

Abstract
Colorectal cancer is the third most common cancer diagnosed in the United States with up to 3% of cases being attributable to a hereditary polyposis syndrome. Established diagnostic and/or testing criteria exist for many of the recognized polyposis syndromes and are an important tool in guiding physicians in the identification of individuals who may benefit from referral to a cancer genetics service for hereditary cancer risk assessment. A formal hereditary cancer risk assessment supports fulfillment of obligations for standard of care, as well as minimizes the negative outcomes that may occur in the absence of informed consent for genetic testing. The implications of a diagnosis may extend beyond the individual patient to include at-risk relatives, and as such, much emphasis should be placed on identifying the most informative individual in a family in which to initiate testing. Advances in our understanding of genes associated with hereditary polyposis and the increasing use of testing that relies on next-generation sequencing technologies may lead to the increased likelihood of a genetic diagnosis; however, in those individuals without a genetic diagnosis whose histories remain concerning for hereditary polyposis, knowledge of family history may inform strategies for early detection and prevention.
Keywords: polyposis, genetic testing, germline, hereditary cancer, next-generation sequencing
Colorectal cancer (CRC) is the third most common cancer in the United States, and it accounts for approximately 8% of all cancer diagnoses.1 CRC related to a hereditable mutation in a single gene, also known as hereditary CRC, is estimated to account for approximately 5% of all cases of the disease. There are several recognized hereditary CRC syndromes, including both hereditary polyposis and nonpolyposis syndromes, each with implications for specific cancer risks in the gastrointestinal (GI) system, and possibly extraintestinal malignancies. The benefits in terms of reduction in morbidity and mortality provided by CRC screening have been well established, and as such, it is important to identify those at an increased risk for CRC due to a hereditary CRC predisposition syndrome that may benefit from early detection and prevention. This article touches on essential elements of a hereditary cancer risk assessment and the importance of genetic counseling in that process. In addition, it reviews genetic testing for the established hereditary polyposis syndromes, current testing technology, likelihood of a genetic diagnosis, and newer genes associated with polyposis syndromes.
Hereditary Cancer Risk Assessment and Genetic Counseling
In its most recently released version of practice guidelines for genetic testing and management of hereditary GI cancer syndromes, the American College of Gastroenterology (ACG) outlines its recommendations for the care of these patients. Among the recommendations put forward by the ACG are key components of making a preliminary determination of the possibility of familial risk to develop cancer, standards for minimal cancer family history assessment, and informed consent that should be adopted in gastroenterology practices.2
Important components of a hereditary cancer risk assessment include evaluation of a patient's personal and/or family history for features concerning for a cancer predisposition syndrome via a targeted four-generation pedigree; development of a differential diagnosis; discussion of available genetic testing options if indicated; outlining a testing strategy that can efficiently identify the familial mutation; and recommendations for management, on-going cancer surveillance, and prevention.3 4 These components are conducive with ensuring that adequate informed consent has been obtained prior to testing. In the event a provider is unable to adequately adhere to these standards, consideration should be given to making a referral to a cancer genetic consultation service for hereditary cancer risk assessment. Recognizing when a referral may be warranted is important not only in terms of meeting obligations for standard of care but also to reduce the likelihood of the occurrence of negative outcomes that might occur in the absence of informed consent. These negative outcomes may include misinterpretation of genetic testing results, inappropriate medical management, unintended psychosocial outcomes, and costly, unnecessary genetic testing.3
Discussions regarding the possibility of hereditary risk for cancer and availability of genetic testing may routinely be undertaken by nongenetics providers, but a thorough hereditary cancer risk assessment routinely includes the involvement of a medical geneticist and/or genetic counselor. A list of Web sites with resources for locating a genetics provider can be found in Table 1. Genetic counseling is an established profession with its own certification body, accredited graduate-level training programs, and code of ethics. Genetic counselors typically work as members of a health-care team, providing risk assessment; interpreting genetic testing; and providing supportive counseling, education, and support to patients and their families, as well as to other medical providers. Collaboration between a genetic counselor and a physician ensures that the complex aspects of hereditary risk assessment have been addressed and helps physicians ensure that recommended standards regarding risk assessment and informed consent for genetic testing have been achieved.
