

SUMMARY
Transcriptional responses to external stimuli remain poorly understood. Using GRO-Seq and PRO-Seq, we show that CDK8 kinase activity promotes RNA polymerase II pause release in response to IFN-γ, a universal cytokine involved in immunity and tumor surveillance. The Mediator kinase module contains CDK8 or CDK19, which are presumed to be functionally redundant. We implemented cortistatin A, chemical genetics, transcriptomics, and other methods to de-couple their function while assessing enzymatic vs. structural roles. Unexpectedly, CDK8 and CDK19 regulated different gene sets via distinct mechanisms. CDK8-dependent regulation required its kinase activity, whereas CDK19 governed IFN-γ responses through its scaffolding function (i.e. kinase-independent). Accordingly, CDK8, not CDK19, phosphorylates the STAT1 transcription factor (TF) during IFN-γ stimulation, and CDK8 kinase inhibition blocked activation of JAK-STAT pathway TFs. Cytokines such as IFN-γ rapidly mobilize TFs to “reprogram” cellular transcription; our results implicate CDK8 and CDK19 as essential for this transcriptional reprogramming.
Keywords: transcription, Mediator kinase, cortistatin A, CDK8, CDK19, interferon, promoter-proximal pausing, RNA polymerase II, STAT1, eRNA
Graphical Abstract
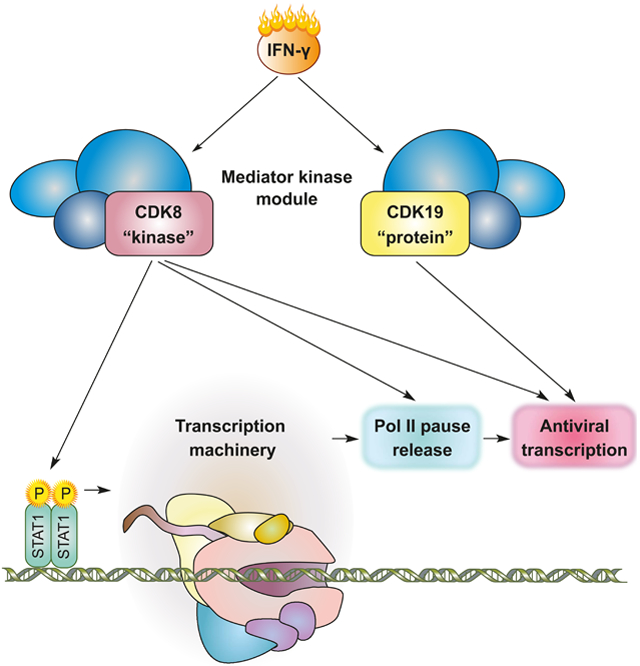
eTOC blurb:
Steinparzer et al. discover that the Mediator kinases CDK8 and CDK19 are functionally and mechanistically distinct transcription regulators in the IFN-γ antiviral program: CDK8 and CDK19 activate distinct gene sets, and they do so in kinase-dependent (CDK8) and kinase-independent (CDK19) ways. Further, CDK8 kinase activity promotes RNAPII pause release.
INTRODUCTION
Understanding how pathway-specific transcriptional responses are controlled remains an important but challenging endeavor. The transcriptional response to the ubiquitously acting cytokine IFN-γ protects against bacterial and viral infection, and is indispensable for tumor surveillance (Schneider et al., 2014). Whereas the basic signaling components of the IFN-γ pathway are defined, the chromatin-associated processes that adjust the transcriptional output to physiological needs are incompletely understood.
Upon IFN-γ binding to its receptor, the tyrosine kinases JAK1 and JAK2 phosphorylate the transcription factor (TF) signal transducer and activator of transcription 1 (STAT1) causing its nuclear translocation (Schneider et al., 2014). STAT1 is tightly controlled to prevent pathologies associated with hyper- or hypo-activation. An important control mechanism targets the JAKs in the cytoplasm (Schneider et al., 2014); another layer of control occurs in the nucleus. For example, STAT1 promoter occupancy is limited by negative feedback that initiates once productive transcription complexes have been established (Wiesauer et al., 2015). Chromatin-associated STAT1 is also regulated by phosphorylation of its activation domain (AD) at residue S727 (Sadzak et al., 2008; Wen et al., 1995). This physiologically important modification adjusts the transcriptional output in gene-specific ways (Bancerek et al., 2013).
The RNA polymerase II (RNAPII) enzyme transcribes all protein-coding and most non-coding RNAs in the human genome, and Mediator appears to be required to activate RNAPII function, genome-wide (Allen and Taatjes, 2015). The mammalian Mediator complex consists of 26 subunits and a 600 kDa, four-subunit kinase module, consisting of MED12, MED13, CCNC, and CDK8 (or CDK19), that can reversibly associate with Mediator (Allen and Taatjes, 2015). Consequently, CDK8 and CDK19 are considered Mediator-associated kinases. CDK8 and CDK19 are highly similar (77% amino acid sequence identity) paralogs, and each appears to associate in a mutually exclusive fashion with the Mediator kinase module (Galbraith et al., 2013).
The transcriptional effects of Mediator kinases are generally cell type- and context-specific (Galbraith et al., 2013; Johannessen et al., 2017). These characteristics are consistent with the fact that sequence-specific, DNA-binding TFs represent a major class of proteins that are targeted by CDK8 and CDK19 (Poss et al., 2016), and CDK8 and/or CDK19-dependent phosphorylation of these TFs has been shown to alter TF activity in a few well-studied cases (Bancerek et al., 2013; Nitulescu et al., 2017). Notably, STATs appear to represent a more common target of Mediator kinases, as they have been shown to be phosphorylated by CDK8 and/or CDK19 in diverse cell lineages (Bancerek et al., 2013; Nitulescu et al., 2017; Poss et al., 2016). These findings suggest that Mediator kinase regulation of STAT TF function may be a common theme in biology.
Prior work has implicated Mediator kinases as promising targets for therapeutic manipulation of cytokine responses (Bancerek et al., 2013; Chen et al., 2017; Johannessen et al., 2017; Nitulescu et al., 2017). These studies have focused on CDK8 and used knockdown approaches. Thus, the role of CDK19 vs. CDK8 was not assessed and the requirement for the kinase activity per se (i.e. in contrast to the physical presence of CDK8) remained undefined. Among the Mediator kinase inhibitors that have been characterized (Dale et al., 2015; Johannessen et al., 2017; Koehler et al., 2016), the natural product cortistatin A (CA) stands out based upon its potency and high selectivity for CDK8 and CDK19 (Pelish et al., 2015). Kinome-wide screens revealed essentially no off-target kinase inhibition, even at concentrations that were orders of magnitude higher than its in vitro- measured Kd (0.2 nM)(Pelish et al., 2015). Here, we used CA in combination with chemical genetics and conditional knockout or knockdown methods to thoroughly evaluate the regulatory roles of CDK8 and CDK19 in the context of IFN-γ signaling and anti-viral responses. Our results define kinase-specific and kinase-independent roles for each, and establish CDK8 and CDK19 as essential but non-redundant regulators of IFN-γ transcriptional responses.
RESULTS
Mediator kinase activity is a gene-selective regulator of IFN-γ-stimulated transcription
Our previous study demonstrated that CDK8 controlled transcriptional responses to IFN-γ (Bancerek et al., 2013), but it remained unclear whether its effects were kinase-dependent or kinase-independent. To address this question, we employed cortistatin A (CA), a potent and highly selective inhibitor of the Mediator kinases CDK8 and CDK19 (Pelish et al., 2015). As expected, IFN-γ-stimulated phosphorylation of STAT1 at Y701 was not affected by CA treatment, whereas CA inhibited IFN-γ-induced phosphorylation of STAT1 S727 (Figure 1A), consistent with previous findings in human cells (Bancerek et al., 2013; Nitulescu et al., 2017; Pelish et al., 2015).
Figure 1. Mediator kinase inhibition impairs IFN-γ-stimulated transcription in gene-selective ways.

(A) CA inhibits IFN-γ-induced STAT1 AD phosphorylation at S727. WT MEFs (± 100 nM CA, 1 h pre-treatment) were subjected to 45 min IFN-γ stimulation followed by Western blot against phosphorylated STAT1 at S727 (pS727) or Y701 (pY701) and total STAT1, CDK8, CDK19 or tubulin. STAT1 bands correspond to STAT1 α or STAT1 β isoforms.
(B) Gene expression changes (mRNA) in WT MEFs upon 6 h IFN-γ treatment (blue, padj < 0.05; red, padj > 0.05). Genes with padj < 0.05, log2FoldChange (lfc) ≥ 1, FPKM stimulated ≥ 1 were regarded as IFN-γ-induced (221 genes).
(C) Overlap of genes induced after 6 h IFN-γ at mRNA vs. pre-mRNA levels.
(D and E) Effects of CA on expression of IFN-γ-induced genes. WT MEFs (± 100 nM CA, 1 h pre-treatment) were stimulated with IFN-γ (6 h) followed by RNA-Seq. The numbers of differentially expressed (upregulated: lfc > 0, padj < 0.05; downregulated: lfc < 0, padj < 0.05) IFN-γ-induced genes (defined in B) at mRNA (D) and pre-mRNA (E) level are shown.
(F, G and H) Mediator kinases act in part through STAT1 S727 phosphorylation. (F) Gene expression (mRNA) changes in S727A MEFs upon 3 h IFN-γ treatment. (G) Effects of STAT1 S727A mutation on IFN-γ-induced genes: genes induced by IFN-γ in WT MEFs are upregulated (55 genes, lfc > 0, padj < 0.05), down-regulated (78 genes, lfc < 0, padj < 0.05) or unaffected (32 genes) in S727A MEFs. (H) Overlap of IFN-γ-induced genes downregulated (lfc < 0, padj < 0.05) by CA (100 nM CA, 1 h pre-treatment) in WT MEFs vs. S727A MEFs.
To investigate the effects of CDK8 and CDK19 kinase activity on the transcriptome, mouse embryo fibroblasts (MEFs) were treated for 1 h with /CA (100 nM, or DMSO control) followed by stimulation with IFN-γ for 6 h. Ribosomal RNA-depleted total RNA from these cells was subjected to RNA-Seq; expression (FPKMs) for exonic (i.e., mRNA) and intronic (i.e., pre-mRNA) reads were calculated separately, as described (Madsen et al., 2015), with modifications (see Methods). Principal component analysis (PCA) and analysis of counts normalized for library size and composition (Love et al., 2014) revealed that replicate 1 of an IFN-γ-stimulated triplicate was an outlier (Figures S1A, left panel and S1B) and was omitted from subsequent analyses.
Treatment of MEFs for 6 h with IFN-γ increased the mRNA levels of 221 genes (log2-fold-change, lfc, ≥ 1; padj < 0.05; FPKM ≥ 1) and downregulated mRNA levels of only 3 genes (lfc ≤ −1; padj < 0.05; FPKM > 1; Figure 1B; Table S1A). Similar numbers and largely overlapping sets of genes were induced by IFN-γ at the pre-mRNA level (Figures 1C and S1C; Table S1B) indicating that the increased mRNA levels did not result from changes in mRNA stability, but rather from transcriptional stimulation by IFN-γ, as reported previously (Bancerek et al., 2013; Dolken et al., 2008). Of the 221 genes induced by IFN-γ, 38 were less induced (lfc < 0, padj < 0.05) and 7 more strongly induced (lfc > 0, padj < 0.05) in cells treated with CA (Figure 1D; Table S1A). Comparable results were obtained at the pre-mRNA level (31 and 5, respectively; Figure 1E; Table S1B). As expected, CA treatment did not affect cell viability within the time frame of these experiments (Figure S1D). Together, these results indicated that inhibition of Mediator kinase activity primarily reduced IFN-γ-stimulated transcription, but had gene-selective effects.
Mediator kinases act in part through STAT1 S727 phosphorylation
STAT1 S727 is an established CDK8 kinase target (Bancerek et al., 2013; Pelish et al., 2015). To find out whether the effects of CA on induction of IFN-γ-regulated genes were dependent on STAT1 S727 phosphorylation by Mediator kinases, we compared responses of MEFs derived from STAT1 S727A knock-in mice (Bancerek et al., 2013) with those expressing wild-type STAT1. Stimulation of S727A MEFs with IFN-γ (3 h) induced a robust response (Figures 1F and S1E; Table S2). Compared to WT MEFs, the induction of many IFN-γ target genes was altered in S727A MEFs (Figure 1G; Table S2), consistent with previous studies (Bancerek et al., 2013). CA treatment predominantly decreased induction in both WT and S727A MEFs (Figures S1F and S1G; Table S2), and most of the CA-sensitive genes were similarly affected in both WT and S727A MEFs (Figure 1H; Table S2). These data indicate that Mediator kinases act not only through STAT1, but also other proteins, to help regulate the transcriptional response to IFN-γ.
Mediator kinase inhibition increases RNAPII pausing at IFN-γ-regulated genes
Given that CA treatment modulated IFN-γ-stimulated transcription (Figure 1), we asked whether CA effects could be linked to any specific stage of transcription. We employed global nuclear run-on followed by sequencing (GRO-Seq), a well-tested method that detects actively transcribing polymerases and measures nascent transcription, genome-wide (Core et al., 2008). A short stimulation (30 min) with IFN-γ allowed us to assess the effects of Mediator kinase activity on the primary (i.e., STATI-driven) IFN-γ response.
We completed biological replicate GRO-Seq experiments in MEFs following 30-minute treatment with IFN-γ upon pre-treatment with CA (100 nM, 1 h, or DMSO control), as with mRNA analyses. Gene Set Enrichment Analysis (GSEA) confirmed induction of expected IFN-γ-responsive gene sets, with significant enrichment (FDR q-value < 0.05) of IFN-γ and JAK-STAT signaling pathways (Figure 2A; Table S3A). The total number of genes induced after 30-minute IFN-γ stimulation was 200 (Ifc > 0; padj < 0.05; Table S4A), similar to the number of genes induced at the mRNA level after 6 h (Figure 1B). The overlap between IFN-γ-induced genes in GRO-Seq (30 min IFN-γ) and RNA-Seq (6 h IFN-γ) experiments comprised 51 genes (Figure S2A). CA treatment resulted in reduced induction of a large proportion of IFN-γ target genes (Figure 2B, Table S4B), consistent with CA effects on mRNA levels during 6 h stimulation with IFN-γ (Figures 1D and 1E).
Figure 2. Mediator kinase inhibition increases RNAPII pausing.

(A – F) MEFs pre-treated with CA (100 nM, 1 h) or DMSO (Ctrl) were stimulated with IFN-γ (30 min; IFN30) or unstimulated (IFN0), and subjected to GRO-Seq.
(A) Moustache plot of false discovery rate (FDR) vs. normalized enrichment score (NES) based upon GSEA of GRO-Seq data for IFN30 vs. IFN0. Dashed line: 0.05 FDR. Only positively enriched gene sets are found at FDR<0.05. Gene sets for IFN-γ and JAK-STAT pathways are highlighted.
(B) Effects of CA on induction of IFN-γ target genes (lfc ≥ 1, padj < 0.05).
(C and D) Plot of GRO-Seq reads (pooled replicates) at Irf1 locus for IFN0.Ctrl and IFN30.Ctrl (C) as well as IFN30.Ctrl and IFN30.CA (D). Pausing indices (PI) and transcripts per million reads (TPM; nt 501 to transcript end) are indicated.
(E) ECDF plot of PI distribution (transcriptome-wide) under IFN30.Ctrl (red) and IFN30.CA (blue) conditions. Kolmogorov-Smirnov Test p-value < 2.2e-16.
(F) Median PI and statistical assessment of PI changes for all expressed genes (RefSeq). Median PI value shown for each condition (red = highest). Mann-Whitney U test, p-value ns ≥ 0.05; * < 0.05; ** < 0.01; *** < 0.001.
(G - J) HCT116 WT and CDK8as cells were stimulated with IFN-γ (IFN) for 45 min (or unstimulated, PBS) and simultaneously treated with 10 μM 3MB-PP1 (3MB) or DMSO, and subsequently subjected to PRO-Seq.
(G) GSEA for IFN-γ response of WT HCT116 cells (DMSO.IFN vs. DMSO.PBS).
(H) GSEA for CDK8 inhibition in IFN-γ-stimulated HCT116 cells (CDK8as 3MB.IFN vs. WT.3MB.IFN).
(I) Effects of CDK8 inhibition (CDK8as, 3MB) on expression of IFN-γ-induced genes compared to WT cells. Both IFN-γ-stimulated (IFN) and -unstimulated (PBS) as well as 3MB-and control (DMSO)-treated cells were analyzed. IFN-γ-induced genes (N=83): padj < 0.1, lfc > 1 for WT DMSO.IFN vs. WT DMSO.PBS using gene body (+301 to end) counts.
