

Abstract
Many viral factors manipulate the host post-translational modification (PTM) machinery for efficient viral replication. In particular, phosphorylation and SUMOylation can distinctly regulate the activity of the human cytomegalovirus (HCMV) transactivator immediate early 2 (IE2). However, the molecular mechanism of this process is unknown. Using various structural, biochemical, and cell-based approaches, here we uncovered that IE2 exploits a cross-talk between phosphorylation and SUMOylation. A scan for small ubiquitin-like modifier (SUMO)-interacting motifs (SIMs) revealed two SIMs in IE2, and a real-time SUMOylation assay indicated that the N-terminal SIM (IE2–SIM1) enhances IE2 SUMOylation up to 4-fold. Kinetic analysis and structural studies disclosed that IE2 is a SUMO cis-E3 ligase. We also found that two putative casein kinase 2 (CK2) sites adjacent to IE2–SIM1 are phosphorylated in vitro and in cells. The phosphorylation drastically increased IE2–SUMO affinity, IE2 SUMOylation, and cis-E3 activity of IE2. Additional salt bridges between the phosphoserines and SUMO accounted for the increased IE2–SUMO affinity. Phosphorylation also enhanced the SUMO-dependent transactivation activity and auto-repression activity of IE2. Together, our findings highlight a novel mechanism whereby SUMOylation and phosphorylation of the viral cis-E3 ligase and transactivator protein IE2 work in tandem to enable transcriptional regulation of viral gene.
Keywords: host–pathogen interaction, post-translational modification (PTM), viral protein, SUMO-interacting motif (SIM), sumoylation, phosphorylation, nuclear magnetic resonance (NMR), enzyme kinetics, transcription co-activator, viral transcription
Introduction
The immediate early (IE)2 genes are the first viral genes transcribed during infection. The IE gene products are essential to optimize the host cell environment for replication of the viral genome and the transcription of early and late genes. IE1 and IE2 are the two predominant IE proteins in human cytomegalovirus (HCMV), which are splicing products of the major immediate early promoter (MIEP) (1). Although IE1 is essential only at a low multiplicity of infection, IE2 is strictly indispensable for viral replication and growth (2).
IE2 regulates HCMV growth at multiple levels. It arrests the cell cycle in “pseudo-S” phase at the G1-S boundary to facilitate viral replication over host genome replication (3). IE2 is an essential factor for replication complex assembly (4). Finally, IE2 works as a transactivator for early/late viral genes and various host genes (5). IE2 transactivates TATA box-containing promoters with the help of basal transcription factors (TBP, TFIIB, and TAFs) (5, 6). Interestingly, IE2 regulates its promoter MIEP by binding to cis-regulatory sequence downstream of the transcription start site (7). Apart from transcription factors, IE2 also functions with transcription co-activators and chromatin modifiers to regulate the host transcriptional machinery (6, 8–10).
IE2 is a pleiotropic regulator. Hence, its expression and activity are tightly regulated transcriptionally, post-transcriptionally, and also by post-translational modifications (PTMs) like SUMOylation and phosphorylation (7, 11, 12). SUMOylation is the covalent addition of protein small ubiquitin-like modifier (SUMO) to the lysine side chain of a substrate (13). SUMOylation occurs through an enzymatic cascade involving sequential action of an E1-activating enzyme, an E2-conjugating enzyme (UBC9), and a few E3 ligases. SUMOylation of IE2 occurs at the two lysine residues Lys-175 and Lys-180 (12, 14). The cytomegalovirus with SUMOylation-deficient IE2 has severe growth defects due to impaired initiation of gene expression, replication compartment assembly, and MIEP autoregulation (15, 16). Proteins with a SUMO-interacting motif (SIM) can identify SUMO or SUMOylated proteins via the noncovalent SIM–SUMO interaction (17). IE2 also contains a SIM at its N terminus, which is essential for its localization to nuclear puncta and transactivation activity (18). The SIM recruits SUMOylated transcription factors (e.g. TAF12) to facilitate transactivation. Moreover, deletion of the SIM decreases IE2 SUMOylation (15, 18). Despite the functional importance of IE2/SUMO noncovalent interaction, its molecular details are unknown. Additionally, the molecular mechanism underlying the role of SIM in SUMOylation of IE2 is unclear.
IE2 is phosphorylated by several host kinases (19). Phosphorylation by ERK2 (a mitogen-activated protein kinase (MAPK)) inhibits its transactivation activity without affecting auto-repression of MIEP (20, 21). IE2 is also phosphorylated by the kinase CK2, which is intriguing because CK2 is a part of the viral tegument (22). During infection, the uncoating of the HCMV tegument releases the tegument CK2 in the host cell to activate MIEP expression (23). CK2 phosphorylates IE2 at the serine-rich region (aa 258–275) and in the so-called fragment 5B region (aa 180–252, Fig. 1A) (19). Phosphorylation of the serine-rich region depletes the transactivation activity of IE2 (19). However, the sites of phosphorylation within fragment 5B and the effect of this phosphorylation on the activity of IE2 are unknown.
Figure 1.

Interactions between IE2–SIM1 and SUMO. A, schematic of the domains in IE2. The three predicted SIMs and the two SUMOylation site Lys-175 and Lys-180 are shown. Yellow circles with S denote SUMO. The transactivation domains are shown as blue boxes with TAD. The fragment 5B, serine-rich region, and DBA-binding domain are shown. B, overlay of the 15N-edited HSQC spectra of free 15N-SUMO1 (red) with different stoichiometric ratios of IE2–SIM1 as given in the top left-hand side of the spectra. C, two regions of the spectra are expanded to show a shift of SUMO1 resonances during titration. D, CSPs in SUMO1 upon binding to IE2–SIM1. The CSPs between the free and the bound form are calculated as CSP = ((δHfree − δHbound)2 + (δNfree − δNbound)/5)2)1/2, where δH and δN are the chemical shift of the amide hydrogen and nitrogen, respectively. The yellow and red dashed lines indicate 1× S.D. and 2× S.D., respectively. The secondary structure alignment of SUMO1 against its sequence is provided above the plot. The residues with CSPs significantly above the dashed lines are present at the interface of the SUMO1/IE2–SIM1 complex. E, significant CSPs are mapped onto the SUMO1 structure. The residues with CSP above the yellow and red lines are colored in yellow and red, respectively. F, CSPs in SUMO2 upon binding to IE2–SIM1. G, significant CSPs mapped on the SUMO2 structure. The residues with CSP above yellow and orange lines are colored in yellow and orange, respectively.
We carried out a study of the predicted SIMs in IE2 to uncover a new SIM at the C terminus of IE2 by NMR. NMR also confirmed the previously known N-terminal SIM (IE2–SIM1). The titrations indicate that IE2–SIMs bind to both SUMO1 and SUMO2 with similar affinity. Adjacent to the N-terminal SIM (IE2–SIM1) are the two SUMOylation sites Lys-175 and Lys-180. We developed a fluorescence-based real-time SUMOylation assay, which confirmed that the IE2–SIM1 enhances SUMOylation of IE2. However, the assay also indicated paralog specificity in SUMOylation of IE2. Kinetic analysis revealed that IE2 functions as a SUMO cis-E3, where the IE2–SIM1 reduces the Km value between UBC9∼SUMO and the IE2 SUMOylation sites to increase the rate of IE2 SUMOylation.
We further carried out a comprehensive study of IE2 phosphorylation in cellular conditions. We report that two putative CK2 phosphorylation sites Ser-203 and Ser-205 in fragment 5B are indeed phosphorylated in cells, as well as in vitro by CK2. Ser-203 and Ser-205 are adjacent to the IE2–SIM1. Phosphorylation of Ser-203 and Ser-205 increases the SUMO/IE2–SIM1 affinity by 8-fold. Structures of the complex between SUMO1/2 and phosphorylated IE2–SIM1 attribute the increased affinity to additional salt-bridges formed between the positively-charged residues in SUMO1/2 and the negatively-charged phosphate moiety in the phosphoserines. The drastic increase in SIM–SUMO affinity also enhances the SUMO cis-E3 activity, transactivation activity, and auto-repression activity in IE2. Together, the HCMV protein IE2 uniquely exploits a cross-talk of two post-translational modifications, phosphorylation and SUMOylation, to regulate transcription of the viral genes and ensure a productive infection.
Results
N- and C-terminal SIMs in IE2 interact with SUMO1 and SUMO2
Bioinformatics analysis using JASSA (24) predicted three SIMs in IE2: SIM1, 199–202; SIM2, 410–413; and SIM3, 501–504, as shown in Fig. 1A. Among these, SIM1 was previously identified as a bona fide SIM by pulldown assays in cells (15). However, the rest of the SIMs was not investigated before. The affinity of the SIM–SUMO interaction is typically weak, which is difficult to capture by pulldown experiments. Alternatively, NMR spectroscopy can detect interaction over a broad range of affinities, including weak interactions (25). We designed peptides corresponding to the putative SIMs and tested the binding of the SIMs to SUMO1 by NMR (Table 1). Perturbations due to the altered chemical environment upon ligand binding are reflected in a shift of the backbone amide resonances in the 15N-edited HSQC spectra. The IE2–SIM1 peptide was titrated into a sample of 15N-isotope–labeled SUMO1, and a series of 15N-edited HSQC experiments monitored its effect on 15N-SUMO1. An overlay of the HSQCs is given in Fig. 1B, and two expanded areas of the HSQC are plotted in Fig. 1C. A subset of SUMO1 peaks shifted consistently with increasing concentrations of IE2–SIM1. The chemical shift perturbations (CSP) against SUMO1 residues, given in Fig. 1D, indicated that the maximum perturbations occurred in the residues 35–55, which include the β-strand β2, α-helix α1, and the loop between them (Fig. 1E). The interface corresponds to the canonical interface observed in other SUMO/SIM complexes (17). The same titration experiments were repeated for IE2–SIM2 and IE2–SIM3 domains in separate experiments. IE2–SIM3, but not IE2–SIM2, bound to SUMO1 (Fig. S1A). The pattern of CSP in SUMO1 upon binding IE2–SIM3 is similar to IE2–SIM1, indicating that both the SIMs bind at the same interface.