Table 1. Resources for locating genetics providers.
| Organization | Web site |
|---|---|
| National Society of Genetic Counselors (NSGC) | http://www.nsgc.org |
| American College of Medical Genetics (ACMG) | https://www.acmg.net |
| National Cancer Institute (NCI) Cancer Genetics Services Directory | http://www.cancer.gov/about-cancer/causes-prevention/genetics/directory |
| Canadian Association of Genetic Counsellors (CAGC) | https://www.cagc-accg.ca |
Impacts of Genetic Testing on Care
Identification of those individuals who have a heritable polyposis syndrome can have significant benefit to the proband (index patient), as a genetic diagnosis can lead to initiation of a surveillance strategy that addresses the associated risks with a given polyposis syndrome, and may possibly lead to consideration of prophylactic surgery or more extensive surgery in the event of a cancer diagnosis.2 Initiation of an appropriate management strategy can have a significant impact on reduction in mortality due to colorectal and/or extracolonic GI cancers, and those individuals at highest risk based on genetic susceptibility could gain decades of life through appropriate management and intervention.5
The diagnosis of an inherited CRC syndrome can also have significant implications for management of a patient's immediate and extended family, and as such, identification of the most informative individual in a family in whom to initiate testing cannot be overstated in its importance (Fig. 1). A widely held assumption among patients and providers is that a negative genetic test result is an indicator of the absence of a hereditary component to disease.5 This may not be the case, however, particularly when testing is performed based on family history in an individual who does not have a personal history suggestive of polyposis and/or an associated cancer. A negative test result can be the outcome of several possible explanations, including limitations in current testing technology/methodology; limitations in knowledge of causative genes or an identifiable mutation is present in a gene for which the individual was not tested (e.g., although one for which clinical testing is available); or absence of a potentially identifiable familial mutation in the individual in which testing was performed. In any case, alterations in risk status cannot be made without first identifying a mutation in the family. Once a mutation in a family has been identified, testing in subsequent relatives can be achieved at a significant reduction in the initial cost and with highly informative results.
Fig. 1.
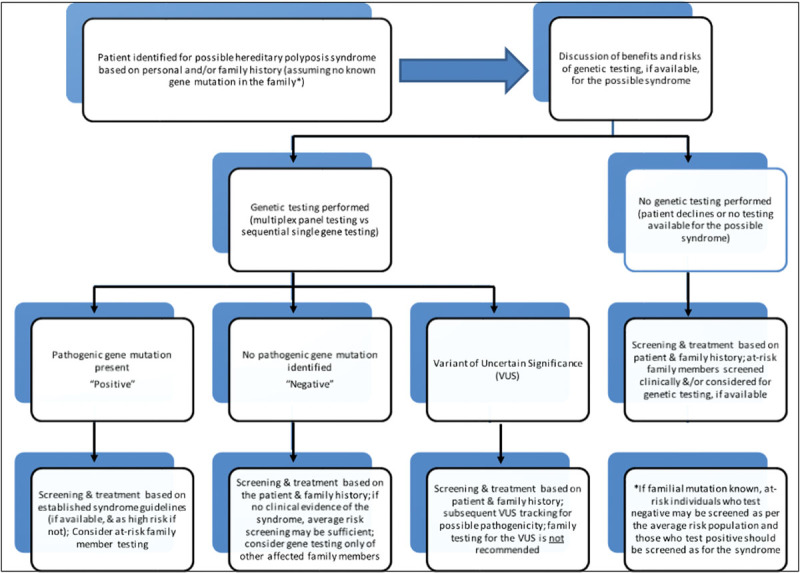
Algorithm for genetic assessment for a patient with a personal or family history of a hereditary polyposis syndrome.
In the absence of a confirmed pathogenic mutation in a family, it may not be possible to delineate individuals in a family who carry the germline mutation (“true positive,” who are in turn at an increased risk for associated malignancies/findings) from those non–mutation carriers (“true negative”) who are likely not at an increased risk over that of the general population. As such, until the presence of a mutation can be confirmed in a family, intensified surveillance may be recommended to all close relatives of an individual with a clinical diagnosis. In addition, until a relative with an identifiable mutation is found, knowledge of family history of cancer and/or polyp history may be useful in risk assessment guidelines as well as empiric cancer screening recommendations.6
Approaches to Genetic Testing for Hereditary Polyposis Syndromes
Germline mutations in one of at least eight genes have been implicated in hereditary polyposis syndromes. A list of these causative genes and a summary of associated hereditary polyposis syndromes can be found in Table 2. The current approach to testing for the presence of a mutation in one of these genes begins with the selection of the gene(s) in which a mutation is most likely to be found, followed by DNA analysis facilitated by a blood or buccal sample. Automated DNA sequencing has been available for over 20 years via methods developed by Frederick Sanger7 in 1977. Until more recently, the approach to genetic testing for the polyposis syndromes has most often been performed through sequential gene testing via the Sanger sequencing and/or multiplex ligation probe assay (MLPA) methods of analysis. The Sanger method has been the dominant approach and long considered the gold standard for DNA analysis8; however, criticisms of this method of analysis include that it is a relatively inefficient, time-consuming, and a fairly expensive method of testing per gene analyzed.9
Table 2. Hereditary polyposis syndromes and their associated genes/characteristics.
