

Human cytomegalovirus (HCMV) is a common pathogen that asymptomatically infects most people and establishes a lifelong infection. However, HCMV can cause end-organ disease that results in death in the immunosuppressed and is a leading cause of birth defects. HCMV infection depends on host metabolism, including lipid metabolism. However, the viral mechanisms for remodeling of metabolism are poorly understood. In this study, we demonstrate that the viral UL37x1 protein (pUL37x1) is important for infection-associated increases in lipid metabolism, including fatty acid elongation to produce very-long-chain fatty acids (VLCFAs). Furthermore, we found that HCMV infection results in a significant increase in phospholipids, particularly those with VLCFA tails (PL-VLCFAs). We found that pUL37x1 was important for the high levels of fatty acid elongation and PL-VLCFA accumulation that occur in HCMV-infected cells. Our findings identify a viral protein that is important for changes in lipid metabolism that occur following HCMV infection.
KEYWORDS: cytomegalovirus, human herpesviruses, lipidomics, metabolism, metabolomics
ABSTRACT
Human cytomegalovirus (HCMV) replication requires host metabolism. Infection alters the activity in multiple metabolic pathways, including increasing fatty acid elongation and lipid synthesis. The virus-host interactions regulating the metabolic changes associated with replication are essential for infection. While multiple host factors, including kinases and transcription factors, important for metabolic changes that occur following HCMV infection have been identified, little is known about the viral factors required to alter metabolism. In this study, we tested the hypothesis that pUL37x1 is important for the metabolic remodeling that is necessary for HCMV replication using a combination of metabolomics, lipidomics, and metabolic tracers to measure fatty acid elongation. We observed that fibroblast cells infected with wild-type (WT) HCMV had levels of metabolites similar to those in cells infected with a mutant virus lacking the UL37x1 gene, subUL37x1. However, we found that relative to WT-infected cells, subUL37x1-infected cells had reduced levels of two host proteins that were previously demonstrated to be important for lipid metabolism during HCMV infection: fatty acid elongase 7 (ELOVL7) and the endoplasmic reticulum (ER) stress-related kinase PERK. Moreover, we observed that HCMV infection results in an increase in phospholipids with very-long-chain fatty acid tails (PL-VLCFAs) that contain 26 or more carbons in one of their two tails. The levels of many PL-VLCFAs were lower in subUL37x1-infected cells than in WT-infected cells. Overall, we conclude that although pUL37x1 is not necessary for network-wide metabolic changes associated with HCMV infection, it is important for the remodeling of a subset of metabolic changes that occur during infection.
IMPORTANCE Human cytomegalovirus (HCMV) is a common pathogen that asymptomatically infects most people and establishes a lifelong infection. However, HCMV can cause end-organ disease that results in death in the immunosuppressed and is a leading cause of birth defects. HCMV infection depends on host metabolism, including lipid metabolism. However, the viral mechanisms for remodeling of metabolism are poorly understood. In this study, we demonstrate that the viral UL37x1 protein (pUL37x1) is important for infection-associated increases in lipid metabolism, including fatty acid elongation to produce very-long-chain fatty acids (VLCFAs). Furthermore, we found that HCMV infection results in a significant increase in phospholipids, particularly those with VLCFA tails (PL-VLCFAs). We found that pUL37x1 was important for the high levels of fatty acid elongation and PL-VLCFA accumulation that occur in HCMV-infected cells. Our findings identify a viral protein that is important for changes in lipid metabolism that occur following HCMV infection.
INTRODUCTION
Human cytomegalovirus (HCMV) is a betaherpesvirus that establishes persistent lifelong infection. Infection is common, with most infected people being asymptomatic (1). HCMV is an opportunistic pathogen that can cause severe and life-threatening disease when the immune system is compromised, such as in solid-organ and stem cell transplant patients or those with HIV/AIDS (1, 2). HCMV is also a major cause of birth defects (2). Congenital infection can result in hearing loss, microcephaly, developmental disabilities, and fetal loss (3, 4).
During infection, the expression of HCMV genes is generally classified into three kinetic classes: immediate early, early, and late (1). The viral immediate early UL37x1 gene encodes a protein named pUL37x1. pUL37x1 has multiple functions that support HCMV replication. pUL37x1 traffics from the endoplasmic reticulum (ER) to the mitochondria, where it blocks cell death; as such, it is also named viral mitochondrion-localized inhibitor of apoptosis (vMIA) (5–7). In the mitochondria, pUL37x1 is found on the outer mitochondrial membrane near the mitochondrion-associated ER membranes (8). pUL37x1 triggers stored ER Ca2+ to be released into the cytosol (9, 10). Furthermore, pUL37x1 contributes to HCMV-induced cytomegaly, the characteristic cytopathic effect of cytomegaloviruses (9–11). More recently, pUL37x1 has been linked to intracellular membrane reorganization by HCMV (10). During the late stages of infection, pUL37x1 induces the accumulation of large vesicles within the cytoplasm (10). The formation of these pUL37x1-dependent vesicles requires fatty acid (FA) synthesis, suggesting a link between pUL37x1 and lipid metabolism.
HCMV replication requires de novo FA synthesis, including FA elongation (12–15). Since HCMV does not encode a metabolic network, it relies on the host to provide the energy, materials, and machinery for FA synthesis. In addition to FA metabolism, HCMV increases the metabolic activity in various pathways, including glycolysis, the tricarboxylic acid (TCA) cycle, nucleotide synthesis, and lipid metabolism (16–19). Limiting nutrients or targeting metabolic pathways inhibits HCMV replication (13, 14, 20–23). HCMV infection results in a significant change in host metabolism, altering the concentrations of many metabolites (12–14, 16, 17, 21–28). HCMV infection alters central carbon metabolism and increases the utilization of glucose and glutamine (14, 24, 27, 29–31). Infection increases the flow of carbons from glucose to lipid synthesis (12–14, 28, 32–34), resulting in the synthesis of new lipids that are incorporated into the virus envelope (13, 35).
HCMV-associated metabolic changes involve various host factors. HCMV replication depends on AMPK-dependent metabolic control (25, 36). During infection, HCMV activates AMPK through calmodulin-dependent kinase kinase (CaMKK) activity (36). CaMKK is required for increased glycolysis following infection (26). However, HCMV limits AMPK downregulation of FA synthesis and elongation (15). Additionally, the ER stress-responsive kinase PKR-like ER kinase (PERK) (also known as eukaryotic translation initiation factor 2-alpha kinase 3 [EIF2AK3]) is necessary for lipid synthesis after infection (33). Previously, we demonstrated that carbons from glucose are used for FA elongation to generate very-long-chain fatty acids (VLCFAs) through the action of host fatty acid elongase 7 (ELOVL7) (12, 13). ELOVL7 is required for efficient virus release and virion infectivity (13). HCMV infection increases ELOVL7 expression (12, 13). The viral UL38 protein (pUL38) is partially responsible for inducing ELOVL7 expression after infection (13). Although pUL38 is important for HCMV to induce metabolic changes in host cells, other unidentified viral mechanisms are likely necessary for the reprogramming of host metabolism that occurs during infection (13, 37).
pUL37x1 localizes to the mitochondria and ER and triggers Ca2+ signaling events that may be important for the control of metabolism during infection (16, 18, 19, 26). We tested the hypothesis that pUL37x1 is important for the metabolic remodeling that is necessary for HCMV replication using a mutant virus that lacks the UL37x1 gene (9, 10). Through metabolomic and lipidomic experiments, we found that pUL37x1 is important for a subset of metabolic changes that occur during infection. Moreover, our findings establish that HCMV infection results in a significant increase in phospholipids with VLCFA tails (PL-VLCFAs) and that pUL37x1 is important for the high levels of PL-VLCFAs that are observed in infected cells. FA elongation and the production of saturated VLCFAs were partially dependent on the presence of pUL37x1 during infection. The findings reported here improve our understanding of the virus-host metabolism interactions that occur during HCMV replication. Our study further illustrates that HCMV remodels metabolism to generate a metabolic environment and lipidome that support infection.
(This article was submitted to an online preprint archive [38].)
RESULTS
HCMV replication requires the products of various metabolic pathways. Recently, HCMV pUL38 has been demonstrated to be a viral protein important for the metabolic changes that happen during HCMV infection (13, 37). pUL38 prevents mTOR deactivation and stimulates SREBP maturation and fatty acid elongation (13, 15). pUL38 also alters metabolism independent of mTOR (37). Beyond pUL38, we have a limited understanding of HCMV mechanisms underlying metabolic regulation during infection. We, and others (16, 18, 19, 26), hypothesize that pUL37x1 is important for alteration of host metabolism during HCMV infection. We tested this hypothesis by comparing the levels of metabolites, fatty acids, and lipids in wild-type (WT) virus-infected cells to those in cells infected with subUL37x1, a mutant virus that lacks the UL37x1 gene (9).
Metabolome remodeling following HCMV infection.
