

Conspectus
Dynamic protein-DNA interactions constitute highly robust cellular machineries to fulfill cellular functions. A vast amount of studies have focused on how DNA-binding proteins search for and interact with their target DNA segments and on what cellular cues can regulate protein binding, for which protein concentration is a most obvious one. In contrast, how protein unbinding could be regulated by protein concentration has evaded attention because protein unbinding from DNA is typically a unimolecular reaction and thus concentration independent. Recent single-molecule studies from multiple research groups have uncovered that protein concentration can facilitate the unbinding of DNA-bound proteins, revealing regulation of protein unbinding as another mechanistic paradigm for gene regulation.
In this account, we review these recent in vitro and in vivo single-molecule experiments that uncovered the concentration-facilitated protein unbinding by multiple types of DNA-binding proteins, including sequence-nonspecific DNA-binding proteins (e.g., nucleoid-associated proteins; i.e., NAP), sequence-specific DNA-binding proteins (e.g., metal-responsive transcription regulators CueR and ZntR), sequence-neutral single-stranded DNA-binding proteins (e.g., Replication protein A; i.e., RPA), and DNA polymerases.
For the in vitro experiments, Marko’s group investigated the exchange of GFP-tagged DNA-bound NAPs with non-tagged NAPs in the solution of increasing concentration using single-molecule magnetic-tweezers-fluorescence microscopy. The faster fluorescence intensity decrease with higher non-tagged NAP concentrations suggests that DNA-bound NAPs undergo faster exchange with higher free NAP concentrations. Chen’s group used single-molecule fluorescence-resonance energy-transfer measurements to study the unbinding of CueR from its cognate oligomeric DNA. The average microscopic dwell times of DNA-bound states become shorter with increasing CueR concentrations in the surrounding, demonstrating that free CueR proteins can facilitate the unbinding of the incumbent one on DNA through either assisted dissociation or direct substitution. Greene’s group studied the unbinding of RPAs from single-stranded DNA using total internal reflection fluorescence microscopy and DNA curtain techniques. The fluorescence-intensity-versus-time traces show faster decay with higher wild-type RPA concentrations, indicating that DNA-bound RPAs can undergo a concentration-facilitated exchange when encountering excess free RPA. van Oijen’s group investigated the leading/lagging-strand polymerase exchange events in the bacteriophage T7 and E. coli replication systems using a combination of single-molecule fluorescence microscopy and DNA-flow-stretching assay. The processivity was observed to have larger decrease when the concentration of the Y526F polymerase mutant increases, indicating that the unbinding of the polymerase is also concentration-dependent. Using stroboscopic imaging and single-molecule tracking, Chen’s group further advanced their study into living bacterial cells. They found CueR, as well as its homologue ZntR, shows concentration-enhanced unbinding from their respective DNA-binding sites in vivo.
Mechanistic consensus has emerged from these in vitro and in vivo single-molecule studies that encompass a range of proteins with distinct biological functions. It involves multivalent contacts between protein and DNA. The multivalency enables the formation of ternary complexes as intermediates, which subsequently give rise to concentration-enhanced protein unbinding. As multivalent contacts are ubiquitous among DNA-interacting proteins, this multivalency-enabled facilitated unbinding mechanism thus provides a potentially general mechanistic paradigm in regulating protein-DNA interactions.
Graphical Abstract
1. Introduction
Dynamic protein-DNA interactions give rise to robust cellular machineries that integrate processes such as DNA replication, transcription, repair, packaging, and gene regulation. Regulatory mechanisms of these processes have attracted research interests and initiated a broad range of studies.1,2 A majority of these studies have focused on how proteins search and bind to DNA for function, and many mechanistic insights have been obtained into how cells could alter protein binding to DNA in response to stimuli. Much less is known, however, on how cells manipulate DNA-bound proteins after stimuli are removed or when new stimuli arise.