| Gene | Syndrome | Clinical characteristics | Mode of inheritance |
|---|---|---|---|
| APC | FAP/aFAP syndromes | Colorectal adenomas/cancer • Duodenal adenomas/cancer • Gastric fundic gland polyps/cancer • Thyroid cancers • Hepatoblastoma (in children) • Benign features:  Osteomas Dental anomalies CHRPE: congenital hypertrophy of the retinal pigment epithelium Desmoid tumors (more in “Adenomatous Polyposis Syndromes: Diagnosis and Management” by Drs Mitchem and Hall, on pp. 321–329) Osteomas Dental anomalies CHRPE: congenital hypertrophy of the retinal pigment epithelium Desmoid tumors (more in “Adenomatous Polyposis Syndromes: Diagnosis and Management” by Drs Mitchem and Hall, on pp. 321–329) |
Autosomal dominant |
| MUTYH | MAP syndrome | • Colorectal adenomas/cancer • Gastric and duodenal polyps/cancers • May include extraintestinal features seen in FAP/AFAP (more in “Adenomatous Polyposis Syndromes: Diagnosis and Management” by Drs Mitchem and Hall, on pp. 321–329) |
Autosomal recessive |
| POLE and POLD1 | PPP syndrome | • Colorectal adenomas/microsatellite stable cancers • Limited evidence of extracolonic involvement (knowledge of full clinical spectrum still evolving) |
Autosomal dominant |
| SMAD4 and BMPR1 | JPS | • Hamartomatous/juvenile polyps throughout GI tract • Colorectal/other GI cancers • JPS and HHT seen with SMAD4 mutation (more in “Hamartomatous Polyps and Associated Syndromes” by Dr Cone, on pp. 330–335) |
Autosomal dominant |
| PTEN | CS and BRRS | • GI polyps (various histologies)/cancers • Benign and malignant tumors of: Thyroid Breast Endometrium Macrocephaly• Mucocutaneous findings • Autism/developmental delay • Vascular involvement (arteriovenous malformations, hemangiomas) (more in “Hamartomatous Polyps and Associated Syndromes” by Dr Cone, on pp. 330–335) |
Autosomal dominant |
|
STK11
(LKB1) |
PJS | • PJS-type hamartomatous GI polyps/cancers • Mucocutaneous hyperpigmentation • Variety of malignancies (colorectal, gastric, pancreatic, breast, and ovarian cancers) • SCTAT, Sertoli cell tumors of the testes (more in “Hamartomatous Polyps and Associated Syndromes” by Dr Cone, on pp. 330–335) |
Autosomal dominant |
| Unknown | SPS | • WHO criteria: > 20 serrated polyps throughout the colon 5 serrated polyps proximal to the sigmoid colon with ≥2 polyps >10 mm Any number of serrated polyps proximal to the sigmoid colon in an individual who has a first-degree relative with SPS Extraintestinal features not well defined (more in “Serrated Polyps and Serrated Polyposis Syndrome” by Dr Ashburn et al., on pp. 336–344) |
Unclear (both autosomal dominant and autosomal recessive inheritance have been suggested) |
Abbreviations: aFAP, attenuated FAP; BRRS, Bannayan–Riley–Ruvalcaba syndrome; GI, gastrointestinal; HHT, hereditary hemorrhagic telangiectasia; FAP, familial adenomatous polyposis; JPS, Juvenile polyposis syndrome; MAP, MUTYH-associated polyposis; PJS, Peutz-Jeghers syndrome; PPP, polymerase proofreading-associated polyposis; SCTAT, sex cord tumors with annular tubules; SPS, serrated polyposis syndrome; WHO, World Health Organization.
