

Abstract
Current formulations of combined oral contraceptives (COC) containing ethinylestradiol (EE) have ≤35 μg due to increased risks of cardiovascular diseases (CVD) with higher doses of EE. Low‐dose formulations however, have resulted in increased incidences of breakthrough bleeding and contraceptive failure, particularly when coadministered with inducers of cytochrome P450 enzymes (CYP). The developed physiologically based pharmacokinetic model quantitatively predicted the effect of CYP3A4 inhibition and induction on the pharmacokinetics of EE. The predicted Cmax and AUC ratios when coadministered with voriconazole, fluconazole, rifampicin, and carbamazepine were within 1.25 of the observed data. Based on published clinical data, an AUCss value of 1,000 pg/ml.h was selected as the threshold for breakthrough bleeding. Prospective application of the model in simulations of different doses of EE (20 μg, 35 μg, and 50 μg) identified percentages of the population at risk of breakthrough bleeding alone and with varying degrees of CYP modulation.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE OF THE TOPIC?
☑ Ethinylestradiol is a key component of most combined oral contraceptives and is known to undergo extensive metabolism, with several reports of clinical DDIs resulting in contraceptive failure.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ A PBPK model was developed for EE to describe its disposition as well as aid in the prediction of CYP3A4 mediated DDIs.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ We demonstrated the utility of population based PBPK modeling as a useful tool to investigate the range of exposure of EE following dosing to individuals with varying degrees of CYP modulation.
HOW MIGHT THIS STUDY CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑ This PBPK model can be further developed by linking the estimated PK to the PD, hence it can help to better understand the still existing knowledge gap for the side effects associated with EE.
Despite continuous advocacy for contraceptive use among women as an effective means of birth control, a recent study carried out by the British Pregnancy Advisory Service (BPAS) criticized the apparent inefficacy of contraceptives. More than 50% of over 60,000 women who visited their clinics in 2016 for an abortion reported using at least one form of contraception when they got pregnant.1
Ethinylestradiol (EE) is a key active ingredient used in a variety of combined oral contraceptive (COC) formulations. Since the introduction of COCs in the 1960s, the dose of EE in the pill has been reduced due to increased risk of cardiovascular diseases (CVD), including thromboembolism and myocardial infarction, particularly in women with other predisposing factors such as smoking and obesity. Although the exact mechanism contributing to the increased CVD risk remains unclear, the use of newer low‐dose formulations of COC (EE dose ≤35 μg) has resulted in a significant decrease in the risk of CVD.2 Thus, guidelines issued by the American College of Obstetricians and Gynecologists (ACOG) advocate the safety and non‐CVD risk of COCs containing <50 μg EE in women younger than 35 years including those with preexisting but well‐controlled hypertension, provided no other CVD risk factors are present. It is also recommended as a safe option in older women >35 years until 50–55 years. Caution is however advised in the use of COC formulations in women >35 years who smoke, as well as in women with dyslipidemia, where it has been suggested that an alternative nonhormonal contraceptive be used.3 In fact, a recent US Food and Drug Administration (FDA) guidance contraindicates the use of COCs in smokers >35 years.4
On the down side, decreasing the dose of EE has led to greater incidences of breakthrough bleeding or contraceptive failure, especially when doses are further reduced to ≤20 μg.5 Furthermore, systemic concentrations of EE have been shown to be significantly reduced when it is concomitantly administered with potent drug metabolism enzyme‐inducing compounds such as rifampicin.6 EE is known to undergo extensive metabolism by cytochrome P450 (CYP) mediated hydroxylation as well as conjugation via sulfotransferases (SULTs) and UDP‐glucuronosyltransferases (UGTs). Any of these enzymes could be susceptible to inhibition and/or induction, resulting in undesirable drug interactions.7 The metabolism of EE has been studied extensively in an attempt to elucidate the complex disposition of the drug.8, 9, 10 Despite this, the assignment of the relative contributions of CYP enzymes, SULTs, and UGTs to the metabolism of EE remains problematic due to inconsistencies in the mass balance data from the various published studies.
An analysis of the University of Washington drug interaction database indicates that most of the clinically relevant drug–drug interactions (DDIs) with >20% inhibition or induction, that have so far been reported for EE, involve drugs which either inhibit or induce CYP3A4, either alone or alongside another drug metabolizing enzyme.11 Although CYP3A4 is perceived to play a major role in the metabolism of EE,5 studies with potent CYP3A4 inhibitors indicate that increases in the AUC of EE are less than 2‐fold. Indeed, in a clinical DDI study between EE and the potent CYP3A4 inhibitor ketoconazole, a 1.4‐fold increase in both AUC and Cmax was reported, thereby suggesting that the fraction of the drug metabolized by CYP3A4 (fmCYP3A4) is likely to be much lower than 0.5.12
A growing number of regulatory submissions have utilized physiologically based pharmacokinetic (PBPK) modeling and simulation approaches to quantitatively and qualitatively predict clinical DDIs, which in some cases may preclude the need to conduct a more cost‐intensive clinical trial.13 However, accurate prediction of clinical DDIs is dependent on a good understanding of the disposition of the drug, including elucidation of the fraction of drug metabolized (fm) through the pathway(s) of interest. Thus, the primary aim of this study was to review the relevant in vitro and clinical data relating to EE metabolism and to use these to develop a PBPK model that could describe its disposition. Data obtained from a clinical DDI study with ketoconazole12 was used to refine the contribution of CYP3A4 to the overall clearance of EE, as shown in the workflow described in Figure 1 . The PBPK model was verified by comparing simulations of the DDIs between EE and known CYP3A4 inhibitors (fluconazole and voriconazole) and potent CYP3A4 inducers (carbamazepine and rifampicin) with clinical data. The PBPK model was then applied prospectively to predict the DDI potential of EE with moderate CYP3A4 inducers and under conditions of complete CYP inhibition, in an attempt to understand the exposure limits of EE that would result in efficacious or potentially harmful concentrations.
Figure 1.