Out of many cellular cues, the concentration of a DNA-binding protein itself is perhaps the most obvious one that directly impacts its binding to DNA — with increasing protein concentration, the binding rate of a protein to any DNA site increases linearly in general.3 On the other hand, unbinding of a protein from DNA is typically a unimolecular process; its kinetics is thus independent of protein concentration (and of DNA concentration as well). A few abnormal behaviors were reported, however, in bulk solution experiments,4–9 where some DNA-binding proteins showed faster unbinding rates from DNA when the concentration of DNA (not protein) was increased, giving rise to the proposed facilitated dissociation pathway.
Recent single-molecule studies in vitro have further uncovered protein-concentration-dependent unbinding from DNA, besides the dependence on the DNA concentration. These include sequence-nonspecific nucleoid-associated proteins,10,11 DNA replication enzymes,12–16 sequence-specific metal-responsive transcription regulators,17 and nonspecific single-stranded DNA (ssDNA) binding proteins.18,19 Latest single-molecule-tracking studies showed that the protein-concentration-dependent unbinding also operates in living cells for transcription regulators,20 demonstrating the physiological relevance of this unusual protein unbinding behavior. Mechanistic consensus is emerging from these studies, and it generally involves intermediate ternary complexes enabled by the multivalent contacts of the protein with DNA. This mechanism is further corroborated by theoretical work,21–24 as well as by an earlier experimental observation of a ternary protein2-DNA complex involving a metal-responsive transcription regulator.25
The fact that these proteins belong to unrelated families having distinct functions but all possess this protein-concentration-dependent unbinding suggests a broad relevance of this unusual unbinding behavior across the spectrum of nucleic-acid-interacting proteins. Here we review key single-molecule experiments and experimental observations across these different protein systems, highlight the consensus concept, and point out other cellular cues that can also alter protein unbinding from DNA.
2. Concentration-dependent protein unbinding from DNA in vitro
In this section, we review and discuss the mechanisms of four different classes of DNA-interacting proteins that exhibit protein-concentration-dependent unbinding kinetics, including nucleoid-associated proteins, transcription regulators, ssDNA-binding proteins, and DNA polymerases.10–19
2.1. Sequence-nonspecific DNA-binding proteins: Nucleoid-associated proteins
Nucleoid-associated proteins (NAPs) bind to DNA in a sequence-nonspecific manner26 to compact and prevent DNA from radiation or chemical damages and to affect transcription activities. Four major NAPs are responsible for modifying the compactness of DNA: the factor for inversion-stimulation protein (Fis), the histone-like nucleoid-structuring protein (H-NS), the histone-like protein from Escherichia coli strain U93 (HU), and the integration host factor (IHF) (Figure 1a).
Figure 1.

(a) NAPs interact with bacterial chromosome and shape the DNA structure for different functions. (b) Experimental configuration of the single-molecule magnetic-tweezers-fluorescence microscope. (c) Experimental results of the Fis exchange assays. The intensity decay is due to the replacement of the pre-bound GFP-Fis on DNA by the WT-Fis in solution. The intensity versus time trajectories indicate that the Fis exchange rate gets faster with higher concentrations of WT-Fis. (d) Schematic mechanism for the facilitated unbinding process. Panels b and c reproduced with permission from Reference10. Copyright 2010 Oxford University Press. Panel d reproduced with permission from Reference27. Copyright 2015 Elsevier Ltd.