Our work builds on the well-defined HCMV remodeling of the metabolome of primary human fibroblast cells under fully confluent, serum-free conditions (12–15, 21–23, 26, 27, 36). These conditions allow for the examination of metabolism while limiting potential lipid or metabolite contamination from bovine serum. Under confluent, serum-free conditions, subUL37x1 expresses pUL36 and pUL38 at WT-like levels while ablating pUL37x1 expression (Fig. 1A), similar to conditions with serum (9). The cell rounding and swelling associated with WT infection, e.g., cytopathic effect (CPE), were lessened in subUL37x1-infected cells at 1 to 2 days postinfection (dpi) (Fig. 1B). However, many subUL37x1-infected cells had WT-like CPE at later stages of infection (Fig. 1B, arrows). At 3 to 4 dpi, a larger amount of cellular debris was observed in cells infected with subUL37x1 than in WT-infected cells, consistent with a rise in cell death (Fig. 1B). Furthermore, we observed a 10-fold loss in virus yield with UL37x1 deletion (Fig. 1C). The loss of pUL37x1 also resulted in the release of fewer infectious virions per total released viral particles, i.e., a higher “particle-to-infectious unit” ratio (Fig. 1D). Overall, these observations are similar to those reported in previous studies of UL37x-1-null mutant viruses (9, 39, 40).
FIG 1.

subUL37x1 mutant virus infection under serum-free conditions. (A) Western blot analysis reveals that subUL37x1 fails to express pUL37x1, while the expression of neighboring genes is unaffected. (B) Infection in fully confluent fibroblast cells under serum-free conditions was visually tracked from 0.25 to 4 dpi. Images from uninfected control cells at the 0.25- and 4-dpi time points are included. Arrows point to mutant-virus-infected cells that have a morphology (cytopathic effect) similar to that of WT-infected cells. (C) Infectious virus particles released by cells infected with the WT or the subUL37x1 mutant at an MOI of 3 were measured at 4 dpi. (D) The particle-to-infectious unit ratios, as measured by viral DNA and infectious titer levels, were compared for particles released by WT- and subUL37x1-infected cells at 4 dpi, at an MOI of 3. For panels C and D, the error bars are the standard deviations (SD) from three independent experiments (n = 3). *, P = <0.05 to 0.01; **, P < 0.01 (by a t test).
Next, we tested if pUL37x1 contributes to metabolic changes associated with HCMV infection. We examined the relative concentrations of intracellular metabolites in infected and uninfected cells using liquid chromatography–high-resolution tandem mass spectrometry (LC-MS/MS). Starting at 1 dpi, WT AD169 virus altered the intracellular concentrations of most metabolites measured (Fig. 2A). We observed an increase in glycolytic metabolites, amino acids, and TCA cycle intermediates in cells infected with WT virus (Fig. 2A), as previously described (14, 17, 21, 22, 25, 26). Infection with subUL37x1 mutant virus similarly altered metabolite levels starting at 1 dpi (Fig. 2A). The metabolic profiles of WT- and subUL37x1-infected cells were strongly correlated during mid- and late stages of replication (Fig. 2B). Importantly, glycolytic and TCA metabolites, e.g., hexose-phosphate, pyruvate, malate, α-ketoglutarate, and succinate, were increased in subUL37x1-infected cells compared to uninfected cells (Fig. 2A). However, these metabolites were slightly reduced compared to the levels in WT-infected cells at 2 to 3 dpi, suggesting that pUL37x1 may be important for achieving the overall high metabolite concentrations associated with infection but not required for HCMV to alter central carbon metabolism.
FIG 2.

Metabolomic comparison of WT- and subUL37x1-infected cells. (A) Comparison of intracellular metabolite levels in fibroblast cells infected with the WT or mutant subUL37x1 virus relative to those in uninfected cells, at an MOI of 3. (B) Correlation plots for metabolite levels normalized to values for uninfected cells (y axis, WT; x axis, subUL37x1). Correlation R values are given for each time point from 0.25 to 3 dpi. For both panels A and B, metabolites were measured from technical replicates from 4 independent experiments (n = 4).
pUL37x1 is important for HCMV-induced FA elongation.
In addition to our metabolomic analysis, we quantitatively measured the effect of pUL37x1 on the cellular concentrations of fatty acids. HCMV infection enhances FA synthesis and elongation (12–14). FA synthase (FAS) forms long-chain FAs of up to 16 carbons in length (C16) by connecting carbons, two at a time. The product of FAS is the 16-carbon saturated FA palmitate (C16:0 [the number following the colon represents the number of double bonds in the FA]). Palmitate can be processed further to make longer chains and/or desaturated to introduce double bonds among the carbons. In humans, longer FAs are made by one or more of the seven FA elongases (ELOVL1 to -7). Similar to FAS, ELOVLs also add one 2-carbon unit per reaction cycle. We have previously shown that HCMV infection significantly increases the concentrations of saturated VLCFAs, specifically those with 26 or more carbons (≥C26) (12, 13).
To determine if pUL37x1 is important for HCMV-induced FA elongation, we measured FAs extracted from lipids of HCMV-infected or uninfected cells. In cells infected with WT virus, saturated VLCFAs were increased by as much as 9-fold compared to the levels in uninfected cells, confirming our previous observations (Fig. 3) (12, 13). While the levels of some FAs were slightly higher in cells infected with subUL37x1 virus than in uninfected cells, the concentrations of C26:0 and C28:0 VLCFAs were significantly reduced in subUL37x1-infected cells relative to WT-infected cells at 2 and 3 dpi (Fig. 3).
FIG 3.
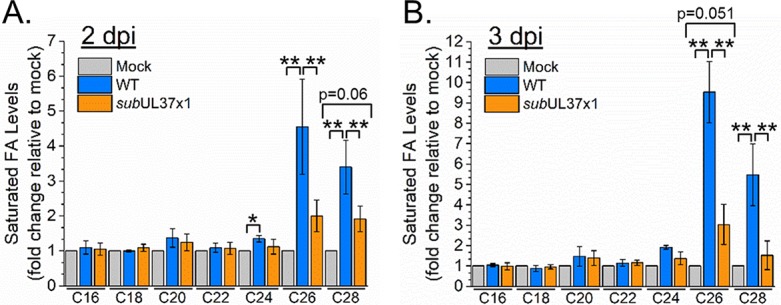
Concentrations of saturated very-long-chain fatty acids (VLCFAs) in WT- and subUL37x1-infected fibroblasts. FA levels relative to those in uninfected cells are shown for cells infected at an MOI of 3. Shown are FA changes at 2 dpi (A) and at 3 dpi (B). All data are represented as means and SD (n = 4). P values for comparison of mock and subUL37x1 infection that are near the 0.05 cutoff are shown. *, P = <0.05 to 0.01; **, P < 0.01 (by one-way analysis of variance [ANOVA] with a Tukey test).
FA elongation can be monitored by measuring the incorporation of 13C from labeled glucose into FAs using LC-MS, i.e., nontandem MS1-only analysis (12, 13, 42). Following a 1-h infection period, we fed cells medium that contained 13C-labeled glucose at positions 1 and 6 ([1,6-13C]glucose). Glycolytic breakdown of this labeled form of glucose will result in pyruvate containing one 13C atom (i.e., 1-labeled pyruvate) (Fig. 4A). Pyruvate can be further metabolized, generating 1-labeled citrate in the mitochondria. Citrate can be exported into the cytoplasm, where it can be converted to 1-labeled acetyl-CoA and unlabeled oxaloacetate. Alternatively, pyruvate may break down into acetate that can be used to generate 1-labeled acetyl-CoA (28, 43). The conversion of acetyl-CoA to malonyl-CoA by acetyl-CoA carboxylase 1 (ACC-1) is the rate-limiting step of FA synthesis and elongation. In our labeled cells, this will result in one 13C atom per 2-carbon unit used for FA synthesis and elongation.
FIG 4.

Synthesis of VLCFAs in WT- and subUL37x1-infected cells. (A) Diagram depicting the flow of carbons from glucose and acetate to FA synthesis and elongation. Carbon atoms are represented by circles; red and orange circles represent 13C-labeled carbons, while black circles depict unlabeled atoms. (B) Incorporation of labeled atoms from [1,6-13C]glucose into saturated FAs. The data are presented as percentages of labeled to unlabeled saturated FAs. (C) Percent labeling of saturated FAs from [1,2-13C]acetate. (D and E) Labeling pattern of C26:0 from labeled glucose (D) and labeled acetate (E). All data are represented as means and SD (n = 3). *, P = <0.05 to 0.01; **, P < 0.01 (by one-way ANOVA with a Tukey test).
Using LC-MS to measure the overall percentage of FA tails that have at least one 13C atom, we observed that uninfected cells have minimal labeling of FAs after correcting for the natural isotopic abundance of 13C. In WT-infected cells, FAs were labeled, and the greatest labeling occurred in longer FAs (e.g., 57% ± 2% of C26:0 was labeled) (Fig. 4B). Relative to uninfected cells, the labeling in subUL37x1-infected cells was enhanced (e.g., 26% ± 6% of C26:0 was labeled in subUL37x1-infected cells, compared to only 2% ± 3% in uninfected cells) (Fig. 4B). Moreover, the observed labeling in subUL37x1-infected cells was decreased relative to that in WT-infected cells (26% ± 6% compared to 57% ± 2% for C26:0; P < 0.01).