(J) PI distribution during IFN-γ response upon CDK8 inhibition (CDK8as 3MB.IFN, blue) vs. uninhibited control (WT 3MB.IFN, red). Distribution shown for genes downregulated by CDK8 inhibition (N=956, padj < 0.1, log2FoldChange < 1 for CDK8as 3MB.IFN vs. WT 3MB.IFN).
The GRO-Seq read counts across Irf1 (RefSeq NM_008390), a STAT1 target gene (Bancerek et al., 2013), revealed RNAPII pausing prior to IFN-γ stimulation, as evident from pausing index (PI) of 2.03 calculated from pooled replicates (see Methods) (Figure 2C; Table S4C). Overlay of reads confirmed similar changes in both replicates (Figure S2B). Paused RNAPII was released upon IFN-γ stimulation, as revealed by almost evenly distributed read counts across the Irf1 gene and concomitant reduction in PI (PI = 0.69; Figures 2C and S2B). However, Mediator kinase inhibition with CA blocked this process (PI = 1.78, Figures 2D and S2B; Table S4C). The CA-mediated increase in PI correlated with lower induction of Irf1 transcripts (199.5 vs. 117.5 TPM, Figure 2D) and IRF1 protein (Figure S2C). Similar regulation was observed also for Tap1, another STAT1 target gene (Bancerek et al., 2013): PI = 5.6 for unstimulated control, PI = 1.82 for 30 min IFN-γ, PI = 2.83 for 30 min IFN-γ plus CA (Table S4C).
The CA-mediated PI increase at Irf1 suggested that Mediator kinase inhibition prevented release of RNAPII pausing during IFN-γ stimulation. In agreement, transcriptome-wide analysis of CA-treated IFN-γ-stimulated samples vs. IFN-γ-stimulated DMSO controls revealed a shift to higher PIs in CA-treated cells (Figures 2E and S2D; Table S4C). Analysis of PIs transcriptome-wide by the Mann-Whitney U test (Team, 2017) also confirmed that CA treatment significantly increased PIs during IFN-γ stimulation (PI = 5.31 vs. 3.82, Figure 2F). PI analysis of the gene group induced by IFN-γ revealed decreased pausing upon stimulation (Figure S2E), consistent with PI changes at Irf1 (Figure 2C). Interestingly, 19 of the 38 IFN-γ-induced genes whose mRNA levels were down-regulated by CA (Figure 1D) showed a corresponding increase in PI with CA treatment (Table S4D). These data demonstrate Mediator kinase-dependent RNAPII pause release during IFN-γ-induced transcription.
Chemical genetics confirms RNAPII pause regulation by CDK8 kinase
We next employed a CDK8 analog-sensitive (CDK8as) HCT116 human cell line (Galbraith et al., 2017), which enabled us to define CDK8-specific effects while testing in a different model system (i.e. human vs. mouse). WT and CDK8as cells were induced with IFN-γ for 45 min, followed by nuclei isolation for PRO-Seq analysis (Kwak et al., 2013). In addition to unstimulated controls, we completed experiments in the presence/absence of the CDK8as inhibitor, 3MB-PP1 (Galbraith et al., 2017). As expected, IFN-γ induced IFN-responsive gene sets in HCT116 cells (Figure 2G; Table S3B). The CDK8as cells behave as hypomorphs (Galbraith et al., 2017); consistently, IFN-γ response in CDK8as cells was diminished compared to WT cells (Figure S2F; Table S3B). Furthermore, inhibition of CDK8 kinase activity with the ATP analog 3MB-PP1 strongly reduced IFN-γ response in CDK8as cells as compared to WT cells treated with 3MB-PP1 (Figure 2H; Table S3B). Reduced induction of IFN-γ response genes with inhibition of CDK8 was similar to the effects in IFNγ-stimulated MEFs treated with CA. Heat maps from the PRO-Seq data demonstrated that 3MB-PP1 treatment did not impair the induction of IFN-γ response genes in WT cells; however, 3MB-PP1 caused reduction of the hypomorph IFN-γ response in CDK8as cells (Figure 2I; Table S5A and S5B).
In IFN-γ stimulated MEFs, we observed a correlation between increased pause index (PI) and reduced gene expression in CA-treated cells (Table S4D). Consistent with these results, a PI analysis from the PRO-Seq data in IFN-γ stimulated HCT116 cells showed a similar trend upon CDK8 kinase inhibition during IFN-γ response (Figures 2J and S2G; Table S5C). In the absence of IFN-γ stimulation, CDK8 inhibition did not increase PI in this group of genes (Figure S2H; Table S5C). Taken together, the data summarized in Figure 2 suggest that (i) Mediator kinase activity contributes to release of paused RNAPII at IFN-γ-induced genes, (ii) inhibition of Mediator kinases increases RNAPII pausing in human cells and MEFs, and (iii) reduced induction of IFN-γ target genes upon Mediator kinase inhibition correlates with defects in RNAPII pause release.
Enhancer RNA (eRNA) transcription implicates specific TFs and Mediator kinases in the IFN-γ response
Enhancer-associated transcription appears to represent the most rapid transcriptional response to a stimulus (Arner et al., 2015). Enhancer activity correlates with the expression of unstable, bidirectional transcripts defined as enhancer RNAs (eRNAs). Although the function of eRNAs remains unclear, their abundance can infer TF activity (Azofeifa et al., 2018). For instance, bidirectional eRNAs originate around sites of TF binding, and if a consensus sequence is defined, these sites can reliably predict active TFs (or TFs being repressed) at the time of the analysis (Azofeifa et al., 2018).
The eRNA profile associated with IFN-γ response has not been addressed, nor has the role of Mediator kinases in eRNA expression. To this end, we measured TF motif displacement (MD) across eRNAs in MEFs, as described (Azofeifa et al., 2018), with minor improvements (see Methods). Quantifying TF activity associated with the IFN-γ response (no IFN-γ vs. 30 min IFN-γ), we observed a significant increase in motif displacement score (MD Score) of IFN-related TF motifs (STAT1, STAT5B; Figure 3A, S3A; Table S6A). The data indicated that eRNA transcripts originating from these TF motifs were induced following IFN-γ treatment. In contrast, eRNAs associated with these same TF motifs (i.e. STAT1, STAT5) were not induced by IFN-γ if Mediator kinase activity was inhibited by CA (Figure 3B, S3A; Table S6B). These results suggest that IFNγ-responsive STAT TFs are activated, at least in part, through Mediator kinase function. In agreement, induction of IFN-γ response genes is reduced in CA-treated MEFs (Figure 1).
Figure 3. eRNA transcription predicts activation of specific TFs and role of Mediator kinases in the IFN-γ response.

(A – D) Motif displacement (MD) score during IFN-γ (30 min) response in MEFs (A and B) and HCT116 cells (C and D), derived from GRO-Seq (MEFs) and PRO-Seq (HCT116) data.
(A) MD score difference for TFs in IFN-γ-stimulated (IFN30) vs. unstimulated (IFN0) MEFs (IFN30 vs. IFN0). STAT1 and STAT5b motifs are significantly enriched upon IFN-γ stimulation.
(B) Effect of CA treatment on MD score during IFN-γ response (IFN30.CA vs. IFN0). STAT1 and STAT5b motifs enriched in (A) are not enriched upon CA treatment.
(C) MD score difference for TFs in IFN-γ-stimulated (IFN) vs. unstimulated (PBS) WT HCT116 cells with 3MB-PP1 treatment (3MB.IFN vs. 3MB.PBS). TF motifs for IFN and JAK-STAT pathways (IRF1, IRF2, IRF3, STAT2) are significantly enriched upon IFN-γ stimulation.
(D) Effect of CDK8 inhibition on MD score during IFN-γ response (CDK8as 3MB.IFN vs. 3MB.PBS). TF motifs for IFN and JAK-STAT pathways (IRF1, IRF2, IRF3, STAT2) enriched in (C) are not enriched upon CDK8 inhibition.
Similar to results in MEFs, PRO-Seq data collected in control or IFNγ-stimulated (45 min) HCT116 cells revealed activation of TFs known to be involved in the IFN response (e.g. STAT1/2, IRF1–3; Figures 3C, S3B, Table S6C); moreover, eRNAs associated with these TFs were not induced by IFN-γ in CDK8-inhibited cells (Figures 3D, S3B, Table S6D). These results suggest that Mediator kinase function during IFN-γ response is conserved in mouse and human cells and specifically identifies a role for CDK8 kinase activity in human cells. In contrast to MEFs, numerous other TFs (i.e. beyond STATs and IRFs) were significantly activated with IFN-γ treatment in HCT116 cells, and these also lacked evidence of activation upon CDK8 kinase inhibition (Figures 3C and 3D; Table S6C and S6D). Such results suggest novel roles for these TFs in the IFN-γ response. Collectively, the data from Figure 3 i) implicate a set of TFs that are rapidly activated upon IFN-γ stimulation in mouse and human cells, and ii) suggest that Mediator kinase activity is required for proper activation of these IFN-γ-induced TFs.
CDK8, not CDK19, is the major IFN-γ-activated STAT1 AD kinase
CA revealed Mediator kinase-dependent effects on IFN-γ-induced genes (Figures 1 and 2) and experiments with CDK8as cells confirmed the involvement of CDK8 kinase activity (Figure 2); however, the role of CDK19 remained unclear because CA inhibits both CDK8 and CDK19 (Pelish et al., 2015). To address the individual contribution of CDK8 and CDK19, we used immortalized MEFs derived from CreERT2-Cdk8fl/fl animals. These cells endogenously express CDK8 and CDK19 and allow conditional depletion of each (see below).
To address the role of CDK8, CreERT2-Cdk8fl/fl MEFs (referred to as CDK8fl-MEF) were treated with 4-hydroxytamoxifen (4OHT) for 3 h followed by 48 h recovery without 4OHT. This resulted in inducible Cdk8 knockout (CDK8-iKO) (Figure S4A) and loss of the CDK8 protein (Figure 4A). As expected, CDK8fl-MEFs treated with 4OHT exhibited decreased IFN-γ-induced S727 phosphorylation of STAT1 (Figure 4A), whereas STAT1 Y701 phosphorylation was not affected, consistent with previous results (Pelish et al., 2015). CDK19 protein levels remained unchanged in Cdk8 deleted cells (Figure 4A), suggesting it was not effectively compensating for CDK8 as a STAT1 AD kinase.
Figure 4. CDK8, not CDK19, is the major STAT1 AD kinase in the IFN-γ response.
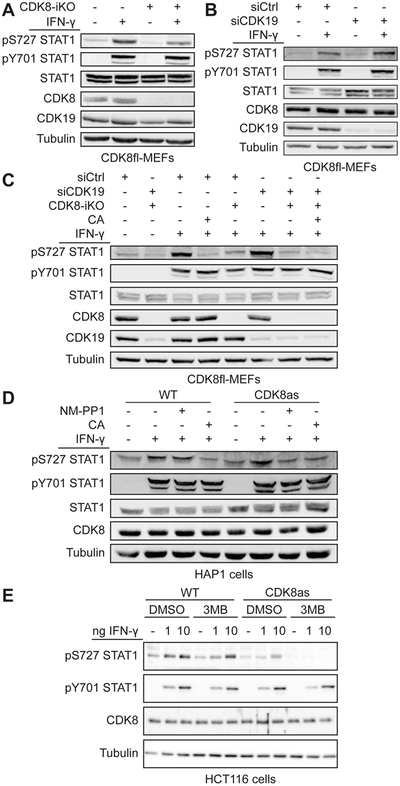
(A) Inducible CDK8 knockout (CDK8-iKO) in CDK8fl-MEFs using 4-hydroxytamoxifen (4OHT) treatment. Cells were 4OHT-treated to activate CreERT2 or control-treated, followed by IFN-γ stimulation (45 min) and subsequent Western analysis using antibodies against phosphorylated STAT1 (pS727 or pY701 STAT1) and total STAT1, CDK8, CDK19 and tubulin.
(B) siRNA knockdown of CDK19 (siCDK19). CDK8fl-MEFs were treated with siCDK19 or non-targeting siCtrl followed by IFN-γ stimulation and immunoblotting as in (A). Quantitative evaluation of blot is shown in Figure S4C.
(C) Effects of inducible CDK8 knockout (CDK8-iKO), CDK19 knockdown (siCDK19), and Mediator kinase inhibition (CA) on IFN-γ-induced STAT1 S727 phosphorylation. Treatments and immunoblotting as in (A, B). Note siCDK19 had no effect on IFN-γ-induced STAT1 S727 phosphorylation (lane 6 vs. lane 3).
(D and E) Effects of CDK8 inhibition (analog-sensitive mutant CDK8as) on IFN-γ-induced STAT1 S727 phosphorylation. (D) HAP1 cells expressing WT or CDK8as from the endogenous locus were treated with NM-PP1 (10 μM, 4 h), CA (100 nM, 1 h) or control-treated before stimulation with IFN-γ (45 min). Extracts analyzed as in (A). Note inhibition of IFN-γ-induced STAT1 S727 phosphorylation by NM-PP1 was comparable to that by CA. (E) HCT116 cells expressing WT or CDK8as from the endogenous locus were simultaneously treated with 3MB-PP1 (or DMSO control) and IFN-γ for 45 min. Extracts were analyzed as in (A).
The role of CDK19 in IFN-γ response was assessed via siRNA-mediated Cdk19 knockdown in CDK8fl-MEFs. This approach was adopted instead of Cdk19 knockout for several reasons. First, it allowed the same (i.e. isogenic) cells to be used for CDK8 knockout and CDK19 depletion, thereby avoiding differences arising from undefined genetic heterogeneities in different cell lines. Second, this inducible depletion system minimized risks of long-term effects (e.g. adaptation and compensation) in cell populations with stable deletions (Rossi et al., 2015). Third, this strategy was justified by our attempts to generate MEFs bearing a stable Cdk19 deletion; in some cases, deletion of Cdk19 in CDK8fl-MEFs caused elevation of CDK8 protein levels (Figure S4B). By contrast, such compensatory effects were not observed if cells were only transiently depleted of CDK19 (Figure 4B).
Knockdown of Cdk19 by treatment of CDK8fl-MEFs with CDK19-siRNA (referred to as siCDK19) resulted in >90% reduction of CDK19 protein levels (Figures 4B and S4C). Notably, IFN-γ-induced STAT1 S727 phosphorylation was not reduced by Cdk19 knockdown, suggesting that CDK19 plays no role in phosphorylation of STAT1 AD in the presence of CDK8 (Figure 4B). To more directly address a potential redundancy of CDK8 and CDK19, CDK8fl-MEFs were treated with 4OHT, siCDK19, or both, and IFN-γ-induced STAT1 S727 phosphorylation was examined (Figure 4C). Similar to experiments shown in Figure 4A and B, 4OHT (i.e. CDK8-iKO), but not siCDK19 treatment, consistently reduced IFN-γ-induced S727 phosphorylation (Figure 4C; replicate and quantitation in Figure S4D). Treatment with both 4OHT and siCDK19 was only slightly more efficient in inhibition of IFN-γ-induced S727 phosphorylation, similar to treatment CA (Figure 4C and S4D). Residual S727 phosphorylation is likely caused by kinases not relevant for IFN-γ pathway (e.g. p38 MAPK, (Kovarik et al., 1999)). Neither the inducible deletion of Cdk8 nor knockdown of Cdk19 caused major changes in the expression of other subunits of the Mediator kinase module (i.e., MED12, MED13 and CCNC) (Figure S4E).
The importance of CDK8 compared to CDK19 in IFN-γ-induced STAT1 S727 phosphorylation was further examined using two different human cell lines expressing analog sensitive CDK8 (CDK8as; F97G) instead of wild-type CDK8: HCT116-CDK8as (Galbraith et al., 2017) and a HAP1-CDK8as cell line (see Methods). The CDK8as version in HAP1 cells was sensitive to the ATP analog NM-PP1 (Figure S4F and G), similar to the reported inhibition of CDK8as in HCT116 cells by 3MB-PP1 (Galbraith et al., 2017). As expected, treatment of HAP1-CDK8as and HCT116-CDK8as cells with different inhibitory ATP analogs (NM-PP1 and 3MB-PP1, respectively) blocked IFN-γ-induced STAT1 S727 phosphorylation, whereas the analogs had no effect in WT cells (Figures 4D, 4E and S4H). Taken together, the data in Figure 4 implicate CDK8, but not CDK19, as the STAT1 AD kinase in the IFN-γ pathway, in both mouse and human cells.
