are inversely correlated with patient outcomes, including overall survival (Provenzano et al., 2008), Interestingly, MDA-MB-231 breast cancer cells seeded in CDMs generated by Hic-5-expressing CAFs are less likely to migrate with an amoeboid phenotype and have decreased directional persistence compared to those seeded in CDMs from Hic-5 knockout CAFs (Goreczny et al., 2017). These changes in migration phenotype are likely one of the contributing factors to the decrease in lung metastases observed in Hic-5 knockout mice.
As noted above, there is growing evidence for crosstalk between the actin and IF cytoskeletons, which likely plays an important role in controling cell shape and motility (Burgstaller et al., 2010; Huber et al., 2015; Osmanagic-Myers et al., 2015).This, in turn, is likely important in myofibroblasts as they organize the surrounding stromal matrix. Evidence of a key role for Hic-5 in connecting vimentin organization with focal adhesions and, therefore, the actin cytoskeleton, was recently described. A collapse of the vimentin cytoskeleton was observed in Hic-5 knockout CAFs and normal lung fibroblasts (Vohnoutka et al., 2019). In contrast, depletion of paxillin did not cause a similar perturbation of the vimentin cytoskeleton. Hic-5 knockout CAFs also exhibit a reduction in centrally localized F-actin stress fibers (an actin “hole”), which could be directly linked to this vimentin phenotype as both are rescued in knockout CAFs by inhibition of the Rho GTPase Cdc42, and phenocopied by pharmacological inhibition of formins in Hic-5 expressing CAFs (Vohnoutka et al., 2019). Together, Hic-5-mediated regulation of Rho GTPase and formin activity may co-ordinately affect both the actin and vimentin cytoskeletons to allow disease-relevant changes in fibroblast phenotype. For instance, low Hic-5 and a collapsed vimentin cytoskeleton may be necessary for a pro-migratory fibroblast phenotype, while high Hic-5 and robust actin and vimentin cytoskeletons are necessary for a highly contractile phenotype, where cells deposit and remodel the surrounding ECM (Fig. 5). Although these observations were made in CAFs, it is interesting to speculate whether a similar phenotype may occur in cancer cells depending on the level of Hic-5 expression and, if so, whether a collapsed vimentin network can still protect against nuclear rupture or facilitate cancer cell amoeboid motility.
5.2. Tumor neovascularization
The tumor stroma also includes blood vessels which support the growing tumor by providing oxygen and nutrients and removing carbon dioxide and waste (Nishida et al., 2006). From early in tumorigenesis, tumor cells promote neovascularization of the tumor stroma by secreting factors which activate an “angiogenic switch,” including vascular endothelial growth factor-A (VEGF-A) and fibroblast growth factor (FGF) (Hanahan and Weinberg, 2011). As tumor progression proceeds, this neovascularization is modulated by both tumor and stromal cells. The process of angiogenesis involves activation of endothelial cells by pro-angiogenic factors, production of proteases to degrade the matrix, cell migration/invasion and proliferation, tube formation, synthesis of new basement membrane, and recruitment of supporting cells including smooth muscle cells and pericytes (Rajabi and Mousa, 2017). The degree of neovascularization varies by cancer type, but most solid tumors are highly vascularized with aberrant, convoluted vessels that support the tumors’ high metabolic requirements. The success of anti-angiogenic drugs such as bevacizumab and cabozantinib in treating a variety of cancers highlights the importance of neovascularization for tumor growth and survival (Rajabi and Mousa, 2017).
Hic-5 is highly expressed in endothelial cells and vascular smooth muscle cells (Kim-Kim-Kaneyama et al., 2011, 2012). Depletion of Hic-5 reduces endothelial sprouting and lumen formation in a 3D in vitro assay, suggesting Hic-5 may play a role in regulating vascularization (Dave et al., 2016). Additionally, inducing sprouting with pro-angiogenic factors including VEGF, FGF, and sphingosine 1-phosphate also increases protein complex formation between Hic-5, FAK, and the matrix metalloproteinase MT1-MMP, which is a membrane-anchored protease critical for endothelial cell invasion during sprouting (Dave et al., 2016). The interaction with MT1-MMP is mediated by Hic-5’s LIM2 and LIM3 domains. While Hic-5 knockdown does not alter MT1-MMP activity, it does reduce its localization at the membrane during endothelial sprouting, which results in impaired invasion.
Hic-5 also plays a role in vascular remodeling after injury due to its mechanosensitive properties (Kim-Kaneyama et al., 2008, 2011). When undisturbed, the femoral arteries of Hic-5-null mice appear similar to those of wild-type mice (Kim-Kaneyama et al., 2008). However, after surgical injury by a wire, the arteries in Hic-5-null mice have delayed recovery and higher numbers of apoptotic cells in the vascular walls. When mechanically stretched in vitro, smooth muscle cells derived from the Hic-5-null mice exhibit an increased cytoplasmic localization of vinculin, another mechanosensing focal adhesion protein that also binds actin (Atherton et al., 2020; Humphrey et al., 2014; Kim-Kaneyama et al., 2005). This provides further evidence that stable focal adhesions help protect against stretch-induced apoptosis and that Hic-5 is required for stable integration of vinculin and other focal adhesion proteins, as previously discussed in the context of mesenchymal tumor cell morphology. This idea is well-supported by other studies and is an important consideration in cells in the vascular wall, which are constantly experiencing mechanical changes (Katsumi et al., 2005; Kim-Kaneyama et al., 2005). Although these studies don’t examine vascular injury in a cancer-related context, the processes of normal vascular wound healing and angiogenesis bear many similarities, suggesting that Hic-5 may be an interesting target to pursue when investigating tumor vascularization (Dabiri et al., 2008; Kareva et al., 2016; Kim-Kaneyama et al., 2012).
Although less well-studied, paxillin also plays a role in remodeling the vasculature. The angiogenic cytokine angiopoietin-1 requires paxillin expression for effective promotion of endothelial cell polarization, migration, and sprouting (Boscher et al., 2019). Angiopoietin-1 binding induces PAK2-mediated paxillin phosphorylation which is necessary for activation of the Rho GTPase Cdc42 at the leading edge and recruitment of the polarity protein Par3. Paxillin also mediates endothelial permeability through control of Rho GTPase signaling, which may be important in allowing tumor cell intravasation and extravasation (Birukova et al., 2009; García-Román and Zentella-Dehesa, 2013; Gawlak et al., 2014).
5.3. Immune cell function
A healthy and intact immune system is a critical defence measure against cancer development (Gonzalez et al., 2018). Accordingly, tumors often demonstrate highly dysregulated immune environments with suppression of normal immune pathways that would destroy the tumor, and activation of inflammatory conditions that promote tumor progression (Pandya et al., 2016). While an in-depth analysis of immune dysregulation is beyond the scope of this review, it is important to note that paxillin has been shown to modulate immune cell signaling, and that a healthy and properly-regulated immune response can impair tumor cell migration and invasion (Gauthier et al., 2017;Herreros et al., 2000; Robertson and Ostergaard, 2011).
In a mouse orthotopic colon cancer model, down-regulation of paxillin expression in macrophages indirectly inhibited tumor growth through effects on the immune system (Zhang et al., 2018). Macrophages are phagocytic immune cells that play roles in both the innate and adaptive immune response, and the M2 class of tumor-associated macrophages (TAMs) are anti-inflammatory, promote tumor growth and progression, and correlate with negative patient prognoses (Aras and Zaidi, 2017). Paxillin expression is upregulated during M2 macrophage activation (Zhang et al., 2018). Furthermore, paxillin depletion inhibits M2 polarization and reduces macrophage invasion and proliferation in vivo, consistent with its role in epithelial cell polarization (Dubois et al., 2017; Xu et al., 2019). Co-injection of colon cancer cells with either wild-type or paxillin-depleted macrophages in a nude mouse model results in significantly decreased tumor volume in mice with paxillin-depleted macrophages, suggesting that paxillin can modulate the immune response in this circumstance to promote tumor growth (Zhang et al., 2018).
Multiple studies have addressed a role for paxillin in T-cells, an important class of cytotoxic cells in the adaptive immune system that are responsible for killing epithelial tumor cells (Thommen and Schumacher, 2018). Cytotoxic T-cells interface with their target cells at immunologic synapses, which are supramolecular clusters of several transmembrane and membrane-associated proteins. When one of these immunologic synapse proteins, CD103 (the α subunit of integrin αEβ7), binds to a tumor-specific antigen, paxillin is phosphorylated and binds to the CD103 cytoplasmic tail at the immunologic synapse (Gauthier et al., 2017). If paxillin expression is knocked down by shRNA or its phosphorylation is inhibited by the Src kinase inhibitor saracatinib, T-cell spreading and effector activities are impaired in vitro, including their abilities to lyse tumor cells and release the anti-tumor cytokine INFγ (Gauthier et al., 2017).
Interestingly, another role for paxillin in T-cells is related to its ability to regulate the positioning of the MTOC, as discussed earlier in a different context. When a T-cell binds to a target cell, paxillin is recruited to both the immunologic synapse and the MTOC (Herreros et al., 2000; Robertson and Ostergaard, 2011). Importantly, paxillin contributes to reorientation of the MTOC toward the target cell. This is a crucial step in T-cell activation because it allows for directed delivery of cytokines to the plasma membrane for secretion to enhance target cell killing (Martín-Cófreces et al., 2008). These and other studies suggest that paxillin expression in T-cells is important for effective anti-tumor immunity, potentially through HDAC6-mediated changes in MT acetylation and MTOC polarization.
While paxillin is expressed in most circulating and tissue-resident immune cells, Hic-5 exhibits only slightly enhanced RNA expression in basophils and minimal expression in any other immune cells, suggesting it has little direct impact on immune function (paxillin and Hic-5 expression data from proteinatlas.org, v19.3) (Thul et al., 2017). Similarly, although leupaxin expression was first identified in leukocytes, there a lack of subsequent research indicating an important role for this protein in cancer-related immunity (Lipsky et al., 1998).
6. Regulation of gene expression
Acute and long-term changes in gene expression are also important in cancer progression and metastasis. Expression of paxillin family members can be repressed through the action of microRNA, and both paxillin and Hic-5 can act in the nucleus to regulate gene transcription, similar to several other focal adhesion proteins including FAK and zyxin (Kadrmas and Beckerle, 2004; Kasai et al., 2003; Kleinschmidt and Schlaepfer, 2017; Shibanuma et al., 2003).
6.1. Regulation of paxillin expression by miRNA
Paxillin expression can be post-transcriptionally regulated by miRNAs during cancer invasion and metastasis. miRNAs are a class of 19–22 nt single strand non-coding RNAs that typically act by binding to target mRNAs, thus inducing the degradation of the mRNAs or inhibition of their translation (Bartel, 2009; Kusenda et al., 2006). miRNAs are highly involved in tumor progression processes such as tumor cell adhesion, migration, invasion, and proliferation (Hayes et al., 2014; Peng and Croce, 2016). miR-218 negatively regulates paxillin translation (Wu et al., 2010). Accordingly, low miR-218 levels in non-small cell lung cancer correlate with high paxillin expression and, furthermore, with decreased patient survival and increased relapse rates (Wu et al., 2010). A similar miR-218-dependent, post-transcriptional regulation of paxillin has been observed in oral cavity squamous cell carcinoma (Wu et al., 2014b). miR-137 inhibits paxillin translation in colorectal cancer, so low miR-137 levels result in high paxillin expression and reduced survival, (Chen et al., 2013). However, miR-137-mediated paxillin inhibition in lung cancer cells results in reduced proliferation and migration, suggesting that the result of paxillin down-regulation is again, context-specific (Bi et al., 2014). Several other studies in cancer cell lines have shown that paxillin expression can be directly inhibited by other miRNAs, thus impairing the ability of cancer cells to migrate and invade (Li et al., 2015; Matsuyama et al., 2016; Qin et al., 2015; Tao et al., 2016). For example, miR-145 interacts with paxillin mRNA in human colorectal cancer cells to suppress its translation (Qin et al., 2015), and miR-27b inhibits paxillin expression and integrin-mediated cell adhesion (Matsuyama et al., 2016).