Advances in technological capabilities have led to the development of next-generation sequencing (NGS) technologies and sequence enrichment methods which are becoming routinely employed in the search for meaningful somatic mutations in sporadic cancers in addition to the identification of known and novel germline mutations involved in familial cancer syndromes.10 This technology uses a similar principle to Sanger sequencing in that it involves analyzing fragments of DNA, but it exceeds traditional sequencing methods in its ability to identify single nucleotide variants, small insertions and deletions, large copy number variants, and mosaicism which would not be detectable by Sanger sequencing alone.9 NGS-based testing has been recognized as a much more efficient process than Sanger sequencing, with higher throughput and significant reduction in time and cost.
Simultaneous testing of multiple genes using NGS technologies, also known as multiplex, or panel testing is increasingly being used as the initial method of analysis in individuals/families being evaluated for hereditary cancer syndromes. Multiple commercial genetic testing laboratories now offer panel testing, which may range from more expansive “pan-cancer” panels that include multiple genes associated with varying levels of risk for a range of hereditary cancer susceptibility genes to those that consist of analysis of multiple genes associated with hereditary susceptibility based on site of tumor origination (e.g., hereditary CRC) or tumor type (i.e., hereditary paraganglioma). Benefits of utilizing panel testing as a first-tier approach are that patients who may be missed by diagnostic criteria for a known cancer susceptibility syndrome and those with an ambiguous presentation and/or unknown family history can be tested simultaneously for multiple hereditary cancer syndromes. This ideally increases the likelihood of identifying a germline mutation and decreasing the time to molecular diagnosis and appropriately tailored familial testing and clinical surveillance.10 11 12
The use of panel testing is not without its challenges, which include technical issues, increased rate of detection of uncertain findings, and limitations in the ability to translate the finding of a deleterious mutation into clinically meaningful use, particularly in genes in which the risks for cancer/polyps are not known of have not yet been well defined. Technical challenges may include uneven sequencing across the genes of interest resulting in poor sequence coverage in some regions of the gene, affecting both the sensitivity and specificity of variant detection.10 The use of panel testing also increases the likelihood of identification of variants of uncertain significance (VUS), a term used when gene analysis results in the identification of a genetic variant in which pathogenicity can neither be confirmed nor excluded. Identification of a VUS should not be used in clinical decision making unless the specific finding has been better characterized in terms of biological and clinical significance and deemed to have a deleterious impact on gene functioning. Finally, testing via a cancer panel may include analysis of genes in which the range of cancer risks are not yet known, or not yet well defined, limiting the ability of the clinician to use the finding in alterations in screening or familial testing.
Although panel tests for hereditary cancer syndromes are widely available through several commercial laboratories, single-gene analysis via Sanger sequencing and/or MLPA is still the recommended first-tier approach in those individuals/families meeting diagnostic criteria for a known polyposis syndrome, or with a known familial mutation.
Likelihood of Genetic Diagnosis
Our increasing understanding of genes associated with polyposis syndromes, as well the methods of analysis needed to identify potentially causative germline mutations, has facilitated an increased likelihood of genetic diagnosis in those meeting diagnostic and/or testing criteria for a given polyposis syndrome. In spite of these advances, there are a significant portion of individuals who meet criteria, or have a significant history, who will remain without a genetic diagnosis, unfortunately. This then impacts the care of the patient and their family in varying ways.
Familial Adenomatous Polyposis and MUTYH-Associated Polyposis
As discussed in previous articles in this issue, familial adenomatous polyposis (FAP) is an autosomal dominantly inherited disease, most often resulting from a mutation in the APC gene, and is the second most common inherited colorectal cancer syndrome.13 An individual may receive a clinical diagnosis of FAP in the presence of at least 100 colorectal adenomas, or less than 100 adenomas when there is a known family history of FAP. An individual may receive a diagnosis of attenuated FAP when the polyp burden is less severe, typically ranging from 10 to 100 adenomas, and age of onset of development of polyps is later than classic FAP.14 The clinical presentation of FAP may overlap with that of MUTYH-associated polyposis, an autosomal recessive condition resulting from biallelic mutations in the MUTYH gene, which also results in an increased risk for adenomatous polyps.15
The likelihood of identification of a causative mutation in an individual presenting with multiple adenomas increases with polyp count. In a study performed to determine the prevalence of pathogenic APC and MUTYH mutations in 7,225 individuals with colorectal adenomas who underwent genetic testing, Grover et al observed APC and biallelic MUTYH mutations in 80 and 2%, respectively, of individuals with more than 1,000 adenomas; 56 and 7 of individuals with 100 to 999 adenomas; 10 and 7% of individuals with 20 to 99 adenomas, and 5 and 4% of individuals with 10 to 19 adenomas.16 The outcome of this study suggests that the prevalence of APC and biallelic MUTYH mutations is similar in those with attenuated polyposis and highlights the need for germline analysis of both genes, particularly in those individuals without a significant family history, which may be explained by biallelic MUTYH mutations or a de novo (new germline gene mutation) APC mutation.