A strategy to de-couple the effects of CDK8 vs. CDK19; kinase function vs. protein presence.
The data in Figure 4 revealed distinct roles for CDK8 vs. CDK19 in STAT1 S727 phosphorylation. We next addressed whether we could de-couple CDK8- vs. CDK19- dependent effects on IFN-γ-induced gene expression. We used the strategy summarized in Figure 5A, which enabled inducible knockout of CDK8 (CDK8-iKO) by 4OHT treatment, knockdown of CDK19 (siCDK19), or both (CDK8-iKO + siCDK19). CA treatment was implemented as needed to de-couple protein presence (CDK8 and/or CDK19) from enzymatic function. As before, CA treatment (or DMSO controls) occurred 1 h prior to IFN-γ stimulation. For these experiments, IFN-γ treatment lasted 3 hours prior to analysis.
Figure 5. CA has no effects on IFN-γ-regulated transcription in the absence of CDK8 and CDK19.

(A) Experimental overview. RNA-Seq experiments were completed using 3 replicates for each condition (8 conditions total; siCtrl: control condition).
(B) IFN-γ-induced gene expression changes (mRNA) in siCtrl cells (siCtrl 3 h IFN vs. siCtrl 0 h IFN).
(C) GSEA of IFN-γ-induced changes in siCtrl cells (siCtrl 3 h IFN vs. siCtrl 0 h IFN). Gene sets representing IFN and JAK-STAT pathways are highlighted.
(D) GSEA of CA effects on IFN-γ-induced (3 h) changes in siCtrl cells (siCtrl CA IFN vs. siCtrl IFN).
(E) CA effects on gene expression changes (mRNA) upon IFN-γ stimulation (3 h) in siCtrl cells (siCtrl CA IFN vs. siCtrl IFN).
(F) CA effects on gene expression changes (mRNA) in absence of CDK8 and CDK19 during IFN-γ stimulation (3 h) (siCDK19 CDK8-iKO CA IFN vs. siCDK19 CDK8-iKO IFN). Note that no genes significantly (blue) up- or down-regulated by CA.
After treatments, mRNA and pre-mRNA levels (three biological replicates) were determined by RNA-Seq (Table S7), as described for 6 h IFN-γ treatment (Figure 1). IFN-γ treatment almost exclusively up-regulated gene expression, with induction of 274 genes (Ifc ≥ 1; padj < 0.05; FPKM in IFN-γ stimulated samples ≥ 1) and only 4 genes downregulated (lfc ≤ 1; padj < 0.05; FPKM in IFN-γ stimulated samples ≥ 1) (Figure 5B). The numbers of genes regulated at the pre-mRNA level were similar and largely overlapping with mRNA gene sets implicating IFN-γ effects at the transcriptional level rather than post-transcriptional regulation (Figure S5A). GSEA for IFN-γ-stimulated vs. unstimulated datasets confirmed induction of IFN-γ and JAK-STAT pathways (Figure 5C; Table S3C). Moreover, the gene sets revealed by GSEA (normalized enrichment score, NES > 1.5; FDR < 0.05) in IFN-γ-stimulated vs. unstimulated cells showed a large overlap between GRO-Seq (t = 30 min IFN-γ) and RNA-Seq (t = 3 h IFN-γ) experiments, and consisted mostly of IFN-γ or IFNγ-related pathways (e.g., JAK-STAT, IL-2, IL-4, IL-6) (Figure S5B; Table S3D).
CA does not affect transcription in the absence of CDK8 and CDK19
To test whether CA effects on transcription were dependent on the Mediator kinases, we analyzed CA effects during the IFN-γ response in MEFs devoid of both CDK8 and CDK19 (siCDK19 CDK8-iKO) compared to control MEFs (siCtrl). As expected, CA negatively affected gene sets associated with the IFN-γ and JAK-STAT pathways and altered the genome-wide IFN-γ response in control MEFs (Figures 5D and 5E; Table S3E). In contrast, CA did not significantly change the transcriptome in the absence of CDK8 + CDK19, with or without IFN-γ stimulation (Figures 5F and S5C). Similar results were seen in CDK8 knockout cells (see below). These results reflect the high degree of CA selectivity (Pelish et al., 2015) and also imply that CA does not alter the biological function of other (i.e. non-kinase) transcriptional regulatory proteins.
CDK8 and CDK19 are non-redundant and mechanistically distinct transcriptional regulators of the IFN-γ response
To analyze effects of CDK8 and CDK19 during IFN-γ response and to distinguish kinase-dependent vs. -independent functions, we generated RNA-Seq comparisons among IFN-γ-induced genes from IFNγ-stimulated control (siCtrl) cell populations. Comparisons were completed between the experiments summarized in Figure 5A. A heat map depicting genes (N = 178) induced by IFN-γ in control cells (siCtrl) (lfc ≥ 1; padj < 0.05; FPKM in IFN-γ stimulated samples ≥ 1) that were differentially expressed (padj < 0.05) in at least one of the conditions is shown in Figure 6A. The data revealed a similar, but not identical, pattern of gene expression changes for Mediator kinase inhibition in control cells (siCtrl CA vs. siCtrl, Figure 6A, line 1) and in the absence of CDK19 (siCDK19 CA vs. siCDK19, Figure 6A, line 2); furthermore, similar gene expression changes were seen in the absence of CDK8 (siCtrl CDK8-iKO vs. siCtrl, Figure 6A, line 3). These results (Figure 6A, lines 1-3) suggested that Mediator kinase activity regulated IFN-γ-induced transcription specifically through CDK8 not CDK19. The lack of significant effect of Mediator kinase inhibition in the absence of CDK8 (siCtrl CDK8-iKO CA vs. siCtrl CDK8-iKO; Figure 6A, line 4) further supports this conclusion.
Figure 6. Transcriptional response to IFN-γ is predominantly executed by kinase-dependent effects of CDK8 and kinase-independent effects of CDK19.

(A - F) CDK8fl-MEFs were treated as described in Figure 5A and assessed by differential mRNA expression (3 biological RNA-Seq replicates each).
(A) Heat map summarizing mRNA expression changes caused by CA, CDK8 knockout and CDK19 knockdown in group of IFN-γ-induced genes. Genes induced by IFN-γ (lfc ≥ 1, padj < 0.05, FPKM stimulated ≥ 1) in siCtrl-treated CDK8fl-MEFs (siCtrl) were analyzed under the following conditions: CA treatment (CDK8+CDK19 inhibition), lane 1; CA treatment in absence of CDK19 (i.e., CDK8 inhibition), lane 2; inducible CDK8 knockout (CDK8-iKO), lane 3; CA treatment in absence of CDK8 (i.e., CDK19 inhibition) lane 4; CDK19 knockdown, lane 5. Only genes that changed (padj < 0.05) in at least one of the 5 conditions are shown.
(B - F) Volcano plots corresponding to treatments shown in (A). Panel order (B - F) corresponds to lane order in (A). No significant (padj < 0.05) gene expression changes upon CA treatment of CDK8 knockout cells (CDK19 inhibition: siCtrl CDK8-iKO CA vs. siCtrl CDK8-iKO) (E).
The pattern for CDK19 depletion was markedly different from other conditions (siCDK19 vs. siCtrl, Figure 6A, line 5), suggesting that CDK19 plays distinct roles in regulation of transcriptional responses to IFN-γ. This was supported by limited overlap of IFN-γ-induced genes downregulated by CDK8 deletion vs. CDK19 knockdown (Figure S5D). Volcano plots corresponding to heat map comparisons are shown in Figure 6B-F.
The distinct transcriptional effects of CDK19 knockdown (siCDK19) on the IFN-γ transcriptional response were apparent also from GSEA comparisons (Table S3E). Genes downregulated by CDK8-iKO (18 gene sets with NES < −1.5) vs. siCDK19 (19 gene sets) shared only 2 gene sets (Figure S5E; Table S3E). Whereas IFN-γ and JAK-STAT pathway gene sets were identified in the CDK8-iKO (Figure S5F) and CA-treated data sets (Figure 5D), these pathways were not observed in siCDK19 gene sets – these gene sets included metabolic and other inflammatory pathways (Figure S5G; Table S3E). Thus, in agreement with the gene expression analyses summarized in Figure 6, GSEA implicated CDK19 in the regulation of different gene sets in IFN-γ-treated cells. We note, however, that expression of several well-known IFN-γ target genes such as Irf1 and Gbp2 were similarly affected by CDK8-iKO, siCDK19, or CA (Table S7).
The data summarized in Figures 5 and 6 revealed that the IFN-γ response was differentially regulated by CDK8 and CDK19, by distinct mechanisms. Regulation by CDK8 depended primarily on its kinase activity whereas the effects of CDK19 appeared to be kinase-independent.
CDK19 is a kinase-independent driver of the IFN-γ anti-viral response
CCNC is required for Mediator kinase activity (Knuesel et al., 2009; Li et al., 2014; Turunen et al., 2014), and we previously established CCNC as important for cellular response to viral infection (Bancerek et al., 2013). Because CCNC interacts with CDK8 or CDK19 in a mutually exclusive manner (Galbraith et al., 2013), these results did not distinguish between CDK8 and CDK19. However, the data shown in Figures 4 and S4 revealed that STAT1 AD phosphorylation was mediated through CDK8. Because STAT1 AD phosphorylation is required for the induction of anti-viral state by IFN-γ (Bromberg et al., 1996; Horvath and Darnell, 1996), this implicates CDK8 as an essential activator of anti-viral response, which is further supported by CDK8-dependent effects on IFNγ-induced genes (Figures 6A and S5F).
To test the role of CDK19 in the anti-viral response, we used methods similar to those used previously for CCNC (Bancerek et al., 2013); specifically, we infected siCDK19 or siCtrl MEFs (pre-stimulated for 24 h with serial dilutions of IFN-γ) with vesicular stomatitis virus (VSV) and assessed cell viability. Cells depleted for CDK19 reproducibly displayed increased sensitivity to VSV infection compared to siCtrl-treated samples (Figure S6A). Calculation of the EC50 (IFN-γ needed to prevent 50% cell death) revealed that cells depleted of CDK19 were 5-fold more sensitive to VSV infection compared to controls (EC50 0.925 vs. 0.181; Figure 7A), demonstrating an important role for the CDK19 protein in the antiviral response.
Figure 7. CDK19 drives the IFN-γ anti-viral response in kinase-independent ways.

(A) IFN-γ-dependent antiviral response in the absence of CDK19. siCDK19 and siCtrl cells were pre-treated with various concentrations of IFN-γ followed by infection with vesicular stomatitis virus (VSV). Percentages of surviving cells and EC50 values (IFN-γ concentration needed to prevent 50% cell death) are shown (as means of duplicate experiments).
(B) Assessment of IFN-γ-dependent antiviral response of CDK19-KO cells rescued with CDK19-WT or CDK19-KDead. CDK19-KO, CDK19-WT and CDK19-KDead cells were IFN-γ-treated and VSV-infected and analyzed as in (A).
(C and D) Overlap of IFN-γ-induced genes upregulated (C) or downregulated (D) upon rescue of CDK19-KO cells with CDK19-WT or CDK19-KDead. Data derived from RNA-Seq of CDK19-KO, CDK19-WT and CDK19-KDead stimulated with IFN-γ for 3 h.
(E) Heat map summarizing expression changes of IFN-γ-induced genes (lfc ≥ 1, padj < 0.05, FPKM stimulated ≥ 1) upon rescue of CDK19-KO cells with CDK19-WT or CDK19-KDead.
To further probe kinase vs. protein functions of CDK19, we generated CDK19 knockout MEFs (CDK19-KO) using CRISPR-Cas9 in CDK8fl-MEFs, and “rescued” them (without clonal selection) with expression of wild-type CDK19 (CDK19-WT) or kinase-dead CDK19 (CDK19-KDead). Expression levels of CDK19-WT and CDK19-KDead were comparable in rescue cells (Figure S6C); importantly, the cell pools did not correspondingly up-regulate CDK8.
VSV infection assays showed that rescue expression of CDK19-WT or CDK19-KDead had comparable effects, with ~30-fold better IFN-γ-dependent survival compared with CDK19-KO MEFs (EC50 0.118, 0.111 and 3.161, respectively; Figures 7B and S6B). Consistent with the VSV infection assays, RNA-Seq data (Table S8) showed similar effects of CDK19-WT and CDK19-KDead rescue expression on IFN-γ-induced genes, as depicted in Venn diagrams (Figures 7C and 7D), heat maps (Figure 7E), and PCA plots (Figure S6C). These data further support a kinase-independent, structural/scaffolding role for the CDK19 protein. Combined with the gene expression data summarized in Figures 5 and 6, these results demonstrated that CDK8 and CDK19 are essential but non-redundant regulators of IFN-γ responses; moreover, unlike CDK8, CDK19 drives the IFN-γ-induced anti-viral defense by a kinase-independent mechanism.
DISCUSSION
The IFN-γ signaling pathway is relevant in most mammalian cell types. The Mediator kinase CDK8 has been shown to regulate IFN-γ-stimulated transcription partially through phosphorylation of the STAT1 AD at S727 (Bancerek et al., 2013; Nitulescu et al., 2017). Potential functions for the highly conserved CDK8 paralog CDK19, however, have not been thoroughly addressed. The development of selective inhibitors of CDK8 and CDK19 has enabled a delineation of kinase-specific functions (Dale et al., 2015; Johannessen et al., 2017; Koehler et al., 2016; Pelish et al., 2015). These studies have established that the transcriptional effects of Mediator kinase inhibition can be distinct from CDK8 or CDK19 knockdown (Poss et al., 2016), which reflects basic structural or scaffolding roles for the kinase protein itself. In this study, we set out to define and de-couple the 1) regulatory roles of CDK19 vs. CDK8 and the 2) effects of their enzymatic activity vs. the structural/scaffolding function for each kinase. To our knowledge, this IFN-γ-focused study represents the most thorough analysis of Mediator kinase structural and enzymatic function to date.
The GRO-Seq and PRO-Seq data have solidified a role for CDK8 in RNAPII pause regulation, which could be inferred from ChIP-Seq data in human cells (Galbraith et al., 2013; Pelish et al., 2015). Inhibition of Mediator kinase activity increased pausing at hundreds of genes in MEFs and human HCT116 cells. This effect was prominent at IFN-γ-induced genes, and we noted a correlation between increased PI and genes negatively regulated by CA. These results point to a general role for Mediator kinase activity in RNAPII pausing and/or pause release. Although the kinase substrates for CDK8 and CDK19 have not been identified in IFN-γ-stimulated cells, experiments in unstimulated HCT116 cells identified high-confidence targets that may contribute to Mediator kinase-dependent RNAPII pause regulation, including AFF4, NELFA, and POLR2M (Poss et al., 2016). CDK8-dependent STAT1 S727 phosphorylation may also impact RNAPII promoter-proximal pausing upon IFN-γ stimulation. Future experiments will seek to delineate phospho-site specific regulatory roles; however, we anticipate that many CDK8 substrates contribute to transcriptional regulation upon activation of IFN-γ signaling cascades.
The most rapid transcriptional response to a stimulus appears to be expression at enhancers (Arner et al., 2015). The MD Score is an unbiased means to assess eRNA transcriptional changes, and mapping changes (positive or negative) to consensus TF binding motifs can reliably infer TF activity (Azofeifa et al., 2018). Using the MD Score method, we identified eRNAs that were strongly induced by IFN-γ (t = 30 min); as expected, the “epicenters” of the induced eRNAs mapped to consensus motifs of JAK-STAT pathway TFs (e.g. STAT1, IRF1) in both mouse and human cells. Notably, Mediator kinase inhibition blocked eRNA induction at these loci, suggesting reduced IFN-γ-responsive TF activity. Whereas the MD Score data showed expected TF induction upon IFN-γ stimulation, eRNAs associated with other TFs were identified as well. This may reflect uncharacterized biological roles for these factors (e.g. ESR2, MEF2) in the IFN-γ response, which could be explored in future studies.
In the context of IFN-γ stimulation, we completed comparative analyses in a cell line (CDK8fl-MEFs) that allowed inducible deletion or depletion of either CDK8 or CDK19, thereby avoiding compensatory effects that commonly arise from clonal selection of knockout cell lines (Rossi et al., 2015). Although both CDK8 and CDK19 were shown to govern the IFN-γ transcriptional response, they regulated distinct sets of genes via distinct mechanisms. The impact of CDK8 derived primarily from its kinase activity. Using a combination of approaches, we observed that phosphorylation of the STAT1 AD at S727 is mediated by CDK8 but not CDK19, extending previous results in IFN-γ-stimulated MEFs (Bancerek et al., 2013). In contrast to CDK8, we observed that the kinase activity of CDK19 was largely inconsequential. Rather, a structural role was evident for CDK19, as its inducible knockdown triggered stark transcriptional effects during IFN-γ stimulation that were not affected by CA treatment.