In addition to direct binding to paxillin mRNA, miRNAs can also regulate paxillin through indirect mechanisms. For example, the miR-200 family/ZEB1 axis potentiates signaling through integrin β1 in response to binding extracellular collagen I, which promotes FAK-dependent paxillin phosphorylation and thereby increases lung cancer cell invasion and metastasis (Ungewiss et al., 2016). In ovarian cancer, miR-708 inhibits the GTPase activating protein Rap1B, which is a regulator of integrin-based adhesion signaling (Hattori and Minato, 2003). This inhibition results in reduced paxillin phosphorylation at focal adhesions and decreased lung metastases (Lin et al., 2015).
6.2. Regulation of gene expression by Hic-5 and paxillin
As mentioned above, Hic-5 is also known as androgen receptor coactivator 55kDa protein (ARA55) (Fujimoto et al., 1999). In addition to its localization at focal adhesions, Hic-5 can also localize to the nucleus as a coactivator for the androgen receptor transcription factor (Heitzer and DeFranco, 2006a; Shibanuma et al., 2003), and studies have shown that it can regulate expression of numerous genes by promoting the formation of a transcriptional complex with the transcriptional coactivator p300 and transcription factors Sp1 and SMAD3 in a variety of cell types (Heitzer and DeFranco, 2006b; Shibanuma et al., 2012; Yang et al., 2000). One particularly relevant study examined Hic-5 responsive gene expression changes in response to glucocorticoid treatment in U2OS osteosarcoma cells (Chodankar et al., 2014). Four of the top 15 gene ontology categories represented in the Hic-5-responsive group of genes were related to cell migration and adhesion, suggesting that Hic-5 may regulate migration indirectly through transcriptional changes.
Hic-5 also plays an important role in transducing mechanical and actin cytoskeleton-related signals to regulate gene expression through pathways including the actin-MRTF-SRF pathway (Varney et al., 2016). MRTFs bind to, and are sequestered by monomeric G-actin, but incorporation of G-actin monomers into F-actin fibers release MRTFs, allowing them to translocate to the nucleus and interact with the transcription factor serum response factor (SRF) (Olson and Nordheim, 2010). Therefore, actin polymerization into stress fibers promotes MRTF-SRF-dependent transcription of genes including several genes involved in contractility, such as smooth muscle actin and calponin (Crider et al., 2011). In fibroblasts, MRTF-SRF signaling promotes Hic-5 expression (Wang et al., 2011), but Hic-5 is also required for TGF-β-dependent nuclear localization of MRTF-A and promotion of smooth muscle actin expression (Varney et al., 2016). These data indicate that Hic-5 promotes fibroblast differentiation via a TGF-β-dependent feed-forward loop that regulates expression of actin cytoskeleton- and contractility-related genes.
Hic-5 may also play a role in other mechanosensitive transcription pathways involving the YAP/TAZ-TEAD (transcriptional enhancer factor-domain) axis. YAP/TAZ nuclear localization and activation correlate with several Hic-5-related phenotypes, including EMT and ECM rigidity-dependent fibroblast activation, although a direct role for Hic-5 in YAP/TAZ signaling has not been reported (Calvo et al., 2013; Shao et al., 2014). Furthermore, YAP/TAZ activity is modulated by several pathways, including a mechanosensitive regulation similar to MRTF-SRF signaling: high ECM rigidity and/or cellular contractility promotes release of YAP/TAZ from cytoplasmic sequestration and allows nuclear translocation (Totaro et al., 2018). YAP/TAZ activity is elevated in many cancers, and results in increased aggressiveness and cell proliferation, as well as induction of expression of several actin cytoskeleton- and focal adhesion-related genes, including formins, integrins, and RhoGEFs like the Dock proteins (Pocaterra et al., 2020; Totaro et al., 2018). Therefore, a role for Hic-5 in mediating YAP/TAZ transcription of proteins relevant to the actin cytoskeleton and cancer cell migration would not be surprising.
Although paxillin contains a nuclear export sequence and localizes most strongly to focal adhesions, several studies have demonstrated that it can also be shuttled into the nucleus to regulate androgen- and MAPK signaling pathway gene transcription (Kasai et al., 2003; Ma and Hammes, 2018). For example, RNAseq analysis in prostate cancer cells produced a panel of over 1000 paxillin-dependent, androgen-responsive genes (Ma et al., 2019). Paxillin expression in these cells increased transcription of pro-proliferative genes, including the CyclinD/Rb/E2F pathway, while it decreased transcription of pro-apoptotic genes, including CASP1 and TNSF10.
Although much of the signaling through paxillin family members occurs directly at focal adhesions, their respective roles in modulating gene expression is clearly also important to consider when studying their impact on tumor progression.
7. Paxillin family member mutations and expression in human cancers
Although somatic mutations of paxillin family members in cancer are generally considered rare, recent evidence indicates that they may play a role in some cancers (Deakin et al., 2012b). A study of lung cancer tissues showed that paxillin had a 9.5% somatic mutation rate, with the most common mutation (A127T) causing increased proliferation in vitro (possibly through binding with the cell survival protein Bcl-2) and increased invasion in a mouse xenograft model (Jagadeeswaran et al., 2008). Even without mutations, expression levels of paxillin family members are often highly dysregulated in a variety of cancers (Fig. 6, table of expression levels) (Deakin et al., 2012b; Rhodes et al., 2004). However, the lack of a consistent increase or decrease in expression—even among cancers of a specific organ—suggests that their role as either oncogenes or tumor suppressors is context-specific and may vary based on tumor type, stage, and other factors. For example, the opposing roles for paxillin and Hic-5 in regulating 3D migratory phenotype suggest that changes in their expression in response to changes in the surrounding ECM environment may promote more effective migration and, therefore, cancer progression (Deakin and Turner, 2011; Gulvady et al., 2018). Additionally, many of the studies which describe up- or down-regulation of paxillin family members use large-scale genomic, proteomic, or transcriptomic analyses and do not always specify whether gene expression is measured in the tumor cells, stromal cells, or both (López-Colomé et al., 2017). While numerous cell-based studies have focused on the modulation of paxillin family member expression in a variety of tumor cell types, few have carefully studied the role of these proteins using patient data. Here, we will briefly present data from studies describing paxillin’s role in gliomas to provide one example of the many contexts in which paxillin family members can affect cancer cell invasion and metastasis.
Fig. 6.
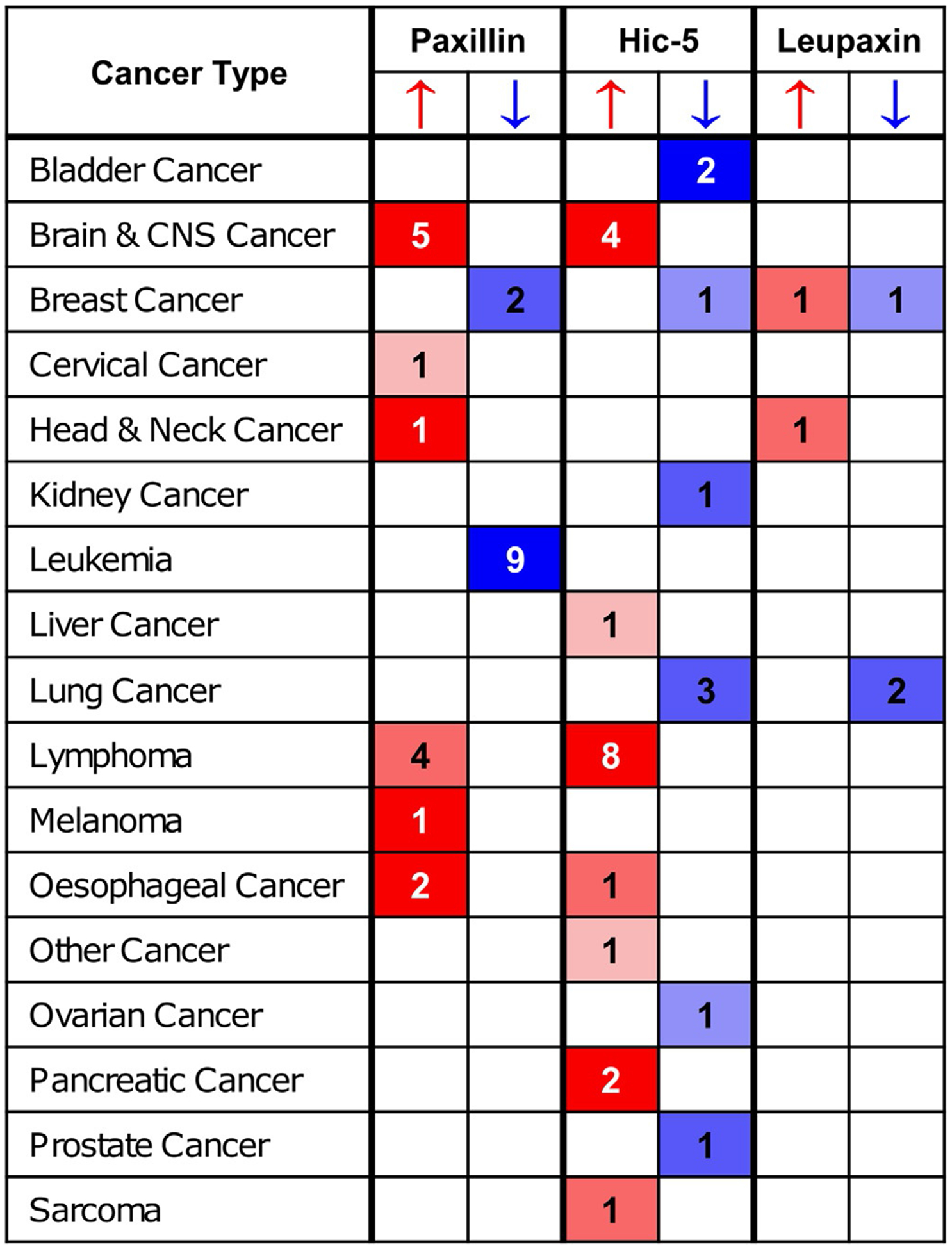
Expression of paxillin family members varies among cancers. Numbers in paxillin, Hic-5, and leupaxin columns indicate how many studies show increased or decreased mRNA expression of that gene in tumor relative to normal tissue (thresholds for inclusion: P <0.0001, fold change ≥ 2, top 10% gene rank). Darker cell color indicates higher gene rank percentile for the studies in that cell. Data were obtained from the Oncomine cancer expression database (Thermo Fisher Scientific Inc., www.oncomine.org).
Paxillin clearly functions as an oncogene in gliomas, and its expression is inversely correlated with patient outcomes (Chen et al., 2016; Sun et al., 2017). RNA sequencing data and immunohistochemical staining both demonstrate increased paxillin expression in glioma tissue relative to normal adjacent tissue, and paxillin expression positively correlates with tumor grade and metastasis (Sun et al., 2017). In a multivariate analysis, where tumors were stratified as either high- or low paxillin expressors, higher-than-median paxillin expression correlated with decreased overall survival with a roughly twofold greater risk of mortality within the study observation period. Gene set enrichment analysis using glioma samples showed that paxillin expression significantly affected gene sets related to cell adhesion, proliferation, and migration. These results were validated using in vitro assays of glioma cell lines (Sun et al., 2017). Ectopic expression of paxillin increased proliferation in clonogenic assays and invasion in transwell assays, while paxillin knockdown decreased both. Similar results have been observed in many other cancers as described throughout this review, suggesting that paxillin family proteins may be useful as both biomarkers and potential treatment targets in cancer.
