

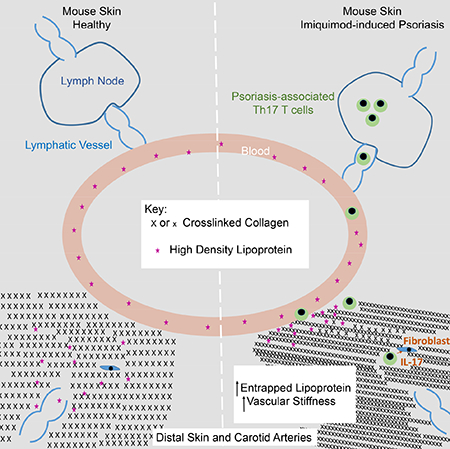
SUMMARY
Lipoproteins trapped in arteries drive atherosclerosis. Extravascular low density lipoprotein (LDL) undergoes receptor uptake, whereas high density lipoprotein (HDL) interacts with cells to acquire cholesterol, and then recirculates to plasma. We developed photoactivatable apoA-I to understand how HDL passage through tissue is regulated. We focused on skin and arteries of healthy mice versus those with psoriasis, which carries cardiovascular risk in man. Our findings suggest that psoriasis-affected skin lesions program interleukin-17-producing T cells in draining lymph nodes to home to distal skin and later to arteries. There, these cells mediate thickening of the collagenous matrix, such that larger molecules including lipoproteins become entrapped. HDL transit was rescued by depleting CD4+ T cells, neutralizing interleukin-17, or inhibiting lysyl oxidase that crosslinks collagen. Experimental psoriasis also increased vascular stiffness and atherosclerosis via this common pathway. Thus, interleukin-17 can reduce lipoprotein trafficking and increase vascular stiffness by, at least in part, remodeling collagen.
Keywords: Autoimmunity, Atherosclerosis, fibrosis, interstitial transport, cytokines, inflammatory disease, artery, skin, Th17 immunity, extracellular matrix, collagen
eTOC Blurb
HDL removes tissue cholesterol and recirculates to the plasma. XXX et al generated a photoactivatable HDL tracking tool, which they applied to psoriasis which has been clinically linked to cardiovascular comorbidity. They find that Th17 autoimmunity promotes collagen deposition in skin and arteries, thus entrapping HDL and LDL in plaques.
Graphical Abstract
INTRODUCTION
Autoimmune diseases like psoriasis, rheumatoid arthritis, and lupus share common features (Theofilopoulos et al., 2017) and possess increased cardiovascular risk that impacts mortality among these patient groups (Armstrong and Krueger, 2016), with risk rising as duration of disease extends (Egeberg et al., 2017). Psoriasis, a disease driven by the cytokines interleukin (IL)-23 and IL-17 (Nestle et al., 2009), is associated with increased incidence of hypertension (Miller et al., 2013a; Takeshita et al., 2015) and atherosclerosis (Harrington et al., 2017). Although links between IL-17 and psoriasis-associated hypertension have not been explored, IL-17 has been identified as a critical mediator in the angiotensin II infusion model of experimental hypertension (Madhur et al., 2010), through adaptive immunity-dependent mechanisms (Guzik et al., 2007) that remain to be fully delineated. It remains uncertain how heightened cardiovascular disease in psoriasis, or any other autoimmune disease, comes about mechanistically and whether treating the noncardiovascular symptoms of autoimmunity (eg., skin lesions in psoriasis) is sufficient to reduce cardiovascular comorbidity.
Atherosclerosis is driven by accumulation of cholesterol in the artery wall that is carried into the subendothelial space on LDL. LDL-cholesterol retention there, followed by uptake in macrophages that in turn leads to the development of atherosclerotic plaque, underlies the disease in general (Gistera and Hansson, 2017). However, statin therapy that lowers plasma LDL has less impact in psoriasis patients compared with the general population (Chodick et al., 2015), suggesting that even though altered lipoproteins are expected to contribute to poor cardiovascular outcomes in psoriasis, effectors in addition to LDL may be key contributors. Another key lipoprotein affecting cardiovascular disease is high density lipoprotein (HDL) that continuously recirculates through all body tissues and accepts cholesterol from cells for export and further clearance through the liver. HDL levels in plasma are especially reduced in patients that go on to develop psoriasis a few years later and throughout the disease (Akkara Veetil et al., 2012). However, mechanistic links between established disease-relevant pathways in psoriasis and lipoprotein biology are not yet defined.
While LDL enters tissues like the artery wall and is thought to drive disease because it is trapped there until taken up by macrophages, LDL is not a lipoprotein particle that typically circulates through multiple organs and tissues, but instead relies on internalization via LDL receptors to carry out its function of delivering cholesterol to cells. By contrast, the fundamental function of HDL as a cholesterol removing particle, rather than a delivery particle, means that it potentially has the capacity to pass from tissue to tissue, removing cholesterol from the tissue in a process called reverse cholesterol transport. During this recirculation, it remains an intact, but constantly remodeling, particle that functions as a vehicle for cholesterol uptake and export. Given this specialized nature of HDL as a molecule that could traffic between cardiovascular tissues and tissues affected by autoimmune disease, we developed an interest in understanding what regulates HDL recirculation and passage through tissues in detail.
It is technically challenging to study HDL transit through the interstitium of tissues and, thus, it remains rather unclear whether HDL trafficking might be altered in disease states. Nonetheless, compelling evidence suggests that oxidization of HDL impairs its clearance from atherosclerotic plaque (Huang et al., 2014) and that loss of adventitial lymphatic vessels promotes atherosclerosis (Vuorio et al., 2014), perhaps through prevention of HDL passage out of tissues (Martel et al., 2013). Several years ago, Rader and Rothblat developed a useful and readily applied technique to track cholesterol egress from tissues by injecting macrophages loaded with [3H]cholesteryl esters into the peritoneal cavity. Cholesterol efflux from the macrophages then allowed appearance and quantification of radioactivity in the plasma, liver, and feces (Zhang et al., 2003). The assay has been pivotal for working out, and verifying in vivo, the roles of a variety of molecules expressed by macrophages in cholesterol efflux. However, the assay is limited largely to work in the peritoneal cavity where the macrophages are usually injected and is most effective for examining the role of molecules expressed by cholesterol effluxing cells (eg., macrophages). Thus, to investigate the trafficking of the HDL particle itself, new tools are essential. Here, we developed a new tool, photoactivatable apoA-I (PGA1), to track the fate of HDL particles in a variety of tissues, such as the skin and arteries presently studied. We go on to apply this tool to the study of cardiovascular comorbidity in psoriasis, where our data reveal a mechanism that leads to the entrapment of HDL and other large molecules, including LDL, in the interstitium of the artery wall as well as skin.
RESULTS
Impaired Passage of HDL through Skin in Experimental Psoriasis
Interstitial fluid and the lymph it subsequently forms is enriched in discoidal HDL (~5–10 nm Stokes radius). The skin is also one of the body’s largest reservoirs of extravascular interstitial fluid (Aukland and Reed, 1993) and contains a large pool of the total body HDL (Randolph and Miller, 2014). We thus wondered how HDL recirculation from the interstitium to lymph and then plasma was affected in skin and whether it was impacted in autoimmune skin disease, like psoriasis. We employed an imiquimod (IMQ)-induced experimental psoriasis model in WT C57BL/6 mice. In this model, daily application of the toll-like receptor 7 agonist imiquimod (IMQ) to ear skin for 14 days was associated with epidermal thickening (Fig. 1A) and local swelling (Fig. 1B). Distal skin sites like the back flank skin did not show histological changes like epidermal thickening or infiltrates (Fig. 1A). There was, nonetheless, altered HDL trafficking in distal back skin (Fig. 1C). Here, a mixture of human albumin and human interstitial HDL, the natural state of endogenous HDL particles, collected from human apoA-I transgenic (Tg-hApoA-I) mice was injected into back skin in a manner so as to minimize interstitial pressure from the injection (Swartz et al., 1999). Then its appearance in plasma, requiring that it first passes through skin tissue and then draining lymphatic vessels, was analyzed using ELISA specific for human albumin or human apoA-I, respectively. The experiment was designed in checkerboard-style, wherein interstitial HDL from IMQ-treated or control mice was injected, separately, into skin of either control or IMQ-treated mice. Albumin traveled with similar efficiency from skin to plasma in either control or IMQ-treated recipient mice (Fig. 1C). However, HDL was delayed in arrivingto plasma when injected into IMQ-treated mice, even though the skin site of injection was remote from the site of IMQ application on the ear where lesions were evident (Fig. 1C). By contrast, interstitial HDL from IMQ-treated mice mobilized normally in naïve recipients (Fig. 1C). These data suggested that systemic changes in the skin micro-environment, including distal nonlesional skin, induced by IMQ negatively affected HDL recirculation.
Figure 1. Characterization of cell and molecular trafficking out of the dermis in experimental psoriasis.

(A) H&E staining of ear and back skin with (right) or without (left) IMQ treatment for 14 days; n=5 mice per group. Scale bar is 10μm. (B) Ear thickness on the left (L) or right (R) ear as a function of whether IMQ was applied for 14 days to that ear or not; n=4–5 per group. (C) Control or IMQ-treated (14 days) recipient mice were intradermally-injected with interstitial HDL + human albumin from control or IMQ-treated (14 days) TghApoA-I donor mice and percent transport of each was assessed; n=3 mice per group. (D) FITC skin painting to assess dendritic cell mobilization to lymph nodes in mice that were treated or not with IMQ (14 days) on both ears; n=7–8 mice per group. (E) Representative frames and graphed summary from intravital imaging to quantify lymph flow. n=5 mice per group. (F) Quantification of insoluble collagen and fragments derived from collagen crosslinks from the mouse back flank skin from mice that were treated with or without IMQ on the ears for 14 days; n=5–8 per group. All data are mean ± SEM. (**P< 0.01; ***P< 0.001).
In contrast to HDL, dendritic cell (DC) migration from the back skin location was normal in IMQ-treated mice (Fig. 1D). Furthermore, intravital imaging of beads taken up into skin-draining lymphatic vessels (Fig. 1E, left; Movie 1), revealed that lymph flow overall was similar in the two conditions (Fig. 1E). Thus, experimental psoriasis impaired HDL recirculation from skin under conditions when DC migration to lymph nodes, fluid flow through lymphatics, and albumin transit from skin to plasma were normal. In keeping with the fact that extracellular matrix molecular arrangement and charge affect molecular transit, insoluble collagen / mg of skin was increased (Fig. 1F) in the absence of measurable changes in soluble collagen (Fig. S1A), hyaluronan (Fig. S1A), or sulfated proteoglycans (Fig. S1A), or loss of skin hydration (Fig. S1B). There was also a significant increase in lysyl pyridinoline residues, confirming an increase in crosslinked collagen in distal, nonlesional skin in IMQ-induced experimental psoriasis (Fig. 1F).
Generation of Photoactivatable ApoA-I Vectors and Knock-in Mice to Study HDL Trafficking
We next aimed to consider whether HDL trafficking in artery walls, a location highly relevant to vascular diseases, might be also impaired in experimental psoriasis. To address this technically challenging question, injection of HDL (such as done above) into thin-walled arteries would not be feasible. Moreover, we aimed to study HDL recirculation using techniques that did not perturb the microenvironment, including in skin, even as the most careful injection may. Thus, we constructed AAV vectors and knock-in mice that encoded the expression of human apoA-I bearing a photoactivatable GFP tag at the N-terminus (PGA1), as detailed in Fig. S2. Specifically, we constructed a technique wherein HDL particles, in whatever form they are found in tissues (Fig. S2A), could be tagged by exposure to light within the tissue microenvironment, and then followed from such interstitial spaces back to plasma (Fig. S2A). PGA1 was functionally active, both in its apoA1 domain and PA-GFP domain (Figs. S2–3) and it reconstituted HDL in WT and apoA1−/− mice (Fig. S3).