Both the kinase-dependent (CDK8-mediated) and kinase-independent (CDK19-mediated) effects appeared to be essential for the IFN-γ response. STAT1 AD phosphorylation (CDK8-dependent) is required for efficient induction of the antiviral state (Bromberg et al., 1996; Horvath and Darnell, 1996). In this study, we demonstrated that the CDK8 paralog CDK19 is a similarly essential component of the IFN-γ-induced antiviral program. Importantly, CDK19 kinase-independent function was verified in rescue experiments; expression of either WT or kinase-dead CDK19 in a null background restored gene expression and VSV resistance toward WT levels. A structural/scaffolding role for CDK19 (i.e. kinase-independent) was also observed during p53 response in human osteosarcoma cells (Audetat et al., 2017), suggesting that CDK19 mediates structural interactions not shared by CDK8. Because CDK8 and CDK19 are mutually exclusive subunits of the Mediator kinase module (Galbraith et al., 2013), these results suggest the assembly of distinct CDK8- and CDK19-Mediator complexes at select genomic loci during IFN-γ stimulation.
Because IFN-γ almost exclusively up-regulated gene expression, this served as a means to study Mediator kinase-dependent effects on transcription activation. We observed that CA had minimal impact on gene expression in unstimulated cells, whereas CA suppressed induction of dozens of genes activated by IFN-γ. These findings have parallels with work from the Roninson group, in which they noted that Mediator kinase inhibition (with Senexin A) suppressed transcriptional activation by NF-κB but had little effect on basal expression (Chen et al., 2017). Similarly, Johannessen et al. noted that Mediator kinase inhibition did not grossly perturb the transcriptome of quiescent macrophages (Johannessen et al., 2017). Given the selective effects on inducible (i.e. not basal) gene expression, the Roninson group concluded that CDK8 and/or CDK19 may represent key mediators of transcriptional reprogramming (Chen et al., 2017). In this context, transcriptional reprogramming refers to the initial stimulus response and is distinct from reprogramming associated with cell differentiation.
Rapid transcriptional changes are important for immune system activation during infection and likely contribute to longer-term shifts in gene expression patterns (e.g. through epigenetic changes (Ivashkiv, 2018)). Our results implicate Mediator kinases in these processes but future study is needed to characterize their impact across longer time frames. It is notable that CDK8 or CDK19 knockdown or CDK8/CDK19 inhibition is not generally cytotoxic under normal growth conditions (Donner et al., 2007; Galbraith et al., 2013; Pelish et al., 2015), implicating Mediator kinase function as more important for adaptive transcriptional responses (i.e. reprogramming). Regulation of interferon responses has broad physiological relevance, ranging from inflammation to aging to tumor cell clearance. Our results reveal that the Mediator kinases activate distinct transcriptional programs – through distinct mechanisms – in response to the ubiquitous inflammatory cytokine IFN-γ, suggesting that separately targeting CDK8 kinase activity or CDK19 protein levels (e.g. with PROTACs) may have diverse biomedical applications.
STAR METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Pavel Kovarik (pavel.kovarik@univie.ac.at).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
MEF (mouse embryonic fibroblasts) cell lines established in this study:
Conditional CDK8 knockout MEFs
MEFs allowing inducible deletion of CDK8 were derived from CreERT2-Cdk8fl/fl mice. Cdk8fl/fl mice (allele Cdk8tm1c(EUCOMM)Hmgu, provided by Yann Herault, IGMBC, Illkirch, France) on C57BL/6 background were crossed with Rosa26CreERT2 mice (Hameyer et al., 2007) to generate CreERT-Cdk8fl/fl mice. Primary MEFs from CreERT2-CDK8fl/fl mice were isolated on day 13.5 and immortalized via the 3T3 method (Todaro and Green, 1963). Activation of the CreERT2 recombinase led to the excision of the loxP-flanked exon 5 resulting in a frameshift and nonsense mediated decay. CDK19 KO MEFs: CDK19 knockout cells were generated in CDK8fl-MEFs using CRISPR/Cas9 (described in detail in the Method details section).
CDK19 KO MEFs expressing CDK19-WT or CDK19-KDead protein
CDK19 KO cells were reconstituted with either CDK19-WT or CDK19-KDead expression constructs as described in the Method details section.
METHOD DETAILS
Cell culture
Mouse embryonic fibroblasts expressing wildtype STAT1 (WT MEFs) were described previously (Bancerek et al., 2013). MEFs allowing inducible deletion of CDK8 were generated by immortalization of primary MEFs derived from CreERT2-Cdk8fl/fl mice. Briefly, Cdk8fl/fl mice (allele Cdk8tm1c(EUCOMM)Hmgu, provided by Yann Herault, IGMBC, Illkirch, France) on C57BL/6 background were crossed with Rosa26CreERT2 mice (Hameyer et al., 2007) to generate CreERT-Cdk8fl/fl mice. Primary MEFs from CreERT2-CDK8fl/fl mice were isolated on day 13.5 and immortalized via the 3T3 method (Todaro and Green, 1963). Activation of the CreERT2 recombinase led to the excision of the loxP-flanked exon 5 resulting in a frameshift and nonsense mediated decay. MEFs were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal calf serum (FCS) and penicillin-streptomycin. Wildtype HAP1 cells (Carette et al., 2011) and CRISPR-Cas9 generated CDK8 knockout HAP1 cells (clone 325-1) were purchased from Haplogen (Austria). HAP1 cells were grown in Iscove’s modified Dulbecco’s medium (IMDM) supplemented with 10% FCS, 200 nM L-glutamine and penicillin-streptomycin. HCT116 cells were grown in McCoy’s media (Gibco, 16600082) with Gibco 100x Antibiotic-Antimycotic (Fisher Scientific, 15240062) penicillin-streptomycin and 10% fetal bovine serum (FBS) supplementation.
Cytokines and inhibitors
Murine IFN-γ (eBioscience) was used at a concentration of 10 ng/ml for stimulation of MEFs. Human IFN-γ (kind gift from James E. Darnell, Rockefeller University, US) was used for stimulation of HAP1 cells at 10 ng/ml. HCT116 cells were treated with 10 ng/ml IFN-γ (Fisher Scientific, #PHC4031). Cortistatin A (kindly provided by Matthew Shair, Harvard University, Cambridge, USA) was applied at a concentration of 100 nM one hour before IFN-γ stimulation. NM-PP1 (Calbiochem, 529581) used for inhibition of analog-sensitive CDK8 mutant (CDK8as) was applied at a concentration of 10 μM 4 h before IFN-γ stimulation. The ATP analog 3MB-PP1 (Cayman Chemical, 56025–83-5) was applied at 10 μM for 45 minutes (simultaneously with IFN-γ).
RNA-Seq
The RNA isolation was done as described (Audetat et al., 2017). In brief cells were seeded with 70% confluency on 15 cm and 6 cm dishes, respectively. For total RNA isolation, 7 ml (15 cm dish) or 2 ml (6 cm dish) Qiazol Lysis Reagent (Qiagen, 79306) were added. The samples were mixed thoroughly before taking 1 aliquot for Chloroform extraction. RNA was precipitated with Isopropanol and Sodium chloride, followed by DNase treatment using RNase-free DNase Set (Qiagen, 79254) and clean up using RNeasy Mini Kit (Qiagen, 74104). For library preparation the NEBNext rRNA Depletion Kit (NEB E6310S) or the NEBNext Poly(A) mRNA Magnetic Isolation Module (NEB E7490S), together with the NEBNext Ultra II RNA Library Prep Kit from NEB (NEB E7770S) were used according to the manufacturer’s protocol. The RNA quality was assessed using Agilent RNA 6000 Nano Assays (5067-1511) that were analyzed on an Agilent 2100 Bioanalyzer. The library quality check and Solexa sequencing was performed at the VBCF NGS Unit (www.viennabiocenter.org/facilities). Single-end fragment libraries (50 bp) were sequenced on the Illumina HiSeq 2500 platform. Processing of raw reads and mapping were done as described for GRO-seq experiment. Quantitation of RNA-Seq data for WT MEFs stimulated with IFN-γ for 6 h +/− CA treatment (Figure 1) was carried out using HTSeq (Anders et al., 2015). Reads mapped to exons and exon-exon junctions were defined as mRNA reads, whilst reads mapped to introns and intron-exon junctions were defined as pre-mRNA reads. The raw as well as processed data are accessible via the NCBI’s Sequence Read Archive (SRA) database (accession number PRJNA542065). Differential expression analysis was performed based on read counts using DESeq2 (Love et al., 2014). Principal component analysis (PCA) and normalization of read counts to library size and composition (using DESeq2) revealed that replicate 1 of sample 6 h IFN-γ without CA was an outlier. This replicate was removed from the subsequent analysis. IFN-γ-induced genes were defined by log2-fold-change (lfc) ≥ 1, padj < 0.05 and FPKM stimulated ≥ 1. Analysis of RNA-Seq data for siCtrl, CDK8-iKO and siCDK19 in the presence or absence of CA with or without 3 h IFN-γ stimulation (Figure 5 and 6) revealed more than 95% uniquely mapped reads in each sample. Sample integrity was analyzed by gene body coverage plots using RSeQC. Transcripts were quantified using Mix2 RNA-Seq data analysis software (Lexogen). Differential expression analysis was performed using DESeq2 version 1.18.1. Exploratory data analysis and visualizations were performed in R-project version 3.4.2 (Team, 2017) (Foundation for Statistical Computing, Vienna, Austria, https://www.R-project.org/) with Rstudio IDE version 1.0.143, ggplot2 (2.2.1), dplyr (0.7.4), readr (1.1.1), gplots (3.0.1). GSEA was completed as described for GRO-Seq experiments. Genes were required to have FPKM > 0.5 in all replicates and conditions.
GRO-Seq
Cells were seeded on three 15 cm dishes (4×106 per dish) for each time point or treatment 16 h prior to the experiments (~70% confluency at the time of experiment). Cells were stimulated or treated as desired, washed twice with cold PBS and detached using 2 ml Trypsin/EDTA per dish. Cells were re-suspended in 10 ml cold PBS and collected by centrifugation at 270 g for 5 minutes. For lysis, cells were incubated for 10 minutes in 10 ml lysis buffer (10 mM Tris-HCl, 5 mM MgCl2, 10 mM NaCl, 0.5% NP-40, 1 mM DTT, 1 mM sodium metabisulfite, 1 mM benzamidine, 0.025 mM PMSF, 4 U/ml SUPERase-In) on ice. Nuclei were collected by centrifugation at 170 g for 10 minutes, washed once with 1 ml reaction buffer (20 mM Tris-HCl, 10 mM MgCl2, 150 mM KCl, 20% Glycerol, 4 U/ml SUPERase-In), and re-suspended in 50 μl reaction buffer and counted. Total 5×106 nuclei in 100 μl reaction buffer were used per run-on reaction. The Run-on reaction and capturing of the labelled RNA was done as described previously (Allen et al., 2014). Briefly, 28.9 μl reaction buffer, 5 μl of rATP, rCTP, rGTP and 5-Bromo-UTP (10 mM each), 0.1 μl DTT, 1 μl RNase-In and 50 μl 2% sarkosyl were added per run-on reaction. The samples were incubated at 30°C for 5 minutes, followed by RNA isolation using TRIzol reagent (Invitrogen, 15596026). RNA precipitation was done using Isopropanol and GlycoBlue. RNA was fragmented by incubation with Ambion Fragmentation Reagents (AM8740) at 7ΰC for 10 minutes, run over an Illustra MicroSpin G-25 column (GE Healthcare, 27532501) according to the manufacturer's protocol and DNase treated (Promega, M6101) for 10 minutes at 37°C. To capture the labelled RNA, two rounds of bead binding were performed. Therefore the samples were incubated for 1 h with Anti-BrU agarose beads (Santa Cruz, sc-32323 AC), followed by extensive washing and elution. Eluted RNA was phenol/chloroform extracted and ethanol precipitated. After the second RNA precipitation samples were immediately used for library preparation. The sequencing library was prepared using the NEBNext Ultra Directional RNA Library Prep Kit (NEB, E7420S) following the manufacturer’s protocol for highly degraded RNA with RNA integrity number (RIN) ≤ 2. The library quality check and sequencing was performed at the Genomics and Microarray Core Facility at the University of Colorado Anschutz Medical Campus and at the BioFrontiers Sequencing Facility at the University of Colorado Boulder. Single-end fragment libraries (50 bp) were sequenced on the Illumina HiSeq 4000 platform. Raw sequencing reads were demultiplexed, and after barcode, adaptor and quality trimming with cutadapt (http://dx.doi.org/10.14806/ej.17.1.200), quality control was performed using FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). The remaining reads were mapped to the GRCm38/mm10 mouse genome assembly using genomic short-read RNA-Seq aligner STAR version 2.5 (Dobin et al., 2013). We obtained at least 65% uniquely mapped reads in each sample. Data analysis and visualizations were performed in R-project version 3.4.2 with Rstudio IDE version 1.0.143. Reads were counted in 2 intervals per transcript (relative to transcription start site, interval 1: (−500;500), interval 2: (501:end)) using featureCounts (Liao et al., 2014). Pausing index was calculated as ratio of read counts in interval 1 to read counts in interval 2 normalized to the length of intervals. Inhibitor response was calculated as ratio of pausing index in the presence of inhibitor to pausing index in the control. Calculation of transcripts per million reads (log2TPM) excluded the first 500 bases downstream the transcriptional start side to minimize effects of RNAPII pausing. PI was calculated both for individual replicates and pooled replicates (by summing up counts). For gene set enrichment analysis (GSEA) the GSEA preranked module on the GenePattern server (Reich et al., 2006) was used, with log2-fold-change values for all detected genes for the indicated comparisons as the ranking metric. Genes were required to have TPM > 0.5 in all replicates and conditions.
The GRO-Seq data are accessible via the NCBI’s Sequence Read Archive (SRA) database (accession number PRJNA542065).
PRO-Seq
Nuclei Preparation
HCT116 cells (WT or CDK8as) were seeded on three 15 cm dishes (1×107 cells/dish), 24 h prior to the experiments (~70% confluency at time of experiment). Cells were treated simultaneously with 10 ng/ml IFN-γ and/or 10 μM 3MB-PP1 for 45 min, washed 3x with ice cold PBS, and then treated with 10 ml (per 15 cm plate) ice-cold lysis buffer (10 mM Tris-HCl pH 7.4, 2 mM MgCl2, 3 mM CaCl2, 0.5% NP-40, 10% glycerol, 1 mM DTT, 1x Protease Inhibitors (1 mM Benzamidine (Sigma B6506-100G), 1 mM Sodium Metabisulfite (Sigma 255556-100G), 0.25 mM Phenylmethylsulfonyl Fluoride (American Bioanalytical AB01620), and 4 U/ml SUPERase-In) and scraped from the plates. Cells were centrifuged 1000 g for 15 min at 4°C. Supernatant was removed and pell et was resuspended in 1.5 ml lysis buffer to a homogenous mixture by pipetting 20-30X before adding another 8.5 ml lysis buffer. Suspension was centrifuged with a fixed-angle rotor at 1000 g for 15 min at 4°C. Supernatant was removed and pellet was resuspe nded in 1 ml of lysis buffer and transferred to a 1.7 ml pre-lubricated tube (Costar cat. No. 3207). Suspensions were then pelleted in a microcentrifuge at 1000 g for 5 min at 4°C. Next, supernatant was removed and pellets were resuspended in 500 μl of freezing buffer (50 mM Tris pH 8.3, 40% glycerol, 5 mM MgCl2, 0.1 mM EDTA, 4 U/ml SUPERase-In). Nuclei were centrifuged 2000 g for 2 min at 4°C. Pellets were resuspended in 100 μl freezing buffer. To determine concentration, nuclei were counted from 1 μl of suspension and freezing buffer was added to generate 100 μl aliquots of 10×106 nuclei. Aliquots were flash frozen in liquid nitrogen and stored at −80°C.
Nuclear run-on and RNA preparation
Nuclear run-on experiments were performed as described (Mahat et al., 2016) with the following modifications: the final concentration of non-biotinylated CTP was raised from 0.25 μM to 25 μM, and the final library clean-up and size selection was accomplished using 1X AMPure XP beads (Beckman).