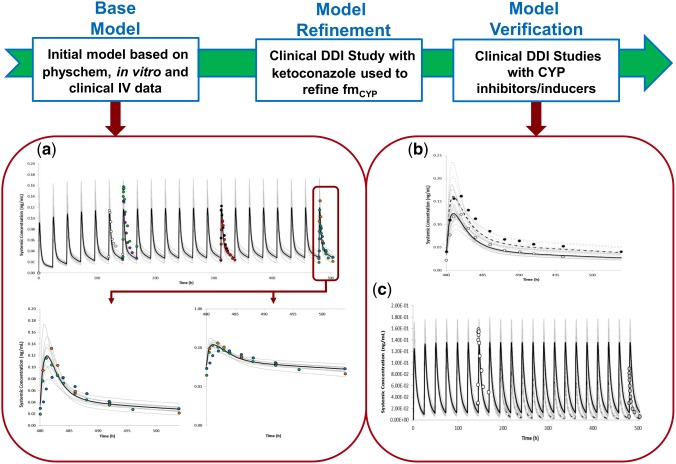
Workflow of EE model development. The base model was developed using a mixture of a bottom‐up and top‐down (i.e., a middle‐out) approach incorporating physicochemical data, in vitro metabolism data, and data from an intravenous study. The estimated fmCYP from in vitro data was refined using a clinical DDI study with ketoconazole. The refined model was independently verified using clinical studies with other CYP inhibitors and inducers. Simulated and observed mean plasma concentration–time profile of EE are shown above after (a) multiple oral doses of 35 μg q.d. administered alone for 21 days; (b) multiple oral doses of 35 μg q.d. administered alone for 21 days and in the presence of voriconazole (strong CYP3A4 inhibitor) given as 400 mg b.d. on Day 18 followed by 200 mg b.d. given on Days 19–21; and (c) multiple oral doses of 35 μg q.d. administered alone for 7 days and In the presence of rifampicin (strong CYP3A4 inducer) given as 600 g q.d. on Days 12–21. The dark lines represent the mean plasma concentration–time profiles, while the gray lines represent the predictions from individual trials of (a) 10 trials × 10 female HV, 20–50 years; (b) 10 trials × 16 female HV, 18–40 years; and (c) 10 trials × 12 female HV, 23–44 years. The black dashed lines represent the mean plasma concentration–time profiles of simulations done in the presence of a perpetrator drug, while the gray dashed lines represent the predictions from individual trials of simulations done in the presence of a perpetrator drug. The different circles in (a) are data points from observed data: open circles,48 purple circles,28 red circles,35 black circles,49 blue circles,24 green circles,26 and brown circles25; while the data points in (b,c) are from references 26 and 29, respectively. [Color figure can be viewed at http://cpt-journal.com]
RESULTS
First‐pass metabolism of EE in the gut and liver
Although EE is well absorbed from the gut, its oral bioavailability is less than 50% due to extensive metabolism in the gut and/or liver during first pass.14, 15 In vitro incubation of EE in intestinal mucosal cells identified only conjugated metabolites, which constituted 38% of the incubated material. Approximately 90% of the metabolites were sulfo‐conjugates, while the rest were identified as glucuronides.16 Results of an in vivo mass balance study showed that 30% of the drug was recovered in the feces 2 weeks after oral administration, of which ∼80% was identified as unconjugated EE.8 The recovery of a high proportion of unconjugated EE in the feces does not necessarily contradict the in vitro study, given that deconjugation of previously conjugated drug by enterocytic microbes and shed enterocytes can also occur in the gut, especially when considering the duration of fecal collection.
A follow‐up in vivo study was performed to assess the extent of EE metabolism in the intestine vs. the liver during first pass by simultaneous sampling from the portal vein and the systemic circulation after oral administration. The results of the study confirmed that conjugation of EE in the gut wall plays a major role in its presystemic metabolism with a gut extraction ratio (EG) of 0.44, while that in the liver (EH) was 0.25.15 Although this study only measured conjugated metabolites, it was preferentially considered for estimating the fraction escaping gut metabolism (FG) given that the afore‐mentioned in vitro study identified conjugated metabolites as the main product of intestinal metabolism. Based on these data, an FG of 0.56 was assumed for EE with the estimated unbound intrinsic clearance for the gut back‐calculated using the “QGut” model17; of which ∼70% was assigned to sulfation, ∼20% to hydroxylation, and the rest to glucuronidation (see details in the Methods).
Relative contributions of metabolic routes in the liver
A total intrinsic hepatic metabolic clearance (CLint) of 275.49 μl/min/mg protein was back‐calculated from a mean intravenous (i.v.) clearance of 16.47 L/h18, 19, 20 via the well‐stirred liver model, using the retrograde calculator within the Simcyp simulator after subtracting renal clearance. Based on the study carried out by Reed et al.,8 the percentage of unchanged drug excreted in the urine is reported as 6% of an oral dose5; hence a value of ∼2.1 L/h was estimated for renal clearance from a mean oral clearance of 34.6 L/h.20, 21
A combination of bottom‐up (using scaled‐up in vitro data) and top‐down (using clinical data) approaches was used to assign the contributions of the respective enzymes to the systemic clearance of EE and hence obtain estimates of fm. Assignment of the scaled‐up hepatic metabolic CLint of 66.77 μl/min/mg protein due to 2‐hydroxylation (which constitutes greater than 90% of the hydroxylated metabolites) of EE from the in vitro study carried out by Shiraga et al. in human liver microsomes (HLM),22 resulted in a contribution of ∼0.2 to the overall systemic clearance of EE (fmCYP). Half of this (0.1) was assigned to CYP3A4, with the remainder apportioned to CYP2C9, 2C8, and 1A2, as indicated in the study with recombinant CYP enzymes.23 Based on the HLM study by Shiraga et al., the metabolic intrinsic clearance for the UGT1A1‐mediated metabolism of EE was calculated to be 21.26 μl/min/mg protein, resulting in an fmUGT1A1 of about 0.05. It should be noted that sulfation and the minor 4‐hydroxylation pathway were not specifically accounted for in the model. Thus, the remaining unassigned hepatic metabolic CLint of 187.46 μl/min/mg protein (275.49‐66.77‐21.26) representing an fm of about 0.57 was entered in the simulator as additional HLM clearance representing other EE hepatic metabolic pathways.
Although 9% of the drug has been reported as eliminated unchanged in the feces,5 due to the difficulty in separating out the amount of drug that remains unabsorbed from that which undergoes deconjugation after absorption and subsequent secretion/metabolism into the gastrointestinal tract, the exact amount of the parent drug eliminated via biliary clearance could not be estimated and is thus lumped with the non‐CYP clearance.
Simulated profiles using the PBPK model for EE
Simulated plasma concentration–time profiles using the developed model were able to reasonably recover observed data from both single doses of 50 μg EE administered i.v. and orally (Figure 2 a–d); as well as multiple doses of 35 μg EE administered orally (Figure 1 a). The predicted population mean (range of trial means) Cmax and AUC(0‐24) for the simulation of 100 individuals (10 trials × 10 female healthy volunteers (HV); 20–50 years) after multiple oral doses of 35 μg EE q.d. for 21 days (Figure 1 a) were 0.125 (0.08–0.154) ng/ml and 1.14 (0.989–1.308) ng/ml.h, respectively. Corresponding observed Cmax and AUC(0‐24) values on day 21 from different clinical studies were 0.087 ng/ml and 1.08 ng/ml.h24; 0.143 ng/ml and 1.199 ng/ml.h 25; and 0.117 ng/ml and 1.062 ng/ml.h.26
Figure 2.
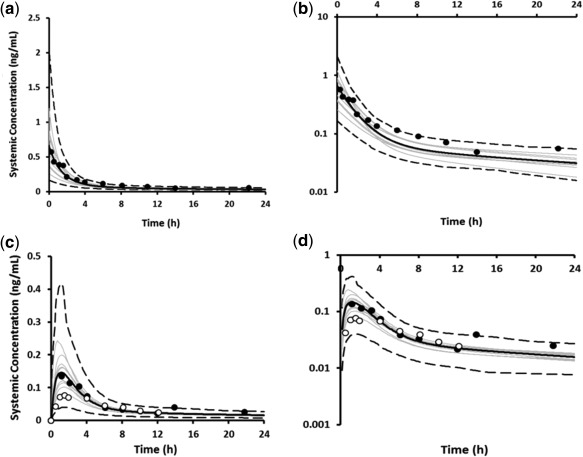
Simulated (black line) and observed (data points) mean plasma concentration–time profiles of EE after a single dose of 50 μg administered intravenously on a linear (a) and logarithmic scale (b); and administered orally on a linear (c) and logarithmic scale (d). The gray lines represent predictions from individual trials (10 trials × 6 female HV; 21–23 years) for the i.v. dosing; and (10 trials × 10 female HV; 20–50 years) for the oral dosing. Dashed lines represent the 5th and 95th percentiles. Observed data were obtained from Ref. 14 (black circles) and Ref. 50 (open circles).