With this new tool in hand, we quantified HDL transit out of skin and into plasma. WT mice were infected with PGA1 vectors. Then 3 weeks later, a region of the hind flank was shaved to expose approximately 4-cm2 area of skin for subsequent photoactivation with 405 nm light generated from a handheld laser. PGA1 emerged in plasma by 1 h after photoactivation, reaching a plateau by 2 h (Fig. 2A). Simultaneous analysis of interstitial fluid revealed clearance of fluorescent PGA1 from the skin occurred steadily during the first 2 h (Fig. 2A). We also similarly light-activated WT mice that did not receive vector to ensure specificity (Figure 2B). We selected 2 h as the optimal time point at which to quantify transit of HDL from skin to plasma. Because reverse cholesterol transport depends on intact lymphatic vasculature in skin, as skin drains to plasma via intervening lymphatic vessels (Lim et al., 2013; Martel et al., 2013), we assessed PGA1 transport from skin of Chy mice compared with littermate controls (Figure 2C). The appearance of plasma fluorescence occurred by 2 h after skin photoactivation in PGA1-expressing WT mice, but not in PGA1-expressing Chy mice (Figure 2C), verifying that the increase in plasma fluorescence was dependent on an intact lymphatic passageway from skin.
Figure 2. PGA1 is useful to study HDL trafficking from skin to plasma.

(A) The hind flank of mice was shaved and photoactivated. Fluorescence of plasma, with blank plasma background subtracted, is plotted from cohorts of mice that included 405 nm light-treated WT mice with or without AAV8-PGA1 infection. (B) Fluorescence of plasma is shown over a time course after photoactivating back skin; n=11–13 per group. (C) The fluorescence of plasma 2 h post photoactivation; n=3–23 per group. Data are mean ± SEM. (*P< 0.05; **P<0.01).
We then generated a mouse line in which PGA1 followed by a stop codon was inserted in front of the first exon of mouse apoA-I. Direct sequencing of PCR-amplified 5’ and 3’ junction fragments verified the success of the PGA1 sequence insertion (Fig. S4, top left). PGA1 protein was robustly observed in PGA1KI/+ mouse plasma, comparable to Tg-hApoA-I mice (Jax 001927) (Fig. S4A). Interstitial fluid PGA1 values were 20% of those observed in plasma, consistent with the established extent of reduction in apoA-I of human skin lymph compared with plasma (Miller et al., 2013b). N PGA1 expression supported generation of plasma HDL particles (Fig. S4B) bearing human apoA-I protein (inset, Fig. S4B). Photoactivation (Fig. S4C) led to elevated fluorescence in the same fractions, indicating that PGA1 was intact and functional in those fractions.
Experimental Psoriasis Traps Photoactivatable HDL in Skin
Having generated and validated use of the PGA1KI/+ mice or PGA1 vectors, we returned to the condition of experimental psoriasis to determine if PGA1 might be useful to assess impaired HDL trafficking, as observed in HDL adoptive transfer experiments (Fig. 1). At 2 h after photoactivation of the nonlesional flank skin distal to the ear where IMQ was applied, fluorescent HDL failed to appear in plasma (Fig. 3A), with a much greater impairment than observed in the earlier injection approach. Remarkably, recovery of interstitial fluid from the sites of light activation revealed a strong retention of fluorescent PGA1 in the skin interstitial fluid (Fig. 3B), indicating that HDL was unable to efficiently pass through the skin microenvironment. An extended time course confirmed that appearance of fluorescent PGA1 in plasma after photoactivation in skin of IMQ-treated mice was greatly suppressed and delayed (Fig. 3C). Area under the curve corresponding to plasma-associated fluorescent HDL during 8 h following photoactivation reached only 40% of control values in IMQ-treated mice (Fig. 3C). Impaired skin to plasma trafficking of HDL took time to develop after IMQ treatment was initiated. Trafficking of HDL from nonlesional back skin to plasma was far more modestly impacted at one week after IMQ treatment on the ear compared with the profound inhibition observed at 2 weeks (Fig. 3D). Collagen accumulation in skin was not increased at week 1 after IMQ treatment, but was increased at week 2 (Fig. 3E).
Figure 3. IMQ-induced experimental psoriasis in mice inhibits HDL transport from skin and prevents its recirculation to distal tissues.

WT pCAG-PGA1 mice with or without IMQ treatment applied to ears for 14 days were photoactivated on their back flank skin, and the fluorescence of plasma (A), or interstitial fluid (B) from the area where photoactivation occurred, 2 h post photoactivation was measured; n=12–28 mice per group for (A); n=4–6 per group for (B). (C) Fluorescence of plasma from PGA1KI/+ mice with or without IMQ treatment for 14 days were compared 2, 4, and 8 h post photoactivation (n=4–6 mice per group). (D-E) Photoactivation experiments as in panel A, but with analysis of a time course of 1 versus 2 weeks of daily IMQ treatment on both ears (D). Insoluble collagen was assessed at each endpoint (E); N= 9–14 in these two panels, carried out over the course of 4 independent experiments. (F) At the end of experiments shown in panel C, following photoactivation of PGA1KI/+ skin at 4 or 8 h, interstitial fluid from the heart or skeletal muscle was collected and fluorescence intensity above baseline values in WT mice was plotted; n=4–6 mice per group.
To address the concept that HDL passage from skin to plasma might then allow passage from plasma to other target organs, we monitored the appearance of fluorescence in interstitial fluid prepared from the heart and skeletal muscle at the time of euthanasia. Fluorescence was diluted in these targets compared with plasma, as expected, but was nonetheless detected in the heart and skeletal muscle and was increased in these organs in mice not treated with IMQ as compared with those that were treated (Fig. 3F).
Experimental Psoriasis Negatively Affects Vascular Stiffness, Arterial Collagen Accumulation, HDL Transit through the Artery, and Atherosclerosis: Role of IL-17
Collagen increases earlier documented in skin were also observed in the heart tissue, with collagen levels in the heart returning to baseline after IMQ treatment ceased (Fig. 4A). Arterial collagen also increased remarkably, albeit with some delay, as arterial collagen was primarily elevated during a 2-week chase period following the initial 2-week IMQ treatment (Fig. 4A). This increase was primarily in the adventitia (Fig. 4A, micrograph panels). Consistent with these findings, IMQ treatment for two weeks on, then two weeks chase, promoted vascular stiffness, as assessed by a rise in the augmentation pressure after placement of a cardiac catheter (Fig. 4B).
Figure 4. Effect of experimental psoriasis on arterial stiffness, collagen accumulation, HDL trafficking through the artery wall, and atherosclerosis.

(A) Quantification of insoluble collagen of the heart or the artery from mice that were treated with or without IMQ on the ears for 14 days, or 14 days followed by another 14-day chase; n=5–9 hearts per group. Right panels show trichrome staining of glycolmethacrylate-embedded sections of the carotid artery wall. Scale bar, for both images, is 50 μm. (B) Augmentation pressure was dynamically assessed after surgical placement of a catheter in the carotid artery of mice treated or not with IMQ (with or without chase). N=5–10 mice per condition. (C) The right common carotid artery of PGA1KI/+ mice was photoactivated and plasma fluorescence determined 2 h after photoactivation; some PGA1KI/+ mice were treated with IMQ on both ears for 14 days, with or without another 14 days chase prior to photoactivation. Some groups also received anti-IL-17 neutralizing mAb during the experiment. N=8–14 mice per group. (D-E) apoE−/− mice were treated or not (baseline) with IMQ daily for 3 weeks after having received anti-IL17 neutralizing mAb or isotype control mAb (panel D) or BAPN or vehicle control (panel E). Aortic arch disease was assessed by the percent of en face aorta that was oil red O positive. N=5–10 mice per group. For all panels, data are mean ± SEM. (*P< 0.05; **P<0.01, ***P<0.005).
Because the cytokine IL-17, produced by T cells, promotes vascular stiffness and accumulation of vascular collagen in angiotensin-induced hypertension (Guzik et al., 2007; Wu et al., 2014), we wondered if the arteries in IMQ-treated mice might, like skin, display impaired HDL trafficking and, if so, whether such impairment would be governed by IL-17. To that end, we treated mice with neutralizing anti-IL-17 mAb or isotype-matched control antibody during the IMQ treatment on skin to produce experimental psoriasis. Simultaneously, HDL trafficking from the carotid artery was assessed by shining light on surgically exposed carotid arteries in PGA1KI/+ mice. When we photoactivated the carotid artery in PGA1KI/+ mice to assess the influence of experimental psoriasis on HDL passage through the artery wall, we examined the plasma first within minutes after carotid photoactivation to ensure that we were not directly photoactivating HDL in the blood and then hourly thereafter (Fig. S5). We also examined the impact of IMQ treatment on this assessment of HDL trafficking from the artery wall. HDL trafficking was indeed suppressed (Fig. 4C), in the same time course that led to increased collagen deposition and augmentation pressure, the two-week on/two-week chase design (Fig. 4A; B). Neutralization of IL-17 during the experiment remarkably abrogated development of impaired HDL transit from the artery wall (Fig. 4C).
These findings in the artery wall prompted us to explore whether the conditions that promote sluggish HDL trafficking might be associated with increased cardiovascular disease. Thus, we studied atherosclerosis in apoE−/− mice fed a cholesterol-enriched diet for 8 weeks, in which during the last 3 weeks of this treatment, some of the mice were treated on their ears daily with IMQ cream. As previously reported (Xie et al., 2017), this treatment also led to significantly reduced plasma cholesterol, lowering VLDL cholesterol in particular (Fig. S6). However, the effect was less dramatic than the effect we observed with the PCSK9 AAV vector. It is noteworthy that a recent study using IMQ treatment on mouse skin concluded that murine atherosclerosis was not enhanced by experimental psoriasis (Madsen et al., 2018). However, this study provided short (5 day), intermittent treatments with IMQ, which lead to dramatic skin inflammation, but may not allow for the increase in tissue collagen associated with HDL trapping, which only occurs over time (Fig. 4A). We would predict such collagen changes would be critical for affecting cardiovascular disease. Indeed, despite the reduction in VLDL observed in apoE−/− mice (Fig. S6), which would favor disease protection, atherosclerosis was nonetheless enhanced in mice treated 3 weeks with IMQ on the ear (Fig. 4D).
Since HDL trafficking from the artery wall and from skin was restored to normal after loss of functional IL-17, we wondered if disease burden would likewise be reduced. Thus, we neutralized IL-17 during the 3 week period that we also administered IMQ to some of the mice. Such neutralization does not alter plasma cholesterol (Xie et al., 2017). Indeed, for this 3-week period of treatment, neutralizing IL-17 abrogated the extent of enhanced atherosclerosis increased by imiquimod (Fig. 4D), correlating with the impact of anti-IL-17 on preserving HDL trafficking through the artery wall (Fig. 4C).
Finally, considering that these changes in the artery wall correlated with collagen accumulation, we next set out to directly address whether inhibiting the crosslinking of any newly synthesized collagen by blocking lysyl oxidase activity would affect atherosclerosis in the context of experimental atherosclerosis. Indeed, the lysyl oxidase inhibitor and widely recognized anti-fibrotic agent beta-aminopropionitrile (BAPN) (Lampi and Reinhart-King, 2018) effectively blocked the enhanced disease associated with IMQ treatment, but did not impact baseline disease (Fig. 4E).