In addition to a reduction in the percentage of labeling, the FAs isolated from cells infected with the subUL37x1 mutant virus had fewer 13C atoms than those from WT-infected cells (Fig. 4D). In WT-infected cells, we were able to observe up to 4 to 5 labeled atoms in C26:0 (i.e., the incorporation of 8 to 10 new carbons). This observation was similar to that for cells fed uniformly labeled [13C]glucose, where two 13C atoms are added per 2-carbon unit (12, 13). However, in subUL37x1-infected cells, only 1 to 2 labeled atoms were observed in C26:0, indicating that only one to two 2-carbon units were added (Fig. 4D).
Our glucose labeling strategy measured the carbon contributions of both citrate and acetate that is derived from glucose to FA elongation. Since HCMV infection increases the flow of carbons from acetate into lipid synthesis (28), we measured the contribution of acetate to HCMV-induced FA elongation using 13C-labeled acetate. In this case, both atoms of the 2-carbon unit were labeled. Again, cells were fed labeled acetate following a 1-h infection period. Similar to the labeled-glucose tracer studies, only minimal labeling of FAs from acetate was observed in uninfected cells (Fig. 4C). Infection increased acetate-derived atoms incorporated into VLCFAs (Fig. 4C). In WT-infected cells, 30% ± 1% of C26:0 was labeled from [13C]acetate. Additionally, up to four labeled atoms from two 2-carbon units were incorporated into C26:0 in WT-infected cells, a larger amount than in uninfected cells (Fig. 4E). Infection with the subUL37x1 mutant virus enhanced FA elongation from acetate compared to that in uninfected cells (Fig. 4C). However, the percent acetate labeling of C26:0 was reduced in subUL37x1-infected cells relative to WT-infected cells (20% ± 6% versus 30% ± 1%; P < 0.05) (Fig. 4C). Additionally, fewer 13C atoms from acetate were incorporated into C26:0 in subUL37x1-infected cells than in WT-infected cells (Fig. 4E). In summary, pUL37x1 is important for the high levels of FA elongation that are observed in HCMV-infected cells.
pUL37x1 expression enhances HCMV-induced ELOVL7 protein levels.
During infection, ELOVL7 produces saturated VLCFAs (13). HCMV infection increases ELOVL7 mRNA and protein expression (13). We examined if pUL37x1 impacts HCMV-induced ELOVL7 expression. In human foreskin fibroblast (HFF) cells, infection with WT virus enhanced ELOVL7 expression by 2 dpi (Fig. 5A), similar to our observations in MRC-5 primary fibroblasts (13). ELOVL7 protein levels were higher in subUL37x1-infected cells than in uninfected cells (Fig. 5A and B). However, the levels of ELOVL7 protein were reduced by approximately 40% in subUL37x1-infected cells relative to WT-infected cells (Fig. 5B and C).
FIG 5.

ELOVL7 fatty acid elongase protein accumulation during HCMV infection. (A) ELOVL7 protein expression was determined by Western blotting from 2 to 4 dpi. (B) Further independent analysis of ELOVL7 expression at 3 dpi. (C) Quantification of ELOVL7 levels at 3 dpi. ELOVL7 levels were normalized to tubulin levels and are shown relative to WT levels. All data are represented as means and SD (n = 3). *, P < 0.05 (by a t test).
We further investigated if pUL37x1 may affect other proteins important for FA elongation. HCMV infection increases the protein expression of ACC-1 (15), the enzyme producing the malonyl-CoA substrate for the FA elongation reaction (Fig. 6A). We found that ACC-1 levels were higher in subUL37x1-infected cells than in uninfected cells (Fig. 6B). Since FA labeling from [13C]glucose was reduced in subUL37x1-infected cells relative to WT-infected cells, we examined ATP citrate lyase (ACLY). ACLY converts citrate to acetyl-CoA in the cytosol. It was previously demonstrated that ACLY protein levels are slightly higher in infected cells than in uninfected cells at late time points (32). We found that ACLY levels were similar in WT- and subUL37x1-infected cells at 2 to 4 dpi (Fig. 6B). ACSS2 synthesizes acetyl-CoA from acetate. At 3 dpi, the cells infected with subUL37x1 virus had ACSS2 protein levels similar to those in WT-infected cells (Fig. 6C and D).
FIG 6.

Impact of HCMV infection on proteins involved in the synthesis of VLCFAs. (A) Schematic of the glucose-to-FA synthesis pathway. Enzymes involved in metabolizing glucose carbons to FAs are shown in blue boxes. (B) Western blotting of proteins involved in glucose metabolism and FA synthesis and their regulation. (C) Western blotting at 3 dpi showing ACSS2 expression levels. (D) Quantification of ACSS2 protein levels from three independent experiments. Protein loading was normalized to the tubulin signal. Data are represented as means and SD.
HCMV-induced metabolic remodeling requires AMPK activity, and infection increases AMPK protein levels (25, 36). Cells infected with subUL37x1 had levels of the catalytic subunit of AMPK, AMPKα, that were similar to the levels in WT-infected cells (Fig. 6B). AMPK reduces ACC-1 activity through phosphorylation. Cells infected with either WT virus or subUL37x1 virus have a larger amount of phosphorylated ACC-1 than uninfected cells (Fig. 6B), suggesting that pUL37x1 effects on FA elongation are mediated through an AMPK-independent mechanism.
PERK is important for HCMV-induced lipid metabolism (33). PERK protein levels are increased by HCMV infection (33, 44–46). We tested if pUL37x1 affects HCMV-induced PERK protein levels. At 3 dpi, the levels of PERK were increased by WT infection (Fig. 7A). However, the levels of PERK were 2-fold lower in subUL37x1-infected cells than in WT-infected cells (Fig. 7A and B).
FIG 7.

PERK protein levels in WT-infected and subUL37x1-infected cells. (A) Western blotting at 3 dpi showing PERK expression levels in human fibroblast cells infected with WT or subUL37x1 virus at an MOI of 3. (B) Quantification of PERK protein levels from three independent experiments. Protein loading was normalized to the tubulin signal. Data are represented as means and SD. *, P < 0.05 (using a one-sample t test).
HCMV infection remodels the host lipidome.
One function of FAs is to act as hydrophobic tails in lipids. Given our observations in Fig. 3 and 4, we hypothesized that pUL37x1 may help support the synthesis of lipids with VLCFA tails following HCMV infection. To test this hypothesis, we developed LC-MS/MS lipidomic methods to examine phospholipids, including those that contain ≥C26 VLCFAs. We found that infection with WT virus markedly increased phospholipid concentrations, especially for those with VLCFA tails (Fig. 8A, left). Of the various classes of phospholipids examined, phosphatidylcholines (PCs) were among the lipid species most altered by infection (Fig. 8A and B). The concentrations of several PC species containing tails with a total of 44 to 48 carbons were significantly increased following HCMV infection (Fig. 8B). Of the top six PC species whose concentrations were increased by HCMV infection, their levels were higher in infected cells than in uninfected cells by 19- to 60-fold for PC(44:1) and by as high as 270- to 1,400-fold for PC(46:1). Since phospholipids contain two fatty acid tails, we examined the tail compositions of these six PC species. All contained a ≥C26 tail (Fig. 8D, left). We found evidence of five different PC(44:1) species, with tail combinations of C14:0 plus C30:1, C16:0 plus C28:1, C16:1 plus C28:0, C18:0 plus C26:1, and C18:1 plus C26:0. PC(46:2) also contains five lipid species with tails ranging from C16 to C30 in length (Fig. 8D). For both PC(44:1) and PC(46:2), C16-C18 FAs were the most common tails observed in combination with ≥C26 VLCFA tails. In this study, C30 FAs were the longest tails observed.
FIG 8.

Phospholipid levels 3 days after infection with HCMV. (A, left) LC-MS/MS phospholipidomic analysis of WT-infected cells compared to uninfected cells. (Right) The same type of lipidomic analysis showing the levels of phospholipids in subUL37x1-infected relative to WT-infected cells. Both heat maps are organized going from smallest to largest changes for subUL37x1 relative to the WT. (B) Phosphatidylcholine (PC) lipids with VLCFA tails. (C) Similar plots showing phosphatidylserine (PS) lipids. (D) The FA tails in PC and PS lipids shown in panels B and C were identified by MS/MS. In some cases, only one of the two tails was observed. The unobserved tails are marked by ^. All data are from day 3 postinfection for cells infected at an MOI of 3 (n = 3). *, P = <0.05 to 0.01; **, P < 0.01 (by one-way ANOVA with a Tukey test). Box plots show the means plus 25 to 75% variance, while the whiskers show the SD. Individual data points are also shown. PA, phosphatidic acid; PC, phosphatidylcholine; PE, phosphatidylethanolamine; PG, phosphatidylglycerol; PI, phosphatidylinositol; PS, phosphatidyserine; LPC, lysoPC (lysophosphatidylcholine); LPE, lysoPE (lysophosphatidylethanolamine).