Sequencing
Sequencing of PRO-Seq libraries was performed at the BioFrontiers Sequencing Facility (UC-Boulder). Single-end fragment libraries (75 bp) were sequenced on the Illumina NextSeq 500 platform (RTA version: 2.4.11, Instrument ID: NB501447), demultiplexed and converted BCL to fastq format using bcl2fastq (bcl2fastq v2.20.0.422); sequencing data quality was assessed using FASTQC (v0.11.5) (https://www.bioinformatics.babraham.ac.uk/proiects/fastqc/) and FastQ Screen (v0.11.0, https://www.bioinformatics.babraham.ac.uk/proiects/fastq_screen/). Trimming and filtering of low-quality reads was performed using BBDUK from BBTools (v37.99) and FASTQ-MCF from EAUtils (v1.05) (Kechin et al., 2017). Alignment to the human reference genome (GRCh37/hg19) was carried out using Hisat2 (v2.1.0) (Kim et al., 2015) in unpaired, no-spliced-alignment mode with a GRCh37/hg19 index, and alignments were sorted and filtered for mapping quality (MAPQ>10) using Samtools (v1.5) (Li et al., 2015). Gene-level count data for transcription start site (TSS, −30 to +300) and gene body (+301 to end) regions were obtained using featureCounts from the Subread package (v1.6.2) (Liao et al., 2013) with custom annotation files for single unique TSS and gene body regions per gene. Custom annotation files with single unique TSS and gene body regions per gene were generated as follows: 1) hg19 RefSeqCurated transcript-level annotation was downloaded from the UCSC genome table browser (09-07-2018), transcripts shorter than 1500 bp and non-standard chromosome were removed, and only transcripts with unique start/stop coordinates per gene were retained; 2) Sense and anti-sense counts were tabulated and each candidate TSS region was ranked by sense and antisense reads to obtain a single ‘most-active’ TSS per gene; 3) Finally, per gene, the TSS was combined with the shortest gene body to avoid the influence of alternative transcription termination/polyadenylation sites. Differential expression analysis of gene body regions was assessed using the DESeq2 package (v1.22.1) (Love et al., 2014) with a custom R script (R v3.5.1 / RStudio v1.1.453 / Bioconductor v3.7) with cutoffs as described in text and figure legends. Analysis of RNAPII pausing was carried out using a custom R script (R v3.5.1 / RStudio v1.1.453) with the ggplot2 package (v3.1.0) used for visualizations. Gene level TSS and gene body counts were normalized by counts-per-million and by region length (cpm/bp), and Pausing Index (PI) calculated as the ratio of normalized reads in the TSS (cpm/bp) to normalized reads in the gene body (cpm/bp). Genes with < 0.5 cpm in all samples were excluded from analysis. Means of replicate values were used for plots and Wilcoxon/Mann-Whitney U tests. For genome browser snapshots, aligned reads were downsampled to the lower aligned read count per replicate using Samtools, to ensure equal contributions from each replicate, followed by merging of replicates and generation of coverage tracks in the bedgraph format using HOMER (v4.9.1) (Heinz et al., 2010) Genome browser snapshots were then generated from the bedgraph files using a custom R script (R v3.5.1 / RStudio v1.1.453 / Bioconductor v3.7) and the Gviz package (v1.26.4) (Hahne and Ivanek, 2016). PRO-Seq data are accessible via the NCBI’s Gene Expression Omnibus (GEO) database (accession number GSE129501).
Modified motif displacement score (MD Score) analysis
We performed the motif displacement (MD) analysis as described (Azofeifa et al., 2018) with the top 20% differentially transcribed enhancer RNAs (eRNAs) quantified by DE-Seq2. This modification was made to improve the signal-to-noise ratio of the MD analysis. For the analysis of TF motifs associated with eRNA transcription we used the hand-curated database of TF binding motif models HOCOMOCO (Kulakovskiy et al., 2013). We note that within the HOCOMOCO database for mouse TF binding motifs, the principal binding model for STAT1 (motif ID: STAT1_MOUSE.H11MO.0.A) corresponds to IRF binding motifs whereas the alternate binding model (motif ID: STAT1_MOUSE.H11MO.1.A) corresponds to the canonical STAT1 motif (Decker et al., 1997; Mancino and Natoli, 2016). We have included this information in the table containing MD scores (Table S6) and used the correct designation in the MD score figure (Figure 3A and B).
Inducible CDK8 knockout
Inducible CDK8 knockout in CDK8fl-MEFs was induced by 4-hydroxytamoxifen (4OHT) treatment (3 h, 250 nM) in low fetal calf serum medium (2% FCS), followed by recovery (2 days). CDK8 knockout was validated by genotyping using the primers CDK8 Intron4/Exon5 fwd 5’-AATAGGTGTGTATCTTATGGCTTCC-3’ and CDK8 Intron4/Exon5 rev 5’- ATTTTTACTCTTCCTCGCTCAGGAC-3’ and by Western blotting.
Knockdown of CDK19
Silencing was performed as described (Bancerek et al., 2013). In brief, approximately 7×104 cells were seeded on a 6 cm dish and incubated 7 h followed by transfection with 100 pmol ON-TARGET plus™ SMART pool siRNA targeting CDK19 (Dharmacon, L-059630-00-0010) or non-targeting control (Dharmacon, D-001810-10-20) using Lipofectamine RNAiMAX Reagent (Invitrogen, 13778-150) in Opti-MEM I (Gibco, 31985070) for 48 h.
Knockout of CDK19
CDK19 knockout was generated in CDK8fl-MEFs using CRISPR/Cas9. Guide RNA (gRNA) sequence (5’-GTACAGCAGTGATTTAACCATGG-3’) targeting exon 4 of CDK19 was designed using the CRISPOR tool (Haeussler et al., 2016). The gRNA together with the purified Cas9 protein was delivered into CDK8fl-MEFs by electroporation. Single cells were grown and screened for the successful knockout using TIDE (Brinkman et al., 2014), sequencing and Western blotting.
HAP1 cells expressing analog-sensitive CDK8 (CDK8as)
CRISPR/Cas9-mediated homologous recombination was used to engineer an ATP analog-sensitive CDK8 mutant (CDK8as) in HAP1 cells. To introduce the F97G mutation in the exon 3 of CDK8 a guide RNA (gRNA; 5’-TGTTTCTGTCTCATGCTGAT-3’) was designed using the Broad Institute sgRNA designer (https://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design). A homology-directed repair template (HDRT) harboring the exon 3 with phenylalanine 97 changed to glycine (F97G) (nt change CTG TTT to CTC GGG), a mutated PAM site (silent mutation), a PGK-neomycin phosphotransferase (Neo) gene flanked by flippase recognition target (FRT) sites and flanking homology arms up- and downstream of exon 3 was generated. The HDRT was cloned together with the gRNA into the pSpCas9(BB)-2A-Puro (PX459) plasmid (Addgene #48139, (Ran et al., 2013)). Transfected cells were selected using 1 μg/ml puromycin for 24 h followed by 3 mg/ml G418 (InvivoGen) treatment on day 6 post-transfection. Surviving clones were analyzed for successful knock-in by PCR-based genotyping and sequencing. In the obtained positive knock-in clones the neomycin selection cassette was removed by transient expression of flippase (pOG44 Flp-Recombinase expression vector, Thermo Scientific, V600520) followed by a negative selection treating the cells with 3 mg/ml G418 (InvivoGen).
CDK19-WT expression construct
Total RNA from WT MEF cells was isolated with the Trizol – Isopropanol method (QIAzol, #79306, Qiagen), followed by DNA-digestion (DNase I #4716728001, Merck), acidic-phenolchloroform clean-up, isopropanol RNA precipitation and reverse transcription with RevertAid Reverse Transcriptase (#EP0442, Thermo Scientific) and oligo dT18 primers (Eurofins Genomics) according to manufacturer's protocol. Nested PCR strategy was applied to amplify and modify the coding sequence of CDK19 for cloning. First, the cDNA was amplified with Q5® High-Fidelity DNA Polymerase (#M0491L, NEB) and target specific primers (Forward: 5’-GAGGAGGCGGGACTGTAGAT-3’, Reverse: 5’- TTTGCATGGTGTCAGTCTTCATTC-3’) followed by gel purification (Monarch DNA gel extraction kit #T1020L, NEB). Primers of the second PCR were designed in a way that they flank the CDS, remove the stop codon and introduce a 5’ XbaI and 3’ XhoI NotI cleavage site (Forward: 5’-CATTCTAGACCGAGGAGTCCCTTGCTGAA-3’, Reverse: 5’- TATGCGGCCGCTATCTCGAGTACCGGTGGGTCTGGTGAGAT-3’). After gel purification and sequence validation, the PCR product and the PiggyBac-EF1-MCS-IRES-Neo cDNA Cloning and Expression Vector (#PB533A-2, SBI System Biosciences) were double digested using XbaI (#FD0684, Thermo Scientific) and NotI (#FD0594, Thermo Scientific) followed by gel purification, T4 DNA Ligase (#EL0011, Thermo Scientific) mediated ligation, transformation into chemically competent DH10B and ampicillin selection (100 pg/ml, #A0839, AppliChem). Colonies were PCR-screened (Forward: 5’- CAATTGAACGGGTGCCTAGAG-3’, Reverse: 5’-CCTTGTTGAATACGCTTGAGGAGA-3’) and plasmids were isolated by using plasmid mini prep kit (QIAprep, #27106, Qiagen). C-terminal triple-Flag-tag was introduced into double-digested (XhoI, #FD0694 Thermo Scientific and NotI in the presence of alkaline phosphatase, FastAP, #EF0651 Thermo Scientific) plasmid using double strand oligonucleotide (purchased from Integrated DNA Technologies as single strand oligonucleotides: +strand 5' TCG Agagac tacaaagaccatgacggtgattataaagatcatgacatcgattacaaggatgacgatgacaagTAGC, -strand 5' GGCCGCTActtgtcatcgtcatccttgtaatcgatgtcatgatctttataatcaccgtcatggtctttgtagtctC). Sequence of final plasmids (pB_EF1_CDK19_IRES_Neo) was validated with PCR and sequencing.
CDK19-KDead expression construct
Kinase-dead CDK19 (CDK19-KDead) was generated by mutation of aspartate 151 to alanine in the consensus active site. The D151A mutation was introduced in the parental plasmid (pB_EF1_CDK19_IRES_Neo) using Gibson Assembly site-directed mutagenesis. Briefly, PCR-amplified fragments (PCR_A: Forward: 5’- agctgtgaccggcgcctactctagaCTAGATGGGGGAAGCAGACAATGG-3’, Reverse: 5’- ctggtttcagTGCCCTGTGGAGCACCCA-3’; PCR_B: Forward: 5’- ccacagggcaCTGAAACCAGCAAATATCC-3’, Reverse: 5’-taggggggggggagggagaggggcgcggccgcGCTACTTGTCATCGTCATC-3’) were purified with column DNA Clean-up kit (Monarch® PCR & DNA Cleanup Kit, # T1030L, NEB) and mixed with Xhol/Notl double digested and gel purified plasmid in a molar ratio of insert to plasmid of 1:2 (0.02pmol for the 6,886bp parental plasmid, 0.04pmol of the 508bp PCR_A and of the 1,168bp PCR_B, respectively), followed by addition of 10 μl 2x Gibson Assembly Master Mix (E2611L, NEB) and incubation at 50°C for 90 min. 5 pl of the reaction mix were used for transformation of 100 μl chemically competent DH10B.
Rescue of CDK19-KO cells with CDK19-WT or CDK19-KDead
CDK19-KO cells (8×105) were electroporated with 1 μg piggyBac transposase plasmid (Cadinanos and Bradley, 2007) and 10 ng CDK19-WT plasmid or 50 ng CDK19-KDead plasmid. Cells were selected for integration of the transposon by G418 treatment (400 μg/μl) for 10 days and subsequently analyzed for CDK19 expression by qRT-PCR and Western blotting.
Generation of CDK8as mutant for in vitro kinase assays
N-terminally glu-tagged CDK8 in a pKozak plasmid (Knuesel et al., 2009) was used to generate the analog-sensitive CDK8 mutant (CDK8as) by mutation of the F97 codon (TTT) to a glycine (GGG) using site-directed mutagenesis. The mutated glu-CDK8as was transformed into DH5α E. coli, and individual clones were selected, grown, purified, and sequenced. Sequence-verified glu-CDK8as was cloned into the Baculovirus transfer vector pACEBac1. The resulting vector was transformed into DH5α E. coli and colonies were selected, purified, and sequenced. Sequence-verified pACEBac1 containing glu-CDK8as was used to transfer glu-CDK8as plus gentamycin resistance into the baculoviral genome using Tn7 transposition. Successful integration was assessed by blue/white screening plus gentamycin resistance. Bacmid DNA was prepared from selected clones and used to transfect insect cells for protein production.
In vitro kinase assays
Wild-type (WT CDK8) and analog-sensitive CDK8 (CDK8as) modules (containing CDK8, CCNC, MED12, and MED13) were purified and assembled as described (Knuesel et al., 2009). Reactions were performed at 300 for 45 min utes in kinase buffer (25 mM Tris pH 7.9, 100 mM KCl, 300 μM ATP, 10 mM MgCl2, and 2 mM DTT). WT CDK8 and CDK8as module, STAT1 substrate (2μL), 2.5 μCi [y-32P]ATP and increasing concentrations of NM-PP1 ATP analog (1 to 100 μM) were added. SDS-PAGE was used to separate proteins and the gels were subsequently stained with coomassie, dried at 55°C for 60 min, exposed on a phosphor-imager screen for 72 hours, and imaged using a Typhoon 9400 scanner. Quantitation of auto-rad bands was performed using ImageJ.
Whole cell extracts and Western blotting
The procedures for whole cell extracts and immunoblotting were described (Sadzak et al., 2008). In brief, whole cell extracts were prepared by lysing the cells for 5 minutes in Frackelton buffer (10mM Tris-HCl, 30 mM Na4P2O2, 50mM NaCl, 50 mM NaF, 1% Triton X-100, 1 mM DTT, 1 mM vanadate and 1x protease inhibitor (Roche, 11836145001)). Lysates were cleared by centrifugation at 13200 rpm and 4°C . SDS loading buffer was added in a 2:1 ratio (lysate:loading buffer) and boiled for 5 minutes. Antibodies against pSer727 STAT1 (Kovarik et al., 1998; Cell signaling, 9177), pY701 STAT1 (Cell Signaling, 9167S and 7649), STAT1 (Santa Cruz, sc-346), CDK8 (Santa Cruz, sc-1521; Cell signaling, 4101S), CDK19 (Sigma Aldrich, HPA007053), IRF1 (Cell Signaling 8478T), tubulin (Cell signaling, 2144S; Sigma Aldrich, T9026), MED12 (Bethyl, A300-774A), MED13 (Santa Cruz, sc-515557) and CCNC (Bethyl, A301-989A) were used for Western blotting.
Cytotoxicity assay
Cells were seeded on a 96well plate with 9000 cells per well. The next day the cells were treated with 100 nM CA for 2, 4, 6 and 8 hours or left untreated. The amount of living cells was assessed by crystal violet staining (0.1% crystal violet, 2% methanol in H2O) for 1 hour in the dark. Cells were then washed twice with PBS, air dried and subsequently incubated with 100 μl solubilization buffer (50:50 mixture of 0.1 M NaH2PO4, pH = 4.5 and 50% ethanol) per well. Crystal violet intensity, that was proportional to the number of alive cells, was determined at 595 nm using a microplate reader (BioTek, Synergy H1). n = 8 per condition, statistical testing was done using One-way ANOVA testing in Prism 6 (GraphPad Software).
Vesicular stomatitis virus (VSV) infection assay
VSV infection and survival assays was performed as described (Bancerek et al., 2013). Briefly, 3×104 cells were seeded on 6-well plates. 4 hours after seeding the medium was exchanged to medium containing siRNA against non-targeting control or CDK19 (50 pmol). After 48 h cells were re-seeded on 96well plates, 3500 cells per well. Four hours after seeding the medium was replaced with new medium that was supplemented with fresh siRNA and IFN-γ in two-fold serial dilutions starting at 10 units. After 24 hours the medium was replaced with medium without siRNA and without IFN-γ, and VSV was added at a multiplicity of infection (MOI) of 0.1. In case of CDK19-KO, CDK19-WT and CDK19-KDead cells 4000 cells were seeded on 96well plates. Four hours after seeding medium was replaced with medium supplemented with IFN-γ in two-fold serial dilutions starting at 10 units. After 24 hours the medium was replaced with medium without IFN-γ, and VSV was added at a multiplicity of infection (MOI) of 0.1. After incubation of cells with VSV for 39 h, cells were washed twice with PBS and stained with crystal violet (0.1% crystal violet, 2% methanol in H2O) for 1 h in the dark. After 2 additional washes, cells were incubated with 100 μl solubilization buffer (50:50 mixture of 0.1 M NaH2PO4, pH = 4.5 and 50% ethanol) per well. Crystal violet intensity, that was proportional to number of surviving cells, was determined at 595 nm using a microplate reader (BioTek, Synergy H1). EC50 calculations were calculated using the AAT-Bioquest EC50 (https://www.aatbio.com/tools/ec50-calculator), with minimum response set to zero.