Table 1.
Analysis of possible SUMO interaction motifs in IE2
| SIMs | Residue no. | Sequence |
Kd (μm) |
|
|---|---|---|---|---|
| SUMO1 | SUMO2 | |||
| IE2-SIM1 | 199–202 | NKIIDTAGCIVISDSEEEQG | 58.2 (± 9.2) | 53.5 (± 8.3) |
| IE2-SIM2 | 410–413 | TMQVNNKGIQIIYTRNHEVK | NB | NB |
| IE2-SIM3 | 501–504 | 1IIHAATPVDLLGALNLC | 368.3 (± 29.1) | 319.85 (± 40.4) |
| IE2-ppSIM1 | 199–202 | NKIIDTAGCIVIpSDpSEEEQG | 7.1 (±0.4) | 7.2 (± 1.6) |
Underline residues are putative SIMs. NB, no binding.
We repeated the titration experiments with IE2–SIMs against 15N-labeled SUMO2 and monitored the effects using 15N-edited HSQC spectra of 15N-SUMO2. Similar to SUMO1, a significant subset of SUMO2 peaks shifted upon titration with IE2–SIM1 as shown in Fig. S1, B and C. The most perturbed region is between β2 and α1 of SUMO2, which is the known SUMO2/SIM interface (Fig. 1, F and G). Additionally, IE2–SIM3, but not IE2–SIM2, interacted with SUMO2 (Fig. S1D).
The CSPs observed during titration were fit against peptide/protein concentration to yield the dissociation constant (Kd) of the complex (Figs. S2 and S3 and Table 1). The Kd value of the N-terminal IE2–SIM1 was 6-fold lower than the C-terminal IE2–SIM3, indicating that the N-terminal SIM has a higher affinity for SUMO than the C-terminal SIM. Both IE2–SIM1 and IE2–SIM3 do not have paralog specificity and have similar affinities for SUMO1 and SUMO2. Taken together, although IE2–SIM1 and IE2–SIM3 interacted with both SUMO1 and SUMO2, the interaction between IE2–SIM1 and SUMO1/2 was stronger. Hence, the IE2–SIM1 was studied further.
Casein kinase 2 phosphorylates two serines adjacent to IE2–SIM1
IE2 phosphorylation modulates its transactivation activity (19). Previous phosphorylation studies have focused on specific sites or domains in IE2 (19–21). A comprehensive report of phosphorylation sites in the cellular condition is missing. Thus, we studied the sites of IE2 phosphorylation in HEK293T cells taking a proteomics approach (Fig. 2A, Fig. S4). Interestingly, in addition to various newly identified phosphorylation sites, two serines adjacent to IE2–SIM1, Ser-203 and Ser-205, were phosphorylated in cells (Fig. 2A). Ser-203 and Ser-205 are putative CK2 target phosphorylation sites. An in vitro phosphorylation assay of IE2–SIM1 by CK2 was carried out to confirm whether these sites are indeed modified by CK2 (Fig. 2B). IE2–SIM1 peptide does not include any serines apart from Ser-203 and Ser-205. It consists of a threonine, which is not the putative CK2 phosphorylation site. Phosphorylation of IE2–SIM1 was carried out using γ-ATP, resolved on SDS-PAGE, and detected by autoradiography. As shown in Fig. 2C, phosphorylated IE2–SIM1 was observed in the reaction with active CK2, whereas the heat-inactivated CK2 could not phosphorylate IE2–SIM1. IE2–ppSIM1 was further analyzed by MALDI-TOF to find out whether it was phosphorylated at a single serine or both the serines (Fig. 2D). Although mass (m/z) for IE2–SIM1 was 2098 Da, the same for phosphorylated IE2–SIM1 was 2258 Da. The difference of 160 Da corresponds to the addition of two phosphate groups, indicating that indeed CK2 phosphorylates IE2–SIM1 at Ser-203 and Ser-205.
Figure 2.

CK2 phosphorylates IE2. A, phosphorylation of IE2 detected in cells by mass spectrometry-based proteomics. All vertical black lines denote detected phosphorylation sites in IE2. Green and red vertical lines denote detected phosphorylation sites in IE2 that are predicted MAPK and CK2 sites, respectively. The region around SIM1 is expanded to show that only two serines immediately adjacent to SIM1 are phosphorylated. B, schematic of IE2–SIM1 and IE2-ppSIM1. C, IE2–SIM1 was incubated with CK2 and γ-ATP, run on SDS-polyacrylamide gel, and analyzed using autoradiography. CP is the control peptide that is a known substrate of CK2. In-CK2 in the last lane is inactivated CK2. The higher molecular weight band corresponds to CK2, which auto-phosphorylates itself. This band is not observed in the heat-inactivated lane. D, mass spectra of IE2–SIM1, and IE2–SIM1 incubated with CK2 (IE2-ppSIM1). The spectra of synthesized IE2–ppSIM1 is given below as a reference. The lines with an asterisk are coming from impurities.
Phosphorylation increases the affinity between IE2–SIM1 and SUMO1/2
Phosphorylation of SIMs may regulate the SUMO–SIM interaction (26). NMR titrations were repeated using a synthetic peptide of IE2–SIM1, where the two serines Ser-203 and Ser-205 are phosphorylated (IE2-ppSIM1, Table 1), to examine the effect of Ser-203/205 phosphorylation on its interaction with SUMO1/2. The pattern of CSPs observed in SUMO1 upon titration with IE2–ppSIM1 is similar to that observed during interaction with unphosphorylated IE2–SIM1 (Fig. S5A), indicating that the interface of binding is identical. However, several peaks went into an intermediate exchange during the titration, suggesting that either the affinity between SUMO1 and the IE2–SIM1 has increased upon phosphorylation and/or phosphorylation of IE2–SIM1 gave rise to dynamic exchange processes at the binding interface. Fitting of NMR chemical shifts against ligand/protein concentration yielded the dissociation constant to be 7.1 (±0.4) μm, which is 8-fold lower than unphosphorylated IE2–SIM1 (Fig. S5B and Table 1). When IE2–ppSIM1 was titrated to SUMO2, the interface of interaction was similar to IE2–SIM1 (Fig. S6A). The Kd of interaction with SUMO2 was 7.2 (±1.6) μm, confirming a significantly tighter binding upon phosphorylation (Fig. S6B). In summary, phosphorylation increased the interaction between IE2–SIM1 and SUMO1/2.
It was important to determine the structure of the complex between SUMO and IE2–ppSIM1 to understand that the molecular mechanism of phosphorylation induced a tighter SUMO/IE2–SIM1 interaction. A 13C,15N-filtered (F1) and 13C,15N-edited (F2) NOESY HSQC were acquired on a sample of 13C,15N-SUMO1/IE2–ppSIM1 at the stoichiometric ratio of 1:1.5 (SUMO1/IE2-ppSIM1) (Fig. 3A). This experiment exclusively detects the intermolecular NOEs between SUMO1 and IE2-ppSIM1. 1H-1H TOCSY and 1H-1H NOESY experiments on free IE2–ppSIM1 provided the proton chemical shifts of IE2-ppSIM1. The chemical shifts of SUMO1 in the complex were assigned by comparing the 15N-1H–edited HSQC and the 13C-1H–edited HSQC of 13C,15N-SUMO1/IE2–ppSIM1 complex with the assignments of free SUMO1. Using the intermolecular NOEs as distance restraints, the structure of the SUMO1/IE2–ppSIM1 complex was solved by HADDOCK (Table 2 provides the structural statistics) (27). Fig. S7A shows the 20 lowest energy structures, which superposed well with an r.m.s.d. of 0.5 Å. In the structure, the hydrophobic residues Ile-200 and Ile-202 packed into the hydrophobic patch between the β2-strand and α1-helix (Fig. 3B). Two hydrogen bonds between Asp-204 in IE2 and Lys-46 in SUMO1 also stabilized the interface. The two salt bridges between the side-chain phosphate oxygen atoms of the phosphorylated serines pSer-203/pSer-205 in IE2–ppSIM1 and Lys-39 of the β2-strand in SUMO1 are responsible for the higher affinity between IE2–ppSIM1 and SUMO1 (Fig. 3C).
Figure 3.

Molecular basis of the enhanced interaction between SUMO and IE2-ppSIM1. A, selected strips from the 13C,15N-filtered (F1), 13C,15N-edited (F2), and NOESY HSQC spectra depicting intermolecular NOEs between 13C-bonded protons of 13C,15N-labeled SUMO1 and unlabeled IE2-ppSIM1. 13C and 1H assignment of SUMO1 atoms are given on the right and left of the strips, respectively. The protons of IE2–ppSIM1 that show NOEs to SUMO1 are assigned. B and D highlights the hydrophobic interactions in the SUMO1/IE2–ppSIM1 and SUMO2/IE2–ppSIM1 complexes, respectively. The SUMO1/2 surface is colored white, except the hydrophobic patches are colored green. The IE2–ppSIM1 backbone is shown as an orange ribbon. The side chains of central hydrophobic residues CIVI are shown as yellow spheres. C and E shows the hydrogen bonds between phosphorylated side chains of IE2–SIM1 with SUMO1 and SUMO2, respectively. The hydrogen bonds are shown as black lines. The two phosphoserines and the residues in SUMO1/2 that form hydrogen bonds are shown. Nitrogen atoms are colored in blue; oxygen atoms are colored in red; and phosphorus atoms are colored in yellow.