When we examined phospholipids from subUL37x1-infected cells, we observed a significant decrease in lipid levels compared to those in WT-infected cells (Fig. 8A, right). The most notably decreased lipids were PC lipids whose levels were greatly elevated in WT-infected cells. The levels of all six of the PC species discussed above were significantly decreased in subUL37x1-infected cells relative to WT-infected cells (Fig. 8B). However, the levels of five of the six PC lipids highlighted in this study were elevated in subUL37x1-infected cells relative to uninfected cells (Fig. 8B). Infection with the mutant virus raised the concentration of these lipids by 4- to 80-fold (Fig. 8B). Only PC(48:5) was not statistically significantly increased following infection with subUL37x1 (Fig. 8B).
The relative levels of some lipids were higher in subUL37x1-infected cells than in WT-infected cells (Fig. 8A). The levels of some phosphatidylserine (PS) lipids were increased by as much as 4-fold in subUL37x1-infected cells relative to WT-infected cells (Fig. 8C). None of these PS lipids contained a tail longer than C22 (Fig. 8D, right). Furthermore, none of these species were increased following infection with WT virus, and one, PS(36:2), was slightly reduced in WT-infected cells compared to uninfected cells (Fig. 8C).
Inhibition of cell death does not alter lipid changes.
Since pUL37x1 is important for limiting cell death, the loss of PC-VLCFAs in subUL37x1-infected cells could be due to an increase in cell death. We tested this possibility by suppressing the cell death that occurs following subUL37x1 infection by treating cells with Nα-tosyl-l-lysine chloromethyl ketone (TLCK). TLCK is a serine protease inhibitor that blocks apoptosis. TLCK treatment at concentrations of up to 100 μM has previously been demonstrated to block cell death associated with UL37x1-null mutant viruses without affecting virus replication (47). Treatment with 35 μM TLCK restored the survival of subUL37x1-infected cells to WT-like levels at 3 dpi (Fig. 9A). At 3 dpi, most lipids were unaltered in TLCK-treated cells (Fig. 9B). However, treatment reduced the levels of some PC lipids with 44 carbons or more in their tails in WT-infected cells (Fig. 9B, left). The levels of PCs in subUL37x1-infected cells were largely unaltered by TLCK treatment (Fig. 9B, right). Similar to WT-infected cells, TLCK treatment in subUL37x1-infected cells decreased the levels of some PCs containing ≥C44 tails. The levels of most PS lipids were also unaltered by TLCK treatment (Fig. 9C). These observations demonstrate that preventing the cell death associated with the loss of UL37x1 fails to restore lipid levels.
FIG 9.

Phosphatidylcholine and phosphatidylserine levels in HCMV-infected cells treated with an apoptosis inhibitor. (A) At 3 dpi, cell survival was determined for cells treated with DMSO or 35 μM Nα-tosyl-l-lysine chloromethyl ketone (TLCK). (B) The abundances of PC lipids at 3 dpi were measured in WT- or subUL37x1-infected cells treated with TLCK relative to DMSO. (C) Abundances of PS lipids under the same conditions (n = 3). *, P = <0.05 to 0.01; **, P < 0.01 (by a t test). Box plots show the means plus the 25 to 75% quartile groups, while the whiskers show the SD. Individual data points are also shown.
pUL37x1 has a limited ability to induce lipid synthesis without infection.
We next investigated if pUL37x1 is sufficient to alter lipid levels. We stably expressed pUL37x1 in uninfected fibroblasts using a lentivirus system. In these cells, pUL37x1 is downstream of the EF1α promoter. As a control, we used the same lentivirus system to express green fluorescent protein (GFP) using the EF1α promoter. We independently generated two pUL37x1-expressing cell lines (Fig. 10A). In this case, the second clone, UL37x1-c.2, had a slightly higher level of pUL37x1 expression. First, we examined PERK and ELOVL7 protein levels in these cells. The levels of both PERK and ELOVL7 were unaltered by the expression of pUL37x1 (Fig. 10A). Lipidomic analyses of the pUL37x1-expressing cells revealed that the levels of some PC and PS lipids were statistically increased in UL37x1-c.2 cells relative to those in GFP-expressing cells (Fig. 10B to E). However, the concentrations of lipids in cells expressing a lower level of pUL37x1 were similar to those in the GFP control cells. These results suggest that high levels of pUL37x1 expression may be sufficient to induce some lipid synthesis.
FIG 10.

Levels of lipids in uninfected fibroblast cells expressing pUL37x1. (A) Western blot analysis of uninfected fibroblasts stably expressing pUL37x1. Two independent clones were analyzed and compared to cells stably expressing GFP. (B) PC lipid analysis of pUL37x1-expressing cells relative to GFP control cells. For clarity, the means and standard errors (SE) measured are shown. (C) Box plot of selected PC lipids from panel B. (D) PS lipid analysis of pUL37x1-expressing cells compared to GFP control cells. Shown are the means and SE. (E) Box plot of selected PS lipids (n = 3). *, P < 0.05 (using one-way ANOVA with a Tukey test). Box plots show the means plus the 25 to 75% quartile groups, while the whiskers show the SD. Individual data points are also shown.
DISCUSSION
Many diverse viruses induce changes in the metabolic activity of their host cells (reviewed in references 16–19 and 48–51). Like HCMV, infection with Kaposi’s sarcoma-associated herpesvirus (KSHV) increases FA synthesis (52, 53). Murine gammaherpesvirus 68 (MHV68) replication is suppressed by type I interferon limitations on fatty acid and cholesterol metabolism (54). These studies, along with those for HCMV and herpes simplex virus (HSV) (described in references 16, 18, and 19), demonstrate that virus-metabolism interactions are essential for herpesvirus infection and can be targeted to limit replication. The metabolic reprogramming associated with HCMV infection requires various host kinases, including PERK (33), mTOR (13), CaMKK (26), and AMPK (25, 36), and transcription factors, including SREBPs (13, 15, 32) and chREBP (55). Our previous observation that mTOR activity is required for HCMV-induced FA elongation led to the finding that pUL38 is required for FA elongation (13). Recently, pUL38 was demonstrated to be important for HCMV-induced glycolysis and amino acid metabolism independent of mTOR (37).
In this study, we tested the hypothesis that a second viral protein, pUL37x1, is important for HCMV to alter host metabolism. We demonstrate that HCMV infection increases the abundance of PL-VLCFAs and that pUL37x1 is important for the high levels of FA elongation that produce VLCFAs following infection. The results described in this study support our hypothesis by demonstrating that pUL37x1 impacts the remodeling of host lipid metabolism that occurs during infection.
pUL37x1 performs multiple functions during infection, including controlling the flux of Ca2+ from the ER to the cytosol and preventing premature cell death by targeting the outer membrane of mitochondria (5, 8, 9, 56, 57). This study is unable to discern if the observed changes in fatty acid elongation and cellular lipids are dependent on or independent of pUL37x1 mitochondrial or Ca2+ flux functions. However, premature cell death or a loss in virus replication in subUL37x1-infected cells is unlikely to solely explain our observed changes in lipid metabolism. A loss in cell number or virus replication would likely result in a general decrease in metabolites and lipids altered by HCMV infection. However, we observed a decrease in only a subset of metabolites and lipids in subUL37x1-infected cells relative to WT-infected cells, and we observed that some lipids are increased by subUL37x1 infection (Fig. 2, 3, and 8). Second, restoring survival of cells infected with the subUL37x1 mutant virus failed to rescue lipid metabolism (Fig. 9). Finally, pUL37x1 is sufficient to increase the concentrations of some lipids when expressed at high levels in uninfected cells (Fig. 10).
Although the levels of lipids observed in uninfected pUL37x1-expressing cells were higher than in GFP-expressing control cells, the levels in pUL37x1-expressing cells were reduced relative to those in WT-infected cells. For example, the level of PC(44:1) was 1.4-fold higher in UL37x1c.2 cells than in control cells (Fig. 10C), whereas in WT-infected cells, it was 35-fold higher than in uninfected cells (Fig. 8B). These observations suggest that other viral factors are also necessary to drive the significant changes in lipid synthesis that are observed for HCMV infection. Our results, together with those for pUL38 (13, 37), suggest that pUL37x1 and pUL38 may synergistically coordinate HCMV-induced metabolic changes or may be part of a larger group of gene products important for remodeling of host metabolism.