Marko’s group investigated the unbinding kinetics of DNA-bound NAPs as a function of the concentration of surrounding NAPs using a single-molecule magnetic-tweezers-fluorescence microscope (Figure 1b).10 HU, Fis, and NHP6A (non-histone protein 6A, a NAP analog in yeast) were chosen as model systems. With one end fixed onto a glass capillary tube and the other end attached to a magnetic bead, DNAs were first stretched in the imaging plane using magnetic tweezers (Figure 1b). Green fluorescence protein (GFP) tagged Fis proteins were incubated with the tethered DNA; the wild-type Fis (WT-Fis), WT-HU, or WT-NHP6A were then introduced. GFP fluorescence signal served as the readout of the DNA-bound GFP-Fis, which decreases over time due to protein unbinding from DNA (Figure 1c). Surprisingly, they observed that the fluorescence intensity decreases faster when there is non-tagged protein in the surrounding. They concluded that DNA-NAPs complexes are more stable in the absence of free proteins in the flow cell, but bound NAPs could undergo exchange with free proteins in solution regardless of its identity (i.e., the bound Fis could be exchanged by Fis, HU, or NHP6A). Consistently, the exchange rate gets faster with higher protein concentrations in the solution (Figure 1c), supporting that the free NAPs can facilitate the unbinding of bound ones off the DNA. Facilitated Fis and NHP6A unbinding were similarly observed when DNA segments or isolated bacterial chromosome were included in the solution.27,28
Marko and coworkers proposed a multiple binding site mechanism29 and a “direct transfer” model27 to explain the protein- and DNA-segment-facilitated dissociation, respectively (Figure 1d). Both models invoke a partial-releasing state (i.e., part of the protein detaches from DNA, vacating a partial-binding site). In the protein-facilitated pathway, the vacated partial binding site on DNA is subsequently bound by a competitor protein, eventually leading to the complete unbinding of the original protein (Figure 1d, upper). In the DNA-segment-facilitated pathway, the partially detached protein uses the vacated binding domain to interact with another DNA, subsequently leading to the detachment of the original DNA (Figure 1d, lower). Both of these pathways give rise to an apparent unbinding of the original protein, and both of their kinetics are faster with increasing concentrations of the surrounding protein or DNA.
Very recently, Marko’s group further studied NAP unbinding from a single DNA binding site30. A single binding site was achieved using a short DNA that is capable of interacting with only one helix-turn-helix domain of the dimeric Fis and monomeric NHP6A proteins. Observations of facilitated dissociation for both Fis and NHP6A proteins indicate that facilitated dissociation can occur without the need for two DNA-binding domains for a protein-DNA complex as long as the transcription factor-DNA complex can partially unbind and expose part of the binding site on DNA to invasion by competitors, essentially with each binding domain acting as a “multivalent DNA binder”.
2.2. Sequence-specific DNA-binding proteins: metal-responsive transcription regulators
In E. coli, the copper-efflux regulator (CueR), a MerR-family metal-responsive regulator, responds to Cu+ and Ag+ ions to activate genes that help cells defend against these toxic metals. Transcription regulation of CueR operates through a DNA-distortion mechanism, in which the apo-repressor and holo-activator forms bind to DNA, causing different DNA structure changes to invoke repression or activation (Figure 2a).31 Little was known, however, about how the transcription regulation switches facilely between the activated state (bound holo-protein) and the repressed state (bound apo-protein), as both apo- and holo-forms bind to the same operator site tightly and metal dissociation from the holo-protein is likely slow.17
Figure 2.

(a) DNA-distortion mechanism of CueR. Apo- and holo-CueR bind to DNA to repress and activate the transcription of metal-defense genes. (b) SmFRET experimental design to study the binding and unbinding events between CueR molecules and DNA. (c) (Upper) single-molecule EFRET trajectory of an immobilized Cy3-DNA interacting with Cy5-labeled apo-CueR shows three EFRET states and microscopic dwell times of each state. (Lower) same as the upper panel but Cy3-DNA interacts with a mixture of Cy5-labeled apo-CueR and holo-CueR, in which Cy5 on holo-CueR is at a different location from that of apo-CueR. The blue arrows denote the transitions from the holo-protein-bound states to the apo-protein-bound states, and the black arrows denote the reverse transitions; these transitions report the direct substitution of a DNA-bound holo-protein by an apo-protein or the reverse. (d) The average dwell time (〈τ2〉) gets shorter with increasing [CueR], reflecting the direct-substitution and assisted-dissociation pathways of CueR-DNA interactions. (e) Proposed mechanism involving a ternary CueR2-DNA complex as an intermediate for direct-substitution and assisted-dissociation pathways to a CueR bound at a specific DNA-binding site. Reproduced with permission from Reference32 and reference17. Copyright 2013 American Chemical Society and 2012 National Academy of Sciences.