Table 2.
NMR and refinement statistics of the SUMO/IE2-ppSIM1 complexes
| SUMO1/IE2-ppSIM1 | SUMO2/IE2-ppSIM1 | |
|---|---|---|
| NMR restraints | ||
| Unambiguous restraints (intermolecular NOEs) | 61 | 52 |
| Dihedral restraints (φ, ψ) of IE2-ppSIM1 | 18 | 20 |
| Haddock parameters | ||
| Cluster size | 200 | 200 |
| Haddock score | −60.6 (±11.5) | −79.8 (±7.5) |
| van der Waals energy | −32.9 (±7.3) | −36.6 (±4.2) |
| Electrostatic energy | −285.3 (±36.6) | −323.7 (±25.6) |
| Restraints violation energy | +3.6 (±1.6) | +0.8 (±0.5) |
| Buried surface area | +1365.7 (±50.0) | +1187.1 (±27.9) |
| All backbone | 0.4 | 0.3 |
| All heavy atoms | 0.6 | 0.6 |
| r.m.s.d.a | ||
| Bond angles | 0.7° | 0.6° |
| Bond lengths | 0.004 | 0.004 |
| Molprobity Clashscoreb | 6.4 | 6.5 |
| Ramachandran statisticsa | ||
| Most favored regions (%) | 96.1 | 99.0 |
| Allowed regions (%) | 3.8 | 1.0 |
| Disallowed regions (%) | 0.1 | 0.0 |
a Data were calculated for an ensemble of the 20 lowest-energy structures.
b Data were calculated for the lowest-energy structure.
The structure of the SUMO2/IE2–ppSIM1 complex was studied to determine whether a similar mechanism operates between phosphorylated IE2–SIM1 and SUMO2. We prepared a sample of 13C,15N-labeled SUMO2/IE2–ppSIM1 at the stoichiometric ratio of 1:1.5 (SUMO2/IE2-ppSIM1). 13C,15N-Filtered (F1) and 13C,15N-edited (F2) NOESY HSQC of this sample detected several intermolecular NOEs between SUMO2 and IE2–ppSIM1 (Fig. S7B), which were used to determine the SUMO2/IE2–ppSIM1 structure. The 20 lowest energy structures superimposed with a low r.m.s.d. of 0.4 Å (Fig. S7C). Similar to SUMO1, the IE2–Ile-200 and Ile-202 side chains are buried in the hydrophobic patch between β2 and α1 of SUMO2 (Fig. 3D). The oxygen atoms of phosphate groups of pSer-203 and pSer-205 form salt bridges with Lys-33, Lys-35 in β2 and His-17 in β1 (Fig. 3E). The additional salt bridges between the phosphoserines in IE2–ppSIM1 and SUMO explain the higher affinity of the IE2–SUMO noncovalent interaction upon phosphorylation of IE2.
IE2–SIM1 enhanced SUMOylation of IE2
SIMs often regulate the SUMOylation of a protein in-cis (28). The identified SUMOylation sites in IE2 are Lys-175 and Lys-180, which are adjacent to IE2–SIM1. In vitro SUMOylation assays were carried out to assess the effect of IE2–SIM1 on the SUMOylation of IE2. A FITC was attached to the N-terminal region of IE2 (aa 172–210), which we termed the IE2 N-terminal domain (IE2–NTD, Fig. 4A). IE2–NTD includes both the SUMOylation sites Lys-175 and Lys-180 as well as the IE2–SIM1. Sumoylation reactions were carried out using IE2–NTD as the substrate. Robust and rapid poly-SUMOylation of IE2–NTD with SUMO1 could be observed in the reaction (Fig. 4B). The reaction was repeated with a SIM-mutated IE2–NTD (IE2–NTDm), where the central hydrophobic residues CIVI were mutated to AAAA. IE2–NTDm showed a significantly reduced rate of SUMOylation (Fig. 4C). The efficiency of SUMOylation was estimated by the decay rate of unmodified or apo IE2–NTD signal against time. Faster decay reflected a higher rate of SUMOylation. Apo IE2–NTD signal decayed rapidly compared with IE2–NTDm, which indicated efficient SUMOylation of IE2–NTD due to the presence of IE2–SIM1 (Table 3 and Fig. 4D). When SUMOylation of IE2–NTD was repeated with SUMO2, the rate of SUMOylation was again higher in IE2–NTD than IE2–NTDm (Fig. 4E, Fig. S8, A and B, and Table 3), indicating that the IE2–SIM1 promotes IE2 SUMOylation.
Figure 4.

In vitro SUMOylation of IE2–NTD. A, FITC fluorophore-labeled IE2–NTD used as a substrate in SUMOylation assays. The substrate lysines and SIM1 are shown. B, products of the SUMOylation reaction with SUMO1 and IE2–NTD as the substrate is resolved on the SDS-polyacrylamide gel and imaged with a filter at 519 nm corresponding to FITC fluorescence. Bands of free IE2–NTD or conjugated with one, two, or multiple (n) SUMO1s are marked. The time points are given at the top of the gel. C, same as B except that IE2–NTDm (CIVI to AAAA) was used as the substrate. D and E, fraction of free IE2–NTD is plotted against time for SUMOylation reactions using SUMO1 and SUMO2, respectively. F, experimental design to monitor SUMOylation of IE2–NTD in real time. G, change of fluorescence anisotropy with time for IE2–NTD and IE2–NTDm in a SUMOylation reaction using SUMO1. H, same as in G using SUMO2 instead of SUMO1.
Table 3.
The rate of SUMOylation in IE2-NTD
| Decay of unmodified IE2-NTD signal | ||
| SUMO1 | Fit | |
| IE2-NTD | 8.0 × 10−3/min | Exponential |
| IE2-NTDm | 6.6 × 10−7/min | Exponential |
| SUMO2 | Fit | |
| IE2-NTD | 2.7 × 10−2/min | Exponential |
| IE2-NTDm | 1.4 × 10−2/min | Exponential |
| Fluorescence anisotropy | ||
| SUMO1 | Fit | |
| IE2-NTD | 3.5 × 10−4/min | Linear |
| IE2-NTDm | 0.8 × 10−4/min | Linear |
| SUMO2 | Fit | |
| IE2-NTD | 1.2 × 10−2/min | Exponential |
| IE2-NTDm | 0.7 × 10−2/min | Exponential |
The poly-SUMOylation of IE2–NTD would increase its size and anisotropy. Hence, the SUMOylation of the substrate IE2–NTD can be monitored in real time by measuring the change in fluorescence anisotropy of IE2–NTD (Fig. 4F). As a control, a reaction without ATP was carried out, where the absence of SUMOylation did not change the anisotropy (Fig. S8C). In the SUMOylation reaction using SUMO1 and IE2–NTD/IE2–NTDm as the substrate, the rate of increase in fluorescence anisotropy was higher for IE2–NTD than IE2–NTDm, confirming that the IE2–SIM1 increased SUMOylation of IE2–NTD. When the reaction was repeated with SUMO2, the rate of SUMOylation was higher than SUMO1 (Table 3). This could be because SUMO2 has a consensus SUMOylation motif at Lys-11, which enhances the rate of poly-SUMOylation. Nevertheless, IE2–SIM1 increased the rate of SUMOylation (Fig. 4G and Table 3). Together, the IE2–SIM1 enhanced the SUMOylation of IE2 significantly.
Mechanism of SIM enhanced IE2 SUMOylation
In principle, IE2–SIM1 could increase the SUMOylation of IE2 either by reducing Km (increasing affinity) between the substrate IE2 and the enzyme UBC9∼SUMO conjugate or by increasing the activity (Kcat) of the enzyme UBC9∼SUMO. To determine the exact effect of IE2–SIM1, we carried out a kinetic study of the IE2 SUMOylation. IE2–NTD is poly-SUMOylated rapidly, which made quantification of the SUMOylated species difficult. We used the mutant of SUMO2 (K11R-SUMO2) that is deficient in poly-SUMOylation, which allowed us to effectively monitor the mono-SUMOylated IE2 (Fig. 5A and Fig. S9). Kinetic analysis of IE2 SUMOylation revealed that IE2–SIM1 reduced the Km of IE2 by 2.5-fold (Fig. 5B) but did not alter the Vmax (Kcat) significantly (Fig. 5C). Therefore, IE2 SIM1 increases the specificity constant (Kcat/Km) by decreasing the Km value of the SUMOylation reaction (Table 4). The presence of IE2–SIM1 significantly increased the SUMOylation of IE2 in cellular conditions (Fig. 5, D and E).
Figure 5.