Simulation of the clinical DDI with ketoconazole: refinement of fmCYP3A4 for EE
The simulated increases in plasma concentration–time profiles of multiple doses of 20 μg EE after coadministration with ketoconazole, a potent CYP3A4 inhibitor, underpredicted the observed data by more than 2‐fold. The predicted increase in AUC was 18% vs. 40% in the clinical study, while the predicted increase in Cmax was 12% vs. the observed value of 39%.12 The in vitro CYP3A4 metabolic CLint was therefore optimized to recover the DDI with ketoconazole, resulting in an increase in fmCYP3A4 from 0.11 to 0.22. By retaining the in vitro‐derived fm for the different CYP enzymes, the in vitro CLint values for the other CYP enzymes were proportionally increased relative to that of CYP3A4 to give an increased fmCYP of ∼0.4, while the fraction assigned to the additional HLM elimination pathway was reduced to account for the increased fmCYP. It should be noted that despite the refinement of fmCYP3A4 to the overall systemic clearance of EE, the simulated concentration–time profiles remained consistent with the observed data, as shown in Figure 2 a–d. A summary of the disposition of EE after oral administration, utilizing the available published data and the final mean contributions to the systemic clearance of EE in the optimized PBPK model are shown in Figure 3 a,b, respectively.
Figure 3.
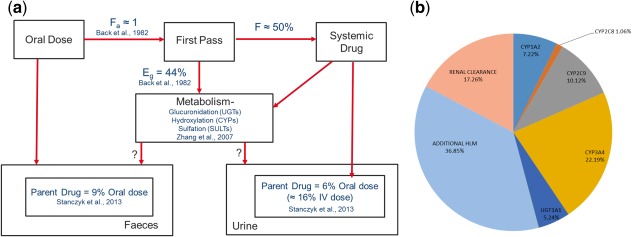
(a) A schematic of the disposition of ethinylestradiol after oral administration based on published data, showing the fraction absorbed (fa), fraction extracted in the gut during first‐pass metabolism (EG), and the fraction of the drug that reaches the systemic circulation (F); as well as the different routes of systemic metabolism of the drug; and (b) predicted mean contribution of metabolic and renal clearance to the systemic elimination of ethinylestradiol using the optimized PBPK model (10 trials × 10 female HV; 20–50 years).
Simulation of DDIs with other CYP3A4 inhibitors/inducers: verification of the fmCYP3A4
Simulated and observed Cmax and AUC ratios of EE following administration of two CYP3A4 inhibitors (fluconazole and voriconazole) as well as two CYP3A4 inducers (rifampicin and carbamazepine) were all reasonably consistent with observed data, as summarized in Table 1. Simulated plasma concentration profiles of multiple doses of 35 μg EE in the presence and absence of voriconazole and rifampicin are shown in Figure 1 b,c, respectively. Simulated plasma concentration profiles of EE in the presence and absence of fluconazole and carbamazepine are shown in Supplementary Figure S1A,B. The predictive ratios for both the Cmax and AUC for all the DDI simulations were within 1.25‐fold of the observed data (Table 1). The simulated trial designs are summarized in Table S1.
Table 1.
Summary of clinical DDI simulations of various CYP perpetrator drugs on ethinylestradiol
| Observed | Predicted (trial range)a | Predicted/observed | ||||||
|---|---|---|---|---|---|---|---|---|
| Reference | EE dosing | Perpetrator dosing | Cmax ratio | AUC ratio | Cmax ratio | AUC ratio | Cmax ratio | AUC ratio |
| 27 | 35 µg QD × 21 days | Voriconazole 400 mg BD Day 18, 200 mg BD Days 19–21 | 1.34 | 1.57 | 1.28 (1.25–1.34) | 1.40 (1.36–1.49) | 0.95 | 0.89 |
| 28 | 35 µg QD × 7 days | Fluconazole 300 mg SD on day 7 | 1.08 | 1.24 | 1.10 (1.08–1.11) | 1.13 (1.11–1.16) | 1.01 | 0.91 |
| 20 µg QD × 21 days | Carbamazepine 300 mg BD × 21 days | 0.65 | 0.56 | 0.66 (0.6–0.71) | 0.61 (0.55–0.66) | 1.02 | 1.09 | |
| 26 | 35 µg QD × 21 days | Carbamazepine 200 mg SD day 1, 200 mg BD days 2–4, 300 mg BD days 5–21 | 0.82 | 0.58 | 0.66 (0.6–0.72) | 0.61 (0.55–0.66) | 0.80 | 1.05 |
| 6 | 35 µg QD × 10 days | Rifampicin 300 mg QD × 10 days (Days 1–10) | 0.58 | 0.36 | 0.51 (0.48–0.54) | 0.4 (0.37–0.44) | 0.88 | 1.11 |
| 29 | 35 µg QD × 21 days | Rifampicin 600 mg QD for 14 days (Days 8–21) | 0.57 | 0.34 | 0.49 (0.44–0.58) | 0.36 (0.31–0.45) | 0.89 | 1.06 |
The clinical DDI simulations were all carried out in a population of female healthy volunteers (10 trials × ‘N’ number of subjects), with the number of virtual subjects (N) and age range for each simulation matched closely to the clinical study. Details of the trial designs are given in Table S1 of the supplementary information.
Assessing the safety and/or toxicity of EE in COC formulations
With current EE formulations, there have been more reports of breakthrough bleeding, particularly when coadministered with CYP inducers due to the relatively low doses of EE, and no reports of CVD when EE is coadministered with potent CYP inhibitors. Despite this, not enough attention has been given to informing the choice and dosage of COC, especially in an era whereby drugs are commonly being coadministered with other medications. We therefore performed an evaluation of reported clinical studies carried out in HV women collated from the University of Washington drug interaction database, showing in vivo induction ≥20% in the pharmacokinetic parameters (AUC and Cmax) of EE. The results of these clinical studies are summarized in Table 2.
Table 2.