Polymerase Proofreading-Associated Polyposis
The term polymerase proofreading-associated polyposis is a more recently established term which has been used to describe individuals presenting with multiple colorectal adenomas which may be attributable to a germline mutation in the exonuclease (proofreading) domain of the POLE and POLD1 genes. Palles et al identified mutations in these genes in families with multiple colorectal adenomas and CRC who were negative for APC and/or MUTYH mutations.17 Given the limited number of cases that have thus far been reported, the clinical characteristics and associated cancer risks of germline mutations in POLE or POLD1 have not yet been well defined. In an effort to add additional information to help define the phenotypic/clinical characteristics of POLE and POLD1, Valle et al analyzed by targeted genotyping analysis for recurrent POLE p.L424V and POLD1 p.S478N mutations in 858 individuals with familial/early onset CRC, of which 191 were polyposis cases. The group identified an apparently de novo p.L424V germline mutation in one female patient diagnosed with CRC and more than 35 colonic polyps at 28 years old.18 Bellido et al built upon these findings by sequencing the complete exonuclease domains of POLE and POLD1 in an overlapping cohort and found a POLD1 p.D316H mutation in a family which included one mutation-positive relative with 13 polyps.19 In a study of 266 unrelated individuals with polyposis, or who met the Amsterdam Criteria for hereditary nonpolyposis syndrome (hereditary nonpolyposis colorectal cancer), Spier et al found that the POLE p.L424V mutation was found to be present in 7% of the individuals with familial polyposis.20 The findings of these groups suggest that a small percentage of polyposis in APC and MUTYH mutation-negative families may be attributable to a mutation in a DNA polymerase gene.
Hamartomatous Polyposis Syndromes
The hamartomatous polyposis syndromes are a group of hereditary polyposis syndromes characterized by the presence of hamartomatous rather than epithelial polyp histology. These disorders account for a significant minority of hereditary colorectal/GI predisposition syndromes, accounting for less than 1% of CRC cases.21 The three most well-defined hamartomatous polyposis syndromes include juvenile polyposis syndrome (JPS),22 Cowden syndrome,23 and Peutz-Jeghers syndrome (PJS).24 These and other hamartomatous polyposis syndromes are discussed in greater detail in this issue.
Juvenile Polyposis Syndrome
A clinical diagnosis of JPS is considered in an individual who meets at least one of the following criteria: (1) at least three to five juvenile polyps of the colon, (2) multiple juvenile polyps throughout the GI tract, and (3) any number of juvenile polyps in the context of a family history of JPS. The two primary genes in which pathogenic mutations have been attributed to JPS include SMAD4 and BMPR1A.21 Mutations in these two genes may be identified in approximately 40 to 45% of individuals meeting clinical criteria for JPS and are believed to account for approximately equal proportions of individuals with JPS.25 26
Cowden Syndrome
Germline mutations in the PTEN gene are causative for a group of diverse conditions which are often collectively referred to as PTEN hamartoma tumor syndromes. These autosomal dominantly inherited conditions include Bannayan–Riley–Ruvalcaba syndrome, PTEN-related Proteus syndrome, and Cowden syndrome, which will be the focus of this section. Cowden syndrome is characterized by an increased risk for breast, thyroid, endometrial, and renal cell cancers. Nonmalignant features include, but are not limited to, autism/developmental delay, mucocutaneous lesions, benign breast disease, benign thyroid disease, GI polyps (of various histologies), vascular malformations, uterine fibroids, and macrocephaly.27 A diagnosis of Cowden syndrome may be considered when a combination of associated features, some of which have been further classified into major and minor criteria, is present. Consensus diagnostic criteria have been established and the National Comprehensive Cancer Network maintains an updated list of these criteria.28 The Lerner Research Institute at Cleveland Clinic also maintains a risk calculator for estimation of PTEN mutation probability (http://www.lerner.ccf.org/gmi/ccscore/).29 Probability of a PTEN mutation is generated from a risk score based on age at diagnosis and presence of associated findings.