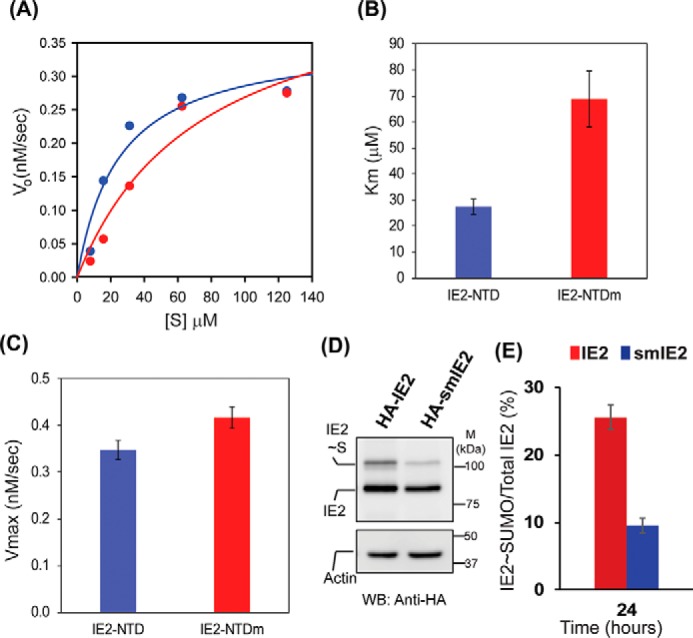
IE2–SIM1 enhances SUMOylation of IE2. A, kinetic data for SUMOylation of IE2–NTD and IE2–NTDm. B and C are the calculated Km and Vmax values, respectively. D, IE2 and SIM mutant IE2 (smIE2) SUMOylation detected in HEK293T cells. E, fraction of IE2∼SUMO over total IE2 is quantified from D and plotted.
Table 4.
Kinetic analysis of IE2 SUMOylation
Temp is temperature.
| Substrate | Vmax | Kcat | Km | Specificity, Kcat/Km | Temp. |
|---|---|---|---|---|---|
| nm/s | 1/s | μm | μm−1 s−1 | °C | |
| IE2-NTD | 0.35 (±0.04) | 8.7 × 10−3 (±5.2 × 10−4) | 27.5 (±3.0) | 3.2 × 10−4 | 25 |
| IE2-NTDm | 0.42 (±0.04) | 10.4 × 10−3 (±5.6 × 10−4) | 68.9 (±10.7) | 1.5 × 10−4 | 25 |
| IE2-NTD | 0.12 (±0.01) | 3.3 × 10−3 (±1.2 × 10−4) | 22.1 (±2.9) | 1.4 × 10−4 | 16 |
| IE2-ppNTD | 0.16 (±0.01) | 3.9 × 10−3 (±0.5 × 10−4) | 6.7 (±0.4) | 6.8 × 10−4 | 16 |
Three mechanisms of IE2–SIM1 enhanced SUMOylation are possible (Fig. 6, A–C). The binding of IE2–SIM1 to the SUMO conjugated at the active site of Ubc9 can increase the rate of SUMOylation (Fig. 6A). Alternatively, SUMO has a noncovalent interaction with UBC9 at its “backside binding” area, and this interaction could also enhance IE2 SUMOylation (Fig. 6B). Moreover, UBC9 is SUMOylated covalently at Lys-14, which could also impact IE2–SUMOylation (Fig. 6C). NMR titration experiments studied the hypothesis in Fig. 6A. 15N labeled WT–UBC9 was titrated with IE2–NTD, and the observed CSPs in UBC9 are plotted in Fig. 6D. The IE2–NTD was truncated from the N-terminal end such that Lys-180 was the sole acceptor lysine present in IE2–NTD. The high CSPs at the α′–α2 and α2–α3 loops indicated the binding site of acceptor Lys-180 and UBC9. The other significant CSPs were observed in the α1–β1 region. The pattern of CSPs was consistent with NMR titrations of UBC9 with SUMO-acceptor sites of p53 and c-Jun (29). Then, C93K–UBC9∼15N-SUMO1 conjugates were purified and titrated with IE2–NTD. The pattern of CSPs on SUMO1 matched with the CSPs observed in isolated SUMO1/IE2–SIM1 complex (Fig. 6E), indicating that the IE2–SIM1 within the IE2–NTD binds to the same interface on the SUMO1 conjugated to UBC9. IE2–NTD does not bind to the UBC9 active site when the active site is mutated to lysine (C93K–UBC9), and hence the binding to the C93K–UBC9∼15N-SUMO1 conjugate could not be studied (Fig. S10A). Nevertheless, combining the NMR data of the IE2–SIM1/SUMO interaction, the IE2–NTD/UBC9∼SUMO1 interaction, and the UBC9∼SUMO1 structure (PDB code 1Z5S), a model of UBC9∼SUMO1/IE2–NTD complex was determined by Xplor-NIH (Fig. 6F and Fig. S11A and Table S1). The structure shows how the IE2–SIM1 binds to SUMO1, whereas the acceptor Lys-180 attacks the active site (Fig. 6I). Interestingly, the essential negatively-charged residue of the ψKX(D/E) motif, which is Glu-182 in this context, forms a salt bridge with the positively charged Lys-101 in the β4α2 loop of UBC9. Lys-101 is essential for the recognition of substrates like p53, PML, and IκB (30). The molecular details provided by the structural model explain the underlying mechanism of substrate recognition by Lys-101 in UBC9.
Figure 6.

Mechanism of cis-SUMO-E3 ligase activity of IE2. The three possible mechanisms are shown in A–C. Cys-93 is the catalytic cysteine in UBC9, which is conjugated to Gly-97 in SUMO1. The critical residue His-20 for noncovalent UBC9–SUMO interaction is shown. The critical residue Lys-14 for covalent UBC9–SUMO interaction is also shown. D, CSPs observed in UBC9 upon titration with IE2–NTD. E, CSPs in SUMO1 within the UBC9∼SUMO1 conjugate, upon titration with IE2–NTD. F, 10 lowest energy model structures of the UBC9∼SUMO1/IE2–NTD complex. UBC9, SUMO1, and IE2–NTD are color-coded as in A. The active site Cys-93 is colored purple; Gly-97 of SUMO1 is colored yellow, and Lys-180 is colored blue. G, same as in F for the SUMO1/UBC9∼SUMO1/IE2–NTD, where SUMO1 is noncovalently bound to UBC9. H, same as in F for the SUMO1–UBC9∼SUMO1/IE2–NTD complex, where SUMO1–UBC9 denotes the SUMO1 covalently linked to Lys-14 of UBC9. I, lowest energy structure of the UBC9∼SUMO1/IE2–NTD complex. J, close-up of the active site shows that Glu-178 forms a salt bridge with Lys-101, and Lys-180 attacks the active site. The inset is shown the complete structure, where the zoomed region is marked with a box. K, gel-shift assay; L, fluorescent anisotropy assay monitored the rate of SUMOylation using either WT, K14R, or H20D UBC9.
A structural model corresponding to Fig. 6B was built using the NMR CSP data, the structure of UBC9∼SUMO1, and the structure of UBC9/SUMO (PDB), where SUMO binds to the “backside” β-sheet of UBC9 (Fig. 6G and Figs. S11B and S12). Again, the Glu-178 forms a salt bridge with Lys-101. The structural model of Fig. 6C was determined using the covalently linked SUMO1–UBC9 structure (PDB code 2VRR, Fig. 6H, and Figs. S11C and S12). In this case, the Glu-178 prefers to form a salt bridge with Lys-74, which is present on the β-strand β4. Lys-74 is vital to identify substrates like RANGAP1 (31). Lys-175 is further away from the IE2–SIM1 than Lys-180 and had more flexibility to access the active site of UBC9 when the IE2–SIM1 binds to either conjugated, covalently linked, or noncovalently linked SUMO. Overall, structural modeling suggested that all three possibilities of SIM-enhanced SUMOylation are possible in IE2–NTD.
SUMOylation assays were performed with appropriate substitutions of UBC9 to delineate the effects of each mechanism. The noncovalent interaction between UBC9 and SUMO involves histidine 20 in UBC9, and the H20D mutation abolishes the interaction (32). Alternatively, The K14R–UBC9 abolishes the covalent SUMO conjugation. The IE2–NTD SUMOylation was studied by initiating a SUMOylation reaction using E1, SUMO1, and WT–UBC9 or H20D–UBC9 or K14R–UBC9. Gel-shift assays or real-time fluorescence anisotropy assay monitored the SUMOylation of IE2–NTD. When the covalent- or noncovalent-mediated interactions between UBC9 and SUMO were abolished individually, the rate of IE2–NTD SUMOylation did not change significantly (Fig. 6, H and I). A possible cause of the result is that the mechanisms involving the covalent or noncovalent UBC9–SUMO interaction do not contribute much to IE2 SUMOylation. However, the three mechanisms could be redundant, so that when one is interrupted, the others compensate.
Phosphorylation of IE2–SIM1 further enhances IE2 SUMOylation
The SUMOylation assays were repeated with IE2–NTD and phosphorylated IE2–NTD (IE2–ppNTD) to find out whether the phosphorylation of IE2–SIM1 enhances the SUMOylation IE2. FITC-tagged IE2–NTD was incubated with CK2 for phosphorylation. The CK2 was subsequently inactivated, and the IE2–NTD was checked by mass spectrometry to ensure that the serines Ser-203 and Ser-205 were phosphorylated. The phosphorylated IE2–NTD (IE2–ppNTD) was later used as a substrate in the SUMOylation assay (Fig. 7A). The rate of SUMOylation was higher in IE2–ppNTD than in IE2–NTD (Fig. 7B). At 60 min, 50% of total IE2–ppNTD was SUMOylated, whereas IE2–NTD was only SUMOylated up to 30% (Fig. 7C).
Figure 7.

Phosphorylation enhanced SUMOylation of IE2–NTD. A, schematic of IE2–NTD and the phosphorylated IE2–NTD. B, SUMOylation of IE2–NTD and IE2–ppNTD against time. The SUMOylation reaction using either IE2–NTD or IE2–ppNTD was run for different times, resolved on the SDS-polyacrylamide gel, and imaged with a filter at 519 nm corresponding to FITC fluorescence. The time of reaction is given at the top. M stands for the marker. C, IE2–NTD∼SUMO conjugate was quantified and plotted against time for IE2–NTD and IE2–ppNTD. D, real-time fluorescence anisotropy measurement of IE2–NTD and IE2–ppNTD SUMOylation. E, SUMOylation of IE2 and phosphorylation mutant pmIE2 observed in HEK293T cells. Cell lysates 48 h post-transfection were separated on SDS-PAGE and blotted with anti-HA. F, ratio of conjugated and total IE2 is quantified from E and plotted against the time of transfection. G, Michaelis-Menten curves for IE2–NTD and IE2–ppNTD. H and I are the calculated Km and Vmax, respectively.