Summary of clinical DDI studies showing >20% in vivo induction of ethinylestradiol
| Reference | EE dose | Perpetrator dosing | N | Cmax (pg/ml) | AUCss (pg/ml.h) | Commentc | ||
|---|---|---|---|---|---|---|---|---|
| Control | Treatment | Control | Treatment | |||||
| 30 | 20 µg | 300 mg H. perforatum(St. John's wort) TID | 16 | 90 (82–98) a | 84 (73–98) a | 994 (874–1118) a | 854 (712–1071) a | Increased incidence of breakthrough bleeding observed in treatment arm (56%) compared to control arm (31%). |
| 31 | 20 µg | 300 mg Carbamazepine (CBZ) BD | 10 | 147 (48) | 95 (23) | 1778 (479) | 986 (192) | Increased progesterone levels, ovulation and breakthrough bleeding in CBZ arm compared to placebo. |
| 24 | 35ug | 800 mg Boceprevir TID | 20 | 87 (78, 97)b | 69 (63, 76)b | 1080 (962, 1220)b | 798 (696, 915)b | No breakthrough bleeding and no meaningful change in mid cycle levels of FSH and LH. |
| 26 | 35ug |
50 mg Topiramate QD100 mg Topiramate QD200 mg Topiramate QD 600 mg CBZ QD |
11 10 12 10 |
139 (46) 129 (56) 130 (34) 117 (34) |
128 (44) 135 (27) 111 (40) 95.5 (41) |
1349 (413) 1113 (429) 1113 (298) 1062 (345) |
1187 (421) 1177 (294) 987 (331) 616 (244) |
Only PK assessments were done. No breakthrough bleeding reported as one of the side effects. |
| 32 | 35ug | Troglitazone 600 mg QD | 15 | 164 (37) | 110 (37) | 1553 (48) | 1052 (36) | Mid cycle bleeding reported in 4 out of 15 subjects in the treatment arm compared to 2 subjects in the control arm. |
| 33 | 35ug | Modafinil 400 mg QD | 18 | 151 (32) | 134 (52) | 1175 (271)b | 965 (532) | Metorrhagia observed in 4 out of 18 subjects in treatment phase but no significant differences in FSH and LH levels. |
| 34 | 35ug | 750 mg Telaprevir TID | 24 | 121 (32) | 90 (28) | 1318 (360) | 958 (297) | Increased incidence of breakthrough bleeding (58%) as well as significantly increased levels of progesterone, FSH and LH in treatment arm compared to control (33%). |
| 25 | 35ug |
30 mg Vicriviroc/100 mg Ritonavir QD Vicriviroc 75 mg BID 100 mg Ritonavir QD |
21 |
143 148 139 |
109 134 124 |
1199 1252 1255 |
846 1217 889 |
Only PK assessments were done. No breakthrough bleeding reported as one of the side effects. |
| 35 | 35ug | Darunavir 600 mg/Ritonavir 100 mg | 11 | 105 (29) | 73 (17) | 1095 (400) | 609 (151) | Reduced decrease in FSH and LH levels within cycle in treatment arm compared to control but no breakthrough bleeding. |
| 29 | 35ug |
Rifampicin 600 mg QD Rifabutin 300 mg QD |
12 |
171 (44) 172 (56) |
98 (32)
156 (52) |
1442 (458) 1435 (347) |
496 (161)
926 (230) |
Increased FSH levels and altered menses with rifampicin but not rifabutin. No ovulation occurred with both treatments shown by low levels of progesterone. |
| 36 | 50ug | Oxcarbazepine titration to 1200 mg QD | 16 | 180 (155–208)b | 117 (97–142) b | 1677 (1355–2086)b | 886 (741–1059) b | Breakthrough bleeding recorded in one subject in treatment arm and none in control arm. |
Observed data are reported as mean (SD) except where indicated.
Observed data is given as median (25th–75th percentile).
Observed data is given as geometric mean (95% confidence intervals).
Cmax and AUC values highlighted in bold are for those studies in which incidences of breakthrough bleeding and/or low levels of FSH and LH were reported.
Correction added on May 7, 2018, after first online publication: In table 2, Reference 25, the numbers in the Cmax columns and the AUC columns weren't aligned with the correct Perpetrator dosing. This has been corrected.
In 8 out of 11 evaluated clinical studies comprising a total of 122 female HVs, there were reported increased incidences of breakthrough bleeding and/or higher levels of follicle stimulating hormone (FSH), luteinizing hormone (LH), or progesterone in the comedication arm compared to the control arm. In the remaining three clinical studies comprising 84 female HVs, no reports of breakthrough bleeding were mentioned, although two out of the three clinical studies had no specific pharmacodynamic (PD) evaluations of either efficacy or toxicity. In all but one of the clinical studies with reported incidences of breakthrough bleeding, coadministration of the perpetrator drug resulted in EE steady‐state mean AUC (AUCss) of less than 1,000 pg/ml.h in the comedication arm of the study.
Although the exact mechanism(s) for the CVD risk of EE is still unclear, most of the CVD effects have been attributed to metabolic changes caused by increased hormonal levels. There is, however, no information regarding what steady‐state systemic concentrations of EE are expected to result in undesirable side effects. Nonetheless, there have been specific bans and/or withdrawals of EE formulations containing >50 μg both in the UK and the US.2, 3 In order to make an assessment of adequate EE dosing required to minimize the risk of both contraceptive failure and CVD, we selected 1,000 pg/ml.h as a lower threshold, given that the AUCss for the comedication arm in the evaluated clinical studies with reported breakthrough bleeding were less than 1,000 pg/ml.h. The mean simulated concentration of 1,675 pg/ml.h observed for the 50 μg oral dose (Table 3) was chosen as the upper threshold associated with a risk for CVD based on the recommended ACOG guidelines. We thereafter determined how frequently within a simulated population of 100 subjects (10 trials of 10 HV women aged between 20 and 50 years), the AUCss for different doses of EE: 20 μg, 35 μg, and 50 μg alone and in the presence of moderate and strong CYP3A4 inhibitors and inducers was below 1,000 pg/ml.h, on the one hand, or above 1,675 pg/ml.h on the other hand.
Table 3.
Simulated (10 trials × 10 female HV) population mean (range of trial means) steady state AUC for different doses of EE (20 μg, 35 μg, 50 μg) alone and in the presence of various degrees of CYP modulation
| EE dosing | Control AUCss (pg/ml) | CYP3A4 inhibitionaAUCss (pg/ml) | Complete CYP inhibitionb AUCss (pg/ml) | Strong CYP3A4 inductionc AUCss (pg/ml) | Moderate CYP3A4 inductiond AUCss (pg/ml) |
|---|---|---|---|---|---|
| 20 µg multiple dosing | 670 (578–737)e | 902 (779–1054)e | 1169 (972–1334) | 244 (205–395)e | 412 (367–437)e |
| N of individuals below lower threshold value | 86 | 59 | 36 | 100 | 99 |
| N of individuals above upper threshold value | 0 | 3 | 7 | 0 | 0 |
| 35 µg multiple dosing | 1172 (1012–1290) | 1579 (1404–1844) | 2046 (1702–2335)f | 427 (310–520)e | 720 (632–765)e |
| N of individuals below lower threshold value | 38 | 13 | 5 | 95 | 90 |
| N of individuals above upper threshold value | 13 | 40 | 64 | 0 | 1 |
| 50 µg multiple dosing | 1675 (1446–1844) | 2256 (1949–2634)f | 2923 (2431–3336)f | 609 (443–743)e | 1029 (902–1161) |
| N of individuals below lower threshold value | 12 | 4 | 2 | 87 | 49 |
| N of individuals above upper threshold value | 41 | 74 | 89 | 1 | 7 |
Simulation was done in the presence of multiple doses of 200mg ketoconazole BD (CYP3A4 Ki = 0.015 µM) as perpetrator.
Simulation was done in the presence of a hypothetical compound as perpetrator with potent inhibition against CYP1A2, CYP2C8, CYP2C9, CYP3A4 (Ki = 0.015 µM) as perpetrator.