QUANTIFICATION AND STATISTICAL ANALYSIS
RNA-Seq experiments were carried out in triplicates; GRO-Seq and PRO-Seq experiments were performed in duplicates. Statistical analysis of RNA-Seq, GRO-Seq and PRO-Seq experiments is described in the corresponding parts of the section METHOD DETAILS.
DATA AVAILABILITY
The datasets generated during this study are available at NCBI Sequence Read Archive (SRA) with the accession number PRJNA542065 (GRO-Seq and RNA-Seq datasets) and at NCBI GEO with the accession number GSE129501 (PRO-Seq dataset).
Unprocessed Western blot images of this study are available at Mendeley under: http://dx.doi.org/10.17632/crj8f3j63z.1
Supplementary Material
Table S1 (related to Figure 1). Effects of Cortistatin A (CA) on expression of IFN-γ-regulated genes (A and B) WT MEFs were stimulated for 6 h with IFN-γ or left unstimulated, with or without 1 h CA pre-treatment. mRNA (A) and pre-mRNA (B) levels were analyzed by RNA-Seq. The tables show genes regulated by IFN-γ stimulation (filtering criteria: log2FoldChange ≤ −1 or ≥ 1; padj < 0.05; FPKM stimulated ≥ 1) and their changes upon CA treatment. log2FoldChange 6vs0 = differential expression analysis of 6 h IFN-γ;-stimulated sample vs. unstimulated sample; padj 6vs0 = adjusted p-value for log2FoldChange 6vs0; log2FoldChange 6CAvs6 = differential expression analysis of 6 h IFN-γ-stimulated, CA-treated sample vs. 6 h IFN-γ-stimulated, untreated sample; padj 6CAvs6 = adjusted p-value for log2FoldChange 6CAvs6
Table S2 (related to Figure 1). Effects of Cortistatin A (CA) on expression of IFN-γ-regulated genes in WT and S727A MEFs WT and S727A MEFs were stimulated for 3 h with IFN-γ or left unstimulated, with or without 1 h CA pre-treatment. mRNA levels were analyzed by RNA-Seq. The table shows genes regulated by IFN-γ stimulation (filtering criteria: log2FoldChange ≤ −1 or ≥ 1; padj < 0.05; FPKM stimulated ≥ 1), their expression in WT and S727A MEFs and their changes upon CA treatment. WT.1.0_WT.0.0-lfc = differential expression analysis of 3 h IFN-γ-stimulated WT vs. unstimulated WT; WT.1.0_WT.0.0-padj = adjusted p-value for log2FoldChange WT.1.0_WT.0.0-lfc; WT.1.1_WT.1.0-lfc = differential expression analysis of 3 h IFN-γ-stimulated and CA-treated WT vs. IFN-γ-stimulated, untreated WT; WT.1.1_WT.1.0-padj = adjusted p-value for log2FoldChange WT.1.1_WT.1.0-lfc; SA.1.1_SA.1.0-lfc = differential expression analysis of 3 h IFN-γ-stimulated and CA-treated S727A vs. IFN-γ-stimulated, untreated S727A; SA.1.1_SA.1.0-padj = adjusted p-value for log2FoldChange SA.1.1_SA.1.0-lfc; SA.1.0_WT.1.0-lfc = differential expression analysis of 3 h IFN-γ-stimulated S727A vs. 3 h IFN-γ-stimulated WT; SA.1.0_WT.1.0-padj = adjusted p-value for log2FoldChange SA.1.0_WT.1.0-lfc
Table S3 (related to Figures 2, S2, 5, S5). Gene set enrichment upon IFN-γ stimulation in GRO-Seq, PRO-Seq and RNA-Seq experiments (A) GSEA for 30 min IFN-γ stimulated vs. unstimulated cells in GRO-Seq experiment. WT MEFs were stimulated for 30 min with IFN-γ or left unstimulated and subsequently analyzed using GRO-Seq. Enriched gene sets were filtered for FDR q-value < 0.05. (B) GSEA for 30 min IFN-γ stimulated vs. unstimulated cells in PRO-Seq experiment. WT and CDK8as HCT116 were pre-treated with 3MB-PP1 of left untreated followed by stimulation with IFN-γ for 30 min and subsequently analyzed using PRO-Seq. Enriched gene sets were filtered for FDR q-value < 0.05. WT DMSO.IFN vs. WT DMSO.PBS = enriched gene sets comparing IFN-γ stimulated WT HCT116 with unstimulated WT HCT116; CDK8as 3MB.IFN vs. WT 3MB.IFN = enriched gene sets comparing IFN-γ-stimulated, 3MB-treated CDK8as HCT116 with IFN-γ-stimulated, 3MB-treated WT HCT116; CDK8as DMSO.IFN vs. WT DMSO.IFN = enriched gene sets comparing IFN-γ-stimulated CDK8as HCT116 with IFN-γ-stimulated WT HCT116. (C) GSEA for mRNA expression in IFN-γ stimulated vs. unstimulated CDK8fl-MEFs. CDK8fl-MEFs were stimulated for 3 h with IFN-γ or left unstimulated and subsequently analyzed using RNA-Seq. Enriched gene sets were filtered for FDR q-value < 0.05. (D) Overlap of GSEA for IFN-γ-stimulated vs. unstimulated cells in GRO-Seq and RNA-Seq experiments. Enriched gene sets were extracted from GSEA shown in (A) and (C). Enriched gene sets were defined by an FDR q-value < 0.05. (E) GSEA for effects of inducible CDK8 knockout, CDK19 knockdown and Mediator kinase inhibition during IFN-γ response. GSEA was carried out based on differential expression analysis (IFN-γ-stimulated, 3 h vs. unstimulated) upon inducible CDK8 knockout (CDK8-iKO), CDK19 knockdown (siCDK19) or CA treatment. The table shows overlapping and distinct gene sets. Gene sets with a cutoff of NES < −1.5 (i.e., negative enrichment) are shown. ES = enrichment score; NES = normalized enrichment score; NOM p-val = nominal p-value; FDR q-val = false discovery rate q-value; FWER p-val = familywise-error rate p-value
Table S4 (related to Figure 2). Effects of Mediator kinase inhibition on RNAPII pausing at IFN-γ-induced genes WT MEFs were stimulated for 30 min with IFN-γ or left unstimulated, with or without 1 h CA pre-treatment and subsequently analyzed by GRO-Seq. (A) The table contains the group of IFN-γ-induced genes with the filtering criteria: lfc > 0; padj < 0.05. Ifn30.ctrl_Ifn0.ctrl_lfc = log2foldchange 30 min IFN-γ stimulated cells vs. unstimulated cells; Ifn30.ctrl_Ifn0.ctrl_padj = adjusted p-value corresponding to Ifn30.ctrl_Ifn0.ctrl_lfc (B) CA decreases gene expression of IFN-γ-induced genes in GRO-Seq. The table contains IFN-γ-induced genes shown in the heat map in Figure 2B. Ifn0_ctrl = unstimulated, untreated cells; Ifn0_CA = unstimulated, CA-treated cells; Ifn30_ctrl = IFN-γ-stimulated, untreated cells; Ifn30_CA = IFN-γ-stimulated, CA-treated cells (C) The table contains counts for pooled replicates at the TSS, within the gene body and the calculated Pausing indices for RefSeq transcripts. Ifn0 = unstimulated sample; Ifn30 = 30 min IFN-γ-stimulated sample; ctrl = untreated sample; CA = CA-treated sample; mean_TSS = counts for pooled replicates at the TSS; mean_geneBody = counts for pooled replicates within the gene body; mean_PI = Pausing index of pooled replicates (D) Correlation of Cortistatin A (CA)-dependent reduction of mRNA levels with increased RNAPII pausing at IFN-γ-induced genes. Data were extracted from Table S1A (RNA-Seq, mRNA) and Table S4C (GRO-Seq). RNA-seq filter: IFN-γ-induced genes (lfc > 1; padj < 0.05; FPKM stim ≥ 1), downregulated by CA (lfc < 0; padj < 0.05); GRO-Seq filter: CA response 30 (= PI IFN30.CA/PI IFN30.Ctrl) > 1.5. log2FoldChange 6CAvs6 = differential expression analysis of 6 h IFN-γ-stimulated CA-treated vs. 6 h IFN-γ-stimulated cells; padj 6CAvs6 = adjusted p-value for log2FoldChange 6CAvs6; PI IFN30.Ctrl = pausing index after 30 min IFN-γ stimulation; PI IFN30.CA = pausing index after 30 min IFN-γ stimulation and CA pre-treatment; CA response 30 = ratio of PI IFN30.CA to PI IFN30.Ctrl
Table S5 (related to Figure 2). Effects of CDK8 kinase inhibition on RNAPII pausing in human cells WT and CDKas HCT116 cells were stimulated for 30 min with IFN-γ or left unstimulated, with or without 3MB-PP1 treatment and subsequently analyzed by PRO-Seq. (A) The table shows differential expression analysis of stimulated WT HCT116 cells (WT_DMSO.IFN) in comparison to unstimulated WT HCT116 cells (WT_DMSO.PBS). (B) The table shows differential expression analysis of stimulated, 3MB-treated CDK8as HCT116 cells (CDK8as_3MB.IFN) in comparison to stimulated, 3MB-treated WT HCT116 cells (WT_3MB.IFN). (C - E) The tables contain counts at the TSS (C), within the gene body (D) and the calculated Pausing index (E) for each condition. WT_DMSO.PBS = unstimulated WT HCT116 cells; WT_3MB.PBS = unstimulated, 3MB-treated WT HCT116 cells; WT_DMSO.IFN = stimulated WT HCT116 cells; WT_3MB.IFN = stimulated, 3MB-treated WT HCT116 cells; CDK8as_DMSO.PBS = unstimulated CDK8as HCT116 cells; CDK8as_3MB.PBS = unstimulated, 3MB-treated CDK8as HCT116 cells; CDK8as_DMSO.IFN = stimulated CDK8as HCT116 cells; CDK8as_3MB.IFN = stimulated, 3MB-treated CDK8as HCT116 cells
Table S6 (related to Figure 3): Effects of Mediator kinase inhibition on eRNA transcription during IFN-γ response in WT MEFs (GRO-Seq) and HCT116 cells (PRO-Seq) (A - D) The tables correspond to Figure 3 and show differences in eRNAs transcription in WT MEFs upon IFN-γ stimulation (A), Mediator kinase inhibition (B) and in HCT116 cells upon IFN-γ stimulation (C) and CDK8 kinase inhibition (D). MEF IFN30 vs IFN0 = 30 min stimulated MEFs vs. unstimulated MEFs; MEF IFN30.CA vs. MEF IFN0 = 30 min stimulated, CA-treated MEFs vs. unstimulated MEFs; WT HCT116 3MB.IFN vs. WT HCT116 3MB.PBS = 30 min stimulated, 3MB-treated WT HCT116 vs. unstimulated, 3MB-treated WT HCT116; CDK8as HCT116 3MB.IFN vs. CDK8as HCT116 3MB.PBS = 30 min stimulated, 3MB-treated CDK8as HCT116 vs. unstimulated, 3MB-treated CDK8as HCT116
Table S7 (related to Figures 5 and 6): Differential expression analysis for effects of inducible CDK8 knockout, CDK19 knockdown and Mediator kinase inhibition (CA) during IFN-γ response (A and B) The tables correspond to Figure 4 and 5, and show genes regulated by 3 h stimulation with IFN-γ at the mRNA (A) and pre-mRNA (B) level (log2FoldChange (lfc) ≤ −1 or 1; padj ≥ 0.05; FPKM stimulated ≥ 1) in siCtrl-treated CDK8fl-MEFs (siCtrl.stim_siCtrl.unst). Differential expression analysis was carried out for the following conditions: CA effects (CA.stim_siCtrl.stim); CA effects in absence of CDK19 (siCDK19.CA.stim_siCDK19.stim); CDK8 knockout effects (CDK8-iKO.stim_siCtrl.stim); CA effects in absence of CDK8 (CDK8- iKO.CA.stim_CDK8-iKO.stim); CDK19 silencing effects (siCDK19.stim_siCtrl.stim); stim = 3 h IFN-γ stimulation; unst = unstimulated; lfc = log2FoldChange; padj = adjusted p-value
Table S8 (related to Figure 7): CDK19 regulates IFN-γ-induced gene expression in kinase-independent ways CDK19-KO, CDK19-WT and CDK19-KDead MEFs were stimulated for 3 h with IFN-γ or left unstimulated. mRNA levels were analyzed by RNA-Seq. The table shows genes regulated by IFN-γ stimulation (filtering criteria: log2FoldChange ≤ −1 or ≥ 1; padj < 0.05; FPKM stimulated 1) and their expression in CDK19-KO, CDK19-WT and CDK19-KDead cells. WT_1- WT_0_lfc = differential expression analysis of 3 h IFN-γ-stimulated CDK19-WT vs. unstimulated CDK19-WT; WT_1-WT_0_padj = adjusted p-value for WT_1-WT_0_lfc; CDK19KO_1-WT_1_lfc = differential expression analysis of 3 h IFN-γ-stimulated CDK19-KO vs. 3 h IFN-γ-stimulated CDK19-WT; CDK19KO_1-WT_1_padj = adjusted p-value for CDK19KO_1-WT_1_lfc; CDK19KDead_1-CDK19KO_1_lfc = differential expression analysis of 3 h IFN-γ-stimulated CDK19-KDead vs. 3 h IFN-γ-stimulated CDK19-KO; CDK19KDead_1-CDK19KO_1_padj = adjusted p-value for CDK19KDead_1- CDK19KO_1_lfc
Table S9 (relates to Key Resources Table): Oligonucleotides for PCR cloning, genotyping and guide RNA
Key Resource Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| pS727 STAT1 | Kovarik et al., 1998 | N/A |
| pS727 STAT1 | Cell Signaling Technology | Cat#9177; RRID: AB_ 2197983 |
| pY701 STAT1 (58D6) | Cell Signaling Technology | Cat#9167S; RRID: AB_561284 |
| pY701 STAT1 (D4A7) | Cell Signaling Technology | Cat#7649; RRID: AB_10950970 |
| STAT1 p84/p91 (E23) | Santa Cruz Biotechnology | Cat#sc-346; RRID: AB_632435 |
| CDK8 | Santa Cruz Biotechnology | Cat#sc-1521; RRID: AB_2260300 |
| CDK8 | Cell Signaling Technology | Cat#4101S; RRID: AB_1903934 |
| CDK19 | Sigma-Aldrich | Cat# HPA007053; RRID: AB_1846369 |
| Alpha-Tubulin | Cell Signaling Technology | Cat#2144S; RRID: AB_2210548 |
| Alpha-Tubulin | Sigma-Aldrich | Cat#T9026; RRID: AB_477593 |
| MED12 | Bethyl | Cat#A300-774A; RRID: AB_669756 |
| MED13, TRAP240(E-12) | Santa Cruz Biotechnology | Cat# sc-515557 |
| CCNC | Bethyl | Cat#A301-989A; RRID: AB_1576505 |
| IRF1 (D5E4) | Cell Signaling Technology | Cat#8478T; RRID: AB_10949108 |
| Peroxidase-conjugated AffiniPure Goat Anti-Rabbit IgG (H+L) | Jackson Immunoresearch | Cat#111-035-003; RRID: AB_2313567 |
| Peroxidase-conjugated AffiniPure Goat Anti-Mouse IgG (H+L) | Jackson Immunoresearch | Cat#115-035-003; RRID: AB_10015289 |
| Bacterial and Virus Strains | ||
| Vesicular stomatitis virus (VSV), Indiana strain | Laboratory of Birgit Strobl | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Murine Interferon-gamma | eBioscience | Cat#14-8311 |
| Human Interferon-gamma | Laboratory of James E. Darnell | N/A |
| Human Interferon-gamma | Fisher Scientific | Cat#PHC4031 |
| Cortistatin A | Laboratory of Matthew Shair | N/A |
| NM-PP1 | Calbiochem | Cat#529581 |
| 3MB-PP1 | Cayman Chemical | Cat#56025-83-5 |
| 4-Hydroxytamoxifen | Sigma-Aldrich | Cat#T176-10MG |
| G418 (Geneticin) | Invivogen | Cat#ant-gn-1 |
| Lipofectamine RNAiMAX Transfection Reagent | Invitrogen | Cat#13778-150 |
| cOmplete Protease Inhibitor Cocktail | Roche | Cat#11836145001 |
| SUPERase-In | Invitrogen | Cat#AM2694 |
| RQ1 RNase-Free DNase | Promega | Cat#M6101 |
| DNase I | Merck | Cat#4716728001 |
| RevertAid Reverse Transcriptase | Thermo Scientific | Cat#EP0442 |
| Q5® High-Fidelity DNA Polymerase | NEB | Cat#M0491L |
| XbaI | Thermo Scientific | Cat#FD0684 |
| NotI | Thermo Scientific | Cat#FD0594 |
| XhoI | Thermo Scientific | Cat#FD0694 |
| T4 DNA Ligase | Thermo Scientific | Cat#EL001 |
| Ampicillin | AppliChem | Cat#A0839 |
| Fast Alkaline Phosohatase | Thermo Scientific | Cat#EF0651 |
| Critical Commercial Assays | ||
| NEBNext Ultra Directional RNA Library Prep Kit | NEB | Cat# E7420S |
| NEBNext Ultra II RNA Library Prep Kit | NEB | Cat# E7770S |
| NEBNext rRNA Depletion Kit | NEB | Cat# E6310S |
| NEBNext Poly(A) mRNA Magnetic Isolation Module | NEB | Cat# E7490S |
| RNase-Free DNase Set | Qiagen | Cat#79254 |
| RNeasy Mini Kit | Qiagen | Cat#74104 |
| QIAzol Lysis Reagent | Qiagen | Cat#79306 |
| TRIzol Lysis Reagent | Invitrogen | Cat#15596026 |
| RNA 6000 Nano Assays | Agilent | Cat#5067-1511 |
| Ambion Fragmentation Reagents | Ambion | Cat#AM8740 |
| Illustra MicroSpin G-25 columns | GE Healthcare | Cat#27532501 |
| Anti-BrU agarose beads | Santa Cruz Biotechnology | Cat#sc-32323AC |
| Monarch DNA gel extraction kit | NEB | Cat#T1020L |
| Plasmid mini prep kit | Qiagen | Cat#2710 |
| Gibson Assembly Master Mix | NEB | Cat#E2611L |
| Deposited Data | ||
| Raw and analyzed data - RNA-Seq experiments, GRO-Seq experiment | This paper | SRA database: PRJNA542065 https://www.ncbi.nlm.nih.gov/sra |
| Raw and analyzed data - PRO-Seq experiment | This paper | GEO: GSE129501 https://www.ncbi.nlm.nih.gov/geo/ |
| Raw image files - Western blots | This paper | http://dx.doi.org/10.17632/crj8f3j63z.1 |
| Mouse reference genome (GRCm38/mm10) | Genome Reference Consortium | https://www.ncbi.nlm.nih.gov/grc/mouse |
| Human reference genome (GRCh37/hg19) | Genome Reference Consortium | https://www.ncbi.nlm.nih.gov/grc/human |
| HOCOMOCO database | Kulakovskiy et al.,2013 | http://hocomoco11.autosome.ru/ |
| Experimental Models: Cell Lines | ||
| WT MEFs | Bancerek et al., 2013 | N/A |
| CDK8fl-MEFs | This study | N/A |
| CDK19 KO MEFs | This study | N/A |
| CDK19 KO MEFs expressing CDK19-WT | This study | N/A |
| CDK19 KO MEFs expressing CDK19-KDead | This study | N/A |
| HAP1 WT | Haplogen | N/A |
| HAP1 CDK8 KO (clone 325-1) | Haplogen | N/A |
| HAP1 CDK8as | This study | N/A |
| HCT116 WT | Galbraith et al., 2017 | N/A |
| HCT116 CDK8as | Galbraith et al., 2017 | N/A |