The role of pUL37x1 in lipid metabolism may be connected to its known functions in Ca2+ signaling or inhibiting apoptosis. The release of Ca2+ into the cytoplasm from the ER may stimulate metabolism through downstream signaling events (26). Alternatively, pUL37x1-mediated depletion of Ca2+ from the ER may induce changes in the host that are important for HCMV replication. An ER stress response can be triggered when ER-stored Ca2+ is released into the cytosol (58). Consistent with this possibility, we found that PERK protein levels were reduced in subUL37x1-infected cells compared to WT-infected cells (Fig. 7); however, further experiments are needed to determine if Ca2+ flux is needed for the increase in PERK protein levels during infection. Furthermore, FA elongation and the synthesis of multiple lipids occur at the ER and could be sensitive to changes caused by pUL37x1. We also found that PS lipids were increased in subUL37x1-infected cells relative to WT-infected cells (Fig. 8). PS lipids on the outer leaflet of the plasma membrane are an apoptotic signal (59, 60), suggesting that pUL37x1 may exert further antiapoptotic properties by limiting the accumulation of PS lipids. PS synthesis occurs at the mitochondrion-associated ER membrane (61). pUL37x1 traffics to the outer mitochondrial membrane, where it contacts the ER (8), suggesting that it may be possible for pUL37x1 to act directly on the PS synthesis pathway. Further experimental work is needed to determine if the role of pUL37x1 in inhibiting apoptosis or stimulating Ca2+ flux is connected to the metabolic changes observed in this study.
Using our newly developed LC-MS/MS lipidomic methods, we examined lipids with up to 48 total carbons in their tails and found that some lipids had a tail as long as C30 (Fig. 8). A previous lipidomic study demonstrated that cellular PC lipid species with long-chain FA tails were increased by HCMV infection, while those with shorter chains were decreased, and that PC lipids are a major fraction of the total phospholipids found in the purified virus envelope (35). Notably, this previous analysis examined lipids with a total of only 30 to 42 carbons in their tails, and individual FA lengths were not reported. We previously demonstrated that the virus envelope contains saturated VLCFAs (12, 13). Some PC-VLCFAs whose levels were increased by HCMV infection contained saturated VLCFAs (Fig. 8). Relative to WT-infected cells, we observed a reduction in ELOVL activity and FA elongation, as measured by glucose and acetate labeling, in subUL37x1-infected cells (Fig. 3 and 4). Together, these observations suggest that the virus envelope may contain PC-VLCFAs that we observed in this study and that pUL37x1 may contribute to the lipid environment necessary to build the virus envelope.
During late stages of replication, pUL37x1 causes the accumulation of large vesicles (10). These vesicles may form due to the enhanced FA elongation and lipid synthesis described in this study that occur in HCMV-infected cells expressing pUL37x1. Additionally, pUL37x1 is needed to maintain a low particle-to-infectious unit (IU) ratio; the mutant virus ratio was 500:1, compared to ∼100:1 for the WT (Fig. 1D). Viruses produced in ELOVL7 knockdown cells show a similar loss in their particle-to-infectivity ratio (∼400:1 at the same time point) (13). Our observations suggest that at least part of the defect in subUL37x1 replication is due to a loss in lipid metabolism.
Given the reliance of HCMV replication, and viruses in general, on metabolism, it is important to define the viral and host factors required for viral remodeling of metabolism. Our work establishes that pUL37x1 is important for some of the metabolic reprogramming observed during HCMV infection. Furthermore, our results show that HCMV infection significantly induces the accumulation of phospholipids with VLCFA tails and highlight the need to continue to study the role of lipid metabolism during HCMV infection. Based on our lipidomic studies, we conclude that pUL37x1 is important for the overall metabolic phenotype observed in HCMV-infected cells. Our results further support the critical functions of pUL37x1 in HCMV replication and provide a foundation for future studies in defining the mechanisms underlying metabolic remodeling by HCMV.
MATERIALS AND METHODS
Cells, viruses, and experimental setup.
Human foreskin fibroblast (HFF) cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS), 10 mM HEPES, and penicillin-streptomycin. Prior to infection, cells were maintained at full confluence for 3 days in serum-containing growth medium. Cells were switched to serum-free medium (DMEM, HEPES, and penicillin-streptomycin) the day before infection. Cells were infected with either the wild-type (WT) HCMV AD169 strain or subUL37x1, a UL37x1-null virus (9). Unless noted otherwise, infections were performed at a multiplicity of infection (MOI) of 3 infectious units per cell. Both WT and mutant viruses were kindly provided by Thomas Shenk. Both WT and subUL37x1 viruses were grown in HFF cells. Mutant virus was not grown in cells expressing pUL37x1. All virus stocks were made by pelleting virions from the supernatant of infected cells through 20% sorbitol. Viruses were resuspended in serum-free DMEM and stored at −80°C. Infectious virus titers were measured as the 50% tissue culture infectious dose (TCID50) by visualizing infectious centers. Infectious centers were further confirmed by immunofluorescence microscopy. In this case, cells on the TCID50 plate were fixed using methanol, and plaques were identified using an anti-pUL123 (IE1) monoclonal antibody (clone 1B12), followed by anti-mouse Alexa 488 secondary antibody. This method has similarly been used to quantify a mutant HCMV that lacks the UL38 gene (13). The particle-to-IU ratio was determined using UL123-specific primers to measure viral DNA (62).
Metabolic tracer studies were performed using [1,6-13C]glucose and fully labeled [1,2-13C]acetate sodium salt from Cambridge Isotopes (catalog numbers CLM-2717 and CLM-440, respectively). Following a 1-h infection period, cells were washed with warm phosphate-buffered saline (PBS) three times prior to the addition of serum-free DMEM containing either 4.5 g/liter glucose (25 mM) or 0.0035 g/liter acetate (60 μM). For 3-dpi samples, the labeling medium was replenished at 2 dpi.
UL37x1 expression in uninfected cells.
The UL37x1 gene from BAC-AD169 was cloned into a lentiviral pLVX-EF1α-puro vector between the EcoRI and BamHI sites (TaKaRa). Since UL37x1 contains a BamHI site, PCR was used to generate UL37x1 that was flanked by EcoRI and BglII sites. Digestion and ligation destroyed the BamHI site in the final plasmid. The pLVX-EF1α-UL37x1 plasmid was verified by sequencing following cloning. A pLVX-EF1α-puro plasmid expressing GFP was used as a control. Stable expression of pUL37x1 in HFF cells was done using lentiviral transduction as previously described (13). For lipidomic analyses, 8 × 104 cells were seeded per well on a 6-well plate. Cells were grown for 48 h in DMEM containing 10% FBS, HEPES, and penicillin-streptomycin. The cells were washed with PBS prior to lipid extraction as described below.
TLCK treatment.
Nα-Tosyl-l-lysine chloromethyl ketone (TLCK) was purchased from Sigma-Aldrich. Following a 1-h infection or mock infection period, cells were treated with 35 μM TLCK or the dimethyl sulfoxide (DMSO) vehicle. At 2 dpi, the drug was refreshed. At 3 dpi, cell survival was determined using a lactate dehydrogenase (LDH) release cytotoxicity assay kit (Pierce, Thermo Scientific). Lipids were extracted at 3 dpi and analyzed as described below.
Protein analysis.
Proteins were examined by Western blotting of whole-cell lysates. SDS-PAGE was performed with Tris-glycine-SDS running buffer using Mini-Protean TGX anyKD or 4 to 20% gels (Bio-Rad). Proteins were transferred to an Odyssey nitrocellulose membrane (Li-Cor). Western blot analyses were performed using 5% bovine serum albumin (BSA) in Tris-buffered saline with 0.05% Tween 20 (TBS-T) for blocking and antibody incubation, except for analyses of anti-ELOVL7, anti-ATP citrate lyase, and anti-PERK blots, which were performed using 3% milk in TBS-T. The following antibodies were used: mouse monoclonal anti-pUL37x1 clone 4B6-B (9), mouse monoclonal anti-pUL36 clone 10C8 (63), mouse monoclonal anti-pUL38 clone 8D6 (64), rabbit polyclonal anti-ELOVL7 (catalog number SAB3500390; Sigma-Aldrich), rabbit monoclonal anti-AMPKα (catalog number 2603; Cell Signaling), rabbit polyclonal anti-ATP citrate lyase (catalog number PA5-29497; Thermo), rabbit polyclonal anti-ACC-1 (catalog number 3662; Cell Signaling), rabbit polyclonal phospho-ACC-1 Ser79 (catalog number 3661; Cell Signaling), rabbit monoclonal anti-PERK (catalog number 3192; Cell Signaling), rabbit monoclonal anti-ACCS2 (catalog number 3658; Cell Signaling), mouse monoclonal anti-actin (catalog number 929-42212; Li-Cor), and mouse monoclonal anti-α-tubulin (catalog number T6199; Sigma-Aldrich). Blots with mouse monoclonal anti-HCMV, anti-actin, and anti-tubulin antibodies were incubated for 1 h at room temperature. All others were incubated overnight at 4°C. Quantification of Western blots was performed using a Li-Cor Odyssey CLx imaging system.
Metabolomics and metabolite analysis.