Simulation was done in the presence of multiple doses of 600mg rifampicin QD, a strong CYP3A4 inducer as perpetrator (Indmax = 16, IndC50 = 0.32 µM).
Simulation was done in the presence of multiple doses of 600mg efavirenz QD, a moderate CYP3A4 inducer as perpetrator (Indmax = 9.9, IndC50 = 3.8 µM).
Simulations in which the population mean is below the threshold of 1000pg/ml.h.
Simulations in which the population mean is above that of a 50 µg dose of EE.
The predicted population mean AUCss for the 100 individuals at the different doses alone and in the presence of various degrees of CYP modulation are summarized in Table 3 and Figure S2 of the supplementary information. The AUCss of 86 simulated individuals was below 1,000 pg/ml.h for the 20 μg dose. In the presence of CYP3A4 or complete CYP inhibition, the concentration of EE increased as expected, with >50% of the virtual subjects having AUCss above the lower threshold. With the 35 μg dose, the AUCss of close to 50% of the virtual subjects was above the lower threshold. However, the presence of either moderate or strong CYP3A4 induction resulted in simulated concentrations below the lower threshold for up to 90% of the virtual subjects. In the presence of CYP3A4 or complete CYP inhibition, the simulated AUCss of close to 50% of the virtual subjects were above the population mean AUCss of 1,675 pg/ml.h. Finally, with the 50 μg dose, moderate and strong CYP3A4 induction resulted in ∼50% and 90% of the individual subjects respectively having AUCss below the threshold for efficacy.
DISCUSSION
A number of clinical studies have demonstrated the contraceptive inefficacy of EE following coadministration of rifampicin and most of the anti‐epileptic drugs (AEDs), which are known to be inducers of CYP3A4.5 Thus, when drugs in development are identified as CYP3A4 inducers, there is cause for concern and a clinical DDI study with EE is recommended. A PBPK model for EE was developed using publicly available in vitro and mass balance data in an attempt to elucidate the disposition of the drug, particularly with respect to the contribution of CYP3A4 to its metabolism. Despite the perception that EE is mainly metabolized by CYP3A4, in vitro data showed that the enzyme was only responsible for 10% of its systemic clearance. Simulation of the clinical DDI with ketoconazole indicated that in order to recover the observed magnitude of interaction, the fmCYP3A4 had to be increased, but it still remained at less than 25% (Figure 3 b). The contribution was then further verified with simulations involving other CYP3A4 inhibitors and inducers.
It should be noted that for rifampicin, induction of CYP3A4 and 2C9 was considered in the simulations with EE and for carbamazepine, only CYP3A4 induction. Although it is relatively well known that rifampicin can induce non‐CYP enzymes, including UGT1A1 and some SULTs,37, 38 few publications relating to in vitro–in vivo extrapolation (IVIVE) of these enzymes and induction by rifampicin can be found in the public domain. However, the information in Smith et al.38 was used to obtain estimates of parameters describing the UGT1A1‐mediated induction by rifampicin. When used in simulations (data not shown), they were found to have negligible impact on the exposure of EE, probably due to the relatively small contribution of UGT1A1 to the clearance of EE. In the publication by Li et al.,37 in vitro experiments conducted using human hepatocytes indicated that pretreatment with rifampicin had little impact on the formation of glucuronide metabolites, but led to a significant increase in the formation of the EE‐3‐sulfate metabolite. However, the less than 2‐fold increase in sulfate metabolite formation is relatively small when compared with the greater than 10‐fold increase in CYP3A4 activity.39 Nonetheless, if sulfation predominates the remaining unassigned fm of 0.37 (Figure 3 b), in vivo induction of the SULTs can be clinically relevant, particularly for potent SULT inducers.
These observations and the finding that the verified PBPK model for EE was able to predict with reasonable accuracy the reduction in exposure of EE following coadministration of rifampicin and carbamazepine when considering induction of CYP3A4, lends further support to the fact that the fmCYP3A4 is likely to be in the region of 25%. This gives confidence in prospective application of the model for assessment of the CYP3A4 induction potential of drugs in development that have been identified as CYP3A4 inducers in vitro.
Based on the review of the clinically published DDI studies (Table 2) and with the aid of prospective DDI simulations using the verified PBPK model, the potential risks of breakthrough bleeding or contraceptive inefficacy with the different doses of EE present in oral contraceptive formulations, either taken alone or with other CYP‐modulating drugs was assessed. The simulations suggest that formulations containing 20 μg doses are likely to result in steady‐state concentrations lower than 1,000 pg/ml.h, which may result in breakthrough bleeding or contraceptive inefficacy, unless they are coadministered with drugs that completely inhibit some or all of its CYP metabolism. On the other hand, formulations containing 35 μg doses, although suitable for attaining adequate contraception when administered alone, may result in incidences of breakthrough bleeding when coadministered with moderate or strong CYP3A4 inducers. Increasing the dose to 50 μg in such scenarios may still be insufficient in providing adequate contraception in all individuals, as shown in Table 3 and Figure S2 and evidenced in the clinical DDI study with oxcarbazepine.36
Although the developed PBPK model has only been verified against modulators of CYP3A4, the simulated control study for the different commonly prescribed doses of EE gives an indication of the number of individuals whose AUCss would fall outside the suggested therapeutic window. This study therefore highlights the previously unreported but relatively narrow therapeutic index of formulations containing ethinylestradiol, particularly with regard to the lower threshold value, which in turn corroborates the study carried out by the BPAS.
Coadministration of EE with several medications (in cases of poly‐pharmacy), which together induce multiple pathways (including non‐CYP pathways) is expected to further reduce its systemic concentration, thereby increasing the risk of breakthrough bleeding. Doose et al.26 reported lower systemic concentrations of EE in obese individuals compared to nonobese individuals. Thus, physiological conditions that reduce the systemic exposure of EE could also increase the risk of breakthrough bleeding, particularly when coadministered with other enzyme inducers. The FDA guidance on labeling for combined hormonal contraceptives states that “enzyme inducers (e.g., CYP3A4) may decrease their effectiveness or increase breakthrough bleeding.” They advise the use of a back‐up or alternative method of contraception when enzyme inducers are going to be prescribed.4 The modeling work described in this article describes one approach whereby the effects of inducers or inhibitors on the pharmacokinetics of oral contraceptives can be simulated and thus ascertained beforehand.