| Experimental Models: Organisms/Strains | ||
| Cdk8tm1c(EUUOMM)Hmgu | Yann Herault, IGMBC | N/A |
| Rosa26CreERT2 | Hameyer et al., 2007 | N/A |
| CreERT2-CDK8fl/fl | This study | N/A |
| Oligonucleotides | ||
| ON-TARGET plus SMART pool siRNA CDK19 | Dharmacon | Cat#L-059630-00-0010 |
| ON-TARGET plus non-targeting siRNA pool | Dharmacon | Cat#D-001810-10-20 |
| PCR primers and guide RNAs - see Table S9 | ||
| Recombinant DNA | ||
| pSpCas9(BB)-2A-Puro (PX459) | Addgene plasmid | Cat#48139 |
| pOG44 Flp-Recombinase expression vector | Thermo Scientific | Cat#V600520 |
| PB-EF1α-MCS-IRES-Neo cDNA cloning and expression vector | SBI System Biosciences | Cat#PB533A-2 |
| pB_EF1_CDK19_IRES_Neo | This study | N/A |
| piggyBac transposase plasmid | Cadinanos and Bradley, 2007 | N/A |
| pKozak plasmid | Knuesel et al., 2009 | N/A |
| Software and Algorithms | ||
| Image Lab version 5.2.1 | Bio-Rad | |
| Gen5 Microplate Reader Software | BioTek | |
| AAT-Bioquest EC50 | AAT-Bioquest | https://www.aatbio.com/tools/ec50-calculator |
| GraphPad Prism 6 | Graph Pad Software | https://www.graphpad.com |
| HTSeq | Anders et al., 2015 | https://github.com/simon-anders/htseq |
| cutadapt | http://dx.doi.org/10.14806/ej.17.1.200 | |
| FastQC version 0.11.5 | http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ | |
| FastQ Screen version 0.11.0 | https://www.bioinformatics.babraham.ac.uk/projects/fastq_screen/ | |
| STAR version 2.5 | Dobin et al., 2013 | https://code.google.com/archive/p/rna-star/ |
| R-project version 3.4.2 with RStudio IDE version 1.0.143 | Team, 2017 | https://www.R-project.org/ |
| GenePattern server | Reich et al., 2006 | http://software.broadinstitute.org/cancer/software/genepattern/ |
| DESeq2 version 1.18.1 and 1.22.1 | Love et al., 2014 | http://www.biocondutor.org/packages/release/bioc/html/DESeq2.html |
| RSeQC | http://rseqc.sourceforge.net/ | |
| Mix2 RNA-Seq data analysis software | Lexogen | |
| Broad Institute sgRNA designer | https://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design | |
| BBDUK from BBTools (v37.99) | https://sourceforge.net/projects/bbmap/ | |
| FASTQ-MCF from ea-utils (v1.05) | Kechin et al., 2017 | https://expressionanalysis.github.io/ea-utils/ |
| Hisat2 (v2.1.0) | Kim et al., 2015 | https://ccb.jhu.edu/software/hisat2/index.shtml |
| Samtools (v1.5) | Li et al., 2015 | https://sourceforge.net/projects/samtools/files/samtools/1.5/ |
| featureCount (v1.6.2) | Liao et al., 2014 | http://subread.sourceforge.net/ |
| HOMER (v4.9.1) | Heinz et al., 2010 | http://homer.ucsd.edu/homer/ |
| Gviz package (v1.26.4) | Hahne and Ivanek, 2016 | https://bioconductor.org/packages/release/bioc/html/Gviz.html |
| CRISPOR tool | Haeussler et al., 2016 | http://crispor.tefor.net/ |
| TIDE | Brinkman et al., 2014 | https://tide.deskgen.com/ |
| Broad Institute sgRNA designer | https://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design | |
Highlights:
Mediator kinases CDK8 & CDK19 are distinct transcription regulators in IFN-γ pathway
CDK8 acts as kinase while CDK19 functions as scaffold in driving IFN-γ gene profiles
CDK8 and CDK19 activate distinct gene sets
Mediator kinase CDK8 promotes pol II pause release during IFN-γ response
ACKNOWLEDGMENTS
We thank T. Read, K. Audetat and M. Allen for helpful discussions and M. Shair for cortistatin A. C. Larson and A. Vogt are acknowledged for sequencing advice. We thank T.M. Decker and T. Lee for antibody testing with IP-MS and P. Fischer for advice on RNA-Seq library scaling. We thank K. Chylinski and VBCF Protein Technologies Facility (www.viennabiocenter.org/facilities) for help with CRISPR-CAS9 genome editing. This work was supported by the Austrian Science Fund (FWF) grants P27538-B21, SFB-F43 and W1261 to PK, the NIH (GM117370 to DJT, GM008759 to JDR, AG051335 to CBL, GM120109 and CA117907 to JME), the NSF (MCB-1817582 to JME), the Fonfara-LaRose Family Fund (to JME), the FWF grant I2192-B22 ERASE and the European Research Council grant AdG 695214 GameofGates to GS-F. We acknowledge the BioFrontiers Computing Core at UC-Boulder, supported by the NIH (OD012300).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing financial interests
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Allen BL, and Taatjes DJ (2015). The Mediator complex: a central integrator of transcription. Nat Rev Mol Cell Biol 16, 155–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen MA, Andrysik Z, Dengler VL, Mellert HS, Guarnieri A, Freeman JA, Sullivan KD, Galbraith MD, Luo X, Kraus WL, et al. (2014). Global analysis of p53-regulated transcription identifies its direct targets and unexpected regulatory mechanisms. eLife 3, e02200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W (2015). HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics (Oxford, England) 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arner E, Daub CO, Vitting-Seerup K, Andersson R, Lilje B, Drablos F, Lennartsson A, Ronnerblad M, Hrydziuszko O, Vitezic M, et al. (2015). Transcribed enhancers lead waves of coordinated transcription in transitioning mammalian cells. Science 347, 1010–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audetat KA, Galbraith MD, Odell AT, Lee T, Pandey A, Espinosa JM, Dowell RD, and Taatjes DJ (2017). A Kinase-Independent Role for Cyclin-Dependent Kinase 19 in p53 Response. Mol Cell Biol 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azofeifa JG, Allen MA, Hendrix JR, Read T, Rubin JD, and Dowell RD (2018). Enhancer RNA profiling predicts transcription factor activity. Genome research. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bancerek J, Poss ZC, Steinparzer I, Sedlyarov V, Pfaffenwimmer T, Mikulic I, Dolken L, Strobl B, Muller M, Taatjes DJ, and Kovarik P (2013). CDK8 kinase phosphorylates transcription factor STAT1 to selectively regulate the interferon response. Immunity 38, 250–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkman EK, Chen T, Amendola M, and van Steensel B (2014). Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res 42, e168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bromberg JF, Horvath CM, Wen Z, Schreiber RD, and Darnell JE Jr. (1996). Transcriptionally active Stat1 is required for the antiproliferative effects of both interferon alpha and interferon gamma. Proc Natl Acad Sci U S A 93, 7673–7678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadinanos J, and Bradley A (2007). Generation of an inducible and optimized piggyBac transposon system. Nucleic Acids Res 35, e87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carette JE, Raaben M, Wong AC, Herbert AS, Obernosterer G, Mulherkar N, Kuehne AI, Kranzusch PJ, Griffin AM, Ruthel G, et al. (2011). Ebola virus entry requires the cholesterol transporter Niemann-Pick C1. Nature 477, 340–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Liang J, Ji H, Yang Z, Altilia S, Hu B, Schronce A, McDermott MSJ, Schools GP, Lim CU, et al. (2017). CDK8/19 Mediator kinases potentiate induction of transcription by NFkappaB. Proc Natl Acad Sci U S A 114, 10208–10213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Core LJ, Waterfall JJ, and Lis JT (2008). Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science 322, 1845–1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale T, Clarke PA, Esdar C, Waalboer D, Adeniji-Popoola O, Ortiz-Ruiz MJ, Mallinger A, Samant RS, Czodrowski P, Musil D, et al. (2015). A selective chemical probe for exploring the role of CDK8 and CDK19 in human disease. Nat Chem Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decker T, Kovarik P, and Meinke A (1997). GAS elements: a few nucleotides with a major impact on cytokine-induced gene expression. J Interferon Cytokine Res 17, 121–134. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics (Oxford, England) 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolken L, Ruzsics Z, Radle B, Friedel CC, Zimmer R, Mages J, Hoffmann R, Dickinson P, Forster T, Ghazal P, and Koszinowski UH (2008). High-resolution gene expression profiling for simultaneous kinetic parameter analysis of RNA synthesis and decay. RNA (New York, N.Y 14, 1959–1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donner AJ, Szostek S, Hoover JM, and Espinosa JM (2007). CDK8 is a stimulus-specific positive coregulator of p53 target genes. Mol Cell 27, 121–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galbraith MD, Allen MA, Bensard CL, Wang X, Schwinn MK, Qin B, Long HW, Daniels DL, Hahn WC, Dowell RD, and Espinosa JM (2013). HIF1A employs CDK8-mediator to stimulate RNAPII elongation in response to hypoxia. Cell 153, 1327–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galbraith MD, Andrysik Z, Pandey A, Hoh M, Bonner EA, Hill AA, Sullivan KD, and Espinosa JM (2017). CDK8 Kinase Activity Promotes Glycolysis. Cell reports 21, 1495–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeussler M, Schonig K, Eckert H, Eschstruth A, Mianne J, Renaud JB, Schneider-Maunoury S, Shkumatava A, Teboul L, Kent J, et al. (2016). Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome biology 17, 148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahne F, and Ivanek R (2016). Visualizing Genomic Data Using Gviz and Bioconductor. Methods Mol Biol 1418, 335–351. [DOI] [PubMed] [Google Scholar]
- Hameyer D, Loonstra A, Eshkind L, Schmitt S, Antunes C, Groen A, Bindels E, Jonkers J, Krimpenfort P, Meuwissen R, et al. (2007). Toxicity of ligand-dependent Cre recombinases and generation of a conditional Cre deleter mouse allowing mosaic recombination in peripheral tissues. Physiological genomics 31, 32–41. [DOI] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath CM, and Darnell JE Jr. (1996). The antiviral state induced by alpha interferon and gamma interferon requires transcriptionally active Stat1 protein. Journal of virology 70, 647–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivashkiv LB (2018). IFNgamma: signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nature reviews 18, 545–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johannessen L, Sundberg TB, O'Connell DJ, Kolde R, Berstler J, Billings KJ, Khor B, Seashore-Ludlow B, Fassl A, Russell CN, et al. (2017). Small-molecule studies identify CDK8 as a regulator of IL-10 in myeloid cells. Nat Chem Biol 13, 1102–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kechin A, Boyarskikh U, Kel A, and Filipenko M (2017). cutPrimers: A New Tool for Accurate Cutting of Primers from Reads of Targeted Next Generation Sequencing. J Comput Biol 24, 1138–1143. [DOI] [PubMed] [Google Scholar]
- Kim D, Langmead B, and Salzberg SL (2015). HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12, 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knuesel MT, Meyer KD, Donner AJ, Espinosa JM, and Taatjes DJ (2009). The human CDK8 subcomplex is a histone kinase that requires Med12 for activity and can function independently of mediator. Mol Cell Biol 29, 650–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehler MF, Bergeron P, Blackwood EM, Bowman K, Clark KR, Firestein R, Kiefer JR, Maskos K, McCleland ML, Orren L, et al. (2016). Development of a Potent, Specific CDK8 Kinase Inhibitor Which Phenocopies CDK8/19 Knockout Cells. ACS medicinal chemistry letters 7, 223–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovarik P, Stoiber D, Eyers PA, Menghini R, Neininger A, Gaestel M, Cohen P, and Decker T (1999). Stress-induced phosphorylation of STAT1 at Ser727 requires p38 mitogen-activated protein kinase whereas IFN-gamma uses a different signaling pathway. Proc Natl Acad Sci U S A 96, 13956–13961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulakovskiy IV, Medvedeva YA, Schaefer U, Kasianov AS, Vorontsov IE, Bajic VB, and Makeev VJ (2013). HOCOMOCO: a comprehensive collection of human transcription factor binding sites models. Nucleic Acids Res 41, D195–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak H, Fuda NJ, Core LJ, and Lis JT (2013). Precise maps of RNA polymerase reveal how promoters direct initiation and pausing. Science 339, 950–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Zhang B, Xing X, and Wang T (2015). Combining MeDIP-seq and MRE-seq to investigate genome-wide CpG methylation. Methods 72, 29–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Fassl A, Chick J, Inuzuka H, Li X, Mansour MR, Liu L, Wang H, King B, Shaik S, et al. (2014). Cyclin C is a haploinsufficient tumour suppressor. Nat Cell Biol 16, 1080–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2013). The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res 41, e108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics (Oxford, England) 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome biology 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsen JG, Schmidt SF, Larsen BD, Loft A, Nielsen R, and Mandrup S (2015). iRNA-seq: computational method for genome-wide assessment of acute transcriptional regulation from total RNA-seq data. Nucleic Acids Res 43, e40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahat DB, Kwak H, Booth GT, Jonkers IH, Danko CG, Patel RK, Waters CT, Munson K, Core LJ, and Lis JT (2016). Base-pair-resolution genome-wide mapping of active RNA polymerases using precision nuclear run-on (PRO-seq). Nature protocols 11, 1455–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancino A, and Natoli G (2016). Specificity and Function of IRF Family Transcription Factors: Insights from Genomics. J Interferon Cytokine Res 36, 462–469. [DOI] [PubMed] [Google Scholar]
- Nitulescu II, Meyer SC, Wen QJ, Crispino JD, Lemieux ME, Levine RL, Pelish HE, and Shair MD (2017). Mediator Kinase Phosphorylation of STAT1 S727 Promotes Growth of Neoplasms With JAK-STAT Activation. EBioMedicine 26, 112–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelish HE, Liau BB, Nitulescu II, Tangpeerachaikul A, Poss ZC, Da Silva DH, Caruso BT, Arefolov A, Fadeyi O, Christie AL, et al. (2015). Mediator kinase inhibition further activates super-enhancer-associated genes in AML. Nature 526, 273–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poss ZC, Ebmeier CC, Odell AT, Tangpeerachaikul A, Lee T, Pelish HE, Shair MD, Dowell RD, Old WM, and Taatjes DJ (2016). Identification of Mediator Kinase Substrates in Human Cells using Cortistatin A and Quantitative Phosphoproteomics. Cell reports 15, 436–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nature protocols 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich M, Liefeld T, Gould J, Lerner J, Tamayo P, and Mesirov JP (2006). GenePattern 2.0. Nature genetics 38, 500. [DOI] [PubMed] [Google Scholar]
- Rossi A, Kontarakis Z, Gerri C, Nolte H, Holper S, Kruger M, and Stainier DY (2015). Genetic compensation induced by deleterious mutations but not gene knockdowns. Nature 524, 230–233. [DOI] [PubMed] [Google Scholar]
- Sadzak I, Schiff M, Gattermeier I, Glinitzer R, Sauer I, Saalmuller A, Yang E, Schaljo B, and Kovarik P (2008). Recruitment of Stat1 to chromatin is required for interferon-induced serine phosphorylation of Stat1 transactivation domain. Proc Natl Acad Sci U S A 105, 8944–8949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider WM, Chevillotte MD, and Rice CM (2014). Interferon-stimulated genes: a complex web of host defenses. Annual review of immunology 32, 513–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Team, R.C. (2017). R: A Language and Environment for Statistical Computing.