The altered rate of SUMOylation upon SIM phosphorylation was also monitored in real time by the fluorescence anisotropy assay using SUMO1 and IE2–NTD/IE2–ppNTD as the substrate (Fig. 7D). The rate of increase in anisotropy was measured to be 1.2 × 10−4/min for WT IE2–NTD, which almost doubled to 2.1 × 10−4/min for IE2–ppNTD, indicating that phosphorylation of IE2–SIM1 enhances the SUMOylation of IE2–NTD.
SUMOylation of IE2 was measured in HEK293T cells to assess whether the effect of phosphorylation on SUMOylation of IE2 is persistent in full-length IE2 in cellular conditions. HA-tagged IE2 or a double phospho-inactive mutant IE2 pm S203A/S205A was co-transfected with SUMO1, lysed 24/48 h post-transfection, and probed with HA antibody (Fig. 7, E and F). The amount of IE2 pm∼SUMO1 was significantly lower than IE2∼SUMO1, indicating that the phosphorylation of serines near SIM1 enhances SUMOylation of IE2. The enzyme kinetics experiments were repeated with IE2–NTD and IE2–ppNTD. The experiments were carried out at 16 °C to slow down the reaction and effectively measure the kinetics (Fig. 7G and Fig. S13). The kinetic analysis given in Table 4 indicates that the phosphorylation reduces the Km value of the reaction by 3.5-fold but does not change the Vmax significantly (Fig. 7, H and I). Together, phosphorylation of two serines near IE2–SIM1 by CK2 increases the SUMOylation of IE2.
Phosphorylation of IE2–SIM1 increases transactivation by IE2
IE2 functions as a transactivator for various viral promotors and auto-repressors for its promotor. IE2 SUMOylation and IE2 SIM1 are essential for its function. Ser-203 and Ser-205 phosphorylation enhanced both the IE2–SIM1/SUMO interaction and IE2–SUMOylation. The role of phosphorylation on the IE2-mediated transactivation was examined by a luciferase assay, where the luciferase gene is expressed under the IE2-responsive promoter (pUL54-Luc). Consequently, the level of luciferase expression was proportional to the IE2 transactivation activity. A phosphorylation-inactive S203A, S205A double mutant (pmIE2) and a SIM mutant (CIVI to AAAA, smIE2) of IE2 were designed. PUL54-Luc was transfected into HEK293T along with HA-IE2 (WT or mutants) and SUMO1 (Fig. S14). Transactivation activity of pmIE2 is reduced by ∼25% compared with WT–IE2, indicating that phosphorylation of Ser-203 and Ser-205 is vital for transactivation (Fig. 8A). Interestingly, the activity of pmIE2 is comparable with smIE2, indicating that SIM phosphorylation is necessary for the SUMO-dependent transactivation activity of IE2.
Figure 8.

Functional implications of phosphorylation-induced enhanced SIM–SUMO interaction and SUMOylation. A, luciferase transactivation assays were performed in HEK293T cells 36 h post-transfection with or without IE2 or mutants of IE2 and SUMO1. The relative luciferase activity is plotted against the IE2 or its mutants. B same is repeated without transfection of SUMO1. C, luciferase auto-repression activity was monitored using IE2 and its mutants. C denotes the control where IE2 was not transfected. D, model of phosphorylation-induced enhanced IE2 transactivation/auto-repression activity. IE2 is colored in light blue, and the DNA-binding domain (DBD) is colored in dark blue. IE2 binds to the TBP, which binds to the promoter. Phosphorylation increases the interaction between IE2–SIM1 and SUMOylated transcription factors (e.g. TAF12) to enhance the transactivation activity. IE2 can directly bind to the cis-regulatory sequences (crs) for auto-repression via the DBD. Phosphorylation can enhance SUMOylation of IE2 to increase its association with the chromatin modifiers like the HDAC/HMT/CoREST complex and increase auto-repression. HDAC, histone deacetylase, HMT, histone methyltransferase.
The effect of phosphorylation was also checked with co-transfection of only IE2 and pUL54-Luc, but not SUMO. Interestingly, the activity of WT–IE2 with endogenous SUMO reduced by 50% compared with overexpressed SUMO (Fig. 8B). IE2 SUMOylation increases upon overexpression of SUMO (Fig. S15). The increase of both SUMOylation and transactivation activity of IE2 upon overexpression of SUMO underscores the importance of IE2 SUMOylation for its transactivation activity. Alternatively, increased SUMOylation of IE2-associated transcription factors may also increase the IE2-mediated transactivation (Fig. 8D). Nevertheless, the activity of pmIE2 dropped by 40% compared with WT–IE2, emphasizing the significance of SIM phosphorylation when the amount of SUMOylated transcription factors or SUMO is not abundant (Fig. 8B). Additionally, the importance of Ser-203/205 phosphorylation on the auto-repression activity of IE2 was studied using similar luciferase assays where Luc was expressed under MIEP. Expression of luciferase decreases on overexpression of IE2 indicating MIEP repression by IE2. Interestingly, MIEP repression is relieved in pmIE2, indicating the importance of SIM phosphorylation for the auto-repression activity (Fig. 8C). IE2 SUMOylation is important for MIEP repression, and SIM phosphorylation enhances IE2 SUMOylation. Our data suggest that SIM phosphorylation augments SUMOylation-dependent activity of IE2 as an auto-repressor.
Discussion
SUMOylation is an essential component of cell-signaling pathways, and it is unsurprising that the intracellular viruses have co-evolved to exploit the host cell SUMOylation system (33, 34). However, very little is known about the molecular mechanism of how viruses co-opt the machinery. For example, it is known that IE2 binds SUMO noncovalently, and this interaction is indispensable for SUMOylation of IE2 (15). However, the molecular details of IE2–SUMO noncovalent interaction and its role in the SUMOylation of IE2 are unknown. In this work, NMR titration studies determined that apart from the known N-terminal SIM, IE2 includes another C-terminal SIM. Unlike some other SIMs that have paralog specificity, IE2–SIMs binds equally well to both SUMO1 and SUMO2. The C-terminal SIM is located in the DNA-binding domain of IE2, and the noncovalent interaction with SUMO may affect its binding to the cis-regulatory elements in MIEP. The N-terminal SIM is located near the SUMOylation sites and is indispensable for the SUMOylation of IE2. The binding of N-terminal SIM to SUMO is 8-fold tighter than the C-terminal SIM.
The structural studies showed IE2–SIM1 binds to the β2α1 groove of SUMO and forms a parallel β-strand with β2. The four central hydrophobic residues in IE2–SIM 199CIVI202 pack against the hydrophobic interface between β2 and α1. SUMOylation monitored by fluorescence spectroscopy revealed that the IE2–SIM1/SUMO interaction enhances the rate of SUMOylation. The effect is more for SUMO1 than SUMO2, probably because the inherent SUMOylation site in SUMO2 makes the reaction more processive. Kinetic studies indicated that IE2–SIM1 increases the affinity between the substrate (IE2) and enzyme complex (UBC9∼SUMO), but it does not change Kcat of the SUMOylation reaction. Typically, E3s decrease the binding constants of E2∼Ubl for substrate (Km) and increase the turnover rate of E2 (Kcat) to increase the specificity constant (Kcat/Km) of the reaction. In SUMOylation, thioester conjugation of SUMO to UBC9 reduces the flexibility of residues Cys-93, Asp-127, and Pro-128, which help the acceptor lysine of the substrate to attack the active site (35). Consequently, UBC9∼SUMO active site is constitutively primed for SUMOylation, and reducing the Km is sufficient to induce catalysis. IE2 can be considered as a SUMO cis-E3, where the IE2–SIM1 binds to SUMO in the UBC9∼SUMO enzyme complex to bring the complex in the vicinity of SUMOylation sites Lys-175/Lys-180 and to increase the rate of SUMOylation.
The thioester-conjugated SUMO, covalently-bound SUMO, and noncovalently-bound SUMO can potentially interact with IE2–SIM1 to enhance the rate of SUMOylation. Generally, the mechanism used by the enzyme/substrate complex depends on the substrate. For example, although the presence of SIM enhanced SUMOylation of SP100, Daxx, PML, and TDG, covalent binding of SUMO to UBC9 only enhanced SUMOylation of SP100 and Daxx but not of PML and TDG (36). In the case of IE2, modeling studies indicated that sterically all three mechanisms are possible. However, disruption of either covalent or noncovalent SUMO interaction had little effect on the rate of SUMOylation. The thioester-conjugated UBC∼SUMO/IE2–NTD complex could be energetically favorable and the only mechanism at play here. Alternatively, all three mechanisms could be redundant for IE2.