In this study a cutoff value of 1,000 pg/ml.h for EE exposure was used where contraceptive efficacy is achieved with minimal safety concerns. Using a single threshold value for all individuals is a limitation of the study, but as the relationships between the PK and PD of EE in individual subjects are incompletely understood,29 this represents a pragmatic approach with the available information. The pharmacokinetics of EE show high interindividual variability (Table 2) and as such in certain individuals, systemic concentrations of EE greater than 1,000 pg/ml.h may be seen at doses of 20 μg, as shown in the clinical DDI study with carbamazepine.31
METHODS
PBPK model development
All PBPK simulations were carried out using the HV population within the Simcyp simulator v. 17 rel. 1 (Simcyp, Sheffield UK). The structural design and functional capabilities of the simulator as well as equations describing the genetic, physiological, and demographic variables for the population have been described previously.40 Prior metabolic, protein binding, and physicochemical data for EE were collated from the literature and incorporated into a minimal PBPK model with an additional single adjusting compartment (SAC) to recover the biphasic plasma concentration vs. time profile of EE. Input parameters for the SAC compartment were derived using the parameter estimation module in Simcyp and observed data from an i.v. clinical study.41 Oral absorption of the drug is described by a first‐order absorption process with fraction absorbed (fa) and ka predicted from polar surface area (PSA) and number of hydrogen bond donors for the compound.42
In vitro enzyme kinetic parameters for CYPs and UGTs were scaled up using physiological data as described previously.40 The unbound metabolic intrinsic clearance (CLint) estimated from in vitro studies in recombinant CYP3A4, 2C9, 2C8, and 1A2 enzymes expressed in baculovirus‐infected SF21 cells amounted to 11.38 μl/min/mg protein.23 However, this CLint estimate was much lower than that obtained when EE was directly incubated in human liver microsomes.22 Given that Shiraga et al.22 used pooled liver samples from 46 donors, the estimated unbound intrinsic clearance of 66.77 μl/min/mg protein for the 2‐hydroxylation pathway in this study was preferentially selected as input for the hydroxylation pathway in the PBPK simulation after applying the individual fmCYP of 50.6%, 27.5%, 2.3%, and 19.6% for CYP3A4, 2C9, 2C8, and 1A2, respectively, obtained from the study with recombinant enzymes.23 For glucuronidation, the in vitro metabolic CLint of 21.26 μl/min/mg protein derived from HLMs was used as the input and assigned to UGT1A1.22
In the absence of abundance data for SULTs in the simulator to do a direct IVIVE, the total intrinsic hepatic metabolic clearance of 275.49 μl/min/mg protein was back‐calculated from an i.v. clearance of 16.47 L/h via the well‐stirred liver model based on Eq. 2, below. The remaining hepatic intrinsic clearance was assigned as additional HLM clearance in the simulator to account for EE systemic clearance not mechanistically accounted for. A similar approach using the “Qgut” model described in Eq. 2 below was used to apportion the total gut intrinsic clearance using an FG of 0.56, after accounting for the intrinsic clearances due to the gut CYP enzymes (CYP2C9 and CYP3A4) and UGT1A1. The additional intrinsic gut clearance was assigned as intestinal cytosolic clearance in the simulator to represent sulfation.
| (1) |
| (2) |
Where CLH is hepatic blood clearance, obtained by deducting renal clearance from i.v. clearance; QH is hepatic blood flow; fuB is fraction of drug unbound in blood; fugut is fraction of unbound drug in the gut; FG is the fraction of the drug escaping gut metabolism; and Qgut is a composite term of the enterocytic blood flow and the permeability of the drug.17
PBPK model verification
Quantitative predictions of the clinical DDI between EE and ketoconazole, a known CYP3A4 inhibitor, was simulated using the initial model to verify the fmCYP3A4. The automated sensitivity analysis module within the Simcyp simulator was thereafter used to refine the CYP3A4 CLint value to recover the AUC ratio of 1.4 in the clinical DDI study with ketoconazole due to a greater than 2‐fold underprediction of both the AUC and Cmax ratios. Further simulations with other CYP3A4 inhibitors (fluconazole and voriconazole) and inducers (carbamazepine and rifampicin) with the refined model were carried out based on published clinical studies.
The characteristics of the virtual subjects for all the simulations were matched closely with those of the clinical DDI studies: the number of subjects, age range, and gender ratios were replicated. Differential equations describing the kinetics of substrates, inhibitors, and inducers as well as enzyme dynamics with or without inhibition and/or induction have been described previously. The interaction parameters present in perpetrator models (inhibitors and inducers) against specific enzymes and/or transporters are only utilized in simulations when these enzymes/transporters are present in the substrate model.43, 44 Models for all of the compounds used in the simulations except voriconazole are available in the Simcyp v. 17 compound library. The final input parameters used for the EE model are shown in Table 4, while the input parameters used to simulate the kinetics of voriconazole are given in Table S3 of the supplementary information.
Table 4.
Input parameter values used to simulate the kinetics of ethinylestradiol
| Parameter | Value | Method/reference |
|---|---|---|
| Molecular weight (g/mol) | 296.4 | (https://pubchem.ncbi.nlm.nih.gov/compound/5991) |
| Log P | 3.81 | Predicted from ACD 45 |
| Compound type | Diprotic acid | Predicted from ACD |
| pKa | 10.2, 13.1 | Predicted from ACD |
| B/P | 1 | Assumed |
| fu | 0.015 | (46, 47) |
| Main plasma binding protein | Human serum albumin | (46) |
| Absorption | First order absorption | |
| fa | 0.948 | Predicted from Physchem data (PSA/HBD) |
| ka (1/h) | 1.103 | Predicted from Physchem data (PSA/HBD) |
| fugut | 1 | Assumed |
| Qgut (L/h) | 11.74 | Predicted from Physchem data (PSA/HBD) |
| Peff,man (10−4cm/s) | 2.68 | Predicted from Physchem data (PSA/HBD) |
| PSA(Å2)/HBD | 42.7/2 | (45) |
| Distribution model | Minimal PBPK Model | |
| VSS (L/kg) | 4.06 | (14, 21) |
| Kin (L/h) | 0.287 | Optimized 41 |
| Kout (L/h) | 0.096 | Optimized 41 |
| Vsac (L/kg) | 2 | Optimized 41 |
| Elimination | Enzyme kinetics | |
| CYP3A4 CLint (μL/min/pmol) | 0.5 | Optimized with ketoconazole DDI study 12 |
| CYP2C9 CLint (μL/min/pmol) | 0.51 | Optimized (fmCYP2C9 from 23) |
| CYP1A2 CLint (μL/min/pmol) | 0.51 | Optimized (fmCYP1A2 from 23) |
| CYP2C8 CLint (μL/min/pmol) | 0.13 | Optimized (fmCYP2C8 from 23) |
| UGT1A1 Vmax (pmol/min/mg protein) | 408.5 | (22) |
| UGT1A1 Km (μM) | 19.22 | (22) |
| Additional CLint (HLM) (μL/min/mg protein) | 118.83 | Retrograde calculation |
| Additional CLint (HICEL) (μL/min) | 43.92 | Back‐calculated from Qgut and Fg 15 |
| CLR (L/h) | 2.079 | (5) |
FUNDING
No funding was received for this work.
CONFLICT OF INTEREST
All authors are paid employees of Simcyp Limited (A Certara Company).
AUTHOR CONTRIBUTIONS
U.E., S.N., I.G., and K.R.Y. wrote the article; U.E., H.H., M.D., and K.R.Y. performed the research; U.E., S.N., I.G., and K.R.Y. analyzed the data.
Supporting information
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Eleanor Savill for her assistance in the preparation and submission of the article; Farzaneh Salem for most of the information used in developing the voriconazole model; Kim Crewe, Kate Gill, and Ciaran Fisher for useful discussions around compound model development in general.
This article was published online on 27 April 2018. Few errors were subsequently identified in Table 2. This notice is included in the online and print versions to indicate that both have been corrected on 11 May 2018.