Experimental Psoriasis Impedes Recirculation of Multiple Larger Macromolecules in a Manner Dependent upon Increased Collagen
To clarify mechanisms associated with impaired HDL trafficking, we turned back to our studies in skin, where mechanistic work is more tractable. Elevations in collagen density were always observed in skin and arteries with impaired HDL recirculation. We wondered if a reduction in interstitial volume caused by increased collagen accounted for the failure of HDL to recirculate efficiently, in which case we would expect to observe (a) loss of interstitial space not occupied by collagen in dermis affected by impaired HDL trafficking, (b) more broadly impaired trafficking of interstitial molecules based at least roughly on Stokes radius and not specific to HDL, (c) and reversal of impaired trafficking under conditions when increased collagen was prevented.
First, we determined if HDL lipid loading (which affects its overall size) differed enough between control and IMQ-treated groups to affect molecular size, we analyzed HDL particles on nondenaturing gels after interstitial HDL was collected from back skin of PGA1KI/+ mice, treated or not with IMQ for 2 weeks, compared with interstitial fluid from Tg-hApoA-I mice. The latter was observed in a range of sizes similar to that of human plasma HDL (Fig. 5A), all greater than 7 nm Stokes radius indicative of lipid loading. PGA1KI/+ HDL was also observed at a size indicative of lipid loading in vivo (Fig. 5A), consistent with in vitro results (Fig. S2H), and this fusion protein ran as a larger mass overall. However, there was no size shift in HDL particle size associated with IMQ treatment (Fig. 5A).
Figure 5. Role of collagen density influencing molecular transport as a function of molecular mass and interstitial volume.

(A) Nondenaturing gel and immunoblot analysis to assess Stokes radius of interstitial PGA1-derived HDL and the effect of imiquimod treatment, compared with human plasma HDL or interstitial HDL from hApoA1 transgenic mice. Standards marked in right lane. (B-C) FIB-SEM images of dermal back skin from mice treated on the ear without (B) or with (C) IMQ for 14 days. (D) Quantification of interfibrillar volume remaining after collagen fibril volume is accounted for in 3-dimensional analysis of FIB-SEM images. N= 2 mice / group, with 500 random cubes analyzed for interfibrillar volume for each of the two samples, with combined data representing 1000 volume samples and plotted as mean ± SEM. (E) Percent transport of IgA or IgM to plasma after injection of fluorescence-labeled IgA or IgM in the back skin of control or imiquimod-treated mice (14 days on both ears). Similar assay as in panel B for human plasma LDL and albumin. N = 5 per group. (F) Hydroxyproline assay on back skin from PGA1KI/+ mice treated 14 days with or without IMQ on both ears, using vehicle or BAPN as an accompanying treatment during this same period. (G) Fluorescence intensity in plasma 2 h after photoactivation of back skin in mice from panel E; N = 7–8 mice / group for panels (E) and (F). All data are mean ± SEM (* P< 0.05, **P < 0.01, *** P< 0.001).
To examine possible loss of interstitial space between collagen fibrils in skin from IMQ-treated mice that retained HDL, we carried out focused ion beam scanning electron microscopy (FIB-SEM), examining the back skin dermis just beneath the papillary ridges in mice treated or not with IMQ on both ears daily for 2 weeks (Figs. 5B, C). There was a striking change in the arrangement and packing density of collagen fibrils in the nonlesional back skin of IMQ-treated mice (Figs. 5B, C; Supplemental Movies 2), with a clear reduction in the space between collagen fibrils (Fig. 5D). There were also dark rims of contrast around collagen fibrils in replicates of the IMQ-treated skin (Fig. 5C) but not in replicates of control skin (Fig. 5B). This contrast difference may relate to collagen fibril association with other as of yet undefined molecules in the interstitium.
The increased collagen density in nonlesional skin raised the general possibility that typically larger macromolecules (> albumin) might generally be less capable of moving through a tightly woven interstitium with a higher density of collagen. Thus, using the injection approach of different molecules in skin (from Fig. 1), we compared the ability of additional molecules to exit the skin and mobilize to plasma in control and IMQ-treated mice. IgA, with a Stokes radius of ~6.5 nm, below that of even the smallest interstitial HDL observed herein, mobilized from skin similarly in the control and IMQ-treated mice (Fig. 5E), reminiscent of albumin. However, pentameric IgM, at ~12.65 Stokes radius, was reduced in recirculation to plasma if injected into the back skin of IMQ-treated mice (Fig. 5E), consistent with the concept that size is a key determinant in affecting interstitial trafficking in experimental psoriasis. Beyond IgM, few naturally occurring extracellular macromolecules are larger in size than lipoproteins, suggesting that the mechanism we have uncovered might particularly impact trafficking and trapping of lipoproteins. Indeed, LDL mobilization to plasma was also markedly suppressed from the back skin of mice with experimental psoriasis (Fig. 5E).
Finally, we prevented the accumulation of additional dermal collagen in experimental psoriasis by treating with BAPN (Lampi and Reinhart-King, 2018) during the period of IMQ application. The use of this inhibitor did not affect baseline collagen levels but indeed prevented the increase in dermal collagen, quantified by hydroxyproline assessments (Fig. 5F). In PGA1KI/+ mice, photoactivation of the back skin showed that the IMQ-induced impairment in HDL mobilization to plasma was completely reversed by BAPN treatment (Fig. 5G), again with no effect of BAPN on baseline transit of HDL (Fig. 5G). Taken together, these data support the concept that experimental psoriasis alters tissue microenvironment distally, such that due to a loss in interstitial space between matrix fibrils, particularly collagen fibrils, molecules in the size range of lipoproteins are unable to mobilize effectively.
HDL Trafficking and Induction of Procollagen in Experimental Psoriasis is Regulated by CD4+ T Cells and IL-17
Current clinical treatments to combat psoriasis include neutralizing the cytokines IL-23 and IL-17. Depletion of CD4+ T cells or neutralization of IL-17 during the course of IMQ treatment prevented impairment of HDL trafficking from the back skin (Fig. 6A). However, neutralization of the p40 subunit shared by IL-23 and IL-12 was unable to restore HDL trafficking during experimental psoriasis (Fig. 6A). Collagen accumulation in the skin was prevented in the treatments that restored HDL transit (Fig. 6B). Thus, here again, collagen accumulation in the skin correlated with impaired HDL mobilization. Since inhibitors of collagen crosslinking, BAPN or IL-17, abrogated the enhancing effect of IMQ on atherosclerosis (Figs. 4E, 4F), we also tested whether depletion of CD4+ T cells affected atherosclerosis. Indeed, the increase in atherosclerotic plaque observedin response to IMQ was reversed by anti-CD4 T cell depletion, although in the short time frame of depletion (3 weeks), baseline disease was not affected by loss of CD4+ T cells (Fig. S6C-D).
Figure 6. IL-17 and CD4+ T cells govern HDL transport and abrogate procollagen production in distal back skin of IMQ-treated mice.

PGA1 transport was analyzed after photoactivation of back skin from mice treated or not with IMQ and other antibodies. Fluorescence of plasma 2 h post photoactivation (A) or insoluble collagen in the back skin (B); n=8 mice per group. (C) Flow cytometry single-cell suspensions, pregated on CD45+CD4+TCRβ+ cells, from WT back skin, treated with or without IMQ on both ears for 0, 3 or 9 days plus antibodies as indicated; n=3 mice per group. (D) Dermis (top) or back skin (bottom) treated with IMQ for 14 days analyzed for various gene expression by quantitative RT-PCR; n=7–10 mice per group. (E) Back skin dermis was solubilized in acid and immunoblotted with anti-collagen I antibody to identify procollagen I (Procol I) or extractable processed collagen I (Col I). n=5–12 samples per group. (F) Collagen assay (left) and HDL trafficking (right) analyzed in plasma 2 h after photoactivation of the back skin from mice treated or not with IMQ in the presence or absence of FTY720; N=8–11. All data, mean ± SEM (* P< 0.05, **P < 0.01, *** P< 0.001). Color codes defined in legend apply to all panels in the figure.
In skin, by day 9 following IMQ treatment, CD4+ RORγτ+ IL-17-producing T cells were evident at low levels in single cell suspensions of back skin (Fig. 6C). Analysis of skin tissue by quantitative PCR for cytokine mRNA revealed the expected strong induction at the site IMQ application of IL-17A, IL-17F, IL-22, TGFβ, IL-1β, and IFNγ (Fig. 6D). By contrast, in the distal back skin where HDL trafficking was impaired at day 14, IL-17A mRNA was the only cytokine mRNA elevated at day 14, while other cytokine mRNAs were not or were scarcely induced at either day 7 or day 14 (Fig. 6D). Neutralization of IL-12/IL-23 p40 led to a marked reduction in several cytokine mRNAs at the lesional site of topical IMQ application on the ear (Fig. 6D), suggesting that the neutralizing antibody was at least partially efficacious to quell local lesion immunity. However, anti-p40 mAb was unable to abrogate appearance of IL-17 mRNA in the nonlesional distal back skin observed at day 14, in contrast to the loss of CD4+ T cells or IL-17 blockade (Fig. 6D). We then wondered if the role of CD4+ T cells and the cytokine IL-17 in regulating the accumulation of insoluble collagen was mechanistically linked to changes in the fragmentation, or turnover, of collagen or new collagen production in the form of procollagen. While insoluble collagen cannot be reliably extracted in acid, collagen fragments and procollagens are readily extracted in acidic conditions and can be analyzed by immunoblot (Voorhees et al., 2015). This approach revealed little to no evidence of collagen fragments, since no bands smaller than soluble processed collagen I (150 kD) were detected. However, bands of higher molecular weight corresponding to procollagen I were increased in the back skin when IMQ was applied to the ear for 14 days (Fig. 6E), indicating an induction of new collagen synthesis, that upon crosslinking by lysyl oxidase likely accounts for the rise in stable, insoluble collagen. This rise in production of procollagen was prevented by loss of CD4+ T cells or neutralization of IL-17, but was not affected by neutralizing IL-12/23 p40 (Fig. 6E). These data suggest that Th cells producing IL-17 regulate collagen synthesis in distal back skin.
These findings, indicating that CD4+ T cells regulate collagen synthesis and HDL trafficking in back skin remote from the site of IMQ application, raised the possibility that T cells programmed in the lymph nodes draining the site of IMQ application might home to distal skin, as previously demonstrated in IMQ-induced experimental psoriasis for γδ T cells (Ramirez-Valle et al., 2015). Thus, we treated some mice receiving IMQ with the S1P1 agonist FTY720 to impede the ability of lymph node T cells to enter the blood and home to distal sites (Cyster and Schwab, 2012). This treatment blocked the collagen increase in distal skin (Fig. 6F, left graph; Fig. S7) and partially restored HDL trafficking from skin to plasma (Fig. 6F, right graph). These data are consistent with a model in which T cells are not needed for local inflammation at the site of the IMQ-induced psoriasis-like lesion, as previously indicated (Naik et al., 2017), but that nonetheless an induction of a T cell response in lymph nodes allows for the programming of widespread T cell homing back to distal skin, where subtle changes are induced that increase collagen levels without evidence of overt inflammation.
Genetic Absence of T Cells Differentially Affects Local and Distal skin Sequelae in IMQ-induced Psoriasis
Next, we studied RAG1−/− mice and TCRβ−/− × TCRδ−/− mice compared with WT control mice with respect to the impact on lesional ear skin swelling (ear thickness) or HDL trafficking from distal skin. Consistent with other literature finding no necessity for conventional T cells in local lesions of experimental psoriasis (Pantelyushin et al., 2012), histological features of psoriasis were augmented in mice lacking T cells for the entire 14 days of IMQ treatment (Fig. 7A, compare to Fig. 1B), and this was borne out by increases in overall ear skin thickness after 14 days of daily IMQ application (Fig. 7B). Depletion of CD4+ cells at day 7 ameliorated disease but this effect was lost by day 14 (Fig. 7B), when the genetic absence of T cells enhances disease. Neutralizing IL-17, by contrast to loss of T cells, was able to suppress lesional changes like ear thickening even in the 14-day IMQ regimen. These findings are consistent with the presence of local alternative sources of IL-17, such as innate lymphocytes (Fig. 7B).