In vitro, CK2 phosphorylates IE2 at the serine-rich segment and the region fragment 5B (aa 180–252) (19). However, the exact sites of phosphorylation in fragment 5B are unknown. The putative CK2 phosphorylation sites in fragment 5B are Ser-203 and Ser-205. We report that in cellular conditions, Ser-203 and Ser-205 are indeed phosphorylated in IE2. The same residues are also phosphorylated by CK2 in vitro. The region, including Ser-203 and Ser-205, becomes highly negatively-charged upon phosphorylation, and due to technical difficulties with quantitative MS of charged peptide fragments, we could not confirm whether CK2 is the sole kinase that phosphorylates IE2–SIM1. These phosphorylated serines are immediately next to IE2–SIM1 and influence both the covalent and noncovalent binding between SUMO and IE2. The affinity of IE2–SIM1/SUMO noncovalent interaction increases significantly by 8-fold upon phosphorylation of the adjacent serines. Structures of SUMO/IE2–ppSIM1 complexes show that pSer-203 and pSer-205 form additional salt bridges with positively-charged residues in SUMO to tighten the interaction between SIM and SUMO. The stronger noncovalent interaction also impacts the covalent interaction between SUMO and IE2. In vitro SUMOylation of IE2 increases significantly upon phosphorylation of Ser-203 and Ser-205. Kinetic analysis revealed that the phosphorylated SIM reduces the Km value between UBC9∼SUMO and IE2 to increase the specificity of the reaction by 3-fold. Moreover, in cellular conditions, SUMOylation of IE2 was severely affected in the phospho-deficient variant. Hence, phosphorylation by CK2 increases both noncovalent and covalent interaction between IE2 and SUMO.
The covalent interaction between IE2 and SUMO is vital for IE2's auto-repression activity (16). SUMOylated IE2 can recruit chromatin modifiers, e.g. HDAC and HMTs, to repress its promoter. Phosphorylation by CK2 enhances IE2 SUMOylation and, consequently, facilitates auto-repression. The noncovalent interaction with SUMO is essential to recruit SUMOylated transcription factors (e.g. TAF12) during transactivation (18). The functional significance of CK2-mediated phosphorylation of IE2–SIM1 is highlighted by the fact that the transactivation activity of the phospho-deficient mutant pmIE2 decreased by 25% compared with WT–IE2. Although phosphorylation of the serine-rich region depletes the transactivation activity of IE2, phosphorylation of IE2–SIM1 in fragment 5B enhances the transactivation activity, indicating that PTMs regulate the complex functional interplay of IE2 domains.
Intriguingly, the activity of phospho-deficient pmIE2 is comparable with SIM-mutated smIE2, suggesting that the effect of phosphorylation is equivalent to the presence of SIM in cellular conditions. Because the IE2–SIM1/SUMO interaction is weak, the tighter binding upon phosphorylation may be critical to “switch on” the interaction with SUMOylated transcription factors and enable efficient transactivation. Global SUMOylation, including the SUMOylation of transcription factors, increase during HCMV infection (37). The surge in SUMOylated transcription factors and the phosphorylation of viral transactivator IE2 by tegument CK2 may work in tandem to regulate the transcription of the viral genome.
A comparison of Km values obtained from the kinetic analysis indicated that the presence of SIM reduced Km by 2.5-fold, and phosphorylation reduced the Km value by another 3.5-fold. Together, the presence of SIM and its phosphorylation decreased the Km value between the substrate and the enzyme by 8-fold, which drastically enhances the rate of the SUMOylation. Herein, we have uncovered that the HCMV transactivator protein IE2 hijacks a cross-talk between two host PTMs, SUMOylation and phosphorylation. The intriguing mechanism of phosphorylation-enhanced SUMO interaction improves the transactivation and auto-repression activity of the viral protein. A better understanding of the molecular mechanisms underlying viral hijack of the cross-talk between multiple PTMs might provide novel opportunities for intervention.
Experimental procedures
Plasmid and peptides
HCMV IE2 peptides were synthesized from Lifetein. pET28-SUMO1(C-His), pET28-SUMO2(C-His), pET11-AOS1/UBA2, and pET28-ΔN364 SENP2 were gifted by Dr. Christopher Lima, Sloan Kettering Institute, New York. pET28-UBC9 was obtained from Addgene (25213). This construct was used as a template for site-directed mutagenesis to obtain H20D UBC9, K14R UBC9, and C93K UBC9. SUMO1-pQE80L and SUMO2-pET15b were obtained from TIFR, Mumbai, India. Mammalian expression vectors, pDEST-SG5 HA-IE2, pUL54-Luc, and pSG5 Flag-SUMO1, were kind gifts from Dr. Jin-Hyun Ahn, Sungkyunkwan University School of Medicine. The pDEST-SG5 HA-IE2 was used as a template for site-directed mutagenesis to generate CIVI199/200/201/202AAAA HA-IE2 and S203A/S205A HA-IE2.
Protein purification
All the proteins were expressed and purified from BL21 (DE3) cells in 15NH4Cl-M9 medium for labeled proteins and in LB for unlabeled proteins. Labeled proteins were purified in phosphate buffer for NMR, and in Tris buffer for in vitro assays. 15N-Labeled SUMO1/2 were cultured at 37 °C to 0.8 OD600 and induced with 0.5 mm isopropyl 1-thio-β-d-galactopyranoside for 4–5 h. Cells were lysed by sonication in lysis buffer containing 50 mm Na2HPO4, pH 8.0, 20 mm imidazole, and 300 mm NaCl. Lysate was clarified by centrifugation, and supernatant was incubated with pre-equilibrated Ni-NTA beads for 1 h. After incubation, flow-through was collected, and beads were washed with at least 5 column volumes of lysis buffer. Protein was eluted by increasing the concentration of imidazole in lysis buffer. Fractions containing SUMO1/2 were concentrated and were further processed through a gel-filtration column (Superdex 75 16/600) in PBS buffer.
For in vitro assays, E1 (UBA2/AOS1) and E2 (UBC9) were purified with Ni-NTA affinity purification followed by gel filtration as discussed above, but in Tris buffer. Lysis/wash buffer composition was 50 mm Tris, pH 8, 350 mm NaCl, 1 mm phenylmethylsulfonyl fluoride, 1 mm β-mercaptoethanol, 20 mm imidazole. Elution buffer contained 25 mm Tris, pH 8, 150 mm NaCl, 1 mm β-mercaptoethanol, 250 mm imidazole, and gel filtration buffer contained 20 mm Tris, pH 8, 50 mm NaCl, 1 mm β-mercaptoethanol.
For SUMOylation assays, the mature forms of SUMO1 and SUMO2 were obtained by processing CHis-SUMO1 or CHis-SUMO2 with SENP2. After Ni-NTA purification, fractions containing CHis-SUMO1/2 were pooled and incubated with purified His-ΔN 364 SENP2 (1:1000 molar ratio) at room temperature until complete digestion of the C-terminal extension. SENP2 and unprocessed SUMO were removed by passing the reaction mixture through Ni-NTA beads, and flow-through containing mature SUMO was collected, concentrated, and further purified with Superdex 75. ΔN 364 SENP2-pET 28b was expressed in BL21 (DE3) and purified through Ni-NTA affinity purification as mentioned above. The buffer used for SENP2 purification is similar to the buffer used for E1 purification. After Ni-NTA, partially purified SENP2 was concentrated and stored.
In vitro biochemical assays
For SUMOylation of IE2–NTD or IE2–ppNTD or IE2–NTDm, 5 μm peptide and 5 μm SUMO1/2 were incubated with 1 μm E1 and 2.5 μm E2. The reaction was started by adding ATP. SUMOylation buffer contains 20 mm HEPES, pH 7.5, 50 mm NaCl, 5 mm MgCl2, 0.1% Tween 20. The reaction was analyzed either on 12% SDS-PAGE or by a change in anisotropy. Gels were imaged for FITC fluorophore (λex − 495 nm and λem − 519 nm). Although anisotropy was measured using MOS450 fluorimeter (λex − 470 nm and λem − 520–560 nm).
IE2–ppNTD was obtained by phosphorylating IE2–NTD with purified CK2. Human CK2 was obtained from New England Biolabs (P6010S). IE2–NTD was phosphorylated in buffer provided by the manufacturer, which contains 50 mm Tris, pH 7.5, 10 mm MgCl2, 0.1 mm EDTA, 2 mm DTT, and 0.1% Brij35. In a 50-μl reaction, 50 units of CK2 was sufficient to phosphorylate 50 μm IE2 peptide in 2 h at 30 °C. CK2 was heat-inactivated at 65 °C for 15 min after IE2 phosphorylation. IE2–NTD phosphorylation was analyzed using autoradiography or by MALDI-TOF MS. Phosphorylated IE2–NTD (IE2–ppNTD) was used as a substrate for SUMOylation assays.
C93K UBC9-SUMO1 conjugates were made either with 15N-C93K UBC9 or 15N-SUMO1. Conjugation reaction was performed in buffer containing 20 mm CAPS, pH 9.5, 50 mm NaCl, 5 mm MgCl2, 3 mm DTT in the presence of 10 μm E1, 300 μm C93K UBC9, 500 μm SUMO1, and 3 mm ATP. The reaction was performed overnight at 37 °C. Conjugate formation was analyzed on an SDS gel before further purification by Mono Q and size-exclusion chromatography. Conjugation reaction mixture was diluted by 25 mm Tris, pH 8, to reduce salt concentration. The diluted reaction mixture was loaded onto the Mono Q column, and proteins were eluted with increasing concentrations of NaCl (up to 1 m). Fractions containing C93K UBC9-SUMO1 conjugate were pooled and further purified by SD75 in PBS buffer.