- Todaro GJ, and Green H (1963). Quantitative studies of the growth of mouse embryo cells in culture and their development into established lines. The Journal of cell biology 17, 299–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turunen M, Spaeth JM, Keskitalo S, Park MJ, Kivioja T, Clark AD, Makinen N, Gao F, Palin K, Nurkkala H, et al. (2014). Uterine leiomyoma-linked MED12 mutations disrupt mediator-associated CDK activity. Cell reports 7, 654–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen Z, Zhong Z, and Darnell JE Jr. (1995). Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell 82, 241–250. [DOI] [PubMed] [Google Scholar]
- Wiesauer I, Gaumannmuller C, Steinparzer I, Strobl B, and Kovarik P (2015). Promoter Occupancy of STAT1 in Interferon Responses Is Regulated by Processive Transcription. Mol Cell Biol 35, 716–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 (related to Figure 1). Effects of Cortistatin A (CA) on expression of IFN-γ-regulated genes (A and B) WT MEFs were stimulated for 6 h with IFN-γ or left unstimulated, with or without 1 h CA pre-treatment. mRNA (A) and pre-mRNA (B) levels were analyzed by RNA-Seq. The tables show genes regulated by IFN-γ stimulation (filtering criteria: log2FoldChange ≤ −1 or ≥ 1; padj < 0.05; FPKM stimulated ≥ 1) and their changes upon CA treatment. log2FoldChange 6vs0 = differential expression analysis of 6 h IFN-γ;-stimulated sample vs. unstimulated sample; padj 6vs0 = adjusted p-value for log2FoldChange 6vs0; log2FoldChange 6CAvs6 = differential expression analysis of 6 h IFN-γ-stimulated, CA-treated sample vs. 6 h IFN-γ-stimulated, untreated sample; padj 6CAvs6 = adjusted p-value for log2FoldChange 6CAvs6
Table S2 (related to Figure 1). Effects of Cortistatin A (CA) on expression of IFN-γ-regulated genes in WT and S727A MEFs WT and S727A MEFs were stimulated for 3 h with IFN-γ or left unstimulated, with or without 1 h CA pre-treatment. mRNA levels were analyzed by RNA-Seq. The table shows genes regulated by IFN-γ stimulation (filtering criteria: log2FoldChange ≤ −1 or ≥ 1; padj < 0.05; FPKM stimulated ≥ 1), their expression in WT and S727A MEFs and their changes upon CA treatment. WT.1.0_WT.0.0-lfc = differential expression analysis of 3 h IFN-γ-stimulated WT vs. unstimulated WT; WT.1.0_WT.0.0-padj = adjusted p-value for log2FoldChange WT.1.0_WT.0.0-lfc; WT.1.1_WT.1.0-lfc = differential expression analysis of 3 h IFN-γ-stimulated and CA-treated WT vs. IFN-γ-stimulated, untreated WT; WT.1.1_WT.1.0-padj = adjusted p-value for log2FoldChange WT.1.1_WT.1.0-lfc; SA.1.1_SA.1.0-lfc = differential expression analysis of 3 h IFN-γ-stimulated and CA-treated S727A vs. IFN-γ-stimulated, untreated S727A; SA.1.1_SA.1.0-padj = adjusted p-value for log2FoldChange SA.1.1_SA.1.0-lfc; SA.1.0_WT.1.0-lfc = differential expression analysis of 3 h IFN-γ-stimulated S727A vs. 3 h IFN-γ-stimulated WT; SA.1.0_WT.1.0-padj = adjusted p-value for log2FoldChange SA.1.0_WT.1.0-lfc
Table S3 (related to Figures 2, S2, 5, S5). Gene set enrichment upon IFN-γ stimulation in GRO-Seq, PRO-Seq and RNA-Seq experiments (A) GSEA for 30 min IFN-γ stimulated vs. unstimulated cells in GRO-Seq experiment. WT MEFs were stimulated for 30 min with IFN-γ or left unstimulated and subsequently analyzed using GRO-Seq. Enriched gene sets were filtered for FDR q-value < 0.05. (B) GSEA for 30 min IFN-γ stimulated vs. unstimulated cells in PRO-Seq experiment. WT and CDK8as HCT116 were pre-treated with 3MB-PP1 of left untreated followed by stimulation with IFN-γ for 30 min and subsequently analyzed using PRO-Seq. Enriched gene sets were filtered for FDR q-value < 0.05. WT DMSO.IFN vs. WT DMSO.PBS = enriched gene sets comparing IFN-γ stimulated WT HCT116 with unstimulated WT HCT116; CDK8as 3MB.IFN vs. WT 3MB.IFN = enriched gene sets comparing IFN-γ-stimulated, 3MB-treated CDK8as HCT116 with IFN-γ-stimulated, 3MB-treated WT HCT116; CDK8as DMSO.IFN vs. WT DMSO.IFN = enriched gene sets comparing IFN-γ-stimulated CDK8as HCT116 with IFN-γ-stimulated WT HCT116. (C) GSEA for mRNA expression in IFN-γ stimulated vs. unstimulated CDK8fl-MEFs. CDK8fl-MEFs were stimulated for 3 h with IFN-γ or left unstimulated and subsequently analyzed using RNA-Seq. Enriched gene sets were filtered for FDR q-value < 0.05. (D) Overlap of GSEA for IFN-γ-stimulated vs. unstimulated cells in GRO-Seq and RNA-Seq experiments. Enriched gene sets were extracted from GSEA shown in (A) and (C). Enriched gene sets were defined by an FDR q-value < 0.05. (E) GSEA for effects of inducible CDK8 knockout, CDK19 knockdown and Mediator kinase inhibition during IFN-γ response. GSEA was carried out based on differential expression analysis (IFN-γ-stimulated, 3 h vs. unstimulated) upon inducible CDK8 knockout (CDK8-iKO), CDK19 knockdown (siCDK19) or CA treatment. The table shows overlapping and distinct gene sets. Gene sets with a cutoff of NES < −1.5 (i.e., negative enrichment) are shown. ES = enrichment score; NES = normalized enrichment score; NOM p-val = nominal p-value; FDR q-val = false discovery rate q-value; FWER p-val = familywise-error rate p-value
Table S4 (related to Figure 2). Effects of Mediator kinase inhibition on RNAPII pausing at IFN-γ-induced genes WT MEFs were stimulated for 30 min with IFN-γ or left unstimulated, with or without 1 h CA pre-treatment and subsequently analyzed by GRO-Seq. (A) The table contains the group of IFN-γ-induced genes with the filtering criteria: lfc > 0; padj < 0.05. Ifn30.ctrl_Ifn0.ctrl_lfc = log2foldchange 30 min IFN-γ stimulated cells vs. unstimulated cells; Ifn30.ctrl_Ifn0.ctrl_padj = adjusted p-value corresponding to Ifn30.ctrl_Ifn0.ctrl_lfc (B) CA decreases gene expression of IFN-γ-induced genes in GRO-Seq. The table contains IFN-γ-induced genes shown in the heat map in Figure 2B. Ifn0_ctrl = unstimulated, untreated cells; Ifn0_CA = unstimulated, CA-treated cells; Ifn30_ctrl = IFN-γ-stimulated, untreated cells; Ifn30_CA = IFN-γ-stimulated, CA-treated cells (C) The table contains counts for pooled replicates at the TSS, within the gene body and the calculated Pausing indices for RefSeq transcripts. Ifn0 = unstimulated sample; Ifn30 = 30 min IFN-γ-stimulated sample; ctrl = untreated sample; CA = CA-treated sample; mean_TSS = counts for pooled replicates at the TSS; mean_geneBody = counts for pooled replicates within the gene body; mean_PI = Pausing index of pooled replicates (D) Correlation of Cortistatin A (CA)-dependent reduction of mRNA levels with increased RNAPII pausing at IFN-γ-induced genes. Data were extracted from Table S1A (RNA-Seq, mRNA) and Table S4C (GRO-Seq). RNA-seq filter: IFN-γ-induced genes (lfc > 1; padj < 0.05; FPKM stim ≥ 1), downregulated by CA (lfc < 0; padj < 0.05); GRO-Seq filter: CA response 30 (= PI IFN30.CA/PI IFN30.Ctrl) > 1.5. log2FoldChange 6CAvs6 = differential expression analysis of 6 h IFN-γ-stimulated CA-treated vs. 6 h IFN-γ-stimulated cells; padj 6CAvs6 = adjusted p-value for log2FoldChange 6CAvs6; PI IFN30.Ctrl = pausing index after 30 min IFN-γ stimulation; PI IFN30.CA = pausing index after 30 min IFN-γ stimulation and CA pre-treatment; CA response 30 = ratio of PI IFN30.CA to PI IFN30.Ctrl
Table S5 (related to Figure 2). Effects of CDK8 kinase inhibition on RNAPII pausing in human cells WT and CDKas HCT116 cells were stimulated for 30 min with IFN-γ or left unstimulated, with or without 3MB-PP1 treatment and subsequently analyzed by PRO-Seq. (A) The table shows differential expression analysis of stimulated WT HCT116 cells (WT_DMSO.IFN) in comparison to unstimulated WT HCT116 cells (WT_DMSO.PBS). (B) The table shows differential expression analysis of stimulated, 3MB-treated CDK8as HCT116 cells (CDK8as_3MB.IFN) in comparison to stimulated, 3MB-treated WT HCT116 cells (WT_3MB.IFN). (C - E) The tables contain counts at the TSS (C), within the gene body (D) and the calculated Pausing index (E) for each condition. WT_DMSO.PBS = unstimulated WT HCT116 cells; WT_3MB.PBS = unstimulated, 3MB-treated WT HCT116 cells; WT_DMSO.IFN = stimulated WT HCT116 cells; WT_3MB.IFN = stimulated, 3MB-treated WT HCT116 cells; CDK8as_DMSO.PBS = unstimulated CDK8as HCT116 cells; CDK8as_3MB.PBS = unstimulated, 3MB-treated CDK8as HCT116 cells; CDK8as_DMSO.IFN = stimulated CDK8as HCT116 cells; CDK8as_3MB.IFN = stimulated, 3MB-treated CDK8as HCT116 cells
Table S6 (related to Figure 3): Effects of Mediator kinase inhibition on eRNA transcription during IFN-γ response in WT MEFs (GRO-Seq) and HCT116 cells (PRO-Seq) (A - D) The tables correspond to Figure 3 and show differences in eRNAs transcription in WT MEFs upon IFN-γ stimulation (A), Mediator kinase inhibition (B) and in HCT116 cells upon IFN-γ stimulation (C) and CDK8 kinase inhibition (D). MEF IFN30 vs IFN0 = 30 min stimulated MEFs vs. unstimulated MEFs; MEF IFN30.CA vs. MEF IFN0 = 30 min stimulated, CA-treated MEFs vs. unstimulated MEFs; WT HCT116 3MB.IFN vs. WT HCT116 3MB.PBS = 30 min stimulated, 3MB-treated WT HCT116 vs. unstimulated, 3MB-treated WT HCT116; CDK8as HCT116 3MB.IFN vs. CDK8as HCT116 3MB.PBS = 30 min stimulated, 3MB-treated CDK8as HCT116 vs. unstimulated, 3MB-treated CDK8as HCT116
Table S7 (related to Figures 5 and 6): Differential expression analysis for effects of inducible CDK8 knockout, CDK19 knockdown and Mediator kinase inhibition (CA) during IFN-γ response (A and B) The tables correspond to Figure 4 and 5, and show genes regulated by 3 h stimulation with IFN-γ at the mRNA (A) and pre-mRNA (B) level (log2FoldChange (lfc) ≤ −1 or 1; padj ≥ 0.05; FPKM stimulated ≥ 1) in siCtrl-treated CDK8fl-MEFs (siCtrl.stim_siCtrl.unst). Differential expression analysis was carried out for the following conditions: CA effects (CA.stim_siCtrl.stim); CA effects in absence of CDK19 (siCDK19.CA.stim_siCDK19.stim); CDK8 knockout effects (CDK8-iKO.stim_siCtrl.stim); CA effects in absence of CDK8 (CDK8- iKO.CA.stim_CDK8-iKO.stim); CDK19 silencing effects (siCDK19.stim_siCtrl.stim); stim = 3 h IFN-γ stimulation; unst = unstimulated; lfc = log2FoldChange; padj = adjusted p-value
Table S8 (related to Figure 7): CDK19 regulates IFN-γ-induced gene expression in kinase-independent ways CDK19-KO, CDK19-WT and CDK19-KDead MEFs were stimulated for 3 h with IFN-γ or left unstimulated. mRNA levels were analyzed by RNA-Seq. The table shows genes regulated by IFN-γ stimulation (filtering criteria: log2FoldChange ≤ −1 or ≥ 1; padj < 0.05; FPKM stimulated 1) and their expression in CDK19-KO, CDK19-WT and CDK19-KDead cells. WT_1- WT_0_lfc = differential expression analysis of 3 h IFN-γ-stimulated CDK19-WT vs. unstimulated CDK19-WT; WT_1-WT_0_padj = adjusted p-value for WT_1-WT_0_lfc; CDK19KO_1-WT_1_lfc = differential expression analysis of 3 h IFN-γ-stimulated CDK19-KO vs. 3 h IFN-γ-stimulated CDK19-WT; CDK19KO_1-WT_1_padj = adjusted p-value for CDK19KO_1-WT_1_lfc; CDK19KDead_1-CDK19KO_1_lfc = differential expression analysis of 3 h IFN-γ-stimulated CDK19-KDead vs. 3 h IFN-γ-stimulated CDK19-KO; CDK19KDead_1-CDK19KO_1_padj = adjusted p-value for CDK19KDead_1- CDK19KO_1_lfc
Table S9 (relates to Key Resources Table): Oligonucleotides for PCR cloning, genotyping and guide RNA
Data Availability Statement
The datasets generated during this study are available at NCBI Sequence Read Archive (SRA) with the accession number PRJNA542065 (GRO-Seq and RNA-Seq datasets) and at NCBI GEO with the accession number GSE129501 (PRO-Seq dataset).
Unprocessed Western blot images of this study are available at Mendeley under: http://dx.doi.org/10.17632/crj8f3j63z.1