All metabolite levels were measured using liquid chromatography–high-resolution tandem mass spectrometry (LC-MS/MS). At various times after infection, metabolic reactions were rapidly quenched using cold 80% methanol, and extraction was performed as previously described (21, 27). Extracted water-soluble metabolites were dried under nitrogen gas and resuspended in Optima high-performance liquid chromatography (HPLC)-grade water (Fisher Chemical) for reverse-phase liquid chromatography or 50% methanol for hydrophilic interaction chromatography (HILIC). Mass spectrometric analysis was performed by using four Thermo Scientific mass spectrometers: Quantum Ultra triple-quad, Quantum Max triple-quad, Exactive orbitrap, and Q-Exactive Plus orbitrap. Metabolites were measured in both positive and negative modes using reverse-phase or HILIC separation (65, 66). Metabolites were identified using mass spectral and ultra-high-performance liquid chromatography (UHPLC) retention time data that were generated from external standards. Peaks were examined, and metabolites were identified using the Metabolomic Analysis and Visualization Engine (MAVEN) (67, 68). Each metabolite was measured using two technical replicates per experiment. Each metabolic experiment contained controls for determining ionization artifacts and sample degradation. All chemicals and solutions for metabolomics, fatty acid analysis, and lipidomics were of ultrapure HPLC or MS grade.
Fatty acid analysis.
Fatty acid (FA) analyses were performed as previously described (12, 13). Briefly, lipids were extracted from cells lysed using cold 50% methanol containing 0.05 M HCl. Cells were scraped and transferred to glass vials. Lipids were extracted in chloroform, and FA tails were chemically cleaved via saponification at 80°C for 1 h in basic methanol buffer. The FAs were isolated using hexane and dried under nitrogen gas. Following resuspension in a 1:1:0.3 solution of methanol-chloroform-water or a 1:1:1 solution of methanol-chloroform-isopropanol, FAs were analyzed by UHPLC and high-resolution mass spectrometry (LC-MS). FAs were examined using a Luna C8 reversed-phase column (Phenomenex) and an Exactive or Q-Exactive Plus orbitrap mass spectrometer (13). In the latter case, FAs were separated using the LC buffer solutions described in “Lipidomics and lipid analysis” below. FA spectra were collected at a resolution of 140,000 (at m/z 200). For labeling experiments, the instrument was calibrated immediately before the start of the analysis. Experiments using metabolic tracers and FA labeling were additionally analyzed at a resolution of 280,000. The data were analyzed, and FA levels were quantified using MAVEN. For labeling experiments, the data were corrected for naturally occurring carbon 13 using MATLAB (13). Each sample was measured using two technical replicates per experiment.
Lipidomics and lipid analysis.
Similar to the FA analysis, cells were washed with PBS prior to analysis. For lipidomics, cells were lysed in cold 50% methanol, and lipids were extracted by using chloroform. The chloroform was removed by drying the samples under nitrogen gas. Lipids were resuspended in a 1:1:1 solution of methanol-chloroform-isopropanol. For each sample, 3 wells on a 6-well plate were used. Lipids were extracted in parallel from two wells and independently analyzed (e.g., technical duplicates). The cells in the third well were counted. Samples were normalized according to the total number of live cells at the time of lipid extraction. Each sample was resuspended in 125 μl of a 1:1:1 solution per 100,000 cells. In parallel with the biological samples, the extraction procedure was performed on wells that lacked cells. These “no-cell” controls were used to determine contaminants that were removed from the analysis. For initial experiments, 1 μl of Splash Lipidmix lipidomic mass spectrometry standards (Avanti Polar Lipids) was added to 50% methanol prior to extraction. These standards were used to establish our lipidomic methods to determine the extraction efficiency and proper data analyses/interpretations. For the final data included in this paper, the Lipidmix standards were not included in the samples to eliminate the possibility of contamination. After resuspension, the samples were immediately analyzed. During data collection, the samples were stored at 4°C in an autosampler. Lipids were separated using a Kinetex 2.6-μm C18 column (catalog number 00F-4462-AN; Phenomenex) at 60°C on a Vanquish UHPLC system (Thermo Scientific). Two solvents were used for UHPLC: solvent A (40:60 water-methanol plus 10 mM ammonium formate and 0.1% formic acid) and solvent B (10:90 methanol-isopropanol plus 10 mM ammonium formate and 0.1% formic acid). UHPLC was performed at a 0.25-ml/min flow rate using the following gradient conditions: 75% solvent A–25% solvent B for 2 min, 35% solvent A–65% solvent B for 2 min at a curve value of 4, a hold at 35% solvent A–65% solvent B for 1 min, 0% solvent A–100% solvent B for 11 min at a curve value of 4, and a hold at 0% solvent A–100% solvent B for 4 min. The column was briefly washed and reequilibrated before the next run. In total, each run lasted 30 min. All lipids were examined using a Q-Exactive Plus mass spectrometer operating in a data-dependent full MS/dd-MS2 TopN mode. MS1 data were collected at a resolution of either 70,000 or 140,000 (at m/z 200) using an automatic gain control (AGC) target of 1e6 with transient times of 250 ms and 520 ms, respectively. Spectra were collected over a mass range of m/z 200 to 1,600. MS2 spectra were collected using a resolution of 35,000, an a AGC target of 1e5, and a 120-ms maximum injection time. Each sample was analyzed twice, using 10 μl of sample for negative mode and 8 μl of sample for positive mode. In negative mode, a normalized collision energy (NCE) value of 20 was used. In positive mode, the NCE value was increased to 30. All lipids were ionized using a heated electrospray ionization (HESI) source. The following HESI parameters were used in negative mode: sheath gas flow rate of 44, auxiliary gas flow rate of 14, sweep gas flow rate of 2, spray voltage of 3.3 kV, S-lens radio frequency (RF) of 76, auxiliary gas temperature of 220°C, and capillary temperature of 320°C. Similar settings were used in positive mode except that the spray voltage was increased to 3.5 kV and the S-lens RF level was decreased to 65. The instrument was calibrated weekly.
As with metabolites and FA, lipids were measured using two technical replicates per experiment. Lipidomic data were analyzed using MAVEN and Xcalibur (Thermo Scientific). In addition to the Splash Lipidmix, the following lipid standards were used for lipidomic method development and to establish LC retention times and MS2 fragmentation patterns for diacylglycerol (DG)(20:0/18:0), lysophosphatidic acid (lysoPA)(16:0), lysoPA(18:1), lysophosphatidylcholine (lysoPC)(16:0), lysoPC(18:0), lysoPC(22:0), lysophosphatidylethanolamine (lysoPE)(16:0), lysoPE(18:0), lysophosphatidylglycerol (lysoPG)(16:0), lysophosphatidylinositol (lysoPI)(16:0), lysophosphatidyserine (lysoPS)(16:0), lysoPS(18:1), monoacylglycerol (MG)(16:0), MG(18:1), phosphatidic acid (PA)(16:0/18:1), PA(18:0/20:4), PA(18:0/22:6), PC(16:0/18:1), PC(22:1/22:1), PC(24:0/24:0), phosphatidylethanolamine (PE)(16:0/18:1), PE(18:0/18:0), PE(18:0/20:4), phosphatidylglycerol (PG)(16:0/18:1), PG(18:0/20:4), PG(18:0/22:6), phosphatidylinositol (PI)(16:0/18:1), PI(18:0/20:4), PS(16:0/18:1), PS(18:0/20:4), triacylglycerol (TG)(16:0/18:1/16:0), TG(18:0/18:0/18:1), TG(18:1/16:0/18:1), and lipid reference material in human serum from the NIST.
Lipids were initially identified according to their MS1 mass using a 5-ppm cutoff and LC retention times. Lipid identification was confirmed using the following MS2 data: PC fragment of 184.074 (positive mode), PE fragment of 196.038 or the tail plus 196.038 (negative mode) and loss of 141.019 (positive mode), PG fragment of 152.996 plus the identification of the FA tails (negative mode), PI fragment of 241.012 (negative), and PS neutral loss of 87.032 (negative). PC lipids were further confirmed in negative mode using PC lipid ions containing a formic adduct. Both PC and PS tails were identified in negative mode.
Data availability.
The MS data files were deposited in the MetaboLights database (https://www.ebi.ac.uk/metabolights) under accession number MTBLS1073.
ACKNOWLEDGMENTS
We acknowledge Thomas Shenk for providing guidance in our studies and reagents, including the subUL37x1 mutant virus and anti-HCMV monoclonal antibodies. We thank Joshua Rabinowitz for providing advice and assistance with the early stages of these studies. We are grateful to Felicia Goodrum, Jim Alwine, Lynn Enquist, Edward Mocarski, the members of the Goodrum lab, and the other members of the Purdy lab for helpful discussions regarding this work.
This work was funded in part by a grant (ADHS18-198868) from the Arizona Biomedical Research Commission, made available through the Arizona Department of Health Services to J.G.P. Additional funding was provided by the BIO5 Institute, the Department of Immunobiology, University of Arizona College of Medicine—Tucson, and a new investigator award from University of Arizona Health Sciences to J.G.P.