Single turnover reactions were performed for Michaelis-Menten kinetics experiments. UBC9∼K11R SUMO2 conjugates were formed in a 50-μl reaction with 1 μm E1, 10 μm E2, and 10 μm K11R SUMO2 in SUMOylation buffer. The reaction was started by adding E1 and was incubated at 37 °C for 10 min. The reaction was stopped by adding 1450 μl of quenching buffer. Quenching buffer contained 20 mm HEPES, pH 7.5, 50 mm NaCl, 5 mm EDTA, 0.1% Tween 20. IE2–NTD SUMOylation was performed by adding an equal volume of quenched reaction to different concentrations of serially diluted IE2–NTD peptides (so that conjugates and peptides are further diluted by half to give the desired concentration). Aliquots were taken at desired time points, and the reaction was stopped by SDS loading dye. The reaction was resolved on 12% SDS-PAGE in nonreducing conditions and was then transferred onto polyvinylidene difluoride membrane and blotted with SUMO2 antibody. All the biochemical assays were performed in triplicate, and the error bars represent the S.E.
NMR experiments
The NMR spectra of SUMO1 and SUMO2 were recorded at 298 K on 800 MHz Bruker Avance III HD spectrometer with a cryoprobe head, processed with NMRpipe (38) and analyzed with Sparky (39). All the NMR experiments with FHA-Chk2 were performed at 293 K. The SUMO1/2 samples were prepared in PBS buffer, with 5 mm DTT at pH 7.4 and 10% D2O. 1H-1H TOCSY and 1H-1H NOESY were acquired and used to assign IE2–ppSIM. For NMR titration experiments, ∼3 mm peptides were titrated into ∼0.3 mm 15N-SUMO1 or 15N-SUMO2. The titration data were fit in 1:1 protein/ligand model using the equation, CSPobs = CSPmax {([P]t + [L]t + Kd) − ([P]t + [L]t + Kd)2 − 4[P]t[L]t]1/2}/2[P]t, where [P]t and [L]t are total concentrations of protein and ligand at any titration point. The SUMO/IE2–ppSIM1 NMR samples were prepared in PBS buffer, with 5 mm DTT at pH 7.4 and 10% D2O. 13C,15N-filtered (F1) and 13C,15N-edited (F2) NOESY HSQC were collected on a 13C,15N-SUMO1/IE2–ppSIM1 (1:1.5) complex sample with a mixing time of 200 ms to measure intermolecular NOEs between SUMO1 and IE2-ppSIM1. A similar experiment was performed to obtain intermolecular NOEs in the SUMO2/IE2–ppSIM1 complex.
Structure determination and modeling
Unambiguous restraints between the SUMO1 and IE2–ppSIM1 were determined from the intermolecular NOEs observed in the 13C,15N-filtered (F1) and 13C,15N-edited (F2) NOESY HSQC. Dihedral angles of IE2–ppSIM1 were determined from 1H-1H TOCSY and NOESY experiments. The dihedral angles were used to determine an extended structure of IE2-ppSIM1. The solution structure was calculated in HADDOCK (27) using the structure of SUMO1 (PDB code 4WJO) and the extended structure of IE2-ppSIM1. Rigid body energy minimization generated 1000 initial complex structures, and the best 200 by total energy were selected for torsion angle dynamics and subsequent Cartesian dynamics in an explicit water solvent. Default scaling for energy terms was applied. The interface of SUMO1 was kept semi-flexible during simulated annealing and the water refinement steps. Following the standard benchmarked protocol, cluster analysis of the 200 water-refined structures yielded a single clear ensemble. The SUMO2/IE2–ppSIM1 complex was similarly docked, except that intermolecular restraints between SUMO2 and IE2–ppSIM1 were used, and the starting structure of SUMO2 was taken from a crystal structure of free SUMO2 (PDB code 1WM3). The structures of IE2–SIM1/SUMO1 and IE2–SIM1/SUMO2 were deposited in the PDB with accession codes 6K5T and 6K5R.
The model of UBC9∼SUMO1/IE2–NTD was calculated in XPLOR-NIH. The intramolecular and intermolecular distance restraints were calculated from the structure of UBC9∼SUMO1 (PDB code 1Z5S). IE2–NTD was kept flexible throughout the structure calculation. The other restraints used were the SUMO1/IE2–ppSIM1 NOE restraints and UBC9/IE2–NTD CSPs measured in this work. During structure calculation, the distance of acceptor Lys-180 and the active site was restrained to 1.8 Å. A hundred structures were calculated, and the 10 lowest energy structures were used for analysis. The model of SUMO1/UBC9∼SUMO1/IE2–NTD, where an additional SUMO1 molecule is noncovalently bound to UBC9, was calculated similarly, except the SUMO1/UBC9 intermolecular restraints were estimated from the SUMO1/UBC9 structure (PDB: 2UYZ) and the SUMO1/IE2–SIM1 restraints were set between the noncovalent bound SUMO1 and IE2–SIM1. The model of SUMO1–UBC9∼SUMO1/IE2–NTD complex, where the SUMO1 is covalently bound to UBC9, was calculated similarly, except that SUMO1–UBC9 restraints were obtained from the SUMO1–UBC9 structure (PDB code 2VRR), and the SUMO1/IE2–SIM1 restraints were set between the covalently-bound SUMO1 and IE2–SIM1. The refinement statistics are given in Table S1.
Cell culture and transfection
HEK293T cells were maintained in Dulbecco's modified Eagle's medium with 10% serum. For any experiment, cells were seeded into 12-well tissue culture plates. Cells were transfected at 60–80% confluency with 500 ng of FLAG-SUMO1 and 500 ng of HA-IE2 (WT or mutant as mentioned) using Lipofectamine 3000 reagent. Cells were harvested 48 h post-transfection and lysed with 2× SDS loading dye. Lysates were run on a 12% SDS gel and were probed with HA antibody (CST-3724) after blotting.
For transactivation assays, HEK293T cells were seeded into a 12-well plate and were cultured to 70–80% confluency. Cells were transfected with 100 ng of pUL54-Luc, 5 ng of pTK-Renilla (transfection control), and 400 ng of WT–IE2 with or without 400 ng of FLAG-SUMO1. In the case of mutants (S203A/S205A-IE2 and C199A/I200A/V201A/I202A-IE2) transfected plasmid amount was increased to acquire similar expression as WT–IE2. Luciferase concentration was measured 36–40 h post-transfection by Dual-Glo–luciferase kit (Promega).
HA-IE2 was immunoprecipitated from HEK293T for PTM analysis by mass spectrometry. One 100-mm dish was transfected with 10 μg of HA-IE2 plasmid with Lipofectamine 3000. IP was performed 36h post-transfection. HA-tagged Sepharose beads (CST) were used for pulldown. The protocol provided by the manufacturer was followed for the IP. After IP, beads were directly loaded onto reducing SDS-PAGE, and immunoprecipitated proteins were resolved. The band, matching the size of the protein of interest, was excised and analyzed by mass spectrometry.
Author contributions
V. T. and K. S. C. data curation; V. T., K. S. C., and R. D. formal analysis; V. T., K. S. C., and R. D. investigation; V. T. methodology; V. T. and R. D. writing-original draft; V. T., K. S. C., and R. D. writing-review and editing; R. D. conceptualization; R. D. funding acquisition; R. D. validation; R. D. project administration.
Supplementary Material
Acknowledgments
The NMR data were acquired at the NCBS-TIFR NMR Facility. We thank Purushotham Reddy for helping with NMR data acquisition.
This work was supported by intramural grants from the National Center for Biological Sciences and the Tata Institute of Fundamental Research. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S15 and Table S1.
The atomic coordinates and structure factors (codes 6K5R and 6K5T) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- IE
- immediate early
- PTM
- post-translational modification
- HCMV
- human cytomegalovirus
- SIM
- SUMO-interacting motif
- MIEP
- major immediate early promoter
- TBP
- TATA-box–binding protein
- DBD
- DNA-binding domain
- CK2
- casein kinase 2
- Ni-NTA
- nickel-nitrilotriacetic acid
- CAPS
- 3-(cyclohexylamino)propanesulfonic acid
- IP
- immunoprecipitation
- PDB
- Protein Data Bank
- aa
- amino acid
- NTD
- N-terminal domain
- IE2–NTDm
- SIM-mutated IE2–NTD
- SUMO
- small ubiquitin-like modifier
- MAPK
- mitogen-activated protein kinase
- CSP
- chemical shift perturbation
- Luc
- luciferase
- r.m.s.d.
- root mean square deviation
- NOESY
- nuclear Overhauser effect spectroscopy
- HSQC
- heteronuclear single quantum coherence
- TOCSY
- total correlation spectroscopy
- HMT
- histone methyltransferase
- PML
- promyelocytic leukemia.