Pathogenic mutations in PTEN may be detected in 25 to 85% of individuals meeting diagnostic criteria for Cowden. This considerable discrepancy in likelihood of genetic diagnosis may be attributed to the larger number being the result of earlier studies in families who may have met more strict diagnostic criteria, while the lower end of the range is the result of more recent study which included individuals based on more relaxed criteria.29 30
Peutz-Jeghers Syndrome
PJS is an autosomal dominant hamartomatous polyposis syndrome related to a mutation in the STK11 gene, which is characterized by the association between GI hamartomatous polyps (polyps may also occur outside the GI tract), mucocutaneous pigmentation, and increased risk for cancers of the GI tract, reproductive tract, and breast. A clinical diagnosis of PJS is made when an individual presents with two or more of the following features: (1) two or more PJS polyps of the small intestine, (2) any number of PJS polyps in the context of a family history of PJS, (3) characteristic mucocutaneous macules in the context of a family history of PJS, or (4) any number of PJS-type polyps in an individual who also presents with characteristic PJS-type polyps. In an individual with a clinical diagnosis of PJS, germline testing for STK11 mutations may reveal a mutation in 80 to 94% of affected individuals.31
Miscellaneous Hamartomatous Syndromes
Identification of individuals and/or families with a hamartomatous polyposis syndrome may require careful evaluation of a family history, pathology reports, and knowledge of associated extracolonic findings in the clinical presentation, as many individuals present with features that overlap one or more syndrome. In an effort to aid clinicians in the evaluation of individuals who present with moderate polyp load but who do not meet established genetic testing criteria for any one polyposis syndrome, Ngeow et al studied 603 individuals with five or more GI polyps, including at least one hamartomatous or hyperplastic/serrated polyp. Observations from the group included that patients whose polyps included one or more unspecified hamartomatous polyps were more likely to have a germline mutation when compared with those who did not. A mixed polyposis presentation (>3 different histological subtypes of adenoma, hamartoma, lipoma, ganglioneuroma, juvenile, inflammatory polyps) was associated with an increased prevalence of underlying germline mutation, most often in the PTEN gene. The group also reported that 11% of patients not meeting established clinical criteria for a hereditary polyposis syndrome did in fact have an underlying germline mutation, the majority of which did not have a positive family history of CRC.32
Serrated Polyposis Syndrome
Serrated polyposis syndrome (SPS), previously referred to as hyperplastic polyposis syndrome, is a term often used to emphasize the presence of multiple sessile serrated adenomas. This condition has also been discussed in detail earlier in this issue. Proposed diagnostic criteria for SPS include patients fulfilling one or more of the following criteria: (1) at least five serrated polyps proximal to the sigmoid colon with two or more larger than 10 mm; (2) any number of serrated polyps proximal to the sigmoid colon in an individual who has a first-degree relative with SPS; and (3) more than 20 serrated polyps of any size but distributed throughout the colon (cumulative history).33 It has been suggested that this definition may be restrictive, as SPS is likely represented by several phenotypes of which serrated polyps are a defining feature.34 Patients with SPS are at increased risk for CRC with the actual risk having not yet defined, although published case reports suggest that between 10 and 50% of patients meeting SPS criteria have a family history of CRC. SPS has been found to exhibit both autosomal dominant and recessive patterns of inheritance and at this time a causative gene has not been identified and clinical testing is unavailable.34
Conclusion
There are several recognized hereditary CRC syndromes for which a strategy for early detection and prevention may exist for those who have been determined through genetic testing and/or personal and family history to be at risk for a polyposis syndrome. Despite advances in our knowledge of causative genes, and methods of genetic analysis, a significant portion of affected individuals will remain without a genetic diagnosis. Identification of the most informative family member in which to initiate testing may increase the likelihood of diagnosis of the polyposis syndromes for which testing currently exists. A formal hereditary cancer risk assessment that includes the involvement of a clinical genetics provider may aid physicians in the often complex components of a complete risk assessment, counseling, and testing. In the absence of identification of a genetic diagnosis, knowledge of family history may guide the formulation of screening recommendations.