REFERENCES
- 1.Mocarski ES, Shenk T, Griffiths P, Pass RF. 2013. Cytomegaloviruses, p 1960–2014. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Britt W. 2008. Manifestations of human cytomegalovirus infection: proposed mechanisms of acute and chronic disease. Curr Top Microbiol Immunol 325:417–470. [DOI] [PubMed] [Google Scholar]
- 3.Hyde TB, Schmid DS, Cannon MJ. 2010. Cytomegalovirus seroconversion rates and risk factors: implications for congenital CMV. Rev Med Virol 20:311–326. doi: 10.1002/rmv.659. [DOI] [PubMed] [Google Scholar]
- 4.Britt WJ. 2017. Congenital human cytomegalovirus infection and the enigma of maternal immunity. J Virol 91:e02392-16. doi: 10.1128/JVI.02392-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goldmacher VS, Bartle LM, Skaletskaya A, Dionne CA, Kedersha NL, Vater CA, Han JW, Lutz RJ, Watanabe S, Cahir McFarland ED, Kieff ED, Mocarski ES, Chittenden T. 1999. A cytomegalovirus-encoded mitochondria-localized inhibitor of apoptosis structurally unrelated to Bcl-2. Proc Natl Acad Sci U S A 96:12536–12541. doi: 10.1073/pnas.96.22.12536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McCormick AL. 2008. Control of apoptosis by human cytomegalovirus. Curr Top Microbiol Immunol 325:281–295. [DOI] [PubMed] [Google Scholar]
- 7.Brune W, Andoniou CE. 2017. Die another day: inhibition of cell death pathways by cytomegalovirus. Viruses 9:E249. doi: 10.3390/v9090249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bhuvanendran S, Salka K, Rainey K, Sreetama SC, Williams E, Leeker M, Prasad V, Boyd J, Patterson GH, Jaiswal JK, Colberg-Poley AM. 2014. Superresolution imaging of human cytomegalovirus vMIA localization in sub-mitochondrial compartments. Viruses 6:1612–1636. doi: 10.3390/v6041612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sharon-Friling R, Goodhouse J, Colberg-Poley AM, Shenk T. 2006. Human cytomegalovirus pUL37x1 induces the release of endoplasmic reticulum calcium stores. Proc Natl Acad Sci U S A 103:19117–19122. doi: 10.1073/pnas.0609353103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sharon-Friling R, Shenk T. 2014. Human cytomegalovirus pUL37x1-induced calcium flux activates PKCalpha, inducing altered cell shape and accumulation of cytoplasmic vesicles. Proc Natl Acad Sci U S A 111:E1140–E1148. doi: 10.1073/pnas.1402515111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Smith MG. 1956. Propagation in tissue cultures of a cytopathogenic virus from human salivary gland virus (SGV) disease. Proc Soc Exp Biol Med 92:424–430. doi: 10.3181/00379727-92-22498. [DOI] [PubMed] [Google Scholar]
- 12.Koyuncu E, Purdy JG, Rabinowitz JD, Shenk T. 2013. Saturated very long chain fatty acids are required for the production of infectious human cytomegalovirus progeny. PLoS Pathog 9:e1003333. doi: 10.1371/journal.ppat.1003333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Purdy JG, Shenk T, Rabinowitz JD. 2015. Fatty acid elongase 7 catalyzes lipidome remodeling essential for human cytomegalovirus replication. Cell Rep 10:1375–1385. doi: 10.1016/j.celrep.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Munger J, Bennett BD, Parikh A, Feng XJ, McArdle J, Rabitz HA, Shenk T, Rabinowitz JD. 2008. Systems-level metabolic flux profiling identifies fatty acid synthesis as a target for antiviral therapy. Nat Biotechnol 26:1179–1186. doi: 10.1038/nbt.1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Spencer CM, Schafer XL, Moorman NJ, Munger J. 2011. Human cytomegalovirus induces the activity and expression of acetyl-coenzyme A carboxylase, a fatty acid biosynthetic enzyme whose inhibition attenuates viral replication. J Virol 85:5814–5824. doi: 10.1128/JVI.02630-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Purdy JG. 2019. Pathways to understanding virus-host metabolism interactions. Curr Clin Microbiol Rep 6:34–43. doi: 10.1007/s40588-018-0109-7. [DOI] [Google Scholar]
- 17.Rabinowitz JD, Purdy JG, Vastag L, Shenk T, Koyuncu E. 2011. Metabolomics in drug target discovery. Cold Spring Harb Symp Quant Biol 76:235–246. doi: 10.1101/sqb.2011.76.010694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goodwin CM, Xu S, Munger J. 2015. Stealing the keys to the kitchen: viral manipulation of the host cell metabolic network. Trends Microbiol 23:789–798. doi: 10.1016/j.tim.2015.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shenk T, Alwine JC. 2014. Human cytomegalovirus: coordinating cellular stress, signaling, and metabolic pathways. Annu Rev Virol 1:355–374. doi: 10.1146/annurev-virology-031413-085425. [DOI] [PubMed] [Google Scholar]
- 20.Grady SL, Purdy JG, Rabinowitz JD, Shenk T. 2013. Argininosuccinate synthetase 1 depletion produces a metabolic state conducive to herpes simplex virus 1 infection. Proc Natl Acad Sci U S A 110:E5006–E5015. doi: 10.1073/pnas.1321305110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hwang J, Purdy JG, Wu K, Rabinowitz JD, Shenk T. 2014. Estrogen-related receptor alpha is required for efficient human cytomegalovirus replication. Proc Natl Acad Sci U S A 111:E5706–E5715. doi: 10.1073/pnas.1422361112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vastag L, Koyuncu E, Grady SL, Shenk TE, Rabinowitz JD. 2011. Divergent effects of human cytomegalovirus and herpes simplex virus-1 on cellular metabolism. PLoS Pathog 7:e1002124. doi: 10.1371/journal.ppat.1002124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.DeVito SR, Ortiz-Riaño E, Martínez-Sobrido L, Munger J. 2014. Cytomegalovirus-mediated activation of pyrimidine biosynthesis drives UDP-sugar synthesis to support viral protein glycosylation. Proc Natl Acad Sci U S A 111:18019–18024. doi: 10.1073/pnas.1415864111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chambers JW, Maguire TG, Alwine JC. 2010. Glutamine metabolism is essential for human cytomegalovirus infection. J Virol 84:1867–1873. doi: 10.1128/JVI.02123-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Terry LJ, Vastag L, Rabinowitz JD, Shenk T. 2012. Human kinome profiling identifies a requirement for AMP-activated protein kinase during human cytomegalovirus infection. Proc Natl Acad Sci U S A 109:3071–3076. doi: 10.1073/pnas.1200494109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McArdle J, Schafer XL, Munger J. 2011. Inhibition of calmodulin-dependent kinase kinase blocks human cytomegalovirus-induced glycolytic activation and severely attenuates production of viral progeny. J Virol 85:705–714. doi: 10.1128/JVI.01557-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Munger J, Bajad SU, Coller HA, Shenk T, Rabinowitz JD. 2006. Dynamics of the cellular metabolome during human cytomegalovirus infection. PLoS Pathog 2:e132. doi: 10.1371/journal.ppat.0020132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vysochan A, Sengupta A, Weljie AM, Alwine JC, Yu Y. 2017. ACSS2-mediated acetyl-CoA synthesis from acetate is necessary for human cytomegalovirus infection. Proc Natl Acad Sci U S A 114:E1528–E1535. doi: 10.1073/pnas.1614268114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yu Y, Maguire TG, Alwine JC. 2011. Human cytomegalovirus activates glucose transporter 4 expression to increase glucose uptake during infection. J Virol 85:1573–1580. doi: 10.1128/JVI.01967-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yu Y, Clippinger AJ, Alwine JC. 2011. Viral effects on metabolism: changes in glucose and glutamine utilization during human cytomegalovirus infection. Trends Microbiol 19:360–367. doi: 10.1016/j.tim.2011.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Landini MP. 1984. Early enhanced glucose uptake in human cytomegalovirus-infected cells. J Gen Virol 65(Part 7):1229–1232. doi: 10.1099/0022-1317-65-7-1229. [DOI] [PubMed] [Google Scholar]
- 32.Yu Y, Maguire TG, Alwine JC. 2012. Human cytomegalovirus infection induces adipocyte-like lipogenesis through activation of sterol regulatory element binding protein 1. J Virol 86:2942–2949. doi: 10.1128/JVI.06467-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu Y, Pierciey FJ Jr, Maguire TG, Alwine JC. 2013. PKR-like endoplasmic reticulum kinase is necessary for lipogenic activation during HCMV infection. PLoS Pathog 9:e1003266. doi: 10.1371/journal.ppat.1003266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sanchez V, Dong JJ. 2010. Alteration of lipid metabolism in cells infected with human cytomegalovirus. Virology 44:71–77. doi: 10.1016/j.virol.2010.04.026. [DOI] [PubMed] [Google Scholar]
- 35.Liu ST, Sharon-Friling R, Ivanova P, Milne SB, Myers DS, Rabinowitz JD, Brown HA, Shenk T. 2011. Synaptic vesicle-like lipidome of human cytomegalovirus virions reveals a role for SNARE machinery in virion egress. Proc Natl Acad Sci U S A 108:12869–12874. doi: 10.1073/pnas.1109796108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McArdle J, Moorman NJ, Munger J. 2012. HCMV targets the metabolic stress response through activation of AMPK whose activity is important for viral replication. PLoS Pathog 8:e1002502. doi: 10.1371/journal.ppat.1002502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rodriguez-Sanchez I, Schafer XL, Monaghan M, Munger J. 2019. The human cytomegalovirus UL38 protein drives mTOR-independent metabolic flux reprogramming by inhibiting TSC2. PLoS Pathog 15:e1007569. doi: 10.1371/journal.ppat.1007569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Purdy JG, Xi Y, Harwood S, Wise L. 2019. Human cytomegalovirus remodeling of lipid metabolism requires pUL37x1. bioRxiv doi: 10.1101/526228. [DOI] [PMC free article] [PubMed]
- 39.McCormick AL, Meiering CD, Smith GB, Mocarski ES. 2005. Mitochondrial cell death suppressors carried by human and murine cytomegalovirus confer resistance to proteasome inhibitor-induced apoptosis. J Virol 79:12205–12217. doi: 10.1128/JVI.79.19.12205-12217.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang A, Hildreth RL, Colberg-Poley AM. 2013. Human cytomegalovirus inhibits apoptosis by proteasome-mediated degradation of Bax at endoplasmic reticulum-mitochondrion contacts. J Virol 87:5657–5668. doi: 10.1128/JVI.00145-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reference deleted.