Figure 7. Impact of genetic loss of T cells on local skin inflammation and HDL trafficking from distal skin.

Representative histological analysis (H&E) of ear section from RAG1 KO mouse that was treated with IMQ for 14 days (A). Scale bar, 200 m. (B) Ear thickness was assessed in various strains of mice indicated by color codes; n=4–8. (C) Effect of anti-CD4 mAb compared with genetic absence of T cells on HDL trafficking from skin to plasma, measured using 2-h photoactivation from the back skin of WT mice infected with pCAG-PGA1 and treated with IMQ (or not); n=4–8 mice per group. Data are mean ± SEM (*P< 0.05, **P < 0.01, ***P< 0.001). Color codes defined in legend apply to all panels in the figure.
Despite the lack of T cell dependence on the inflammatory features of the lesion site itself, HDL trafficking from the nonlesional back skin did not succumb to IMQ-induced blockade in the genetic absence of T cells (Fig. 7C), fitting with the data obtained using anti-CD4 depleting mAb that CD4+ T cells drive impaired HDL trafficking from nonlesional skin in experimental psoriasis (from Fig. 6). Thus, the role of T cells in this model of experimental psoriasis is more prominent in nonlesional than in lesional skin.
DISCUSSION
Despite significant clinical importance, the links between cardiovascular disease and autoimmunity are poorly understood. It is likely that underlying connections are multifactorial. Here, using a straightforward model of experimental psoriasis, we provide evidence that IL-17, likely derived from circulating CD4+ T cells, governs molecular trafficking of lipoproteins through its propensity to promote collagen accumulation in tissues, even in distal sites where otherwise disease activity is not pronounced. The role of IL-17 in affecting the accumulation of collagen has the potential to contribute to multiple cardiovascular parameters, affecting not only lipoprotein clearance but also vascular stiffness, where IL-17 had already been identified as culprit in models of hypertension (Wu et al., 2014). However, the idea that there might be a common mechanism in psoriasis contributing not just to hypertension but also atherosclerosis in the context of autoimmunity had not been obvious.
We approached the present studies focused on lipoprotein trafficking by generating a fusion protein of apoA-I with photoactivatable GFP (PGA1). We chose to specifically design tools to track HDL, because of its known role in recirculating through tissue to carry out its function. Using this tool, we were surprised to find that the egress of interstitial HDL was inhibited from “nonlesional skin” in experimental psoriasis in a manner that pointed not to a disease-induced change in HDL particles directly but rather a pivotal change in the environment. Given the clinical evidence that nonlesional skin in psoriasis patients exhibits increased collagen synthesis, fibroblast proliferation, and skin optical properties consistent with elevated collagen density (Koivukangas et al., 1995; Portugal-Cohen et al., 2012; Priestley, 1983; Priestley and Adams, 1983), we quantified the overall density of collagen and other extracellular matrix constituents. Indeed, levels of crosslinked collagen in nonlesional skin was increased, making collagen—the most abundant extracellular matrix molecule in skin--a candidate for the environmental factor that slowed HDL clearance. The versatility of the photoactivatable HDL, wherein trafficking of HDL can be studied in any tissue accessible to focused light, allowed us to identify that HDL passage out of not just skin but also the carotid artery wall was decreased in experimental psoriasis. Impaired trafficking of HDL, wherever we observed it, was always associated with a rise in collagen. In the artery wall, adventitial collagen is not only expected to promote arterial stiffness (Wu et al., 2014) but also well positioned to limit molecular passage of HDL to lymphatic vessels, given that lymphatic vessels are also localized in the adventitia (Martel et al., 2013), allowing trapping of HDL in the artery wall. Focused ion beam electron microscopy in the skin revealed that the increase in collagen truly reduced available interstitial space, and use of the lysyl oxidase inhibitor and anti-fibrotic agent BAPN allowed us to directly link this increase in collagen causally to failed HDL transport through the matrix. While we directly implicate collagen in the mechanism of action, we cannot eliminate that other molecules also participate in the changes observe. Electron microscopic analysis is consistent with an altered chemistry at the borders of collagen fibrils, where interactions with other molecules may contribute to the chemistry or overall character of the interstitial space.
In molecular adoptive transfer experiments, larger molecules, including HDL, IgM, and LDL were all strongly reduced in clearing from skin of IMQ-treated mice, suggesting that the role of dense collagen in inhibiting molecular transit was to provide a physical barrier to flux. That LDL trafficking was also impaired raises the possibility that the trapping of LDL in key sites like the artery wall may contribute to the overall negative cardiovascular outcome in psoriasis patients and our atherosclerosis studies, rather than the sole problem being the trapping of HDL. However, it is important to realize that while our experiments with HDL using the PGA1 tool tracked actual interstitial HDL, our experiments with LDL used adoptive transfer of LDL into skin to illustrate a proof of concept about the potential that molecular size explains our results and may extend to LDL entrapment. However, it may not be the case that endogenous LDL is trapped in the skin of these mice, due to the fact that the larger size of LDL also makes it quite reticent to enter tissues from the blood in the first place (Michel et al., 2015). Indeed, even in healthy tissue, like healthy arteries, HDL passes through the interstitial efficiently, but LDL does not disseminate much beyond the intimal endothelium (Nordestgaard et al., 1990). LDL is more likely to enter sites with increased endothelial permeability, as illustrated by direct trafficking of LDL from the blood to the atherosclerosis-prone injured artery wall that exhibits increased permeability and allows access of LDL to interstititium in the first place (Hjelms and Stender, 1992).
Psoriasis is considered an autoimmune disease, although specific antigens have not been identified. There is strong evidence for induction of a Th17 immune response in man and in experimental models, including the one we used herein. Two surprising considerations emerge from our work with respect to the role of adaptive immunity in psoriasis. First, while it has been known that lesional and nonlesional skin, and possibly other organs, are affected in psoriasis, it has not been thought that dependence upon adaptive immunity might be greater in nonlesional tissue. Second, the lesional and nonlesional tissue sites show differential effects regarding blockade of IL-12 p40 and IL-17, with anti-p40 mAb preferentially reducing induction of pro-inflammatory cytokines at the lesional site but having no influence at the nonlesional site where anti-IL17 and depletion of CD4+ T cells was quite effective. While IL-23 receptor engagement is thought to render Th17-polarized T cells pathogenic (Meyer Zu Horste et al., 2016), it is possible that the Th17 cells lacking IL-23 receptor activation participate in remodeling collagen in tissue spaces. It is noteworthy that IL-17 mRNA is elevated in nonlesional skin of psoriasis patients with minimal to no increase in IL-23 or IL-12 (Chan et al., 2006), providing a close clinical correlation to our findings in mice. Furthermore, anti-IL-23 treatment does not appear to relieve the cardiovascular comorbidity in psoriasis (Hu and Lan, 2017).
The idea that IL-17 is key driver in collagen deposition and fibrosis is supported by literature that includes but goes beyond the evidence that IL-17 is a pivotal mediator of vascular stiffness in hypertension. Bleomycin-induced lung fibrosis in mice is dependent upon a cooperative relationship between IL-17 and TGFβ (Wilson et al., 2010). IL-17 signaling has also been linked to collagen production in systemic sclerosis (scleroderma) (Nakashima et al., 2012), well recognized to be driven by dysregulated collagen and debilitating fibrosis. Also, IL-17A levels in humans correlate with the extent of vascular collagen and TGFβ signaling in T cells mediated atherosclerotic plaque fibrosis by inducing IL-17A that was served as a required mediator for collagen accumulation (Gistera et al., 2013). Indeed, the latter study argued for a beneficial role of IL-17A in atherosclerosis because fibrosis of plaques near the vascular lumen guards against plaque rupture (Gistera et al., 2013). Nonetheless, fibrosis is arterial locations like the adventitia promote vascular stiffness and limits access to adventitial lymphatics. One mechanism of action may be direct induction of collagen synthesis in vascular stromal cells by IL-17 itself (Gistera et al., 2013; Wu et al., 2014).
In conclusion, we here report the advent of a new tool to study the transit of HDL from place to place. By combining use of this new tool with a mouse model of experimental psoriasis, and by carrying out studies at the crossroads of immunity and lipoprotein biology, a model that might explain how autoimmunity may promote cardiovascular co-morbidity emerges. The relevance of this paradigm in humans now deserves attention and the extent to which these mechanisms are relevant in other autoimmune conditions like lupus or rheumatoid arthritis, the latter of which is known to accumulate HDL in interstitial fluid (Oliviero et al., 2009), also needs attention. The tool we have developed to track HDL is amenable to application in a wide variety of models. Furthermore, as it is now clear that HDL concentrations in plasma are insufficient to predict cardiovascular risk, our tool may find application in projects designed to uncover scenarios when total plasma HDL levels mask key underlying dysfunctions in HDL, including its trafficking through tissues.
Limitations of study
Limitations of the study include the possibility that BAPN had off target effects. It is also important to keep in mind that PGA1 may not fully recapitulate endogenous HDL. Furthermore, while we suggest that the heighted atherosclerosis observed in concert with experimental psoriasis is due to changes in lipoprotein trafficking, we cannot eliminate other mechanisms at play in the atherosclerosis experiment. While anti-CD4 T cell depletion actually heightened inflammation in the skin following prolonged experimental psoriasis, and thus might be expected to enhance atherosclerosis rather than inhibit it in response to IMQ, we cannot rule out that a major effect of anti-CD4 mAb in the atherosclerosis model, along with the effects of anti-IL-17 and BAPN, were to block inflammation in the artery wall, rather than affect lipoprotein entrapment.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents may be directed to, and will be fulfilled by, the corresponding author Gwendalyn J. Randolph (gjrandolph@wustl.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
WT, apoA-I knock out, transgenic human apoA-I, TCRβδ double knock out, RAG1 knock out, and CD11c-YFP transgenic mice (all C57BL/6 background) were purchased and obtained from the Jackson Laboratories (Bar Harbor, Maine, USA). Males, 8–10 weeks of age, were used in all experiments involving AAV vector infection due to preferential infection with AAV vectors in males versus females (Davidoff et al., 2003). Chy mice bearing a mutation in Flt4 gene, backcrossed onto the C57BL/6 background (Martel et al., 2013), were bred in-house. Heterogeneous PGA1 knock-in mouse (PGA1KI/+) was generated using the CRISPR/Cas9 approach, with the donor plasmid designed, assembled, and validated at the Genome Engineering and iPSC Center at Washington University School of Medicine (Department of Genetics). Single-cell, pronuclear injections were performed in the Micro-injection Core at Washington University School of Medicine (Department of Pathology). gRNAs and Cas9 mRNA were injected at a concentration of 2.5ng/μl and 5ng/μl, respectively. The donor plasmid containing the described insert flanked by 1000 bp homology arms (2000bp total) was injected at a concentration of 10 ng/μl. The following primers were used to detect the success of PGA1 sequence insertion in 5’ junction: SM760.genomic.F 5’ AAACTGGCCACCGTACTCAG 3’; SM760.Junc.R 5’ CTTGCTCACCATTTGCTGCC 3’ (product size: 1383bp), and in 3’ junction: SM760.Junc.F 5’ CCAGCCATCTGTTGTTTGCC 3’; SM760.genomic.R 5’ GGAATTCGTCCAGGTAGGGC 3’ (product size: 1239bp). All experiments described herein were approved by the animal oversight committee at Washington University.