References
- 1. Smyth, C. Contraception Failure Blamed for Abortions. <https://www.thetimes.co.uk/article/contraception-failure-blamed-for-abortions-w8ddnm5qz> (2017). Accessed 7 July 2017.
- 2. Chasan‐Taber, L. & Stampfer, M.J. Epidemiology of oral contraceptives and cardiovascular disease. Ann. Intern. Med. 128, 467–477 (1998). [DOI] [PubMed] [Google Scholar]
- 3. Shufelt, C.L. & Bairey Merz, C.N. Contraceptive hormone use and cardiovascular disease. J. Am. Coll. Cardiol. 53, 221–231 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. U.S. Food & Drug Administration . Labeling for Combined Hormonal Contraceptives: Guidance for Industry. <https://www.fda.gov/ucm/groups/fdagov-public/@fdagov-drugs-gen/documents/document/ucm590673.pdf> (2017). Accessed 1 December 2017.
- 5. Stanczyk, F.Z. , Archer, D.F. & Bhavnani, B.R. Ethinyl estradiol and 17beta‐estradiol in combined oral contraceptives: pharmacokinetics, pharmacodynamics and risk assessment. Contraception 87, 706–727 (2013). [DOI] [PubMed] [Google Scholar]
- 6. LeBel, M. et al Effects of rifabutin and rifampicin on the pharmacokinetics of ethinylestradiol and norethindrone. J. Clin. Pharmacol. 38, 1042–1050 (1998). [DOI] [PubMed] [Google Scholar]
- 7. Zhang, H. et al Pharmacokinetic drug interactions involving 17alpha‐ethinylestradiol: a new look at an old drug. Clin. Pharmacokinet. 46, 133–157 (2007). [DOI] [PubMed] [Google Scholar]
- 8. Reed, M.J. , Fotherby, K. & Steele, S.J. Metabolism of ethynyloestradiol in man. J. Endocrinol. 55, 351–361 (1972). [DOI] [PubMed] [Google Scholar]
- 9. Helton, E.D. , Williams, M.C. & Goldzieher, J.W. Human urinary and liver conjugates of 17alpha‐ethnylestradiol. Steroids 27, 851–867 (1976). [DOI] [PubMed] [Google Scholar]
- 10. Maggs, J.L. & Park, B.K. A comparative study of biliary and urinary 2‐hydroxylated metabolites of [6,7‐3H]17 alpha‐ethynylestradiol in women. Contraception 32, 173–182 (1985). [DOI] [PubMed] [Google Scholar]
- 11. Department of Pharmaceutics & University of Washington . Drug Interaction Database Program. <https://www.druginteractioninfo.org/> Accessed 1 August 2017.
- 12. Wiesinger, H. et al Pharmacokinetic interaction between the CYP3A4 inhibitor ketoconazole and the hormone drospirenone in combination with ethinylestradiol or estradiol. Br. J. Clin. Pharmacol. 80, 1399–1410 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Luzon, E. , Blake, K. , Cole, S. , Nordmark, A. , Versantvoort, C. & Berglund, E.G. Physiologically based pharmacokinetic modeling in regulatory decision‐making at the European Medicines Agency. Clin. Pharmacol. Ther. 102, 98–105 (2016). [DOI] [PubMed] [Google Scholar]
- 14. Back, D.J. et al An investigation of the pharmacokinetics of ethinylestradiol in women using radioimmunoassay. Contraception 20, 263–273 (1979). [DOI] [PubMed] [Google Scholar]
- 15. Back, D.J. et al The gut wall metabolism of ethinylestradiol and its contribution to the pre‐systemic metabolism of ethinylestradiol in humans. Br. J. Clin. Pharmacol. 13, 325–330 (1982). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Back, D.J. et al The in vitro metabolism of ethinylestradiol, mestranol and levonorgestrel by human jejunal mucosa. Br. J. Clin. Pharmacol. 11, 275–278 (1981). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yang, J. , Jamei, M. , Yeo, K.R. , Tucker, G.T. & Rostami‐Hodjegan, A. Prediction of intestinal first‐pass drug metabolism. Curr. Drug Metab. 8, 676–684 (2007). [DOI] [PubMed] [Google Scholar]
- 18. Orme, M. , Back, D.J. , Ward, S. & Green, S. The pharmacokinetics of ethinylestradiol in the presence and absence of gestodene and desogestrel. Contraception 43, 305–316 (1991). [DOI] [PubMed] [Google Scholar]
- 19. Back, D.J. , Grimmer, S.F. , Rogers, S. , Stevenson, P.J. & Orme, M.L. Comparative pharmacokinetics of levonorgestrel and ethinyloestradiol following intravenous, oral and vaginal administration. Contraception 36, 471–479 (1987). [DOI] [PubMed] [Google Scholar]
- 20. Baumann, A. , Fuhrmeister, A. , Brudny‐Kloppel, M. , Draeger, C. , Bunte, T. & Kuhnz, W. Comparative pharmacokinetics of two new steroidal estrogens and ethinylestradiol in postmenopausal women. Contraception 54, 235–242 (1996). [DOI] [PubMed] [Google Scholar]
- 21. Kuhnz, W. , Humpel, M. , Biere, H. & Gross, D. Influence of repeated oral doses of ethinyloestradiol on the metabolic disposition of [13C2]‐ethinyloestradiol in young women. Eur. J. Clin. Pharmacol. 50, 231–235 (1996). [DOI] [PubMed] [Google Scholar]
- 22. Shiraga, T. , Niwa, T. , Ohno, Y. & Kagayama, A. Interindividual variability in 2‐hydroxylation, 3‐sulfation, and 3‐glucuronidation of ethinylestradiol in human liver. Biol. Pharm. Bull. 27, 1900–1906 (2004). [DOI] [PubMed] [Google Scholar]
- 23. Wang, B. , Sanchez, R.I. , Franklin, R.B. , Evans, D.C. & Huskey, S.E. The involvement of CYP3A4 and CYP2C9 in the metabolism of 17 alpha‐ethinylestradiol. Drug Metab. Dispos. 32, 1209–1212 (2004). [DOI] [PubMed] [Google Scholar]
- 24. Lin, W.H. , Feng, H.P. , Shadle, C.R. , O'Reilly, T. , Wagner, J.A. & Butterton, J.R. Pharmacokinetic and pharmacodynamic interactions between the hepatitis C virus protease inhibitor, boceprevir, and the oral contraceptive ethinyl estradiol/norethindrone. Eur. J. Clin. Pharmacol. 70, 1107–1113 (2014). [DOI] [PubMed] [Google Scholar]
- 25. Kasserra, C. , Li, J. , March, B. & O'Mara, E. Effect of vicriviroc with or without ritonavir on oral contraceptive pharmacokinetics: a randomized, open‐label, parallel‐group, fixed‐sequence crossover trial in healthy women. Clin. Ther. 33, 1503–1514 (2011). [DOI] [PubMed] [Google Scholar]
- 26. Doose, D.R. , Wang, S.‐S. , Padmanabhan, M. , Schwabe, S. , Jacobs, D. & Bialer, M. Effect of topiramate or carbamazepine on the pharmacokinetics of an oral contraceptive containing norethindrone and ethinyl estradiol in healthy obese and nonobese female subjects. Epilepsia 44, 540–549 (2003). [DOI] [PubMed] [Google Scholar]