- 42.Kamphorst JJ, Fan J, Lu W, White E, Rabinowitz JD. 2011. Liquid chromatography-high resolution mass spectrometry analysis of fatty acid metabolism. Anal Chem 83:9114–9122. doi: 10.1021/ac202220b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kamphorst JJ, Chung MK, Fan J, Rabinowitz JD. 2014. Quantitative analysis of acetyl-CoA production in hypoxic cancer cells reveals substantial contribution from acetate. Cancer Metab 2:23. doi: 10.1186/2049-3002-2-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Isler JA, Skalet AH, Alwine JC. 2005. Human cytomegalovirus infection activates and regulates the unfolded protein response. J Virol 79:6890–6899. doi: 10.1128/JVI.79.11.6890-6899.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Siddiquey MNA, Zhang H, Nguyen CC, Domma AJ, Kamil JP. 2018. The human cytomegalovirus endoplasmic reticulum-resident glycoprotein UL148 activates the unfolded protein response. J Virol 92:e00896-18. doi: 10.1128/JVI.00896-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xuan B, Qian Z, Torigoi E, Yu D. 2009. Human cytomegalovirus protein pUL38 induces ATF4 expression, inhibits persistent JNK phosphorylation, and suppresses endoplasmic reticulum stress-induced cell death. J Virol 83:3463–3474. doi: 10.1128/JVI.02307-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McCormick AL, Roback L, Mocarski ES. 2008. HtrA2/Omi terminates cytomegalovirus infection and is controlled by the viral mitochondrial inhibitor of apoptosis (vMIA). PLoS Pathog 4:e1000063. doi: 10.1371/journal.ppat.1000063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sanchez EL, Lagunoff M. 2015. Viral activation of cellular metabolism. Virology 479–480:609–618. doi: 10.1016/j.virol.2015.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lagunoff M. 2016. Activation of cellular metabolism during latent Kaposi’s sarcoma herpesvirus infection. Curr Opin Virol 19:45–49. doi: 10.1016/j.coviro.2016.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jordan TX, Randall G. 2016. Flavivirus modulation of cellular metabolism. Curr Opin Virol 19:7–10. doi: 10.1016/j.coviro.2016.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mushtaq M, Darekar S, Kashuba E. 2016. DNA tumor viruses and cell metabolism. Oxid Med Cell Longev 2016:6468342. doi: 10.1155/2016/6468342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Delgado T, Sanchez EL, Camarda R, Lagunoff M. 2012. Global metabolic profiling of infection by an oncogenic virus: KSHV induces and requires lipogenesis for survival of latent infection. PLoS Pathog 8:e1002866. doi: 10.1371/journal.ppat.1002866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bhatt AP, Jacobs SR, Freemerman AJ, Makowski L, Rathmell JC, Dittmer DP, Damania B. 2012. Dysregulation of fatty acid synthesis and glycolysis in non-Hodgkin lymphoma. Proc Natl Acad Sci U S A 109:11818–11823. doi: 10.1073/pnas.1205995109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lange PT, Schorl C, Sahoo D, Tarakanova VL. 2018. Liver X receptors suppress activity of cholesterol and fatty acid synthesis pathways to oppose gammaherpesvirus replication. mBio 9:e01115-18. doi: 10.1128/mBio.01115-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yu Y, Maguire TG, Alwine JC. 2014. ChREBP, a glucose-responsive transcriptional factor, enhances glucose metabolism to support biosynthesis in human cytomegalovirus-infected cells. Proc Natl Acad Sci U S A 111:1951–1956. doi: 10.1073/pnas.1310779111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McCormick AL, Skaletskaya A, Barry PA, Mocarski ES, Goldmacher VS. 2003. Differential function and expression of the viral inhibitor of caspase 8-induced apoptosis (vICA) and the viral mitochondria-localized inhibitor of apoptosis (vMIA) cell death suppressors conserved in primate and rodent cytomegaloviruses. Virology 316:221–233. doi: 10.1016/j.virol.2003.07.003. [DOI] [PubMed] [Google Scholar]
- 57.Hayajneh WA, Colberg-Poley AM, Skaletskaya A, Bartle LM, Lesperance MM, Contopoulos-Ioannidis DG, Kedersha NL, Goldmacher VS. 2001. The sequence and antiapoptotic functional domains of the human cytomegalovirus UL37 exon 1 immediate early protein are conserved in multiple primary strains. Virology 279:233–240. doi: 10.1006/viro.2000.0726. [DOI] [PubMed] [Google Scholar]
- 58.Dejeans N, Tajeddine N, Beck R, Verrax J, Taper H, Gailly P, Calderon PB. 2010. Endoplasmic reticulum calcium release potentiates the ER stress and cell death caused by an oxidative stress in MCF-7 cells. Biochem Pharmacol 79:1221–1230. doi: 10.1016/j.bcp.2009.12.009. [DOI] [PubMed] [Google Scholar]
- 59.Fadok VA, Bratton DL, Frasch SC, Warner ML, Henson PM. 1998. The role of phosphatidylserine in recognition of apoptotic cells by phagocytes. Cell Death Differ 5:551–562. doi: 10.1038/sj.cdd.4400404. [DOI] [PubMed] [Google Scholar]
- 60.Fadok VA, Laszlo DJ, Noble PW, Weinstein L, Riches DW, Henson PM. 1993. Particle digestibility is required for induction of the phosphatidylserine recognition mechanism used by murine macrophages to phagocytose apoptotic cells. J Immunol 151:4274–4285. [PubMed] [Google Scholar]
- 61.Vance JE, Tasseva G. 2013. Formation and function of phosphatidylserine and phosphatidylethanolamine in mammalian cells. Biochim Biophys Acta 1831:543–554. doi: 10.1016/j.bbalip.2012.08.016. [DOI] [PubMed] [Google Scholar]
- 62.Womack A, Shenk T. 2010. Human cytomegalovirus tegument protein pUL71 is required for efficient virion egress. mBio 1:e00282-10. doi: 10.1128/mBio.00282-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Patterson CE, Shenk T. 1999. Human cytomegalovirus UL36 protein is dispensable for viral replication in cultured cells. J Virol 73:7126–7131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Terhune S, Torigoi E, Moorman N, Silva M, Qian Z, Shenk T, Yu D. 2007. Human cytomegalovirus UL38 protein blocks apoptosis. J Virol 81:3109–3123. doi: 10.1128/JVI.02124-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lu W, Clasquin MF, Melamud E, Amador-Noguez D, Caudy AA, Rabinowitz JD. 2010. Metabolomic analysis via reversed-phase ion-pairing liquid chromatography coupled to a stand alone orbitrap mass spectrometer. Anal Chem 82:3212–3221. doi: 10.1021/ac902837x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Brauer MJ, Yuan J, Bennett BD, Lu W, Kimball E, Botstein D, Rabinowitz JD. 2006. Conservation of the metabolomic response to starvation across two divergent microbes. Proc Natl Acad Sci U S A 103:19302–19307. doi: 10.1073/pnas.0609508103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Melamud E, Vastag L, Rabinowitz JD. 2010. Metabolomic analysis and visualization engine for LC-MS data. Anal Chem 82:9818–9826. doi: 10.1021/ac1021166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Clasquin MF, Melamud E, Rabinowitz JD. 2012. LC-MS data processing with MAVEN: a metabolomic analysis and visualization engine. Curr Protoc Bioinformatics Chapter 14:Unit 14.11. doi: 10.1002/0471250953.bi1411s37. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The MS data files were deposited in the MetaboLights database (https://www.ebi.ac.uk/metabolights) under accession number MTBLS1073.