Footnotes
Conflict of Interest The author has no significant relationship with, or financial interest in, any commercial companies.
References
- 1.1. SEER Cancer Statistics Factsheets: Colon and Rectum Cancer National Cancer Institute; Bethesda, MD. Available at: http://seer.cancer.gov/statfacts/html/colorect.html. Accessed 10/2/2015
- 2.Syngal S Brand R E Church J M Giardiello F M Hampel H L Burt R W; American College of Gastroenterology. ACG clinical guideline: Genetic testing and management of hereditary gastrointestinal cancer syndromes Am J Gastroenterol 20151102223–262., quiz 263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hampel H Bennett R L Buchanan A Pearlman R Wiesner G L; Guideline Development Group, American College of Medical Genetics and Genomics Professional Practice and Guidelines Committee and National Society of Genetic Counselors Practice Guidelines Committee. A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: referral indications for cancer predisposition assessment Genet Med 201517170–87. [DOI] [PubMed] [Google Scholar]
- 4.Riley B D, Culver J O, Skrzynia C. et al. Essential elements of genetic cancer risk assessment, counseling, and testing: updated recommendations of the National Society of Genetic Counselors. J Genet Couns. 2012;21(2):151–161. doi: 10.1007/s10897-011-9462-x. [DOI] [PubMed] [Google Scholar]
- 5.Rubinstein W S, Weissman S M. Managing hereditary gastrointestinal cancer syndromes: the partnership between genetic counselors and gastroenterologists. Nat Clin Pract Gastroenterol Hepatol. 2008;5(10):569–582. doi: 10.1038/ncpgasthep1235. [DOI] [PubMed] [Google Scholar]
- 6.Lu K H, Wood M E, Daniels M. et al. American Society of Clinical Oncology Expert Statement: collection and use of a cancer family history for oncology providers. J Clin Oncol. 2014;32(8):833–840. doi: 10.1200/JCO.2013.50.9257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.National Human Genome Research Institute (NHGRI) A brief history of the human genome project Last updated November 8 2012. Available at: http://www.genome.gov/12011239. Accessed September 29, 2015
- 8.Esteban-Jurado C, Garre P, Vila M. et al. New genes emerging for colorectal cancer predisposition. World J Gastroenterol. 2014;20(8):1961–1971. doi: 10.3748/wjg.v20.i8.1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heald B, Church J. Genetic testing for hereditary colorectal cancer syndromes: a significant change in technology and its clinical implications. Colorectal Dis. 2014;16(12):942–946. doi: 10.1111/codi.12792. [DOI] [PubMed] [Google Scholar]
- 10.Ku C S, Cooper D N, Roukos D H. Clinical relevance of cancer genome sequencing. World J Gastroenterol. 2013;19(13):2011–2018. doi: 10.3748/wjg.v19.i13.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Esplin E D, Snyder M P. Genomic era diagnosis and management of hereditary and sporadic colon cancer. World J Clin Oncol. 2014;5(5):1036–1047. doi: 10.5306/wjco.v5.i5.1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Simbolo M, Mafficini A, Agostini M. et al. Next-generation sequencing for genetic testing of familial colorectal cancer syndromes. Hered Cancer Clin Pract. 2015;13(1):18. doi: 10.1186/s13053-015-0039-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Online Mendelian Inheritance in Man OMIM®. Johns Hopkins University, Baltimore, MD MIM Number: 175100: Last edited June 10, 2015. World Wide Web URL: http://www.omim.org/entry/175100
- 14.Valle L. Genetic predisposition to colorectal cancer: where we stand and future perspectives. World J Gastroenterol. 2014;20(29):9828–9849. doi: 10.3748/wjg.v20.i29.9828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Online Mendelian Inheritance in Man OMIM®. Johns Hopkins University, Baltimore, MD MIM Number:608456: Last edited June 10, 2015. World Wide Web URL: http://www.omim.org/entry/608456