We have used single-molecule fluorescence-resonance-energy-transfer (smFRET)17 to study CueR unbinding from its cognate oligomeric DNA, which was surface-immobilized and labeled with a FRET-donor Cy3 (Figure 2b). The free, FRET-acceptor Cy5-labeled CueR interacts with DNA, and the associated FRET efficiency (EFRET) directly reports the protein-DNA interactions (Figure 2c). The EFRET versus time trajectory shows transitions among three different EFRET states: the E0 state corresponds to the free DNA, and E1 and E2 correspond to two different binding orientations of CueR on DNA differentiated by the single Cy5 label (Figure 2c, upper). The average microscopic dwell times of E1 and E2 states (i.e., 〈τ1〉 and 〈τ2〉) in the EFRET trajectory defines the unbinding rate of CueR from the DNA. Strikingly, the average microscopic dwell times of E1 and E2 states become shorter with increasing CueR concentrations in the surrounding, reflecting that free CueR proteins can facilitate the unbinding of the incumbent one on DNA (Figure 2d).
We proposed that the observed facilitated unbinding at the specific binding site on DNA is through a protein2-DNA ternary complex as an intermediate (Figure 2e). In this mechanism, one of the two DNA-binding domains of the homodimeric CueR detaches from half of the binding site momentarily, allowing another CueR molecule in the solution to bind and form the protein2-DNA ternary complex. This ternary complex can then fall apart, leading to an assisted-dissociation pathway (Figure 2e, lower), or undergo a swap of the two proteins on DNA, leading to a direct-substitution pathway (Figure 2e, upper). Both of these pathways would result in an apparent facilitated unbinding of the incumbent protein when the protein concentration in the surrounding increases. Using a mixture of differently labeled CueR molecules, the direct-substitution pathway was directly observed in the EFRET trajectories (Figure 2c, lower). The ternary protein2-DNA complex, enabled by the bivalent contact between CueR and DNA, was also observed kinetically in a separate study of CueR using engineered DNA Holliday junctions,25 as we described.32
2.3. Sequence-nonspecific ssDNA-binding proteins: replication protein A (RPA)
Sequence-nonspecific ssDNA-binding (SSB) proteins are important for DNA replication, recombination, and repair functions. Replication protein A (RPA) is a heterotrimeric enzyme with four sequence-nonspecific SSB motifs.33 RPAs protect ssDNA from enzymatic degradation, remove ssDNA’s secondary structure, which can inhibit DNA repair, and recruit specific proteins to ssDNA for DNA metabolism (Figure 3a). During the initiation of homologous recombination, for example, RPA is displaced from ssDNA by Rad51.34 The tight binding between RPA and ssDNA (Kd ~ 10−9–10−11 M)35 implies that cells must have a mechanism to unbind RPA from ssDNA facilely for DNA metabolism involving ssDNA intermediates.
Figure 3.

(a) Schematic representation of RPA’s function in DNA replication (left) and recombination (right). (b) Experimental device for a double-tethered DNA-curtain assay. (c) The kymograph (upper) and intensity-versus-time traces of eGFP-RPA under various WT-RPA concentrations (lower). The fluorescent signals of eGFP-RPA decreased with time when WT-RPA was adding to the flow cell. The concentration-dependent-RPA-exchange rate reflects a facilitated dissociation (or exchange) behavior. (d) Schematic depiction of the microscopic dissociation mechanism for the facilitated-dissociation/exchange phenomenon of RPA. Panel b reproduced with permission from Reference36. Copyright 2012 National Academy of Sciences. Panel c and d reproduced with permission from Reference18. Copyright 2014 Public Library of Science.
Greene’s group studied the unbinding of yeast and human RPAs from ssDNA at the single-molecule level using total-internal-reflection-fluorescence (TIRF) microscopy and DNA-curtain 18,19,36 techniques (Figure 3b). In this DNA curtain, a large number of ssDNAs were stretched out by solution flow simultaneously and anchored on both ends.37,38 RPAs tagged with enhanced GFP (eGFP-RPA) were flowed in to bind to the ssDNA curtains, allowing many individual eGFP-RPA-ssDNA complexes to be visualized by TIRF microscopy. The intensity-versus-time traces showed that RPA-ssDNA complexes are stable (half-life > two hours) in an RPA-free flowing solution. However, in the presence of WT-RPA, the fluorescence intensity, which is proportional to the number of eGFP-RPA bound to ssDNA, decreased more noticeably, and the decay rate increased with higher WT-RPA concentrations (Figure 3c). This indicates that RPAs can undergo facilitated exchange or concentration-dependent unbinding from the complex when encountering excess RPA. The same result was also observed with another SSB protein, Rad51.