METHOD DETAILS
Experimental Psoriasis Model
To induce experimental psoriasis, IMQ (Imiquimod Cream 5%, supplied with cream on sterile applicator pads, Perrigo. Co.) was applied daily to mice on the right ear or both ears, applying ~5 mg per ear daily for 7–21 days. To deplete mouse CD4+ T cells, or to neutralize IL17 or IL12/IL23p40 cytokines, 250 μg of anti-mouse CD4 mAb (Bioxcell), anti-mouse IL17mAb (Bioxcell), anti-mouse IL12/IL23p40 (Bioxcell), or rat IgG2a isotype control (Bioxcell) was injected i.p. 2 days before IMQ treatment and injected every 4 days during the treatment period. To inhibit the lysyl amine oxidase enzyme activity, either vehicle (PBS) or 300mg/kg BAPN was injected i.p. every other day during the IMQ treatment period.
Cloning of PGA1 cDNA-containing plasmids and PGA1 protein expression in cultured cells or in mice using AAV
Structural models were rendered using MacPyMol v1.8.2.3 and Swiss-PdbViewer v4.1.0. Information for lipid-free apoA-I was from Pollard et al. (Pollard et al., 2016) for lipidated HDL disc from Bhat et al. (Bhat et al., 2007); and GFP 1EMA from the RCSB PDB protein data bank. Structures were treated to reduce steric clashes. The coding sequence of photoactivated GFP (PAGFP) was cloned from PAGFP-C1 vector (Addgene), and the coding sequence for wild-type human apoA-I (hApoA-I) lacking the signal peptide sequence was cloned from pTYB11 vector (Pollard et al., 2013). Both sequences were inserted into the pTXB1 expression vector (New England Biolabs) with PAGFP fused in the N terminus of hApoA-I linked with an extra linker sequence: ggcggcggcgaagccgccgccaaaggcggcggc. The fusion PAGFP-hApoA-I (PGA1) cDNA construct was expressed using the IMPACT system. Briefly, ER2566 E. coli cells were transformed with PGA1 cDNA and then the expressed protein was purified from cell extracts, as previously described (Li et al., 2001) without the lyophilization step. After elution, purified PGA1 was dialyzed and stored in 10 mM ammonium bicarbonate, pH 7.4 containing 3 mM EDTA and 15 uM sodium azide.
To express PGA1 in mammalian cells and to collect the secreted PGA1 in cultured medium, the signal peptide sequence Atgaaagctgcggtgctgaccttggccgtgctcttcctgacggggagccaggctcggcatttctggcagcaa was fused to the N-terminus of PGA1 cDNA sequence. The whole sequence was then cloned and inserted into pcDNA3.1 vector (Invitrogen). Approximately 24 h after plating 293T cells in DMEM medium containing 10% FBS, essential vitamins, 100 U/ml penicillin, 100μg/ml streptomycin, and 2 mM L-glutamine, cell monolayers reached 80% confluence. Cultured medium was changed to FBS-free medium and cells were transfected using a Lipofectamine 3000 (Invitrogen): cDNA ratio of 1:1 (μl/μg). PGA1 protein expression via intracellular analysis or assessment of PGA1 secretion occurred 2 days post transfection. To express PGA1 in mice, PGA1 cDNA including secretory peptide was inserted in pAAV-MCS vectors that contain Alb or CAG promotor and a β-globin intron flanked by two AAV inverted terminal repeats. The constructs in the pAAV vectors were packaged into AAV8 by large-scale transfection of HEK 293T cells, and viral particles were purified by serial CsCl2 centrifugation (Vector Biolabs). Experiments in which HEK 293T were photoactivated were done using the hand-held laser applied to a suspension of cells in a culture tube. Fluorescence was read out by flow cytometry. In other experiments, supernatants from HEK 293T cells were photoactivated with the hand-held laser in a culture tube, with fluorescence intensity readouts measured using the Cytation 5 fluorescence plate reader. In experiments where HEK 293T cells were injected i.p., 10 million cells were injected.
To express PGA1 protein in vivo, mice were i.v. injected with AAV8 virus (1×1012 genome copies) expressing PGA1 under the control of either CAG or Alb promoter (Vector Biosystems) for 3 weeks prior to experiments or other treatments.
Photoactivation
For in vitro photoactivation on PGA1 protein in buffer, in medium, or in cells, low power laser was applied using a Leica SPE confocal microscope or a hand held lower power laser with wavelength 405±10 nm (SOKY, pointer 5mW (FDA), LP405–5) for indicated lengths of time. For photoactivation of live mice, either previously infected with AAVPGA1 vectors or PGA1KI/+ mice, mice were anesthetized and a handheld high-power laser (SOKY, Violet 405nm 500mW (FDA), PL-405–500B) was applied to the depilated mouse skin in the hind flank with wavelength 405 nm±10 and max output 500mW. The diameter of the light beam from the laser measured 0.7 cm. Alternatively, the common carotid artery was surgically exposed in anesthetized mice. A sterile piece of aluminum foil was slipped beneath the carotid artery to protect surrounding tissue from photoactivation, while leaving only the carotid vessel exposed. The laser was held in one location of skin or carotid artery for 10s and then moved to sequential but non-overlapping tissue sites to cover a total area of 4 cm2 of skin, or 5–6 mm of distance along the common carotid artery. Mice remained anesthetized throughout the procedure and plasma was collected at time points during the procedure or at the end point. A single mouse received no more than 2 blood draws before the endpoint. Fluorescence intensity of plasma or solutions including PBS and medium was measured by Cytation 5 Cell Imaging Multi-Mode Reader (BioTek).
Mass Spectrometry
Protein samples were analyzed on a ThermoFisher LTQ-XL mass spectrometer interfaced to a Sciex Eksigent NanoLC 400. Protein samples at 0.2μg/μL were diluted 20:1 in water then 2μL injected onto a 75μm × 15cm Jupiter C5 5μm column having 300 Å diameter pores at a flow rate of 300 nL/min. A binary gradient used water (A) and acetonitrile (B). Elution started at 95% A / 5% B progressing to 30% A / 70% B at 30 min. From 30 to 32 min, the gradient went to 10% A/ 90% B and was then held at this composition for 2 min. The capillary was maintained at 250°C with a spray voltage of 1.7 kV. Scan range was from m/z 500 to 2000. Full scan time was 0.47s. Software control and analysis used Xcalibur™ 2.2.
Collection of Interstitial Fluid
Interstitial fluid was collected by centrifugation of excised tissues as described and validated previously (Wiig et al., 2003). Briefly, excised tissue was placed over a 15-μm nylon mesh with the subcutaneous side facing the mesh and centrifuged at 400 g for 10 min. The isolated interstitial fluid was immediately frozen at −20°C and stored for later analysis.
Western Blot Analysis
To assess the content of collagen I in the insoluble fraction of the back skin, a piece of depilated skin tissue (~30 mg) was collected, immediately snap frozen with liquid nitrogen and then stored at −80°C for later analysis. Protein extraction and immunoblot analysis for collagen I was performed according to (Meschiari et al., 2018). In brief, 30 mg skin tissue was homogenized in 0.48 ml PBS that contained 1X protease inhibitor (Roche) using Tissue Tearor (Biospecific Products). The resulting homogenate was microcentrifuged (Eppendorf 5810R) at 3,800 rpm for 10 mins. The supernatant was removed and the pellet was homogenized again with Protein Extraction Reagent 4 (Sigma). Approximately 10 μg protein was diluted with 2X LDS buffer (Novex), heated to 95 °C for 5 min, and run on a 4–12% Bis-Tris gel (Thermo Fisher). Purified collagen I protein was included as a positive control (Thermo Fisher). Transfer to PVDF membrane (PerkinElmer Life Sciences) was accomplished after the treatment with Tris-glycine, pH 8.3, buffer using a semi-dry blot (Bio-Rad) apparatus for 60 min at 10 V. Membranes were blocked with 5% nonfat dry milk in 15 mM Tris-glycine, pH 7.4 buffer for 30 min. Primary antibody recognizing collagen I (Cedarlane) was diluted 1:5000 and incubated with the membrane at 4°C overnight.
To assess PGA1 or human apoA-I protein content, all sample aliquots were diluted with 2X LDS buffer (Novex) to which solid DTT was added to achieve a 100 mM final concentration, heated to 95 °C for 5 min, and run on the Novex 4–20% Tris-Glycine gel (Invitrogen). Primary antibodies recognizing human apoA-I (EMD Millipore) or GFP (Abcam) were diluted 1:5000 and incubated with the membrane at 4°C overnight.
Blots, after incubating with primary antibody, were washed and then incubated with HRP-conjugated secondary antibody (Bio-Rad) at a 1:5000 dilution for 1 h at room temperature. Blots were washed again and then incubated with SuperSignal West Pico chemiluminescence substrate (Pierce) and developed using autoradiography film BX.
Interstitial HDL Size Analysis by Non-denaturing Gradient Gel Electrophoresis (NDGGE)
The size of interstitial HDL from PGA1KI/+ mice with or without IMQ treatment mice was determined by a comparison with standards of known Stokes diameter (GE Healthcare, range: 17 nm thyroglobulin, 12.2 nm ferritin, 8.2 nm lactate dehydrogenase, 7.1 nm BSA) following separation using 4–20% NDGGE nondenaturing gells (BIO-RAD) and then detected with Western blot analysis.
Dendritic Cell Trafficking from Skin
FITC was dissolved in 1:1 acetone : dibutylphthlate at 8 mg/ml and applied in 25 μl aliquots to shaved mouse back skin as previously described (Platt et al., 2013). Draining lymph nodes including brachial, axillary, and inguinal lymph nodes were harvested 18 h later and FITC+CD11c+ dendritic cells in the lymph node were quantified (Platt et al., 2013).
Back Skin Interstitial Macromolecule Clearance
To quantify macromolecule clearance from back skin, including human plasma HDL, mouse interstitial HDL (Wiig et al., 2003), human plasma LDL (Invitrogen), human albumin (Sigma), mouse IgA-PE (SouthernBiotech), or mouse IgM-FITC (Sigma), we developed a simple assay to monitor macromolecule transport starting from back skin dermis and performed it on mice with or without IMQ treatment on ears for 14 days. Mixture of either mouse interstitial HDL or human LDL (3mg/mL) with human albumin (5mg/mL) in 1:1 ratio of total 20 μl was injected i.d. into back skin with a careful speed control. Briefly, a 27½ gauge needle was inserted intradermally in the back skin of an anesthetized mouse. The needle was connected to 40-cm thin polyethylene tubing, placed and positioned upright to allow for gravitational force to be applied. The tubing held 20 μl solution. The pressure for injection depended on gravity, to minimize any forceful or uneven pressure during injection. ELISA kit was used to measure plasma human apoA-I (Abcam, ab108803), human ApoB (Abcam, ab108807) or human albumin (Abcam, ab108788). In separate experiments of similar design with respect to injection method, the mixture of IgA-PE (SouthernBiotech) and IgM-FITC (Sigma) was used and the fluorescence intensity was measured in a Cytation 5 Cell Imaging Multi-Mode Reader (BioTek). The percent transport was calculated as the final protein concentration in plasma divided by the initial protein mixture concentration (x 100), or for fluorescent antibodies, the final fluorescent intensity in plasma divided by the initial fluorescence intensity from the mixture (x 100).