- 16.Grover S, Kastrinos F, Steyerberg E W. et al. Prevalence and phenotypes of APC and MUTYH mutations in patients with multiple colorectal adenomas. JAMA. 2012;308(5):485–492. doi: 10.1001/jama.2012.8780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Palles C, Cazier J B, Howarth K M. et al. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat Genet. 2013;45(2):136–144. doi: 10.1038/ng.2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Valle L, Hernández-Illán E, Bellido F. et al. New insights into POLE and POLD1 germline mutations in familial colorectal cancer and polyposis. Hum Mol Genet. 2014;23(13):3506–3512. doi: 10.1093/hmg/ddu058. [DOI] [PubMed] [Google Scholar]
- 19.Bellido F, Pineda M, Aiza G. et al. POLE and POLD1 mutations in 529 kindred with familial colorectal cancer and/or polyposis: review of reported cases and recommendations for genetic testing and surveillance. Genet Med. 2016;18(4):325–332. doi: 10.1038/gim.2015.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Spier I, Holzapfel S, Altmüller J. et al. Frequency and phenotypic spectrum of germline mutations in POLE and seven other polymerase genes in 266 patients with colorectal adenomas and carcinomas. Int J Cancer. 2015;137(2):320–331. doi: 10.1002/ijc.29396. [DOI] [PubMed] [Google Scholar]
- 21.Gammon A, Jasperson K, Kohlmann W, Burt R W. Hamartomatous polyposis syndromes. Best Pract Res Clin Gastroenterol. 2009;23(2):219–231. doi: 10.1016/j.bpg.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Online Mendelian Inheritance in Man OMIM®. Johns Hopkins University, Baltimore, MD MIM Number:174900: Last edited August 26, 2008. World Wide Web URL: http://www.omim.org/entry/174900
- 23.Online Mendelian Inheritance in Man OMIM®. Johns Hopkins University, Baltimore, MD MIM Number:158350: Last edited October 10, 2014. World Wide Web URL: http://www.omim.org/entry/158350
- 24.Online Mendelian Inheritance in Man OMIM®. Johns Hopkins University, Baltimore, MD MIM Number:175200: Last edited March 9, 2015. World Wide Web URL: http://www.omim.org/entry/175200
- 25.Aretz S, Stienen D, Uhlhaas S. et al. High proportion of large genomic deletions and a genotype phenotype update in 80 unrelated families with juvenile polyposis syndrome. J Med Genet. 2007;44(11):702–709. doi: 10.1136/jmg.2007.052506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zbuk K M, Eng C. Hamartomatous polyposis syndromes. Nat Clin Pract Gastroenterol Hepatol. 2007;4(9):492–502. doi: 10.1038/ncpgasthep0902. [DOI] [PubMed] [Google Scholar]
- 27.Ngeow J, Eng C. PTEN hamartoma tumor syndrome: clinical risk assessment and management protocol. Methods. 2015;77–78:11–19. doi: 10.1016/j.ymeth.2014.10.011. [DOI] [PubMed] [Google Scholar]
- 28.National Comprehensive Cancer Network (NCCN) NCCN Genetic/Familial High Risk Assessment: Breast and Ovarian Version 2 2015. Available at: http://www.nccn.org/professionals/physician_gls/pdf/genetics_colon.pdf. Accessed October 2, 2015
- 29.Tan M H, Mester J, Peterson C. et al. A clinical scoring system for selection of patients for PTEN mutation testing is proposed on the basis of a prospective study of 3042 probands. Am J Hum Genet. 2011;88(1):42–56. doi: 10.1016/j.ajhg.2010.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eng C. Will the real Cowden syndrome please stand up: revised diagnostic criteria. J Med Genet. 2000;37(11):828–830. doi: 10.1136/jmg.37.11.828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beggs A D, Latchford A R, Vasen H F. et al. Peutz-Jeghers syndrome: a systematic review and recommendations for management. Gut. 2010;59(7):975–986. doi: 10.1136/gut.2009.198499. [DOI] [PubMed] [Google Scholar]
- 32.Ngeow J Heald B Rybicki L A et al. Prevalence of germline PTEN, BMPR1A, SMAD4, STK11, and ENG mutations in patients with moderate-load colorectal polyps Gastroenterology 201314471402–1409., 1409.e1–1409.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rex D K Ahnen D J Baron J A et al. Serrated lesions of the colorectum: review and recommendations from an expert panel Am J Gastroenterol 201210791315–1329., quiz 1314, 1330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guarinos C, Sánchez-Fortún C, Rodríguez-Soler M, Alenda C, Payá A, Jover R. Serrated polyposis syndrome: molecular, pathological and clinical aspects. World J Gastroenterol. 2012;18(20):2452–2461. doi: 10.3748/wjg.v18.i20.2452. [DOI] [PMC free article] [PubMed] [Google Scholar]