Greene et al. introduced a microscopic dissociation18 mechanism to explain their experimental results, in which the protein is partially dissociated from its binding site but not equilibrated with the surrounding solution yet (Figure 3d). For RPA, four domains donate contacts with ssDNA; each of these domains can undergo a rapid unbinding and rebinding process. The low chance of simultaneous dissociation of all four domains results in a stable complex in the absence of free SSB proteins in the surrounding (Figure 3d, left). On the other hand, in the presence of free SSB proteins, they would bind to the individual released ssDNA and consequently cause exchange (Figure 3d, right). Notably, the facilitated exchange of RPA can be stimulated by both RPA and SSB from bacteria18, highlighting that these events are species independent and further supporting that the mechanism is driven by the partial protein dissociation instead of a specific protein-protein contact.
2.4. Multicomponent protein machinery: DNA polymerases
DNA replication is a multi-step process starting from DNA unwinding to RNA primer synthesis, DNA elongation, error checking, and eventually to termination. This intricate procedure is governed by a multiprotein replisome machinery including DNA helicases, primases, DNA polymerases, ssDNA-binding proteins, and other components like clamp, clamp loader or processivity factors (Figure 4a). Replisome was thought to be a stable complex so as to facilitate its function in synthesizing DNA rapidly and continuously. However, rapid exchange of components was recently observed, pointing out the dynamic feature and flexibility of replisome.12–16,39–43
Figure 4.

(a) Schematic diagram and components for the bacteriophage T7 replication system. (b) Experimental design for examining the polymerase exchange during leading strand DNA synthesis. The DNA synthesis rate was calculated by the movement of QD within a given time window. (c) WT and mutant (Y526F) polymerases possess different DNA synthesis rates demonstrated by different slopes in blue and red trajectories (left). The processivity is calculated from the leading-strand DNA synthesis. Results from various mutant concentrations give the titration curve on the right. (d) Experimental design for studying the polymerase exchange during lagging-strand DNA synthesis (upper). Fluctuations in intensity-versus-time trajectory represent polymerase exchange events between different fluorophores (blue and red) (lower). (e) DNA polymerase binds the DNA template (primary binding site) and helicase (secondary binding site). The partial-dissociation state allows free polymerase filling in the opened binding site and consequently displacing the original polymerase. (f) in vitro fluorescence intensity of Pol III* was monitoring with a periodic high laser power pulse (upper). The normalized intensity after pulse was plotted as a function of time with four Pol III* concentrations (lower). Panels a–d reproduced with permission from Reference12,15,39. Copyright 2007, 2011 and 2014 National Academy of Sciences. Panel e reproduced with permission from Reference42. Copyright 2016 Oxford University Press. Panel f reproduced with permission from Reference16. Copyright 2017 eLife Sciences Publications.
van Oijen’s group investigated the T7 polymerase-exchange events in the replisome using a combination of single-molecule fluorescence microscopy and DNA-flow-stretching assays (Figure 4b).12,15 For the leading-strand polymerase study13, a long DNA, onto which a quantum dot (QD) was attached, was tethered to a surface and stretched out by the flow of buffer. Tracking the motion of the QD directly probed the shortening of the DNA construct due to the conversion of the lagging strand into ssDNA and thus provided the processivity information (i.e., amount of DNA synthesis) (Figure 4c, left). The polymerase exchange was examined by introducing unlabeled slow-DNA-synthesis polymerase mutant (Y526F) to the preassembled complex where the labeled T7-DNA polymerase and gp4 helicase/primase were bound to DNA. The processivity was observed to decrease in the presence of the Y526F mutant (Figure 4c, left), and more important, the extent of this decrease got larger when the mutant concentration increased (Figure 4c, right), directly indicating that the unbinding of the polymerase is concentration dependent. For the lagging-strand polymerase study,15 a circular double-stranded DNA molecule was fixed on a coverslip and stretched by a hydrodynamic flow in a microfluidic chamber (Figure 4d, upper). During the rolling-cycle DNA replication, fluorophore-tagged polymerases were detected along with the lagging-strand DNA synthesis by TIRF microscopy. Polymerases exchange was evidenced by the fluorescence signal fluctuations when two color-coded polymerases were present simultaneously in the experiments (Figure 4d, lower). More important, the fluorescence intensity of each spot showed that more than two polymerases could reside in the replisome; the non-DNA-synthesizing polymerases bound with helicases and played as competitors in the dissociation process.