Plasma Lipoprotein Analysis
Blood was collected in EDTA-containing tubes and then centrifuged for 6 min at 6000 rpm. The plasma fraction was collected. Total plasma cholesterol was measured using an enzymatic assay kit (Wako Diagnostics Cholesterol E). For total lipoprotein cholesterol distribution, 100 μl of plasma was applied to a Superose 6 10/300GL column (GE Healthcare) and different fractions of lipoproteins were separated into lipoprotein classes by fast protein liquid chromatography (FPLC). Cholesterol content of each fraction was measured by the enzymatic assay kit. Mouse plasma PGA1 concentration was measured using an enzyme-linked human apoA-I immunosorbent assay (Abcam).
Cholesterol Efflux Assay
Cholesterol efflux was assessed using TOPFLUOR cholesterol loading of macrophages as previously described (Martel et al., 2013). In brief, total bone marrow was obtained from mice by flushing femurs and tibiae with RPMI-1640. Cells obtained were cultured in L929-conditioned medium to generate bone marrow–derived macrophages (BMDMs) over a 6-day culture period. To induce foam cell formation, BMDMs were loaded with cholesterol by incubation with 100 μg/ml oxidized TOPFLOUR cholesterol-LDL for 24 h (Li et al., 2006). After 24 h, cells were washed twice with RPMI-1640, and equilibrated in RPMI-1640 containing 0.2% BSA as a control group lacking cholesterol acceptor. In other groups, purified hApoA-I (Sigma), PGA1, human HDL, or 5% FBS were included to compare cellular cholesterol efflux at different time points. Fluorescence intensity was assessed directly on a single-cell level by flow cytometry, with a reduction in fluorescence correlating with cholesterol efflux.
Preparation of Reconstituted HDL
hApoA-I or PGA1 were purified as described above. Reconstituted HDL particles were prepared by adding 1 mol of either purified hApoA-I or PGA1 to 80 mol of POPC (Avanti Polar Lipids, INC, 850457C) as previously described (Reschly et al., 2002). Briefly, POPC dissolved in chloroform was first dried on the side of a glass tube, and all traces of solvent were removed under vacuum. POPC was suspended in a solution containing sodium deoxycholate at a molar ratio of 2 cholate to 1 POPC by incubation at 37 °C in a shaker water bath for 1 h and then vortexed for 1 h at RT. Sodium deoxycholate was removed by exhaustive dialysis at RT against a buffer containing 10 mM Tris-HCl, pH 7.4, 140 mM NaCl, 1.5 μM sodium azide, and 0.25 μM EDTA. The size of hApoA-I or PGA1 rHDL particle was determined by a comparison with standards of known Stokes diameter (GE Healthcare) following separation using 4–20% non-denaturing gels (BIORAD) and then stained with Coomassie Blue G-250 dye.
Skin Extracellular Matrix Analysis
Soluble collagen, insoluble collagen, glycosaminglycans, and hyaluronan content within depilated mouse back skin) were determined by using extracellular matrix assays (Biocolor Ltd, Belfast, UK) according to the manufacture’s protocol. Mouse back skin of each group was collected and their wet weights were recorded prior to the measurement. Values were calculated by comparing the samples to a standard curve. Data is represented as each extracellular matrix per wet weight. Mouse back skin dry weight was measured after lyophilizing the sample overnight.
Skin Collagen Cross-linking Analysis
Hydroxylysyl pyridinoline (ABIN809022) and lysyl pyridinoline (ABIN773391) levels in skin were quantified by an enzyme-linked immunosorbent assay based on the manufacturer’s instruction.
Hydroxyproline Assay
Hydroxyproline proline measurement was performed according to (Stoilov et al., 2018) In brief, depilated skin tissues (~20 mg each) were hydrolyzed with 6 N hydrochloric acid (Thermo Fisher) at 105°C for 48 h, and then were dried 65°C for 90 min in SpeedVac. Samples were dissolved in 400μl H2O and vortexed vigorously. Then, an aliquot of dissolved sample was oxidized with 1.5 mg of chloramine-T (Sigma) for 20 min at room temperature followed by reaction with 20 mg of p-dimethylaminobezaldehyde (Fluka) that was dissolved in propanol containing 20% perchloric acid (Aldrich) for 10 min at 65°C. Samples were aliquot in 96 wells and read using Cytation 5 Cell Imaging Multi-Mode Reader (BioTek) at 550nm. Hydroxyproline content was determined by extrapolation from a hydroxyproline standard curve (Sigma). Hydroxyproline content per tissue weight was calculated by normalizing with tissue weight.
Blood Pressure Measurement
Blood pressure analysis was performed via the Mouse Cardiovascular Phenotyping Core, Washington University School of Medicine. The catheter was placed into the left ventricle through the right carotid artery to measure aortic pressure. The basal temperatures and heart rates were monitored continuously and maintained steadily.
PCSK9 Adeno-associated Virus induced Atherosclerosis Mouse Model
Eight week-old male mice were give 1×1011 vector genome copies of PCSK9 AAV (Vector Biolabs) through retro-orbital injection. Mice were fed a Western diet (Harlan Teklad) 2 days post virus injection for 5 weeks and then continued on this diet for another 3 weeks, with some being treated with IMQ during this 3-week period. Total plasma cholesterol was measured using the enzymatic assay kit (Wako Diagnostics Cholesterol E).
Atherosclerotic Lesion Assessment
Male apoE−/− mice were bred at Washington University and placed on a cholesterol-enriched diet (Harlan Teklad 88137) at 8 weeks of age for 8 weeks, with some being treated on the ear during the last 3 weeks with IMQ only or IMQ combined with rat IgG2a isotype control (Bioxcell), anti-mouse IL17mAb (Bioxcell), anti-mouse CD4mAb (Bioxcell), or BAPN (Sigma). For aortic sinus analysis, hearts were perfused with PBS, extracted, and fixed in 30% sucrose diluted in 4% paraformaldehyde. The tissue was then embedded in OCT and sectioned on a cryostat (Leica). Sections of 10 μm were collected from the formation to termination of the aortic valves. Samples were stained with Oil Red O to determine lipid deposition. For en face analysis, aortae were fixed with 4% paraformaldehyde, adipose tissue was carefully removed, and the arteries were pinned onto wax dishes. Aortae were pretreated with 100% propylene glycol (sigma #P4347) for 15 minutes, then incubated with Oil Red O (sigma) for 3 hours at room temperature. Aortae were washed with 85% propylene glycol, then washed multiple times in PBS. Total plaque area of aortic sinus or aorta was determined from areas stained with Oil Red O using ImageJ analysis software (NIH).
Common Carotid Artery Collagen Assessment
For preparation of mouse common carotid artery sections, mouse common carotid artery was carefully isolated and fixed in 4% paraformaldehyde, dehydrated in alcohol, embedded in glycol methacrylate (JB-4; Polysciences), sectioned with 2μm thickness and stained with Masson’s Trichrome for collagen assessment.
Skin Histology Analysis
For mouse ear and back skin histological analysis, mouse ear and back skin were isolated and fixed with 4% paraformaldehyde. Paraffin embedded ear or back skin sections were prepared and stained with hematoxylin and eosin by Washington University School of Medicine, The Musculoskeletal Research Center. Ear thickness was measured with a light microscope.
Back Skin Digestion
Depilated back skin was scraped off any excess fat and connective tissue, and digested in RMPI containing 100 μg/mL Liberase TM (Roche Applied Science), 33 μg/mL DNAse I (Sigma) and 1% FCS for 2 h with rotation. Digestion enzymes were quenched by the addition of 5 mM EDTA and 1% serum. All the digests were disaggregated into single cells by passage through a 70-μm strainer and removed dead cells using Ficoll-PLUS (GE Healthcare).
Flow Cytometry
All fluorochrome-conjugated monoclonal antibodies were purchased from either BioLegend or BD Biosciences. Cell suspensions were generated as described above and restimulation was carried out with the following antibodies: anti-CD3 (17A2), anti-CD4 (RM4–5), anti-CD8 (53–6.7), anti-CD19 (6D5), anti-TCRβ (H57–597), anti-CD45.2 (104), anti-IL-17A (TC11–18H10.1), and Anti-RORγt (Q31–378). Briefly, cells were stimulated at 37°C for 5 hours in complete medium s upplemented with 1.0 μl/ml GolgiStop (BD Biosciences) with or without PMA and ionomycin (Sigma). After stimulation, cells were stained with surface antibodies, and then fixed in Cytofix/Cytoperm fixation (BD Biosciences) at 4°C f or 45 minutes before intracellular staining. Representative plots from flow cytometry were all gated on live cells by fixable aqua dead cell staining (Invitrogen).
Intravital 2-Photon Microscopy for Lymphatics and Lymph Flow Measurement
Mice were anesthetized by i.p. injection of ketamine (50 mg/kg) and xylazine (10 mg/kg) and maintained with halved doses administered every hour. Mouse legs were carefully shaved with Nair (Church & Dwight Co.) and washed with 1× PBS to remove the extra lotion. The superficial layer of the skin was removed, and the lymphatic collecting vessels and the movement of fluorescently-labeled 0.5um microspheres (Polysciences, Inc.; CAT#17152) in the popliteal region were visualized via s.c. injection of mixture of 10 to 20 μl of DyLight 488–conjugated tomato lectin (Vector Laboratories) and microspheres into the footpad. The blood vessel vasculature was visualized via i.v. injection 10 to 20ul of Dylight 594-conjuated lectin (Vector Laboratories). During the acquisition, mouse status was closely monitored. Images were collected at Washington University using a customized Leica SP8 2-photon microscope equipped with a ×25/0.95 NA water-dipping objective and a Mai Tai HP DeepSee Laser (Spectra-Physics) tuned to 900 nm. Fluorescence emission was separated by 3 high-efficiency dichroic mirrors cutting at 458, 495, and 560 nm (Semrock) and directly directed to 4 supersensitive external detectors. 3D stacks consisting of between 21 and 31 planes (2.5 μm step size) were captured every 30 seconds. 2-Photon excitation produced a second harmonic signal from collagen within the analyzed tissue. CD11c-YFP reporter mouse was used to image dendritic cells around lymphatics. Diameter of lymphatic vessels, and manual cell tracking were done with Imaris (Bitplane). The distance change per second during the movement of every individual bead was converted to the rate of volume change (Cubic microns / s).
Focused Ion Beam Scanning Electron Microscopy for Back Skin Structure Analysis
Depilated skin was fixed with freshly made and prewarmed (40°C) mixture of solution including 2.5% Glutaraldehyde, 2% Paraformaldehyde, 0.05% Ruthenium Red, 0.2% Tannic Acid in 0.15M Cacodylate, at 37°C. After 5 m in incubation, tissue was changed to fresh fixative for another 1 h. Tissue processing and scanning were performed by WUCCI (Washington University School of Medicine, Center for Cellular Imaging). Images were exported for analysis in Imaris and Fiji software. Pixels identified as collagen based on contrast were identified and remaining pixels were given a mask, transferred to a new channel and visualized in 3D. Quantification of interfibrillar space between collagen fibrils was determined as a fraction of the total tissue volume. Movies were compiled using Adobe Premiere Pro software.
QUANTIFICATION AND STATISTICAL ANALYSIS
GraphPad Prism version 7 was used for statistical analysis. Statistical analyses utilized two-tailed t test or one-way ANOVA followed by analysis of variance with Tukey correction when comparing with multiple groups.