References
- 1. Stenberg R. M., Thomsen D. R., and Stinski M. F. (1984) Structural analysis of the major immediate early gene of human cytomegalovirus. Microbiology 49, 190–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Marchini A., Liu H., and Zhu H. (2001) Human cytomegalovirus with IE-2 (UL122) deleted fails to express early lytic genes. J. Virol. 75, 1870–1878 10.1128/JVI.75.4.1870-1878.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Petrik D. T., Schmitt K. P., and Stinski M. F. (2006) Inhibition of cellular DNA synthesis by the human cytomegalovirus IE86 protein is necessary for efficient virus replication. J. Virol. 80, 3872–3883 10.1128/JVI.80.8.3872-3883.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Colletti K. S., Xu Y., Cei S. A., Tarrant M., and Pari G. S. (2004) Human cytomegalovirus UL84 oligomerization and heterodimerization domains act as transdominant inhibitors of oriLyt-dependent DNA replication: evidence that IE2–L84 and UL84–UL84 interactions are required for lytic DNA replication. J. Virol. 78, 9203–9214 10.1128/JVI.78.17.9203-9214.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hagemeier C., Walker S., Caswell R., Kouzarides T., and Sinclair J. (1992) The human cytomegalovirus 80-kilodalton but not the 72-kilodalton immediate-early protein transactivates heterologous promoters in a TATA box-dependent mechanism and interacts directly with TFIID. J. Virol. 66, 4452–4456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bryant L. A., Mixon P., Davidson M., Bannister A. J., Kouzarides T., and Sinclair J. H. (2000) The human cytomegalovirus 86-kilodalton major immediate-early protein interacts physically and functionally with histone acetyltransferase P/CAF. J. Virol. 74, 7230–7237 10.1128/jvi.74.16.7230-7237.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jupp R., Hoffmann S., Depto A., Stenberg R. M., Ghazal P., and Nelson J. A. (1993) Direct interaction of the human cytomegalovirus IE86 protein with the cis repression signal does not preclude TBP from binding to the TATA box. J. Virol. 67, 5595–5604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lang D., Gebert S., Arlt H., and Stamminger T. (1995) Functional interaction between the human cytomegalovirus 86-kilodalton IE2 protein and the cellular transcription factor CREB. J. Virol. 69, 6030–6037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Reeves M., Murphy J., Greaves R., Fairley J., Brehm A., and Sinclair J. (2006) Autorepression of the human cytomegalovirus major immediate-early promoter/enhancer at late times of infection is mediated by the recruitment of chromatin remodeling enzymes by IE86. J. Virol. 80, 9998–10009 10.1128/JVI.01297-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lee S. B., Lee C. F., Ou D. S., Dulal K., Chang L. H., Ma C. H., Huang C. F., Zhu H., Lin Y. S., and Juan L. J. (2011) Host-viral effects of chromatin assembly factor 1 interaction with HCMV IE2. Cell Res. 21, 1230–1247 10.1038/cr.2011.53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Arend K. C., Lenarcic E. M., and Moorman N. J. (2018) The 5′ untranslated region of the major immediate early mRNA is necessary for efficient human cytomegalovirus replication. J. Virol. 92, e02128–17 10.1128/JVI.02128-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ahn J.-H., Xu Y., Jang W.-J., Matunis M. J., and Hayward G. S. (2001) Evaluation of interactions of human cytomegalovirus immediate-early IE2 regulatory protein with small ubiquitin-like modifiers and their conjugation enzyme Ubc9. J. Virol. 75, 3859–3872 10.1128/JVI.75.8.3859-3872.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gareau J. R., and Lima C. D. (2010) The SUMO pathway: emerging mechanisms that shape specificity, conjugation and recognition. Nat. Rev. Mol. Cell Biol. 11, 861–871 10.1038/nrm3011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hofmann H., Flöss S., and Stamminger T. (2000) Covalent modification of the transactivator protein IE2-p86 of human cytomegalovirus by conjugation to the ubiquitin-homologous proteins SUMO-1 and hSMT3b. J. Virol. 74, 2510–2524 10.1128/JVI.74.6.2510-2524.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Berndt A., Hofmann-Winkler H., Tavalai N., Hahn G., and Stamminger T. (2009) Importance of covalent and noncovalent SUMO interactions with the major human cytomegalovirus transactivator IE2p86 for viral infection. J. Virol. 83, 12881–12894 10.1128/JVI.01525-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Reuter N., Reichel A., Stilp A. C., Scherer M., and Stamminger T. (2018) SUMOylation of IE2p86 is required for efficient autorepression of the human cytomegalovirus major immediate-early promoter. J. Gen. Virol. 99, 369–378 10.1099/jgv.0.001021 [DOI] [PubMed] [Google Scholar]
- 17. Song J., Durrin L. K., Wilkinson T. A., Krontiris T. G., and Chen Y. (2004) Identification of a SUMO-binding motif that recognizes SUMO-modified proteins. Proc. Natl. Acad. Sci. U.S.A. 101, 14373–14378 10.1073/pnas.0403498101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kim E. T., Kim Y.-E., Huh Y. H., and Ahn J.-H. (2010) Role of noncovalent SUMO binding by the human cytomegalovirus IE2 transactivator in lytic growth. J. Virol. 84, 8111–8123 10.1128/JVI.00459-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Barrasa M. I., Harel N. Y., and Alwine J. C. (2005) The phosphorylation status of the serine-rich region of the human cytomegalovirus 86-kilodalton major immediate-early protein IE2/IEP86 affects temporal viral gene expression. J. Virol. 79, 1428–1437 10.1128/JVI.79.3.1428-1437.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Heider J. A., Yu Y., Shenk T., and Alwine J. C. (2002) Characterization of a human cytomegalovirus with phosphorylation site mutations in the immediate-early 2 protein. J. Virol. 76, 928–932 10.1128/jvi.76.2.928-932.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Harel N. Y., and Alwine J. C. (1998) Phosphorylation of the human cytomegalovirus 86-kilodalton immediate-early protein IE2. J. Virol. 72, 5481–5492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Varnum S. M., Streblow D. N., Monroe M. E., Smith P., Auberry K. J., Pas L., Wang D., Ii D. G. C., Rodland K., Wiley S., Britt W., Shenk T., Smith R. D., and Nelson J. A. (2004) Identification of proteins in human cytomegalovirus (HCMV). J. Virol. 78, 10960–10966 10.1128/JVI.78.20.10960-10966.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nogalski M. T., Podduturi J. P., DeMeritt I. B., Milford L. E., and Yurochko A. D. (2007) The human cytomegalovirus virion possesses an activated casein kinase II that allows for the rapid phosphorylation of the inhibitor of NF-κB, IκBα. J. Virol. 81, 5305–5314 10.1128/JVI.02382-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Beauclair G., Bridier-Nahmias A., Zagury J. F., Saïb A., and Zamborlini A. (2015) JASSA: a comprehensive tool for prediction of SUMOylation sites and SIMs. Bioinformatics 31, 3483–3491 10.1093/bioinformatics/btv403 [DOI] [PubMed] [Google Scholar]
- 25. Vaynberg J., and Qin J. (2006) Weak protein-protein interactions as probed by NMR spectroscopy. Trends Biotechnol. 24, 22–27 10.1016/j.tibtech.2005.09.006 [DOI] [PubMed] [Google Scholar]
- 26. Stehmeier P., and Muller S. (2009) Phospho-regulated SUMO interaction modules connect the SUMO system to CK2 signaling. Mol. Cell 33, 400–409 10.1016/j.molcel.2009.01.013 [DOI] [PubMed] [Google Scholar]
- 27. Dominguez C., Boelens R., Bonvin A. M. (2003) HADDOCK: a protein–protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 125, 1731–1737 10.1021/ja026939x [DOI] [PubMed] [Google Scholar]
- 28. Kolesar P., Sarangi P., Altmannova V., Zhao X., and Krejci L. (2012) Dual roles of the SUMO-interacting motif in the regulation of Srs2 sumoylation. Nucleic Acids Res. 40, 7831–7843 10.1093/nar/gks484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lin D., Tatham M. H., Yu B., Kim S., Hay R. T., and Chen Y. (2002) Identification of a substrate recognition site on Ubc9. J. Biol. Chem. 277, 21740–21748 10.1074/jbc.M108418200 [DOI] [PubMed] [Google Scholar]
- 30. Tatham M. H., Chen Y., and Hay R. T. (2003) Role of two residues proximal to the active site of Ubc9 in substrate recognition by the Ubc9·SUMO-1 thiolester complex. Biochemistry 42, 3168–3179 10.1021/bi026861x [DOI] [PubMed] [Google Scholar]
- 31. Bernier-Villamor V., Sampson D. A., Matunis M. J., and Lima C. D. (2002) Structural basis for E2-mediated SUMO conjugation revealed by a complex between ubiquitin-conjugating enzyme Ubc9 and RanGAP1. Cell 108, 345–356 10.1016/S0092-8674(02)00630-X [DOI] [PubMed] [Google Scholar]
- 32. Knipscheer P., van Dijk W. J., Olsen J. V., Mann M., and Sixma T. K. (2007) Noncovalent interaction between Ubc9 and SUMO promotes SUMO chain formation. EMBO J. 26, 2797–2807 10.1038/sj.emboj.7601711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wimmer P., Schreiner S., and Dobner T. (2012) Human pathogens and the host cell SUMOylation system. J. Virol. 86, 642–654 10.1128/JVI.06227-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Everett R. D., Boutell C., and Hale B. G. (2013) Interplay between viruses and host sumoylation pathways. Nat. Rev. Microbiol. 11, 400–411 10.1038/nrmicro3015 [DOI] [PubMed] [Google Scholar]
- 35. Tozluoğlu M, Karaca E., Nussinov R., and Haliloğlu T. (2010) A mechanistic view of the role of E3 in sumoylation. PLoS Comput. Biol. 6, e1000913 10.1371/journal.pcbi.1000913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Knipscheer P., Flotho A., Klug H., Olsen J. V., van Dijk W. J., Fish A., Johnson E. S., Mann M., Sixma T. K., and Pichler A. (2008) Ubc9 sumoylation regulates SUMO target discrimination. Mol. Cell 31, 371–382 10.1016/j.molcel.2008.05.022 [DOI] [PubMed] [Google Scholar]
- 37. Scherer M., Reuter N., Wagenknecht N., Otto V., Sticht H., and Stamminger T. (2013) Small ubiquitin-related modifier (SUMO) pathway-mediated enhancement of human cytomegalovirus replication correlates with a recruitment of SUMO-1/3 proteins to viral replication compartments. J. Gen. Virol. 94, 1373–1384 10.1099/vir.0.051078-0 [DOI] [PubMed] [Google Scholar]
- 38. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., and Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 39. Kneller D. G., and Kuntz I. D. (1993) UCSF Sparky: an NMR display, annotation and assignment tool. J. Cell. Biochem. 53, (S17C), 254 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.