- 27. Andrews, E. et al Pharmacokinetics and tolerability of voriconazole and a combination oral contraceptive co‐administered in healthy female subjects. Br. J. Clin. Pharmacol. 65, 531–539 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hilbert, J. , Messig, M. , Kuye, O. & Friedman, H. Evaluation of interaction between fluconazole and an oral contraceptive in healthy women. Obstet. Gynecol. 98, 218–223 (2001). [DOI] [PubMed] [Google Scholar]
- 29. Barditch‐Crovo, P. et al The effects of rifampin and rifabutin on the pharmacokinetics and pharmacodynamics of a combination oral contraceptive. Clin. Pharmacol. Ther. 65, 428–438 (1999). [DOI] [PubMed] [Google Scholar]
- 30. Murphy, P.A. , Kern, S.E. , Stanczyk, F.Z. & Westhoff, C.L. Interaction of St. John's Wort with oral contraceptives: effects on the pharmacokinetics of norethindrone and ethinyl estradiol, ovarian activity and breakthrough bleeding. Contraception 71, 402–408 (2005). [DOI] [PubMed] [Google Scholar]
- 31. Davis, A.R. , Westhoff, C.L. & Stanczyk, F.Z. Carbamazepine coadministration with an oral contraceptive: effects on steroid pharmacokinetics, ovulation, and bleeding. Epilepsia 52, 243–247 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Loi, C.M. , Stern, R. , Koup, J.R. , Vassos, A.B. , Knowlton, P. & Sedman, A.J. Effect of troglitazone on the pharmacokinetics of an oral contraceptive agent. J. Clin. Pharmacol. 39, 410–417 (1999). [DOI] [PubMed] [Google Scholar]
- 33. Robertson, P., Jr. , Hellriegel, E.T. , Arora, S. & Nelson, M. Effect of modafinil on the pharmacokinetics of ethinyl estradiol and triazolam in healthy volunteers. Clin. Pharmacol. Ther. 71, 46–56 (2002). [DOI] [PubMed] [Google Scholar]
- 34. Garg, V. , van Heeswijk, R. , Yang, Y. , Kauffman, R. , Smith, F. & Adda, N. The pharmacokinetic interaction between an oral contraceptive containing ethinyl estradiol and norethindrone and the HCV protease inhibitor telaprevir. J. Clin. Pharmacol. 52, 1574–1583 (2012). [DOI] [PubMed] [Google Scholar]
- 35. Sekar, V.J. et al Pharmacokinetic interaction between ethinyl estradiol, norethindrone and darunavir with low‐dose ritonavir in healthy women. Antivir. Ther. 13, 563–569 (2008). [PubMed] [Google Scholar]
- 36. Fattore, C. et al Induction of ethinylestradiol and levonorgestrel metabolism by oxcarbazepine in healthy women. Epilepsia 40, 783–787 (1999). [DOI] [PubMed] [Google Scholar]
- 37. Li, A.P. , Hartman, N.R. , Lu, C. , Collins, J.M. & Strong, J.M. Effects of cytochrome P450 inducers on 17alpha‐ethinyloestradiol (EE2) conjugation by primary human hepatocytes. Br. J. Clin. Pharmacol. 48, 733–742 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Smith, C.M. , Faucette, S.R. , Wang, H. & LeCluyse, E.L. Modulation of UDP‐glucuronosyltransferase 1A1 in primary human hepatocytes by prototypical inducers. J. Biochem. Mol. Toxicol. 19, 96–108 (2005). [DOI] [PubMed] [Google Scholar]
- 39. Luo, G. et al CYP3A4 induction by drugs: correlation between a pregnane X receptor reporter gene assay and CYP3A4 expression in human hepatocytes. Drug Metab. Dispos. 30, 795–804 (2002). [DOI] [PubMed] [Google Scholar]
- 40. Jamei, M. , Dickinson, G.L. & Rostami‐Hodjegan, A. A framework for assessing inter‐individual variability in pharmacokinetics using virtual human populations and integrating general knowledge of physical chemistry, biology, anatomy, physiology and genetics: a tale of ‘bottom‐up’ vs ‘top‐down’ recognition of covariates. Drug Metab. Pharmacokinet. 24, 53–75 (2009). [DOI] [PubMed] [Google Scholar]
- 41. Humpel, M. , Nieuweboer, B. , Wendt, H. & Speck, U. Investigations of pharmacokinetics of ethinyloestradiol to specific consideration of a possible first‐pass effect in women. Contraception 19, 421–432 (1979). [DOI] [PubMed] [Google Scholar]
- 42. Winiwarter, S. , Bonham, N.M. , Ax, F. , Hallberg, A. , Lennernas, H. & Karlen, A. Correlation of human jejunal permeability (in vivo) of drugs with experimentally and theoretically derived parameters. A multivariate data analysis approach. J. Med. Chem. 41, 4939–4949 (1998). [DOI] [PubMed] [Google Scholar]
- 43. Rowland Yeo, K. , Jamei, M. , Yang, J. , Tucker, G.T. & Rostami‐Hodjegan, A. Physiologically based mechanistic modeling to predict complex drug‐drug interactions involving simultaneous competitive and time‐dependent enzyme inhibition by parent compound and its metabolite in both liver and gut — the effect of diltiazem on the time‐course of exposure to triazolam. Eur. J. Pharm. Sci. 39, 298–309 (2010). [DOI] [PubMed] [Google Scholar]
- 44. Almond, L.M. et al Prediction of drug‐drug interactions arising from CYP3A induction using a physiologically based dynamic model. Drug Metab. Dispos. 44, 821–832 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Faassen, F. , Kelder, J. , Lenders, J. , Onderwater, R. & Vromans, H. Physicochemical properties and transport of steroids across Caco‐2 cells. Pharm. Res. 20, 177–186 (2003). [DOI] [PubMed] [Google Scholar]
- 46. Kuhnz, W. , Pfeffer, M. & al‐Yacoub, G. Protein binding of the contraceptive steroids gestodene, 3‐keto‐desogestrel and ethinylestradiol in human serum. J. Steroid Biochem. 35, 313–318 (1990). [DOI] [PubMed] [Google Scholar]
- 47. Pacifici, G.M. , Viani, A. , Rizzo, G. & Carrai, M. Plasma protein binding of ethinyloestradiol: effect of disease and interaction with drugs. Int. J. Clin. Pharmacol. Ther. Toxicol. 27, 362–365 (1989). [PubMed] [Google Scholar]
- 48. Sinofsky, F.E. & Pasquale, S.A. The effect of fluconazole on circulating ethinyl estradiol levels in women taking oral contraceptives. Am. J. Obstet. Gynecol. 178, 300–304 (1998). [DOI] [PubMed] [Google Scholar]
- 49. Inglis, A.M. et al Lack of effect of rosiglitazone on the pharmacokinetics of oral contraceptives in healthy female volunteers. J. Clin. Pharmacol. 41, 683–690 (2001). [DOI] [PubMed] [Google Scholar]
- 50. Weber, A. et al Can grapefruit juice influence ethinylestradiol bioavailability? Contraception 53, 41–47 (1996). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material