8. Therapeutic potential
Several current cancer therapeutics work by targeting pathways that involve paxillin and/or Hic-5. Therefore, a better understanding of the role of these proteins may lead to the development of novel therapeutics and diagnostics as well as more effective, targeted application of those already employed in the clinic. Here, we will briefly discuss several examples of chemotherapeutics that are already used clinically and whose mechanism of action is somehow impacted by paxillin or Hic-5 signaling as well as potential future directions in targeting these proteins for treatment
Since paxillin family members have no intrinsic enzymatic activity and belong to an emerging family of proteins containing intrinsically disordered regions (Wright and Dyson, 2014), they are difficult to target directly with small molecule inhibitors (Ambadipudi and Zweckstetter, 2016; Neerathilingam et al., 2016). Intrinsically disordered proteins (IDPs) are proteins which do not fold into consistent, defined, stable structures, but instead fluctuate among a range of 3D conformations (Wright and Dyson, 2014). Post-translational modifications can bias these proteins toward a particular set of conformations, allowing the same protein to favor different interactions in different signaling environments. The interactions between an IDP and a binding partner can often be targeted to prevent signaling, especially if the IDP also contains highly ordered regions, such as the paxillin and Hic-5 LD motifs. This approach has been used to understand the function of the paxillin LD motifs by studying their interactions with FAK (Neerathilingam et al., 2016).
The non-receptor tyrosine kinase, FAK is an important signaling partner of both paxillin and Hic-5, and its activation promotes several pro-tumorigenic pathways, including cell survival, migration, and angiogenesis (Lee et al., 2015; Sulzmaier et al., 2014). Therefore, many small molecule inhibitors have been designed to inhibit its kinase activity and have shown some promise in pre-clinical studies as combinatorial therapies (Kanteti et al., 2016; Lee et al., 2015). However, it is also possible to inhibit FAK signaling by impairing its interaction with paxillin (Deramaudt et al., 2014). Expression of a FAK mutant that cannot bind paxillin results in reduced phosphorylation of FAK and its target substrates, including paxillin, leading to altered adhesion dynamics, reduced cell adhesion, and impaired cell migration. Taking this idea one step further, synthetic antibody inhibitors have been designed against paxillin’s LD2 and LD4 motifs, which are important for its binding to FAK (Nocula-Lugowska et al., 2015; Tumbarello et al., 2002). These synthetic antibodies can act as competitive inhibitors to compete with the focal adhesion targeting domain of FAK and reduce its binding to paxillin (Nocula-Lugowska et al., 2015). The functional outcomes of this inhibition have not yet been studied in detail but based on results seen with FAK mutants deficient in paxillin binding, targeting the paxillin/FAK interaction in this manner may 1 day be an effective therapeutic approach.
Advances in gene therapy may also soon allow modulation of paxillin and Hic-5 expression in vivo by specifically targeting tumor cells with RNAi or CRISPR technologies. For example, siRNAs to paxillin have been be packaged and delivered using carbonate apatite nanocarriers, which accumulate in tumors following intravenous administration in mice (Ashaie et al., 2019). This treatment resulted in significantly decreased tumor volume and weight at all time points in a mouse model of breast cancer relative to those treated with empty carbonate apatite nanocarriers. While CRISPR is currently being used only in clinical trials for gene editing of autologous T-cells (which currently does not involve targeting paxillin or Hic-5), development of delivery vehicles such as these carbonate apatite nanocarriers, polymeric nanoparticles, and liposomes could eventually allow delivery of CRISPR/Cas9 encoding DNA and RNA to tumor cells for in vivo modulation of paxillin expression and its downstream signaling partners (Zhan et al., 2019).
In non-small cell lung cancer (NSCLC), cisplatin is a commonly used treatment that interferes with DNA replication, thereby killing fast-growing cells (Wu et al., 2014a). However, cisplatin resistance is a common problem. Interestingly, when paxillin is overexpressed in lung cancer cell lines, the percentage of apoptotic cells following cisplatin treatment is decreased, and increased doses of cisplatin are required for the same inhibitory effect on cell viability (Wu et al., 2014a). Paxillin has previously been shown to suppress apoptosis through FAK-mediated signaling (Zouq et al., 2009), and this cisplatin-related effect was shown to be mediated by phosphorylation of paxillin at Y31 and Y118, which activates ERK signaling and thereby increases expression of the anti-apoptotic protein Bcl-2 (Wu et al., 2014a). However, cisplatin resistance could be partially overcome in a mouse model by combining cisplatin with an ERK inhibitor. In human NSCLC samples, paxillin phosphorylation positively correlates with ERK phosphorylation and Bcl-2 expression and patients with positive staining for these markers had poorer responses to cisplatin-based chemotherapy than patients with negative staining (Salgia et al., 1999; Wu et al., 2014a). Therefore, ERK inhibitors may serve as a valuable tool to prevent paxillin-mediated signaling and overcome cisplatin resistance in NSCLC.
In addition to targeting downstream kinases, other paxillin-regulated proteins such as MTs are commonly targeted in cancer (Jordan and Wilson, 2004). Two major classes of chemotherapeutic agents—taxanes and vinca alkaloids—bind tubulin and inhibit mitosis by either over-stabilizing microtubules or preventing their assembly, respectively, thereby preventing the assembly of a functional mitotic spindle (Calligaris et al., 2010). Since paxillin also regulates MT stability via acetylation, it would be interesting to explore whether the MT-stabilizing taxanes are more effective in tumors with higher paxillin expression.
Finally, Hic-5 expression may be used as a marker to guide treatment decisions in certain situations. As discussed above, it’s role in ECM deposition and remodeling has now been described in several contexts, including human colorectal cancer and mouse breast cancer. In aggressive human breast cancer subtypes, including Her2-positive, basal-like, and grade three tumors, increased Hic-5 expression correlates with reduced patient survival, suggesting that assessment of tumor stroma Hic-5 levels could be used to guide therapy choices (Goreczny et al., 2018).
9. Conclusion
The paxillin family of proteins are critical players in cancer cell morphology, migration, and invasion. In addition to their well-studied effects on the actin cytoskeleton, their roles in crosstalk with the MT and IF cytoskeletons to modulate cell polarity, trafficking, and ECM remodeling are exciting emerging avenues of investigation. Despite the importance of this crosstalk with other cytoskeletons, the actin cytoskeleton remains a key partner of paxillin, Hic-5, and other focal adhesion proteins in regulating cell migration through dynamic changes in cell morphology and the formation of matrix-degrading structures such as invadopodia. Due to their dysregulation in a wide variety of cancers, targeting paxillin family proteins and their downstream signaling partners may serve as valuable opportunities for diagnostic evaluation and therapeutic intervention in the future.
Funding
Work in the author’s lab was funded by grants from the National Institutes of Health (R35 GM131709, RO1 GM047607, RO1 CA163296) and the Carol M. Baldwin Breast Cancer Research Fund of CNY. We thank the other members of the Turner lab for helpful discussions and apologize to those investigators whose studies we were unable to include in this chapter.
Abbreviations
- CAF
cancer-associated fibroblast
- CDM
cell-derived matrix
- ECM
extracellular matrix
- EMT
epithelial-mesenchymal transition
- ERK
extracellular signal-regulated kinase
- FAK
focal adhesion kinase
- IF
intermediate filament
- MMP
matrix metalloproteinase
- MRTF
myocardin-related transcription factor
- MT
microtubule
- MTOC
microtubule-organizing center
- NSCLC
non-small cell lung cancer
- TGF-β
transforming growth factor beta
- YAP/TAZ
Yes-associated protein/transcriptional coactivator with PDZ-binding motif
References
- Aiello NM, Maddipati R, Norgard RJ, Balli D, Li J, Yuan S, Yamazoe T, Black T, Sahmoud A, Furth EE, Bar-Sagi D, Stanger BZ, 2018. EMT subtype influences epithelial plasticity and mode of cell migration. Dev. Cell 45, 681–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambadipudi S, Zweckstetter M, 2016. Targeting intrinsically disordered proteins in rational drug discovery. Expert Opin. Drug Discov 11, 65–77. [DOI] [PubMed] [Google Scholar]
- Aras S, Zaidi MR, 2017. TAMeless traitors: macrophages in cancer progression and metastasis. Br. J. Cancer 117, 1583–1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashaie MA, Islam RA, Kamaruzman NI, Ibnat N, Tha KK, Chowdhury EH, 2019. Targeting cell adhesion molecules via carbonate apatite-mediated delivery of specific siRNAs to breast cancer cells in vitro and in vivo. Pharmaceutics 11, 309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asthana J, Kapoor S, Mohan R, Panda D, 2013. Inhibition of HDAC6 deacetylase activity increases its binding with microtubules and suppresses microtubule dynamic instability in MCF-7 cells. J. Biol. Chem 288, 22516–22526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atherton P, Lausecker F, Carisey A, Gilmore A, Critchley D, Barsukov I, Ballestrem C, 2020. Relief of talin autoinhibition triggers a force-independent association with vinculin. J. Cell Biol 219, e201903134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badowski C, Pawlak G, Grichine A, Chabadel A, Oddou C, Jurdic P, Pfaff M, Albiges-Rizo C, Block MR, 2008. Paxillin phosphorylation controls invadopodia/podosomes spatiotemporal organization. Mol. Biol. Cell 19, 633–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP, 2009. microRNAs: target recognition and regulatory functions. Cell 136, 215–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battaglia RA, Delic S, Herrmann H, Snider NT, 2018. Vimentin on the move: new developments in cell migration. F1000Research 7, 1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beacham DA, Amatangelo MD, Cukierman E, 2006. Preparation of extracellular matrices produced by cultured and primary fibroblasts. Curr. Protoc. Cell Biol 33, 10.9.1–10.9.21. [DOI] [PubMed] [Google Scholar]
- Bellis SL, Miller JT, Turner CE, 1995. Characterization of tyrosine phosphorylation of paxillin in vitro by focal adhesion kinase. J. Biol. Chem 270, 17437–17441. [DOI] [PubMed] [Google Scholar]
- Bi Y, Han Y, Bi H, Gao F, Wang X, 2014. miR-137 impairs the proliferative and migratory capacity of human non-small cell lung cancer cells by targeting paxillin. Hum. Cell 27, 95–102. [DOI] [PubMed] [Google Scholar]