DATA AND SOFTWARE AVAILABILITY
ADDITIONAL RESOURCES
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER (RRID) |
|---|---|---|
| Antibodies | ||
| Anti-Mouse CD3 (17A2) | BD Biosciences | AB_2732063 |
| Anti-mouse CD4 (RM4–5) | BioLegend | AB_2629698 |
| Anti-mouse TCRγ/δ (GL3) | BioLegend | AB_1731824 |
| Anti-mouse CD19 (6D5) | BioLegend | AB_313654 |
| Anti-mouse TCRβ (H57–597) | BioLegend | AB_313429 |
| Anti-mouse CD45.2 (104) | BioLegend | AB_313445 |
| Anti-mouse IL-17A (TC11–18H10.1) | BioLegend | AB_961384 |
| Anti-mouse RORγt (Q31–378) | BD Biosciences | AB_2687545 |
| Anti-Mouse CD4 mAb | BioXCell | AB_1107636 |
| Anti-Mouse IL17 mAb | BioXCell | AB_10950102 |
| Anti-mouse IL12/IL23p40 | BioXCell | AB_1107698 |
| Rat lgG2a isotype control | BioXCell | AB_1107769 |
| Anti-human ApoA-l | EMD Millipore | AB_93960 |
| Anti-GFP | Abeam | AB_303395 |
| Mouse IgA-PE | Southern Biotech | AB_2732062 |
| Mouse IgM-FITC | Sigma | AB_2732064 |
| Rabbit anti-Rat Collagen I | Cedarlane | AB_10060024 |
| Goat Anti-Rabbit IgG HRP Conjugate | Bio Rad | AB_11125142 |
| Western Diet | Harlan Teklad | CAT# 88137 |
| Bacterial and Virus Strains | ||
| AAV8-PGA1 (pCMV) | This paper | N/A |
| AAV8-PGA1 (pALB) | This paper | N/A |
| AAV8-D377Y-mPCSK9 | Vector Biolabs | CAT# AAV-268246 |
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Imiquimod Cream 5% | Perrigo. Co. | N/A |
| Human plasma LDL | Invitrogen | CAT# L3486 |
| Human albumin | Sigma | CAT# A9511 |
| TopFluor Cholesterol | Avanti Polar Lipids, INC | CAT# 810255 |
| Human apoA-l | Sigma | CAT# A0722 |
| Fluorescently-labeled 0.5um microspheres | Polysciences, Inc. | CAT# 17152 |
| DyLight 488 Labeled Lycopersicon Esculentum (Tomato) Lectin | Vector Laboratories | CAT# DL-1174 |
| DyLight 594 labeled Lycopersicon Esculentum (Tomato) Lectin | Vector Laboratories | CAT# DL-1177 |
| BAPN | Sigma | CAT# A3134 |
| FTY720 | Sigma | CAT# SML0700 |
| Protease inhibitor cocktail | Roche | CAT# 50–720–4060 |
| Reagent 4 | Sigma | CAT# C0356 |
| Pierce Reversible Protein Stain | Thermo Fisher | CAT# 24580 |
| LIVE/DEAD™ Fixable Aqua Dead Cell Stain Kit | Thermo Fisher | CAT# L34957 |
| Hydrochloric acid 6N | Thermo Fisher | CAT# SA56–1 |
| Pickering Protein Hydrolysate standard | PICKERING LABS MS | CAT# 012506H |
| 1-Propanol | J.T.Baker | CAT# 9086–01 |
| Chloramine T | Sigma | CAT# 402868 |
| 4-(dimethylamino)benzaldeyde | Fluka | CAT# 39070 |
| 70% Perchloric Acid | Aldrich | CAT# 311421 |
| Citric acid | Sigma | CAT# 251275 |
| Glacial Acetic Acid | Thermo Fisher | CAT# A38S-212 |
| Sodium Acetate | Thermo Fisher | CAT# BP333–1 |
| Sodium Hydroxide | Thermo Fisher | CAT# SS254–1 |
| Hydroxyproline stock | Sigma | CAT# 418575 |
| Collagen I Rat Protein, Tail | Thermo Fisher | CAT# A1048301 |
| 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine | Avanti Polar Lipids, INC | CAT# 850457C |
| High Molecular Weight Marker | GE Healthcare | CAT# 17044501 |
| Propylene glycol | Sigma | CAT# P4347 |
| Oil Red O | Sigma | CAT# 001516 |
| Pierce ECL Substrate | Thermo Fisher | CAT# 32106 |
| Critical Commercial Assays | ||
| ELISA: human ApoA-I | Abeam | CAT# ab108803 |
| ELISA: human ApoB | Abeam | CAT# ab108807 |
| ELISA: human Albumin | Abeam | CAT# ab108788 |
| ELISA: hydroxylysyl pyridinoline (HP) | antibodies-online | CAT# ABIN809022 |
| ELISA: lysyl pyridinoline (LP) | antibodies-online | CAT# ABIN773391 |
| Sircol-Insoluble Collagen Assay | Biocolor Ltd | CAT# S2000 |
| Blyscan-Glycosaminoglycan Assay | Biocolor Ltd | CAT# B1000 |
| Purple-Jelley-Hyaluronan Assay | Biocolor Ltd | CAT# H1000 |
| Cholesterol enzymatic assay kit | WAKO Life Science INC | CAT# 439–17501 |
| Sircol-Soluble Collagen Assay | Biocolor Ltd | CAT# S1000 |
| Deposited Data | ||
| https://data.mendeley.com/datasets/jp56vt7dcc/1 | ||
| Experimental Models: Cell Lines | ||
| Human: HEK 293T | ATCC | CVCL_0063 |
| Experimental Models: Organisms/Strains | ||
| Mouse: PGA1KI/+ | This paper | N/A |
| Mouse: B6.129P2-Apoa1tm1Unc/J | The Jackson Laboratory | Stock No: 002055 |
| Mouse: C57BL76-Tg(APOA1)1Rub/J | The Jackson Laboratory | Stock No: 001927 |
| Mouse: B6.129S7-Rag1tm1Mom/J | The Jackson Laboratory | Stock No: 002216 |
| Mouse: B6.129P2-Tcrbtm1Mom Tcrdtm1Mom/J | The Jackson Laboratory | Stock No: 002122 |
| Mouse: B6.Cg-Tg(ltgax-Venus)1Mnz/J | The Jackson Laboratory | Stock No: 008829 |
| Mouse: B6.129P2-Apoetm1Unc/J | The Jackson Laboratory | Stock No: 002052 |
| Mouse: B6.Chy | Martel etal.,2013 | N/A |
| Oligonucleotides | ||
| Primer to detect PGA1 gene insertion in 5’ junction. Forward: SM760.genomic.F 5’ AAACTGGCCACCGTACTCAG 3’ | This paper | N/A |
| Primer to detect PGA1 gene insertion in 5’ junction. Reverse: SM760.June.R 5’ CTTGCTCACCATTTGCTGCC 3’ | This paper | N/A |
| Primer to detect PGA1 gene insertion in 3’ junction. Forward: SM760.Junc.F 5’ CCAGCCATCTGTTGTTTGCC 3’ | This paper | N/A |
| Primer to detect PGA1 gene insertion in 3’ junction. Reverse: SM760.genomic.R 5’ GGAATTCGTCCAGGTAGGGC 3’ | This paper | N/A |
| Recombinant DNA | ||
| Plasmid: pAGFP-C1 | Addgene | CAT# 11910 |
| Plasmid: human ApoA-I | Pollard., 2013 Biochemistry | N/A |
| Plasmid: pTXB1-PGA1 | This paper | N/A |
| Plasmid: pcDNA-PGA1 | This paper | N/A |
| Plasmid: pAAV8-pCMV-PGA1 | This paper | N/A |
| Plasmid: pAAV8-pALB-PGA1 | This paper | N/A |
| Software and Algorithms | ||
| MacPyMol v1.8.2.3 | PyMOL | Open source |
| Swiss-PdbViewer v4.1.0 | http://spdbv.vital-it.ch/ | Open source |
| Other | ||
| None | ||
Supplementary Material
Highlights.
New tool created to quantify HDL movement from tissues like artery wall to blood
Experimental psoriasis leads to collagen deposition that traps lipoproteins
Collagen deposition in the artery wall additionally promotes vascular stiffness
Th17 immunity, but not IL-12 or IL-23, drives the unfavorable changes in collagen
ACKNOWLEDGEMENTS
This work was supported by NIH grant R01 HL118206 and DP1DK109668 to GJR. Salary support for LH was supplied by The Leading Technology Group to GJR, 4 T32 AI 7163–39 (Washington University, Division of Immunology), and a Career Development Award from AHA to LH. Additional support was supplied by NIH grant R01HL112276 and R01HL127649 (MGST), American Heart Association 14GRNT20500029 (MJT), 16SDGG30480008 (BHZ), and R37AI496653 to GJR. We are grateful to Mary Wohltmann and Shashi Bala for assistance in the laboratory and to the Genome Engineering and iPSC Center at and Mike White in the Pathology Microinjection Core at Washington University for their help in generating the PGA1 mouse line, to Washington University Center for Cellular Imaging (WUCCI) for FIB-EM and the Mass Spectrometry Facility for Proteomics at Medical College of Wisconsin for mass spectrometry.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no conflicts of interest.