- Bian X, Liang Z, Feng A, Salgado E, Shim H, 2018. HDAC inhibitor suppresses proliferation and invasion of breast cancer cells through regulation of miR-200c targeting CRKL. Biochem. Pharmacol 147, 30–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birukova AA, Cokic I, Moldobaeva N, Birukov KG, 2009. Paxillin is involved in the differential regulation of endothelial barrier by HGF and VEGF. Am. J. Respir. Cell Mol. Biol 40, 99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissell MJ, Radisky DC, Rizki A, Weaver VM, Petersen OW, 2002. The organizing principle: microenvironmental influences in the normal and malignant breast. Differentiation 70, 537–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boateng LR, Huttenlocher A, 2012. Spatiotemporal regulation of Src and its substrates at invadosomes. Eur. J. Cell Biol 91, 878–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochaton-Piallat M-L, Gabbiani G, Hinz B, 2016. The myofibroblast in wound healing and fibrosis: answered and unanswered questions. F1000Research 5, 752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnans C, Chou J, Werb Z, 2014. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol 15, 786–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth AJR, Blanchard GB, Adams RJ, Röper K, 2014. A dynamic microtubule cytoskeleton directs medial actomyosin function during tube formation. Dev. Cell 29, 562–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boscher C, Gaonac’h-Lovejoy V, Delisle C, Gratton J-P, 2019. Polarization and sprouting of endothelial cells by angiopoietin-1 require PAK2 and paxillin-dependent Cdc42 activation. Mol. Biol. Cell 30, 2227–2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brabletz T, Kalluri R, Nieto MA, Weinberg RA, 2018. EMT in cancer. Nat. Rev. Cancer 18, 128–134. [DOI] [PubMed] [Google Scholar]
- Bravo-Cordero JJ, Hodgson L, Condeelis J, 2012. Directed cell invasion and migration during metastasis. Curr. Opin. Cell Biol 24, 277–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MC, Turner CE, 2002. Roles for the tubulin- and PTP–PEST-binding paxillin LIM domains in cell adhesion and motility. Int. J. Biochem. Cell Biol 34, 855–863. [DOI] [PubMed] [Google Scholar]
- Brown MC, Turner CE, 2004. Paxillin: adapting to change. Physiol. Rev 84, 1315–1339. [DOI] [PubMed] [Google Scholar]
- Brown MC, Perrotta JA, Turner CE, 1996. Identification of LIM3 as the principal determinant of paxillin focal adhesion localization and characterization of a novel motif on paxillin directing vinculin and focal adhesion kinase binding. J. Cell Biol 135, 1109–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MC, Curtis MS, Turner CE, 1998. Paxillin LD motifs may define a new family of protein recognition domains. Nat. Struct. Biol 5, 677–678. [DOI] [PubMed] [Google Scholar]
- Burakov A, Nadezhdina E, Slepchenko B, Rodionov V, 2003. Centrosome positioning in interphase cells. J. Cell Biol 162, 963–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgstaller G, Gregor M, Winter L, Wiche G, 2010. Keeping the vimentin network under control: cell-matrix adhesion-associated plectin 1f affects cell shape and polarity of fibroblasts. Mol. Biol. Cell 21, 3362–3375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buschman MD, Bromann PA, Cejudo-Martin P, Wen F, Pass I, Courtneidge SA, 2009. The novel adaptor protein Tks4 (SH3PXD2B) is required for functional podosome formation. Mol. Biol. Cell 20, 1302–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calle Y, Carragher NO, Thrasher AJ, Jones GE, 2006. Inhibition of calpain stabilises podosomes and impairs dendritic cell motility. J. Cell Sci 119, 2375–2385. [DOI] [PubMed] [Google Scholar]
- Calligaris D, Verdier-Pinard P, Devred F, Villard C, Braguer D, Lafitte D, 2010. Microtubule targeting agents: from biophysics to proteomics. Cell. Mol. Life Sci 67, 1089–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calon A, Lonardo E, Berenguer-Llergo A, Espinet E, Hernando-Momblona X, Iglesias M, Sevillano M, Palomo-Ponce S, Tauriello DVF, Byrom D, Cortina C, Morral C, Barceló C, Tosi S, Riera A, Attolini CSO, Rossell D, Sancho E, Batlle E, 2015. Stromal gene expression defines poor-prognosis subtypes in colorectal cancer. Nat. Genet 47, 320–329. [DOI] [PubMed] [Google Scholar]
- Calvo F, Ege N, Grande-Garcia A, Hooper S, Jenkins RP, Chaudhry SI, Harrington K, Williamson P, Moeendarbary EE, Charras G, Sahai E, 2013. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat. Cell Biol 15, 637–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carragher NO, Walker SM, Scott Carragher LA, Harris F, Sawyer TK, Brunton VG, Ozanne BW, Frame MC, 2006. Calpain 2 and Src dependence distinguishes mesenchymal and amoeboid modes of tumour cell invasion: a link to integrin function. Oncogene 25, 5726–5740. [DOI] [PubMed] [Google Scholar]
- Chaffer CL, Weinberg RA, 2011. A perspective on cancer cell metastasis. Science (80-) 331, 1559–1564. [DOI] [PubMed] [Google Scholar]
- Chen WT, Chen JM, Parsons SJ, Parsons JT, 1985. Local degradation of fibronectin at sites of expression of the transforming gene product pp60src. Nature 316, 156–158. [DOI] [PubMed] [Google Scholar]
- Chen DL, Wang DS, Wu WJ, Zeng ZL, Luo HY, Qiu MZ, Ren C, Zhang DS, Wang ZQ, Wang FH, Li YH, Kang TB, Xu RH, 2013. Overexpression of paxillin induced by miR-137 suppression promotes tumor progression and metastasis in colorectal cancer. Carcinogenesis 34, 803–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Xia L, Xu C, Xiao F, Wang Y, 2016. Paxillin functions as an oncogene in human gliomas by promoting cell migration and invasion. Onco. Targets. Ther 9, 6935–6943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H,Tan P,Qian B,Du Y,Wang A,Shi H,Huang Z,Huang S,Liang T,Fu W, 2020. Hic-5 deficiency protects cerulein-induced chronic pancreatitis via down-regulation of the NF-κB (p65)/IL-6 signalling pathway. J. Cell. Mol. Med 24, 1488–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chodankar R, Wu DY, Schiller BJ, Yamamoto KR, Stallcup MR, 2014. Hic-5 is a transcription coregulator that acts before and/or after glucocorticoid receptor genome occupancy in a gene-selective manner. Proc. Natl. Acad. Sci. U. S. A 111, 4007–4012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AG, Vignjevic DM, 2015. Modes of cancer cell invasion and the role of the microenvironment. Curr. Opin. Cell Biol 36, 13–22. [DOI] [PubMed] [Google Scholar]
- Condeelis J, Segall JE, 2003. Intravital imaging of cell movement in tumours. Nat. Rev. Cancer 3, 921–930. [DOI] [PubMed] [Google Scholar]
- Condeelis J, Weissleder R, 2010. In vivo imaging in cancer. Cold Spring Harb. Perspect. Biol 2, a003848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conklin MW, Eickhoff JC, Riching KM, Pehlke CA, Eliceiri KW, Provenzano PP, Friedl A, Keely PJ, 2011. Aligned collagen is a prognostic signature for survival in human breast carcinoma. Am. J. Pathol 178, 1221–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortesio CL, Boateng LR, Piazza TM, Bennin DA, Huttenlocher A, 2011. Calpain-mediated proteolysis of paxillin negatively regulates focal adhesion dynamics and cell migration. J. Biol. Chem 286, 9998–10006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Côté JF, Turner CE, Tremblay ML, 1999. Intact LIM 3 and LIM 4 domains of paxillin are required for the association to a novel polyproline region (Pro 2) of protein-tyrosine phosphatase-PEST. J. Biol. Chem 274, 20550–20560. [DOI] [PubMed] [Google Scholar]
- Crider BJ, Risinger GM, Haaksma CJ, Howard EW, Tomasek JJ, 2011. Myocardin-related transcription factors A and B are key regulators of TGF-B1-induced fibroblast to myofibroblast differentiation. J. Invest. Dermatol 131, 2378–2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cukierman E, Pankov R, Stevens DR, Yamada KM, 2001. Taking cell-matrix adhesions to the third dimension. Science (80-) 294, 1708–1712. [DOI] [PubMed] [Google Scholar]
- Dabiri G, Tumbarello DA, Turner CE, Van De Water L, 2008. Hic-5 promotes the hypertrophic scar myofibroblast phenotype by regulating the TGF-β1 autocrine loop. J. Invest. Dermatol 128, 2518–2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dave JM, Abbey CA, Duran CL, Seo H, Johnson GA, Bayless KJ, 2016. Hic-5 mediates the initiation of endothelial sprouting by regulating a key surface metalloproteinase. J. Cell Sci 129, 743–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Pascalis C, Etienne-Manneville S, 2017. Single and collective cell migration: the mechanics of adhesions. Mol. Biol. Cell 28, 1833–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deakin NO, Turner CE, 2008. Paxillin comes of age. J. Cell Sci 121, 2435–2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deakin NO, Turner CE, 2011. Distinct roles for paxillin and Hic-5 in regulating breast cancer cell morphology, invasion, and metastasis. Mol. Biol. Cell 22, 327–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deakin NO, Turner CE, 2014. Paxillin inhibits HDAC6 to regulate microtubule acetylation, Golgi structure, and polarized migration. J. Cell Biol 206, 395–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deakin NO, Ballestrem C, Turner CE, 2012a. Paxillin and Hic-5 interaction with vinculin is differentially regulated by Rac1 and RhoA. PLoS One 7, e37990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deakin NO, Pignatelli J, Turner CE, 2012b. Diverse roles for the paxillin family of proteins in cancer. Genes Cancer 3, 362–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deramaudt TBTB, Dujardin D, Noulet F, Martin S, Vauchelles R, Takeda K, Ronde P, Rondé P, 2014. Altering FAK-paxillin interactions reduces adhesion, migration and invasion processes. PLoS One 9, e92059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diepenbruck M, Christofori G, 2016. Epithelial-mesenchymal transition (EMT) and metastasis: yes, no, maybe? Curr. Opin. Cell Biol 43, 7–13. [DOI] [PubMed] [Google Scholar]
- Doyle AD, Wang FW, Matsumoto K, Yamada KM, 2009. One-dimensional topography underlies three-dimensional fibrillar cell migration. J. Cell Biol 184, 481–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du X, Xu Q, Pan D, Xu D, Niu B, Hong W, Zhang R, Li X, Chen S, 2019. Hic-5 in cancer-associated fibroblasts contributes to esophageal squamous cell carcinoma progression. Cell Death Dis. 10, 873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubois F, Alpha K, Turner CE, 2017. Paxillin regulates cell polarization and anterograde vesicle trafficking during cell migration. Mol. Biol. Cell 28, 3815–3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumbauld DW, Shin H, Gallant ND, Michael KE, Radhakrishna H, García AJ, 2010. Contractility modulates cell adhesion strengthening through focal adhesion kinase and assembly of vinculin-containing focal adhesions. J. Cell. Physiol 223, 746–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumbauld DW, Lee TT, Singh A, Scrimgeour J, Gersbach CA, Zamir EA, Fu J, Chen CS, Curtis JE, Craig SW, García AJ, 2013. How vinculin regulates force transmission. Proc. Natl. Acad. Sci. U. S. A 110, 9788–9793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy RJ, Weidmann MD, Sharma VP, Condeelis JS, 2017. Tumor cell invadopodia: invasive protrusions that orchestrate metastasis. Trends Cell Biol. 27, 595–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efimov A, Schiefermeier N, Grigoriev I, Brown MC, Turner CE, Small JV, Kaverina I, 2008. Paxillin-dependent stimulation of microtubule catastrophes at focal adhesion sites. J. Cell Sci 121, 196–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emon B, Bauer J, Jain Y, Jung B, Saif T, 2018. Biophysics of tumor microenvironment and cancer metastasis—a mini review. Comput. Struct. Biotechnol. J 16, 279–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etienne-Manneville S, Hall A, 2002. Rho GTPases in cell biology. Nature 420, 629–635. [DOI] [PubMed] [Google Scholar]
- Faix J, Rottner K, 2006. The making of filopodia. Curr. Opin. Cell Biol 18, 18–25. [DOI] [PubMed] [Google Scholar]
- Finak G, Bertos N, Pepin F, Sadekova S, Souleimanova M, Zhao H, Chen H, Omeroglu G, Meterissian S, Omeroglu A, Hallett M, Park M, 2008. Stromal gene expression predicts clinical outcome in breast cancer. Nat. Med 14, 518–527. [DOI] [PubMed] [Google Scholar]
- Fischer KR, Durrans A, Lee S, Sheng J, Li F, Wong STC, Choi H, El Rayes T, Ryu S, Troeger J, Schwabe RF, Vahdat LT, Altorki NK, Mittal V, Gao D, 2015. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 527, 472–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frantz C, Stewart KM, Weaver VM, 2010. The extracellular matrix at a glance. J. Cell Sci 123, 4195–4200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedl P, Alexander S, 2011. Cancer invasion and the microenvironment: plasticity and reciprocity. Cell 147, 992–1009. [DOI] [PubMed] [Google Scholar]
- Friedl P, Wolf K, 2003. Tumour-cell invasion and migration: diversity and escape mechanisms. Nat. Rev. Cancer 3, 362–374. [DOI] [PubMed] [Google Scholar]
- Friedl P, Wolf K, Zegers MM, 2014. Rho-directed forces in collective migration. Nat. Cell Biol 16, 208–210. [DOI] [PubMed] [Google Scholar]
- Fujimoto N, Yeh S, Kang HY, Inui S, Chang HC, Mizokami A, Chang C, 1999. Cloning and characterization of androgen receptor coactivator, ARA55, in human prostate. J. Biol. Chem 274, 8316–8321. [DOI] [PubMed] [Google Scholar]
- García-Román J, Zentella-Dehesa A, 2013. Vascular permeability changes involved in tumor metastasis. Cancer Lett. 335, 259–269. [DOI] [PubMed] [Google Scholar]
- Garcin C, Straube A, 2019. Microtubules in cell migration. Essays Biochem. 63, 509–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauthier L, Corgnac S, Boutet M, Gros G, Validire P, Bismuth G, Mami-Chouaib F, 2017. Paxillin binding to the cytoplasmic domain of CD103 promotes cell adhesion and effector functions for CD8+ resident memory T cells in tumors. Cancer Res. 77, 7072–7082. [DOI] [PubMed] [Google Scholar]
- Gawlak G, Tian Y, O’Donnell JJ, Tian X, Birukova AA, Birukov KG, 2014. Paxillin mediates stretch-induced Rho signaling and endothelial permeability via assembly of paxillin-p42/44MAPK-GEF-H1 complex. FASEB J. 28, 3249–3260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gkretsi V, Stylianopoulos T, 2018. Cell adhesion and matrix stiffness: coordinating cancer cell invasion and metastasis. Front. Oncol 8, 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gobin E, Bagwell K, Wagner J, Mysona D, Sandirasegarane S, Smith N, Bai S, Sharma A, Schleifer R, She JX, 2019. A pan-cancer perspective of matrix meta-lloproteases (MMP) gene expression profile and their diagnostic/prognostic potential. BMC Cancer 19, 581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gönczy P, 2015. Centrosomes and cancer: revisiting a long-standing relationship. Nat. Rev. Cancer 15, 639–652. [DOI] [PubMed] [Google Scholar]
- Gonzalez H, Hagerling C, Werb Z, 2018. Roles of the immune system in cancer: from tumor initiation to metastatic progression. Genes Dev. 32, 1267–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goreczny GJ, Ouderkirk-Pecone JL, Olson EC, Krendel M, Turner CE, 2017. Hic-5 remodeling of the stromal matrix promotes breast tumor progression. Oncogene 36, 2693–2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goreczny GJ, Forsythe IJ, Turner CE, 2018. Hic-5 regulates fibrillar adhesion formation to control tumor extracellular matrix remodeling through interaction with tensin1. Oncogene 37, 1699–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guignandon A, Boutahar N, Rattner A, Vico L, Lafage-Proust MH, 2006. Cyclic strain promotes shuttling of PYK2/Hic-5 complex from focal contacts in osteoblast-like cells. Biochem. Biophys. Res. Commun 343, 407–414. [DOI] [PubMed] [Google Scholar]
- Gulvady AC, Dubois F, Deakin NO, Goreczny GJ, Turner CE, 2018. Hic-5 expression is a major indicator of cancer cell morphology, migration, and plasticity in three-dimensional matrices. Mol. Biol. Cell 29, 1704–1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulvady AC, Forsythe IJ, Turner CE, 2019. Hic-5 regulates Src-induced invadopodia rosette formation and organization. Mol. Biol. Cell 30, 1298–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamidi H, Ivaska J, 2018. Every step of the way: integrins in cancer progression and metastasis. Nat. Rev. Cancer 18, 533–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA, 2011. Hallmarks of cancer: the next generation. Cell 144, 646–674. [DOI] [PubMed] [Google Scholar]
- Harunaga JS, Yamada KM, 2011. Cell-matrix adhesions in 3D. Matrix Biol. 30, 363–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattori M, Minato N, 2003. Rap1 GTPase: functions, regulation, and malignancy. J. Biochem 134, 479–484. [DOI] [PubMed] [Google Scholar]
- Hayes J, Peruzzi PP, Lawler S, 2014. MicroRNAs in cancer: biomarkers, functions and therapy. Trends Mol. Med 20, 460–469. [DOI] [PubMed] [Google Scholar]
- Heitzer MD, DeFranco DB, 2006a. Hic-5, an adaptor-like nuclear receptor coactivator. Nucl. Recept. Signal 4, e019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heitzer MD, DeFranco DB, 2006b. Mechanism of action of Hic-5/androgen receptor activator 55, a LIM domain-containing nuclear receptor coactivator. Mol. Endocrinol 20, 56–64. [DOI] [PubMed] [Google Scholar]
- Herreros L, Rodríguez-Fernández JL, Brown MC, Alonso-Lebrero JL, Cabañas C, Sánchez-Madrid F, Longo N, Turner CE, Sánchez-Mateos P, 2000. Paxillin localizes to the lymphocyte microtubule organizing center and associates with the microtubule cytoskeleton. J. Biol. Chem 275, 26436–26440. [DOI] [PubMed] [Google Scholar]
- Huber F, Boire A, López MP, Koenderink GH, 2015. Cytoskeletal crosstalk: when three different personalities team up. Curr. Opin. Cell Biol 32, 39–47. [DOI] [PubMed] [Google Scholar]
- Humphrey JD, Dufresne ER, Schwartz MA, 2014. Mechanotransduction and extracellular matrix homeostasis. Nat. Rev. Mol. Cell Biol 15, 802–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphries JD, Wang P, Streuli C, Geiger B, Humphries MJ, Ballestrem C, 2007. Vinculin controls focal adhesion formation by direct interactions with talin and actin. J. Cell Biol 179, 1043–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioachim E, Charchanti A, Briasoulis E, Karavasilis V, Tsanou H, Arvanitis D, Agnantis N, Pavlidis N, 2002. Immunohistochemical expression of extracellular matrix components tenascin, fibronectin, collagen type IV and laminin in breast cancer: their prognostic value and role in tumour invasion and progression. Eur. J. Cancer 38, 2362–2370. [DOI] [PubMed] [Google Scholar]
- Ishibe S, Joly D, Liu ZX, Cantley LG, 2004. Paxillin serves as an ERK-regulated scaffold for coordinating FAK and Rac activation in epithelial morphogenesis. Mol. Cell 16, 257–267. [DOI] [PubMed] [Google Scholar]
- Jacob AE, Turner CE, Amack JD, 2016. Evolution and expression of paxillin genes in teleost fish. PLoS One 11, e0165266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagadeeswaran R, Surawska H, Krishnaswamy S, Janamanchi V, Mackinnon AC, Seiwert TY, Loganathan S, Kanteti R, Reichman T, Nallasura V, Schwartz S, Faoro L, Wang YC, Girard L, Tretiakova MS, Ahmed S, Zumba O, Soulii L, Bindokas VP, Szeto LL, Gordon GJ, Bueno R, Sugarbaker D, Lingen MW, Sattler M, Krausz T, Vigneswaran W, Natarajan V, Minna J, Vokes EE, Ferguson MK, Husain AN, Salgia R, 2008. Paxillin is a target for somatic mutations in lung cancer: implications for cell growth and invasion. Cancer Res. 68, 132–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamieson JS, Tumbarello DA, Hallé M, Brown MC, Tremblay ML, Turner CE, 2005. Paxillin is essential for PTP-PEST-dependent regulation of cell spreading and motility: a role for paxillin kinase linker. J. Cell Sci 118, 5835–5847. [DOI] [PubMed] [Google Scholar]
- Janke C, Bulinski JC, 2011. Post-translational regulation of the microtubule cytoskeleton: mechanisms and functions. Nat. Rev. Mol. Cell Biol 12, 773–786. [DOI] [PubMed] [Google Scholar]
- Janke C, Montagnac G, 2017. Causes and consequences of microtubule acetylation. Curr. Biol 27, R1287–R1292. [DOI] [PubMed] [Google Scholar]
- Jansen KA, Donato DM, Balcioglu HE, Schmidt T, Danen EHJ, Koenderink GH, 2015. A guide to mechanobiology: where biology and physics meet. Biochim. Biophys. Acta Mol. Cell Res 1853, 3043–3052. [DOI] [PubMed] [Google Scholar]
- Jordan MA, Wilson L, 2004. Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 4, 253–265. [DOI] [PubMed] [Google Scholar]
- Kadrmas JL, Beckerle MC, 2004. The LIM domain: from the cytoskeleton to the nucleus. Nat. Rev. Mol. Cell Biol 5, 920–931. [DOI] [PubMed] [Google Scholar]
- Kalluri R, 2016. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 16, 582–598. [DOI] [PubMed] [Google Scholar]
- Kanteti R, Batra SK, Lennon FE, Salgia R, 2016. FAK and paxillin, two potential targets in pancreatic cancer. Oncotarget 7, 31586–31601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kareva I, Abou-Slaybi A, Dodd O, Dashevsky O, Klement GL, 2016. Normal wound healing and tumor angiogenesis as a game of competitive inhibition. PLoS One 11, e0166655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasai M, Guerrero-Santoro J, Friedman R, Leman ES, Getzenberg RH, DeFranco DB, 2003. The group 3 LIM domain protein paxillin potentiates androgen receptor transactivation in prostate cancer cell lines. Cancer Res. 63, 4927–4935. [PubMed] [Google Scholar]
- Katsumi A, Naoe T, Matsushita T, Kaibuchi K, Schwartz MA, 2005. Integrin activation and matrix binding mediate cellular responses to mechanical stretch. J. Biol. Chem 280, 16546–16549. [DOI] [PubMed] [Google Scholar]
- Kaverina I, Krylyshkina O, Small JV, 1999. Microtubule targeting of substrate contacts promotes their relaxation and dissociation. J. Cell Biol 146, 1033–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim-Kaneyama J, Suzuki W, Ichikawa K, Ohki T, Kohno Y, Sata M, Nose K, Shibanuma M, 2005. Uni-axial stretching regulates intracellular localization of Hic-5 expressed in smooth-muscle cells in vivo. J. Cell Sci 118, 937–949. [DOI] [PubMed] [Google Scholar]
- Kim-Kaneyama J.r., Wachi N, Sata M, Enomoto S, Fukabori K, Koh K, Shibanuma M, Nose K, 2008. Hic-5, an adaptor protein expressed in vascular smooth muscle cells, modulates the arterial response to injury in vivo. Biochem. Biophys. Res. Commun 376, 682–687. [DOI] [PubMed] [Google Scholar]
- Kim-Kaneyama JR, Takeda N, Sasai A, Miyazaki A, Sata M, Hirabayashi T, Shibanuma M, Yamada G, Nose K, 2011. Hic-5 deficiency enhances mechanosensitive apoptosis and modulates vascular remodeling. J. Mol. Cell. Cardiol 50, 77–86. [DOI] [PubMed] [Google Scholar]
- Kim-Kaneyama J.r., Lei XF, Arita S, Miyauchi A, Miyazaki T, Miyazaki A, 2012. Hydrogen peroxide-inducible clone 5 (Hic-5) as a potential therapeutic target for vascular and other disorders. J. Atheroscler. Thromb 19, 601–609. [DOI] [PubMed] [Google Scholar]
- Kleinschmidt EG, Schlaepfer DD, 2017. Focal adhesion kinase signaling in unexpected places. Curr. Opin. Cell Biol 45, 24–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korsching E, Packeisen J, Liedtke C, Hungermann D, Wülfing P, van Diest PJ, Brandt B, Boecker W, Buerger H, 2005. The origin of vimentin expression in invasive breast cancer: epithelial-mesenchymal transition, myoepithelial histogenesis or histogenesis from progenitor cells with bilinear differentiation potential? J. Pathol 206, 451–457. [DOI] [PubMed] [Google Scholar]