REFERENCES
- Akkara Veetil BM, Matteson EL, Maradit-Kremers H, McEvoy MT, and Crowson CS (2012). Trends in lipid profiles in patients with psoriasis: a population-based analysis. BMC Dermatol 12, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong EJ, and Krueger JG (2016). Lipoprotein Metabolism and Inflammation in Patients With Psoriasis. Am J Cardiol 118, 603–609. [DOI] [PubMed] [Google Scholar]
- Aukland K, and Reed RK (1993). Interstitial-lymphatic mechanisms in the control of extracellular fluid volume. Physiol Rev 73, 1–78. [DOI] [PubMed] [Google Scholar]
- Bhat S, Sorci-Thomas MG, Tuladhar R, Samuel MP, and Thomas MJ (2007). Conformational adaptation of apolipoprotein A-I to discretely sized phospholipid complexes. Biochemistry 46, 7811–7821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan JR, Blumenschein W, Murphy E, Diveu C, Wiekowski M, Abbondanzo S, Lucian L, Geissler R, Brodie S, Kimball AB, et al. (2006). IL-23 stimulates epidermal hyperplasia via TNF and IL-20R2-dependent mechanisms with implications for psoriasis pathogenesis. J Exp Med 203, 2577–2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chodick G, Weitzman D, Shalev V, Weil C, and Amital H (2015). Adherence to statins and the risk of psoriasis: a population-based cohort study. Br J Dermatol 173, 480–487. [DOI] [PubMed] [Google Scholar]
- Cyster JG, and Schwab SR (2012). Sphingosine-1-phosphate and lymphocyte egress from lymphoid organs. Annu Rev Immunol 30, 69–94. [DOI] [PubMed] [Google Scholar]
- Davidoff AM, Ng CY, Zhou J, Spence Y, and Nathwani AC (2003). Sex significantly influences transduction of murine liver by recombinant adeno-associated viral vectors through an androgen-dependent pathway. Blood 102, 480–488. [DOI] [PubMed] [Google Scholar]
- Egeberg A, Skov L, Joshi AA, Mallbris L, Gislason GH, Wu JJ, Rodante J, Lerman JB, Ahlman MA, Gelfand JM, et al. (2017). The relationship between duration of psoriasis, vascular inflammation, and cardiovascular events. J Am Acad Dermatol 77, 650–656 e653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gistera A, and Hansson GK (2017). The immunology of atherosclerosis. Nat Rev Nephrol 13, 368–380. [DOI] [PubMed] [Google Scholar]
- Gistera A, Robertson AK, Andersson J, Ketelhuth DF, Ovchinnikova O, Nilsson SK, Lundberg AM, Li MO, Flavell RA, and Hansson GK (2013). Transforming growth factor-beta signaling in T cells promotes stabilization of atherosclerotic plaques through an interleukin-17-dependent pathway. Sci Transl Med 5, 196–100. [DOI] [PubMed] [Google Scholar]
- Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, Goronzy J, Weyand C, and Harrison DG (2007). Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med 204, 2449–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington CL, Dey AK, Yunus R, Joshi AA, and Mehta NN (2017). Psoriasis as a human model of disease to study inflammatory atherogenesis. Am J Physiol Heart Circ Physiol 312, H867–H873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hjelms E, and Stender S (1992). Accelerated cholesterol accumulation in homologous arterial transplants in cholesterol-fed rabbits. A surgical model to study transplantation atherosclerosis. Arterioscler Thromb 12, 771–779. [DOI] [PubMed] [Google Scholar]
- Hu SC, and Lan CE (2017). Psoriasis and Cardiovascular Comorbidities: Focusing on Severe Vascular Events, Cardiovascular Risk Factors and Implications for Treatment. Int J Mol Sci 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, DiDonato JA, Levison BS, Schmitt D, Li L, Wu Y, Buffa J, Kim T, Gerstenecker GS, Gu X, et al. (2014). An abundant dysfunctional apolipoprotein A1 in human atheroma. Nat Med 20, 193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koivukangas V, Kallionen M, Karvonen J, Autio-Harmainen H, Risteli J, Risteli L, and Oikarinen A (1995). Increased collagen synthesis in psoriasis in vivo. Arch Dermatol Res 287, 171–175. [DOI] [PubMed] [Google Scholar]
- Lampi MC, and Reinhart-King CA (2018). Targeting extracellular matrix stiffness to attenuate disease: From molecular mechanisms to clinical trials. Sci Transl Med 10. [DOI] [PubMed] [Google Scholar]
- Li HH, Thomas MJ, Pan W, Alexander E, Samuel M, and Sorci-Thomas MG (2001). Preparation and incorporation of probe-labeled apoA-I for fluorescence resonance energy transfer studies of rHDL. J Lipid Res 42, 2084–2091. [PubMed] [Google Scholar]
- Li Z, Mintzer E, and Bittman R (2006). First synthesis of free cholesterol-BODIPY conjugates. J Org Chem 71, 1718–1721. [DOI] [PubMed] [Google Scholar]
- Lim HY, Thiam CH, Yeo KP, Bisoendial R, Hii CS, McGrath KC, Tan KW, Heather A, Alexander JS, and Angeli V (2013). Lymphatic vessels are essential for the removal of cholesterol from peripheral tissues by SR-BI-mediated transport of HDL. Cell Metab 17, 671–684. [DOI] [PubMed] [Google Scholar]
- Madhur MS, Lob HE, McCann LA, Iwakura Y, Blinder Y, Guzik TJ, and Harrison DG (2010). Interleukin 17 promotes angiotensin II-induced hypertension and vascular dysfunction. Hypertension 55, 500–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsen M, Hansen PR, Nielsen LB, Cardoso RM, van Eck M, and Pedersen TX (2018). Imiquimod-Induced Psoriasis-Like Skin Lesions Do Not Accelerate Atherosclerosis in Low-Density Lipoprotein Receptor-Deficient Mice. Am J Pathol. [DOI] [PubMed] [Google Scholar]
- Martel C, Li W, Fulp B, Platt AM, Gautier EL, Westerterp M, Bittman R, Tall AR, Chen SH, Thomas MJ, et al. (2013). Lymphatic vasculature mediates macrophage reverse cholesterol transport in mice. The Journal of clinical investigation 123, 1571–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meschiari CA, Jung M, Iyer RP, Yabluchanskiy A, Toba H, Garrett MR, and Lindsey ML (2018). Macrophage overexpression of matrix metalloproteinase-9 in aged mice improves diastolic physiology and cardiac wound healing after myocardial infarction. Am J Physiol Heart Circ Physiol 314, H224–H235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer Zu Horste G, Wu C, Wang C, Cong L, Pawlak M, Lee Y, Elyaman W, Xiao S, Regev A, and Kuchroo VK (2016). RBPJ Controls Development of Pathogenic Th17 Cells by Regulating IL-23 Receptor Expression. Cell Rep 16, 392–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel CC, Nanjee MN, Olszewski WL, and Miller NE (2015). LDL and HDL transfer rates across peripheral microvascular endothelium agree with those predicted for passive ultrafiltration in humans. J Lipid Res 56, 122–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller IM, Skaaby T, Ellervik C, and Jemec GB (2013a). Quantifying cardiovascular disease risk factors in patients with psoriasis: a meta-analysis. Br J Dermatol 169, 1180–1187. [DOI] [PubMed] [Google Scholar]
- Miller NE, Olszewski WL, Hattori H, Miller IP, Kujiraoka T, Oka T, Iwasaki T, and Nanjee MN (2013b). Lipoprotein remodeling generates lipid-poor apolipoprotein A-I particles in human interstitial fluid. Am J Physiol Endocrinol Metab 304, E321–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naik S, Larsen SB, Gomez NC, Alaverdyan K, Sendoel A, Yuan S, Polak L, Kulukian A, Chai S, and Fuchs E (2017). Inflammatory memory sensitizes skin epithelial stem cells to tissue damage. Nature 550, 475–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakashima T, Jinnin M, Yamane K, Honda N, Kajihara I, Makino T, Masuguchi S, Fukushima S, Okamoto Y, Hasegawa M, et al. (2012). Impaired IL-17 signaling pathway contributes to the increased collagen expression in scleroderma fibroblasts. Journal of immunology 188, 3573–3583. [DOI] [PubMed] [Google Scholar]
- Nestle FO, Di Meglio P, Qin JZ, and Nickoloff BJ (2009). Skin immune sentinels in health and disease. Nat Rev Immunol 9, 679–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordestgaard BG, Hjelms E, Stender S, and Kjeldsen K (1990). Different efflux pathways for high and low density lipoproteins from porcine aortic intima. Arteriosclerosis 10, 477–485. [DOI] [PubMed] [Google Scholar]
- Oliviero F, Sfriso P, Baldo G, Dayer JM, Giunco S, Scanu A, Bernardi D, Ramonda R, Plebani M, and Punzi L (2009). Apolipoprotein A-I and cholesterol in synovial fluid of patients with rheumatoid arthritis, psoriatic arthritis and osteoarthritis. Clin Exp Rheumatol 27, 79–83. [PubMed] [Google Scholar]
- Pantelyushin S, Haak S, Ingold B, Kulig P, Heppner FL, Navarini AA, and Becher B (2012). Rorgammat+ innate lymphocytes and gammadelta T cells initiate psoriasiform plaque formation in mice. The Journal of clinical investigation 122, 2252–2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt AM, Rutkowski JM, Martel C, Kuan EL, Ivanov S, Swartz MA, and Randolph GJ (2013). Normal dendritic cell mobilization to lymph nodes under conditions of severe lymphatic hypoplasia. Journal of immunology 190, 4608–4620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard RD, Fulp B, Samuel MP, Sorci-Thomas MG, and Thomas MJ (2013). The conformation of lipid-free human apolipoprotein A-I in solution. Biochemistry 52, 9470–9481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard RD, Fulp B, Sorci-Thomas MG, and Thomas MJ (2016). High-Density Lipoprotein Biogenesis: Defining the Domains Involved in Human Apolipoprotein A-I Lipidation. Biochemistry 55, 4971–4981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portugal-Cohen M, Horev L, Ruffer C, Schlippe G, Voss W, Ma’or Z, Oron M, Soroka Y, Frusic-Zlotkin M, Milner Y, et al. (2012). Non-invasive skin biomarkers quantification of psoriasis and atopic dermatitis: cytokines, antioxidants and psoriatic skin auto-fluorescence. Biomed Pharmacother 66, 293–299. [DOI] [PubMed] [Google Scholar]
- Priestley GC (1983). Hyperactivity of fibroblasts cultured from psoriatic skin: II. Synthesis of macromolecules. Br J Dermatol 109, 157–164. [DOI] [PubMed] [Google Scholar]
- Priestley GC, and Adams LW (1983). Hyperactivity of fibroblasts cultured from psoriatic skin: I. Faster proliferation and effect of serum withdrawal. Br J Dermatol 109, 149–156. [DOI] [PubMed] [Google Scholar]
- Ramirez-Valle F, Gray EE, and Cyster JG (2015). Inflammation induces dermal Vgamma4+ gammadeltaT17 memory-like cells that travel to distant skin and accelerate secondary IL-17-driven responses. Proceedings of the National Academy of Sciences of the United States of America 112, 8046–8051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randolph GJ, and Miller NE (2014). Lymphatic transport of high-density lipoproteins and chylomicrons. The Journal of clinical investigation 124, 929–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reschly EJ, Sorci-Thomas MG, Davidson WS, Meredith SC, Reardon CA, and Getz GS (2002). Apolipoprotein A-I alpha -helices 7 and 8 modulate high density lipoprotein subclass distribution. J Biol Chem 277, 9645–9654. [DOI] [PubMed] [Google Scholar]
- Stoilov I, Starcher BC, Mecham RP, and Broekelmann TJ (2018). Measurement of elastin, collagen, and total protein levels in tissues. Methods Cell Biol 143, 133–146. [DOI] [PubMed] [Google Scholar]
- Swartz MA, Kaipainen A, Netti PA, Brekken C, Boucher Y, Grodzinsky AJ, and Jain RK (1999). Mechanics of interstitial-lymphatic fluid transport: theoretical foundation and experimental validation. J Biomech 32, 1297–1307. [DOI] [PubMed] [Google Scholar]
- Takeshita J, Wang S, Shin DB, Mehta NN, Kimmel SE, Margolis DJ, Troxel AB, and Gelfand JM (2015). Effect of psoriasis severity on hypertension control: a population-based study in the United Kingdom. JAMA Dermatol 151, 161–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theofilopoulos AN, Kono DH, and Baccala R (2017). The multiple pathways to autoimmunity. Nat Immunol 18, 716–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voorhees AP, DeLeon-Pennell KY, Ma Y, Halade GV, Yabluchanskiy A, Iyer RP, Flynn E, Cates CA, Lindsey ML, and Han HC (2015). Building a better infarct: Modulation of collagen cross-linking to increase infarct stiffness and reduce left ventricular dilation post-myocardial infarction. J Mol Cell Cardiol 85, 229–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuorio T, Nurmi H, Moulton K, Kurkipuro J, Robciuc MR, Ohman M, Heinonen SE, Samaranayake H, Heikura T, Alitalo K, et al. (2014). Lymphatic vessel insufficiency in hypercholesterolemic mice alters lipoprotein levels and promotes atherogenesis. Arterioscler Thromb Vasc Biol 34, 1162–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiig H, Aukland K, and Tenstad O (2003). Isolation of interstitial fluid from rat mammary tumors by a centrifugation method. Am J Physiol Heart Circ Physiol 284, H416–424. [DOI] [PubMed] [Google Scholar]
- Wilson MS, Madala SK, Ramalingam TR, Gochuico BR, Rosas IO, Cheever AW, and Wynn TA (2010). Bleomycin and IL-1beta-mediated pulmonary fibrosis is IL-17A dependent. J Exp Med 207, 535–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Thabet SR, Kirabo A, Trott DW, Saleh MA, Xiao L, Madhur MS, Chen W, and Harrison DG (2014). Inflammation and mechanical stretch promote aortic stiffening in hypertension through activation of p38 mitogen-activated protein kinase. Circ Res 114, 616–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X, Zhang L, Lin Y, Wang Y, Liu W, Li X, and Li P (2017). Imiquimod induced ApoE-deficient mice might be a composite animal model for the study of psoriasis and dyslipideamia comorbidity. J Dermatol Sci 88, 20–28. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Zanotti I, Reilly MP, Glick JM, Rothblat GH, and Rader DJ (2003). Overexpression of apolipoprotein A-I promotes reverse transport of cholesterol from macrophages to feces in vivo. Circulation 108, 661–663. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.