- Kramer N, Walzl A, Unger C, Rosner M, Krupitza G, Hengstschläger M, Dolznig H, 2013. In vitro cell migration and invasion assays. Mutat. Res. Rev. Mutat. Res 752, 10–24. [DOI] [PubMed] [Google Scholar]
- Ku H, Meier KE, 2000. Phosphorylation of paxillin via the ERK mitogen-activated protein kinase cascade in EL4 thymoma cells. J. Biol. Chem 275, 11333–11340. [DOI] [PubMed] [Google Scholar]
- Kusenda B, Mraz M, Mayer J, Pospisilova S, 2006. microRNA biogenesis, functionality and cancer relevance. Biomed. Pap 150, 205–215. [DOI] [PubMed] [Google Scholar]
- Lümmermann T, Sixt M, 2009. Mechanical modes of “amoeboid” cell migration. Curr. Opin. Cell Biol 21, 636–644. [DOI] [PubMed] [Google Scholar]
- Langley RR, Fidler IJ, 2011. The seed and soil hypothesis revisited—the role of tumor-stroma interactions in metastasis to different organs. Int. J. Cancer 128, 2527–2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauffenburger DA, Horwitz AF, 1996. Cell migration: a physically integrated molecular process. Cell 84, 359–369. [DOI] [PubMed] [Google Scholar]
- Lawson CD, Burridge K, 2014. The on-off relationship of Rho and Rac during integrin-mediated adhesion and cell migration. Small GTPases 5, e27958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson CD, Ridley AJ, 2018. Rho GTPase signaling complexes in cell migration and invasion. J. Cell Biol 217, 447–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leduc C, Etienne-Manneville S, 2015. Intermediate filaments in cell migration and invasion: the unusual suspects. Curr. Opin. Cell Biol 32, 102–112. [DOI] [PubMed] [Google Scholar]
- Lee BY, Timpson P, Horvath LG, Daly RJ, 2015. FAK signaling in human cancer as a target for therapeutics. Pharmacol. Ther 146, 132–149. [DOI] [PubMed] [Google Scholar]
- Leong HS, Robertson AE, Stoletov K, Leith SJ, Chin CA, Chien AE, Hague MN, Ablack A, Carmine-Simmen K, McPherson VA, Postenka CO, Turley EA, Courtneidge SA, Chambers AF, Lewis JD, 2014. Invadopodia are required for cancer cell extravasation and are a therapeutic target for metastasis. Cell Rep. 8, 1558–1570. [DOI] [PubMed] [Google Scholar]
- Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, Fong SFT, Csiszar K, Giaccia A, Weninger W, Yamauchi M, Gasser DL, Weaver VM, 2009. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 139, 891–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Li Z, Xiong J, Gong B, Zhang G, Cao C, Jie Z, Liu Y, Cao Y, Yan Y, Xiong H, Qiu L, Yang M, Chen H, Jiang S, Yang X, Chen H, 2015. microRNA-212 functions as an epigenetic-silenced tumor suppressor involving in tumor metastasis and invasion of gastric cancer through down-regulating PXN expression. Am. J. Cancer Res 5, 2980–2997. [PMC free article] [PubMed] [Google Scholar]
- Li T, Zhang C, Hassan S, Liu X, Song F, Chen K, Zhang W, Yang J, 2018. Histone deacetylase 6 in cancer. J. Hematol. Oncol 11, 111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin KT, Yeh YM, Chuang CM, Yang SY, Chang JW, Sun SP, Wang YS, Chao KC, Wang LH, 2015. Glucocorticoids mediate induction of microRNA-708 to suppress ovarian cancer metastasis through targeting Rap1B. Nat. Commun 6, 5917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linder S, 2007. The matrix corroded: podosomes and invadopodia in extracellular matrix degradation. Trends Cell Biol. 17, 107–117. [DOI] [PubMed] [Google Scholar]
- Linder S, 2009. Invadosomes at a glance. J. Cell Sci 122, 3009–3013. [DOI] [PubMed] [Google Scholar]
- Lippincott-Schwartz J, Roberts TH, Hirschberg K, 2000. Secretory protein trafficking and organelle dynamics in living cells. Annu. Rev. Cell Dev. Biol 16, 557–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipsky BP, Beals CR, Staunton DE, 1998. Leupaxin is a novel LIM domain protein that forms a complex with PYK2. J. Biol. Chem 273, 11709–11713. [DOI] [PubMed] [Google Scholar]
- Liu YJ, Le Berre M, Lautenschlaeger F, Maiuri P, Callan-Jones A, Heuzé M, Takaki T, Voituriez R, Piel M, 2015. Confinement and low adhesion induce fast amoeboid migration of slow mesenchymal cells. Cell 160, 659–672. [DOI] [PubMed] [Google Scholar]
- Lohmer LL, Kelley LC, Hagedorn EJ, Sherwood DR, 2014. Invadopodia and basement membrane invasion in vivo. Cell Adh. Migr 8, 246–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo T-H, Ng Y-W, Lim L, Manser E, 2004. GIT1 activates p21-activated kinase through a mechanism independent of p21 binding. Mol. Cell. Biol 24, 3849–3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Colomé AM, Lee-Rivera I, Benavides-Hidalgo R, López E, 2017. Paxillin: a crossroad in pathological cell migration. J. Hematol. Oncol 10, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowery J, Kuczmarski ER, Herrmann H, Goldman RD, 2015. Intermediate filaments play a pivotal role in regulating cell architecture and function. J. Biol. Chem 290, 17145–17153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Hammes SR, 2018. Paxillin actions in the nucleus. Steroids 133, 87–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Biswas A, Hammes SR, 2019. Paxillin regulated genomic networks in prostate cancer. Steroids 151, 108463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maller O, DuFort CC, Weaver VM, 2013. YAP forces fibroblasts to feel the tension. Nat. Cell Biol 15, 570–572. [DOI] [PubMed] [Google Scholar]
- Martín-Cófreces NB, Robles-Valero J, Cabrero JR, Mittelbrunn M, Gordón-Alonso M, Sung CH, Alarcón B, Vázquez J, Sánchez-Madrid F, 2008. MTOC translocation modulates IS formation and controls sustained T cell signaling. J. Cell Biol 182, 951–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuyama R, Okuzaki D, Okada M, Oneyama C, 2016. microRNA-27b suppresses tumor progression by regulating ARFGEF1 and focal adhesion signaling. Cancer Sci. 107, 28–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellman I, Nelson WJ, 2008. Coordinated protein sorting, targeting and distribution in polarized cells. Nat. Rev. Mol. Cell Biol 9, 833–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori K, Uchida T, Yoshie T, Mizote Y, Ishikawa F, Katsuyama M, Shibanuma M, 2019. A mitochondrial ROS pathway controls matrix metalloproteinase 9 levels and invasive properties in RAS-activated cancer cells. FEBS J. 286, 459–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouw JK, Ou G, Weaver VM, 2014. Extracellular matrix assembly: a multiscale deconstruction. Nat. Rev. Mol. Cell Biol 15, 771–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy DA, Courtneidge SA, 2011. The “ins” and “outs” of podosomes and invadopodia: characteristics, formation and function. Nat. Rev. Mol. Cell Biol 12, 413–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naslavsky N, Caplan S, 2018. The enigmatic endosome—sorting the ins and outs of endocytic trafficking. J. Cell Sci 131, jcs216499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayal A, Webb DJ, Brown CM, Schaefer EM, Vicente-Manzanares M, Horwitz AR, 2006. Paxillin phosphorylation at Ser273 localizes a GIT1-PIX-PAK complex and regulates adhesion and protrusion dynamics. J. Cell Biol 173, 587–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neerathilingam M, Bairy SG, Mysore S, 2016. Deciphering mode of action of functionally important regions in the intrinsically disordered paxillin (residues 1–313) using its interaction with FAT (focal adhesion targeting domain of focal adhesion kinase). PLoS One 11, e0150153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolopoulos SN, Turner CE, 2000. Actopaxin, a new focal adhesion protein that binds paxillin LD motifs and actin and regulates cell adhesion. J. Cell Biol 151, 1435–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolopoulos SN, Turner CE, 2001. Integrin-linked kinase (ILK) binding to paxillin LD1 motif regulates ILK localization to focal adhesions. J. Biol. Chem 276, 23499–23505. [DOI] [PubMed] [Google Scholar]
- Nishida N, Yano H, Nishida T, Kamura T, Kojiro M, 2006. Angiogenesis in cancer. Vasc. Health Risk Manag 2, 213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiya N, Tachibana K, Shibanuma M, Mashimo J, Nose K, 2001. Hic-5-reduced cell spreading on fibronectin: competitive effects between paxillin and Hic-5 through interaction with focal adhesion kinase. Mol. Cell. Biol 21, 5332–5345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiya N, Shirai T, Suzuki W, Nose K, 2002. Hic-5 interacts with GIT1 with a different binding mode from paxillin. J. Biochem 132, 279–289. [DOI] [PubMed] [Google Scholar]
- Nocula-Lugowska M, Lugowski M, Salgia R, Kossiakoff AA, 2015. Engineering synthetic antibody inhibitors specific for LD2 or LD4 motifs of paxillin. J. Mol. Biol 427, 2532–2547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson EN, Nordheim A, 2010. Linking actin dynamics and gene transcription to drive cellular motile functions. Nat. Rev. Mol. Cell Biol 11, 353–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omoto T, Kim-Kaneyama J, Lei X-F, Orimo A, Ohnishi K, Yoshihara K, Miyauchi A, Li S, Gao L, Umemoto T, Tanaka J, Nakahara K, Takeya M, Ishida F, Kudo S, Haraguchi S, Miyazaki T, Miyazaki A, 2018. The impact of stromal Hic-5 on the tumorigenesis of colorectal cancer through lysyl oxidase induction and stromal remodeling. Oncogene 37, 1205–1219. [DOI] [PubMed] [Google Scholar]
- Osborn M, Weber K, 1983. Tumor diagnosis by intermediate filament typing: a novel tool for surgical pathology. Lab. Investig 48, 372–394. [PubMed] [Google Scholar]
- Oser M, Yamaguchi H, Mader CC, Bravo-Cordero JJ, Arias M, Chen X, DesMarais V, Van Rheenen J, Koleske AJ, Condeelis J, 2009. Cortactin regulates cofilin and N-WASp activities to control the stages of invadopodium assembly and maturation. J. Cell Biol 186, 571–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osmanagic-Myers S, Rus S, Wolfram M, Brunner D, Goldmann WH, Bonakdar N, Fischer I, Reipert S, Zuzuarregui A, Walko G, Wiche G, 2015. Plectin reinforces vascular integrity by mediating crosstalk between the vimentin and the actin networks. J. Cell Sci 128, 4138–4150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padmanaban V, Krol I, Suhail Y, Szczerba BM, Aceto N, Bader JS, Ewald AJ, 2019. E-cadherin is required for metastasis in multiple models of breast cancer. Nature 573, 439–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paget S, 1889. The distribution of secondary growths in cancer of the breast. Lancet 133, 571–573. [PubMed] [Google Scholar]
- Palazzo AF, Eng CH, Schlaepfer DD, Marcantonio EE, Gundersen GG, 2004. Localized stabilization of microtubules by integrin- and FAK-facilitated Rho signaling. Science (80-) 303, 836–839. [DOI] [PubMed] [Google Scholar]
- Pandya PH, Murray ME, Pollok KE, Renbarger JL, 2016. The immune system in cancer pathogenesis: potential therapeutic approaches. J. Immunol. Res 2016, 4273943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankov R, Endo Y, Even-Ram S, Araki M, Clark K, Cukierman E, Matsumoto K, Yamada KM, 2005. A Rac switch regulates random versus directionally persistent cell migration. J. Cell Biol 170, 793–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panzer L, Trü be L, Klose M, Joosten B, Slotman J, Cambi A, Linder S, 2016. The formins FHOD1 and INF2 regulate inter- and intra-structural contractility of podosomes. J. Cell Sci 129, 298–313. [DOI] [PubMed] [Google Scholar]
- Parsons JT, Horwitz AR, Schwartz MA, 2010. Cell adhesion: integrating cytoskeletal dynamics and cellular tension. Nat. Rev. Mol. Cell Biol 11, 633–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastushenko I, Brisebarre A, Sifrim A, Fioramonti M, Revenco T, Boumahdi S, Van Keymeulen A, Brown D, Moers V, Lemaire S, De Clercq S, Minguijón E, Balsat C, Sokolow Y, Dubois C, De Cock F, Scozzaro S, Sopena F, Lanas A, D’Haene N, Salmon I, Marine J-C, Voet T, Sotiropoulou PA, Blanpain C, 2018. Identification of the tumour transition states occurring during EMT. Nature 556, 463–468. [DOI] [PubMed] [Google Scholar]
- Patteson AE, Vahabikashi A, Pogoda K, Adam SA, Mandal K, Kittisopikul M, Sivagurunathan S, Goldman A, Goldman RD, Janmey PA, 2019. Vimentin protects cells against nuclear rupture and DNA damage during migration. J. Cell Biol 218, 4079–4092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul NR, Jacquemet G, Caswell PT, 2015. Endocytic trafficking of integrins in cell migration. Curr. Biol 25, R1092–R1105. [DOI] [PubMed] [Google Scholar]
- Pease JC, Tirnauer JS, 2011. Mitotic spindle misorientation in cancer—out of alignment and into the fire. J. Cell Sci 124, 1007–1016. [DOI] [PubMed] [Google Scholar]
- Peng Y, Croce CM, 2016. The role of microRNAs in human cancer. Signal Transduct. Target. Ther 1, 15004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petit V, Boyer B, Lentz D, Turner CE, Thiery JP, Valles AM, 2000. Phosphorylation of tyrosine residues 31 and 118 on paxillin regulates cell migration through an association with CRK in NBT-II cells. J. Cell Biol 148, 957–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrie RJ, Yamada KM, 2012. At the leading edge of three-dimensional cell migration. J. Cell Sci 125, 5917–5926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrie RJ, Yamada KM, 2016. Multiple mechanisms of 3D migration: the origins of plasticity. Curr. Opin. Cell Biol 42, 7–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrie RJ, Doyle AD, Yamada KM, 2009. Random versus directionally persistent cell migration. Nat. Rev. Mol. Cell Biol 10, 538–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrie RJ, Gavara N, Chadwick RS, Yamada KM, 2012. Nonpolarized signaling reveals two distinct modes of 3D cell migration. J. Cell Biol 197, 439–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petropoulos C, Oddou C, Emadali A, Hiriart-Bryant E, Boyault C, Faurobert E, Pol SV, Kim-Kaneyama JR, Kraut A, Coute Y, Block MR, Albiges-Rizo C, Destaing O, 2016. Roles of paxillin family members in adhesion and ECM degradation coupling at invadosomes. J. Cell Biol 213, 585–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pignatelli J, LaLonde SE, LaLonde DP, Clarke D, Turner CE, 2012a. Actopaxin (α-parvin) phosphorylation is required for matrix degradation and cancer cell invasion. J. Biol. Chem 287, 37309–37320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pignatelli J, Tumbarello DA, Schmidt RP, Turner CE, 2012b. Hic-5 promotes invadopodia formation and invasion during TGF-β-induced epithelial-mesenchymal transition. J. Cell Biol 197, 421–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pocaterra A, Romani P, Dupont S, 2020. YAP/TAZ functions and their regulation at a glance. J. Cell Sci 133, jcs230425. [DOI] [PubMed] [Google Scholar]
- Pollard TD, Borisy GG, 2003. Cellular motility driven by assembly and disassembly of actin filaments. Cell 112, 453–465. [DOI] [PubMed] [Google Scholar]
- Provenzano PP, Keely PJ, 2011. Mechanical signaling through the cytoskeleton regulates cell proliferation by coordinated focal adhesion and Rho GTPase signaling. J. Cell Sci 124, 1195–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provenzano PP, Eliceiri KW, Campbell JM, Inman DR, White JG, Keely PJ, 2006. Collagen reorganization at the tumor-stromal interface facilitates local invasion. BMC Med. 4, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provenzano PP, Inman DR, Eliceiri KW, Knittel JG, Yan L, Rueden CT, White JG, Keely PJ, 2008. Collagen density promotes mammary tumor initiation and progression. BMC Med. 6, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian B, Wei L, Yang Z, He Q, Chen H, Wang A, Yang D, Li Q, Li J, Zheng S, Fu W, 2020. Hic-5 in pancreatic stellate cells affects proliferation, apoptosis, migration, invasion of pancreatic cancer cells and postoperative survival time of pancreatic cancer. Biomed. Pharmacother 121, 109355. [DOI] [PubMed] [Google Scholar]
- Qin J, Wang F, Jiang H, Xu J, Jiang Y, Wang Z, 2015. microRNA-145 suppresses cell migration and invasion by targeting paxillin in human colorectal cancer cells. Int. J. Clin. Exp. Pathol 8, 1328–1340. [PMC free article] [PubMed] [Google Scholar]
- Quail DF, Joyce JA, 2013. Microenvironmental regulation of tumor progression and metastasis. Nat. Med 19, 1423–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raftopoulou M, Hall A, 2004. Cell migration: Rho GTPases lead the way. Dev. Biol 265, 23–32. [DOI] [PubMed] [Google Scholar]
- Rajabi M, Mousa S, 2017. The role of angiogenesis in cancer treatment. Biomedicines 5, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rashid M, Belmont J, Carpenter D, Turner CE, Olson EC, 2017. Neural-specific deletion of the focal adhesion adaptor protein paxillin slows migration speed and delays cortical layer formation. Development 144, 4002–4014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, Barrette T, Pandey A, Chinnaiyan AM, 2004. ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia 6, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridley AJ, 2015. Rho GTPase signalling in cell migration. Curr. Opin. Cell Biol 36, 103–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridley AJ, Schwartz MA, Burridge K, Firtel RA, Ginsberg MH, Borisy G, Parsons JT, Horwitz AR, 2003. Cell migration: integrating signals from front to back. Science (80-) 302, 1704–1709. [DOI] [PubMed] [Google Scholar]
- Robertson LK, Ostergaard HL, 2011. Paxillin associates with the microtubule cytoskeleton and the immunological synapse of CTL through its leucine-aspartic acid domains and contributes to microtubule organizing center reorientation. J. Immunol 187, 5824–5833. [DOI] [PubMed] [Google Scholar]
- Ryan SD, Bhanot K, Ferrier A, De Repentigny Y, Chu A, Blais A, Kothary R, 2012. Microtubule stability, Golgi organization, and transport flux require dystonin-a2-MAP1B interaction. J. Cell Biol 196, 727–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahai E, Marshall CJ, 2003. Differing modes for tumour cell invasion have distinct requirements for Rho/ROCK signalling and extracellular proteolysis. Nat. Cell Biol 5, 711–719. [DOI] [PubMed] [Google Scholar]
- Salgia R, Li J-L, Ewaniuk DS, Wang Y-B, Sattler M, Chen W-C, Richards W, Pisick E, Shapiro GI, Rollins BJ, Chen LB, Griffin JD, Sugarbaker DJ, 1999. Expression of the focal adhesion protein paxillin in lung cancer and its relation to cell motility. Oncogene 18, 67–77. [DOI] [PubMed] [Google Scholar]
- Sanz-Moreno V, Gadea G, Ahn J, Paterson H, Marra P, Pinner S, Sahai E, Marshall CJ, 2008. Rac activation and inactivation control plasticity of tumor cell movement. Cell 135, 510–523. [DOI] [PubMed] [Google Scholar]
- Schaeffer D, Somarelli JA, Hanna G, Palmer GM, Garcia-Blanco MA, 2014. Cellular migration and invasion uncoupled: increased migration is not an inexorable consequence of epithelial-to-mesenchymal transition. Mol. Cell. Biol 34, 3486–3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaller MD, 2001. Paxillin: a focal adhesion-associated adaptor protein. Oncogene 20, 6459–6472. [DOI] [PubMed] [Google Scholar]
- Shamir ER, Ewald AJ, 2014. Three-dimensional organotypic culture: experimental models of mammalian biology and disease. Nat. Rev. Mol. Cell Biol 15, 647–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao DD, Xue W, Krall EB, Bhutkar A, Piccioni F, Wang X, Schinzel AC, Sood S, Rosenbluh J, Kim JW, Zwang Y, Roberts TM, Root DE, Jacks T, Hahn WC, 2014. KRAS and YAP1 converge to regulate EMT and tumor survival. Cell 158, 171–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharifi MN, Mowers EE, Drake LE, Collier C, Chen H, Zamora M, Mui S, Macleod KF, 2016. Autophagy promotes focal adhesion disassembly and cell motility of metastatic tumor cells through the direct interaction of paxillin with LC3. Cell Rep. 15, 1660–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharifi MN, Mowers EE, Macleod KF, 2017. Autophagic degradation of focal adhesions underlies metastatic cancer dissemination. Mol. Cell. Oncol 4, 1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma VP, Eddy R, Entenberg D, Kai M, Gertler FB, Condeelis J, 2013. Tks5 and SHIP2 regulate invadopodium maturation, but not initiation, in breast carcinoma cells. Curr. Biol 23, 2079–2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibanuma M, Mashimo JI, Kuroki T, Nose K, 1994. Characterization of the TGFβ1-inducible hic-5 gene that encodes a putative novel zinc finger protein and its possible involvement in cellular senescence. J. Biol. Chem 269, 26767–26774. [PubMed] [Google Scholar]
- Shibanuma M, Kim-Kaneyama J, Ishino K, Sakamoto N, Hishiki T, Yamaguchi K, Mori K, Mashimo J, Nose K, 2003. Hic-5 communicates between focal adhesions and the nucleus through oxidant-sensitive nuclear export signal. Mol. Biol. Cell 14, 1158–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibanuma M, Kim-Kaneyama JR, Sato S, Nose K, 2004. A LIM protein, Hic-5, functions as a potential coactivator for Sp1. J. Cell. Biochem 91, 633–645. [DOI] [PubMed] [Google Scholar]
- Shibanuma M, Mori K, Nose K, 2012. Hic-5: a mobile molecular scaffold regulating the anchorage dependence of cell growth. Int. J. Cell Biol 2012, 426138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sit S-T, Manser E, 2011. Rho GTPases and their role in organizing the actin cytoskeleton. J. Cell Sci 124, 679–683. [DOI] [PubMed] [Google Scholar]
- Smith MA, Blankman E, Deakin NO, Hoffman LM, Jensen CC, Turner CE, Beckerle MC, 2013. LIM domains target actin regulators paxillin and zyxin to sites of stress fiber strain. PLoS One 8, e69378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiering D, Hodgson L, 2011. Dynamics of the rho-family small GTPases in actin regulation and motility. Cell Adh. Migr 5, 170–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subauste MC, Pertz O, Adamson ED, Turner CE, Junger S, Hahn KM, 2004. Vinculin modulation of paxillin-FAK interactions regulates ERK to control survival and motility. J. Cell Biol 165, 371–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulzmaier FJ, Jean C, Schlaepfer DD, 2014. FAK in cancer: mechanistic findings and clinical applications. Nat. Rev. Cancer 14, 598–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L-H, Yang F-Q, Zhang C-B, Wu Y-P, Liang J-S, Jin S, Wang Z, Wang H-J, Bao Z-S, Yang Z-X, Jiang T, 2017. Overexpression of paxillin correlates with tumor progression and predicts poor survival in glioblastoma. CNS Neurosci. Ther 23, 69–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang K, Boudreau CG, Brown CM, Khadra A, 2018. Paxillin phosphorylation at serine 273 and its effects on Rac, Rho and adhesion dynamics. PLoS Comput. Biol 14, 1–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao WY, Wang CY, Sun YH, Su YH, Pang D, Zhang GQ, 2016. microRNA-34c suppresses breast cancer migration and invasion by targeting GIT1. J. Cancer 7, 1653–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- te Boekhorst V, Friedl P, 2016. Plasticity of cancer cell invasion: mechanisms and implications for therapy In: Advances in Cancer Research, first ed. Elsevier Inc. [DOI] [PubMed] [Google Scholar]
- Theodosiou M, Widmaier M, Böttcher RT, Rognoni E, Veelders M, Bharadwaj M, Lambacher A, Austen K, Müller DJ, Zent R, Fässler R, 2016. Kindlin-2 cooperates with talin to activate integrins and induces cell spreading by directly binding paxillin. Elife 5, e10130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theveneau E, Linker C, 2017. Leaders in collective migration: are front cells really endowed with a particular set of skills? F1000Research 6, 1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thievessen I, Fakhri N, Steinwachs J, Kraus V, McIsaac RS, Gao L, Chen BC, Baird MA, Davidson MW, Betzig E, Oldenbourg R, Waterman CM, Fabry B, 2015. Vinculin is required for cell polarization, migration, and extracellular matrix remodeling in 3D collagen. FASEB J. 29, 4555–4567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JW, Cooley MA, Broome JM, Salgia R, Griffin JD, Lombardo CR, Schaller MD, 1999a. The role of focal adhesion kinase binding in the regulation of tyrosine phosphorylation of paxillin. J. Biol. Chem 274, 36684–36692. [DOI] [PubMed] [Google Scholar]
- Thomas SM, Hagel M, Turner CE, 1999b. Characterization of a focal adhesion protein, Hic-5, that shares extensive homology with paxillin. J. Cell Sci 112, 181–190. [DOI] [PubMed] [Google Scholar]
- Thommen DS, Schumacher TN, 2018. T cell dysfunction in cancer. Cancer Cell 33, 547–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thul PJ, Akesson L, Wiking M, Mahdessian D, Geladaki A, Ait Blal H, Alm T, Asplund A, Björk L, Breckels LM, Bäckström A, Danielsson F, Fagerberg L, Fall J, Gatto L, Gnann C, Hober S, Hjelmare M, Johansson F, Lee S, Lindskog C, Mulder J, Mulvey CM, Nilsson P, Oksvold P, Rockberg J, Schutten R, Schwenk JM, Sivertsson A, Sjöstedt E, Skogs M, Stadler C, Sullivan DP, Tegel H, Winsnes C, Zhang C, Zwahlen M, Mardinoglu A, Pontén F, Von Feilitzen K, Lilley KS, Uhlén M, Lundberg E, 2017. A subcellular map of the human proteome. Science 356, eaa13321. [DOI] [PubMed] [Google Scholar]
- Todd JR, Ryall KA, Vyse S, Wong JP, Natrajan RC, Yuan Y, Tan AC, Huang PH, 2016. Systematic analysis of tumour cell-extracellular matrix adhesion identifies independent prognostic factors in breast cancer. Oncotarget 7, 62939–62953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Totaro A, Panciera T, Piccolo S, 2018. YAP/TAZ upstream signals and downstream responses. Nat. Cell Biol 20, 888–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsubouchi A, Sakakura J, Yagi R, Mazaki Y, Schaefer E, Yano H, Sabe H, 2002. Localized suppression of RhoA activity by Tyr31/118-phosphorylated paxillin in cell adhesion and migration. J. Cell Biol 159, 673–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumbarello DA, Turner CE, 2007. Hic-5 contributes to epithelial-mesenchymal transformation through a RhoA/ROCK-dependent pathway. J. Cell. Physiol 211, 736–747. [DOI] [PubMed] [Google Scholar]
- Tumbarello DA, Brown MC, Turner CE, 2002. The paxillin LD motifs. FEBS Lett. 513, 114–118. [DOI] [PubMed] [Google Scholar]
- Turner CE, 2000a. Paxillin and focal adhesion signalling. Nat. Cell Biol 2, E231–E236. [DOI] [PubMed] [Google Scholar]
- Turner CE, 2000b. Paxillin interactions. J. Cell Sci 113, 4139–4140. [DOI] [PubMed] [Google Scholar]
- Turner CE, Miller JT, 1994. Primary sequence of paxillin contains putative SH2 and SH3 domain binding motifs and multiple LIM domains: identification of a vinculin and pp125 (Fak)-binding region. J. Cell Sci 107, 1583–1591. [DOI] [PubMed] [Google Scholar]
- Turner CE, Glenney JR, Burridge K, 1990. Paxillin: a new vinculin-binding protein present in focal adhesions. J. Cell Biol 111, 1059–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner CE, Kramarcy N, Sealock R, Burridge K, 1991. Localization of paxillin, a focal adhesion protein, to smooth muscle dense plaques, and the myotendinous and neuromuscular junctions of skeletal muscle. Exp. Cell Res 192, 651–655. [DOI] [PubMed] [Google Scholar]
- Turner CE, Brown MC, Perrotta JA, Riedy MC, Nikolopoulos SN, McDonald AR, Bagrodia S, Thomas S, Leventhal PS, 1999. Paxillin LD4 motif binds PAK and PIX through a novel 95-kD ankyrin repeat, ARF-GAP protein: a role in cytoskeletal remodeling. J. Cell Biol 145, 851–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungewiss C, Rizvi ZH, Roybal JD, Peng DH, Gold KA, Shin D-H, Creighton CJ, Gibbons DL, 2016. The microRNA-200/Zeb1 axis regulates ECM-dependent β1-integrin/FAK signaling, cancer cell invasion and metastasis through CRKL. Sci. Rep 6, 18652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valkenburg KC, de Groot AE, Pienta KJ, 2018. Targeting the tumour stroma to improve cancer therapy. Nat. Rev. Clin. Oncol 15, 366–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallés AM, Beuvin M, Boyer B, 2004. Activation of Rac1 by paxillin-Crk-DOCK180 signaling complex is antagonized by Rap1 in migrating NBT-II cells. J. Biol. Chem 279, 44490–44496. [DOI] [PubMed] [Google Scholar]
- van Helvert S, Storm C, Friedl P, 2018. Mechanoreciprocity in cell migration. Nat. Cell Biol 20, 8–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varney SD, Betts CB, Zheng R, Wu L, Hinz B, Zhou J, Van De Water L, 2016. Hic-5 is required for myofibroblast differentiation by regulating mechanically dependent MRTF-A nuclear accumulation. J. Cell Sci 129, 774–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vohnoutka RB, Gulvady AC, Goreczny G, Alpha K, Handelman SK, Sexton JZ, Turner CE, 2019. The focal adhesion scaffold protein Hic-5 regulates vimentin organization in fibroblasts. Mol. Biol. Cell 30, 3037–3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Song K, Krebs TL, Yang J, Danielpour D, 2008. Smad7 is inactivated through a direct physical interaction with the LIM protein Hic-5/ARA55. Oncogene 27, 6791–6805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Hu G, Betts C, Harmon EY, Keller RS, Van De Water L, Zhou J, 2011. Transforming growth factor-β1-induced transcript 1 protein, a novel marker for smooth muscle contractile phenotype, is regulated by serum response factor/myocardin protein. J. Biol. Chem 286, 41589–41599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb DJ, Donais K, Whitmore LA, Thomas SM, Turner CE, Parsons JT, Horwitz AF, 2004. FAK-Src signalling through paxillin, ERK and MLCK regulates adhesion disassembly. Nat. Cell Biol 6, 154–161. [DOI] [PubMed] [Google Scholar]
- Wei SC, Yang J, 2016. Forcing through tumor metastasis: the interplay between tissue rigidity and epithelial-mesenchymal transition. Trends Cell Biol. 26, 111–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei B, Zhou X, Liang C, Zheng X, Lei P, Fang J, Han X, Wang L, Qi C, Wei H, 2017. Human colorectal cancer progression correlates with LOX-induced ECM stiffening. Int. J. Biol. Sci 13, 1450–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West KA, Zhang H, Brown MLC, Nikolopoulos SN, Riedy MC, Horwitz AF, Turner CE, 2001. The LD4 motif of paxillin regulates cell spreading and motility through an interaction with paxillin kinase linker (PKL). J. Cell Biol 154, 161–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson BJ, Allen JL, Caswell PT, 2018. Vesicle trafficking pathways that direct cell migration in 3D matrices and in vivo. Traffic 19, 899–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winer A, Adams S, Mignatti P, 2018. Matrix metalloproteinase inhibitors in cancer therapy: turning past failures into future successes. Mol. Cancer Ther 17, 1147–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf K, Mazo I, Leung H, Engelke K, Von Andrian UH, Deryugina EI, Strongin AY, Bröcker EB, Friedl P, 2003. Compensation mechanism in tumor cell migration: mesenchymal-amoeboid transition after blocking of pericellular proteolysis. J. Cell Biol 160, 267–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood CK, Turner CE, Jackson P, Critchley DR, 1994. Characterisation of the paxillin-binding site and the C-terminal focal adhesion targeting sequence in vinculin. J. Cell Sci 107, 709–717. [PubMed] [Google Scholar]
- Wright PE, Dyson HJ, 2014. Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol 16, 18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Akhmanova A, 2017. Microtubule-organizing centers. Annu. Rev. Cell Dev. Biol 33, 51–75. [DOI] [PubMed] [Google Scholar]
- Wu DW, Cheng YW, Wang J, Chen CY, Lee H, 2010. Paxillin predicts survival and relapse in non-small cell lung cancer by microRNA-218 targeting. Cancer Res. 70, 10392–10401. [DOI] [PubMed] [Google Scholar]
- Wu D-W, Wu TC, Wu JY, Cheng Y-W, Chen YC, Lee MC, Chen CY, Lee H, 2014a. Phosphorylation of paxillin confers cisplatin resistance in non-small cell lung cancer via activating ERK-mediated Bcl-2 expression. Oncogene 33, 4385–4395. [DOI] [PubMed] [Google Scholar]
- Wu DW, Chuang CY, Lin WL, Sung WW, Cheng YW, Lee H, 2014b. Paxillin promotes tumor progression and predicts survival and relapse in oral cavity squamous cell carcinoma by microRNA-218 targeting. Carcinogenesis 35, 1823–1829. [DOI] [PubMed] [Google Scholar]
- Xu W, Gulvady AC, Goreczny GJ, Olson EC, Turner CE, 2019. Paxillin-dependent regulation of apical-basal polarity in mammary gland morphogenesis. Development 146, dev174367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada KM, Sixt M, 2019. Mechanisms of 3D cell migration. Nat. Rev. Mol. Cell Biol 20, 738–752. [DOI] [PubMed] [Google Scholar]
- Yang L, Guerrero J, Hong H, DeFranco DB, Stallcup MR, 2000. Interaction of the τ2 transcriptional activation domain of glucocorticoid receptor with a novel steroid receptor coactivator, Hic-5, which localizes to both focal adhesions and the nuclear matrix. Mol. Biol. Cell 11, 2007–2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye X, Weinberg RA, 2015. Epithelial-mesenchymal plasticity: a central regulator of cancer progression. Trends Cell Biol. 25, 675–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida K, Soldati T, 2006. Dissection of amoeboid movement into two mechanically distinct modes. J. Cell Sci 119, 3833–3844. [DOI] [PubMed] [Google Scholar]
- Yuminamochi T, Yatomi Y, Osada M, Ohmori T, Ishii Y, Nakazawa K, Hosogaya S, Ozaki Y, 2003. Expression of the LIM proteins paxillin and Hic-5 in human tissues. J. Histochem. Cytochem 51, 513–521. [DOI] [PubMed] [Google Scholar]
- Zaidel-Bar R, Kam Z, Geiger B, 2005. Polarized downregulation of the paxillin-p130CAS-Rac1 pathway induced by shear flow. J. Cell Sci 118, 3997–4007. [DOI] [PubMed] [Google Scholar]
- Zhan T, Rindtorff N, Betge J, Ebert MP, Boutros M, 2019. CRISPR/Cas9 for cancer research and therapy. Semin. Cancer Biol 55, 106–119. [DOI] [PubMed] [Google Scholar]
- Zhang LL, Zhang LF, Shi YB, 2018. Down-regulated paxillin suppresses cell proliferation and invasion by inhibiting M2 macrophage polarization in colon cancer. Biol. Chem 399, 1285–1295. [DOI] [PubMed] [Google Scholar]
- Zouq NK, Keeble JA, Lindsay J, Valentijn AJ, Zhang L, Mills D, Turner CE, Streuli CH, Gilmore AP, 2009. FAK engages multiple pathways to maintain survival of fibroblasts and epithelia: differential roles for paxillin and p130Cas. J. Cell Sci 122, 357–367. [DOI] [PMC free article] [PubMed] [Google Scholar]