van Oijen’s group proposed a multisite competitive exchange mechanism15,42 to explain the above phenomenon, such as a model with two binding sites (1 and 2) that simplifies potential scenarios (Figure 4e). The polymerase molecule dissociates from the complex only when it unbinds from both sites. A molecule remains stable when the speed of rebinding to one site is faster than that of unbinding from the other site. Therefore, the dissociation rate depends on the unbinding and rebinding rate of the polymerase from and to the two binding sites in diluted or no-competitive-molecule solution. In the presence of competitors, three steps are required to take place for them to substitute the original molecule: (1) the original molecule unbinds from one site, (2) the competitor binds to the now vacant site before the rebinding occurs, and (3) the new molecule stays for a sufficient amount of time for the original molecule to completely unbind. Simulations using this two-binding-site model illustrate that the strength of the competitor, determined by their microscopic binding rate constant and their concentration, dominates the exchange speed.
Very recently, they and Reyes-Lamothe’s group both examined the exchange events of DNA polymerase III complex from E. coli during replication in live cells.16,44 van Oijen’s group investigated the exchange of Pol III* in live cells by single-molecule fluorescence recovery after photobleaching and tracking (Figure 4f). They quantified the exchange rate in vitro by monitoring the recovery of the fluorescence intensity and observed that the exchange time is shorter when the surrounding Pol III* is at a higher concentration (Figure 4f). Moreover, they showed the exchange of Pol III* in live cells by tracking DNA bound fluorescently tagged Pol III*s (i.e., fused yellow- and red-fluorescent proteins at τ and ε subunit, respectively). Combined with simulations, the cross-correlation analysis of signals from τ and ε units denotes the Pol III exchange in vivo. However, changing the concentration of Pol III* is a very challenging task due to the requirement of simultaneously changing the concentration of all individual subunits in a controllable manner without perturbing other metabolic processes. Data of direct protein-concentration-facilitated exchange in cells remains lacking.
3. Concentration-dependent protein unbinding from DNA in vivo
The above in vitro experiments established that protein unbinding from DNA can be facilitated by itself, as manifested by the dependence on protein concentration. It remained unknown, however, whether this facilitated unbinding would be relevant in a living cell environment. Combining single-molecule super-resolution tracking and genetic engineering, our group20 further investigated the unbinding of CueR, as well as it Zn2+-sensing homolog ZntR, from chromosomal recognition sites in living E. coli cells to probe how bacteria could use this unbinding mechanism for regulating transcription.
To study CueR (and ZntR) interactions with DNA in living E. coli cells, we tagged regulators with the photoconvertible fluorescent protein mEos3.2 at their chromosomal loci, as well as encoded the tagged genes in a plasmid, from which the regulator expressions could be induced. To mimic the holo-activator form, we grew cells in the presence of a high concentration of metal that was known to cause maximal induction of their respective regulons. The apo-repressor forms of CueR and ZntR were made by mutating one of their metal-binding cysteines. Using time-lapse stroboscopic imaging,2,45 we tracked the motions of single photoconverted tagged regulator proteins at tens of nanometer precision until the tag photobleached (Figure 5a). Analysis of single-molecule displacement (r) distributions gave the minimal number of diffusion states and their corresponding diffusion constants (Figure 5b). The state where the regulator is bound specifically to recognition sites could be resolved and has a very small diffusion constant similar to that of chromosomal loci.46 From the resolved distribution of r, one could determine an upper displacement threshold r0 (Figure 5b). Thresholding the single-molecule displacement-versus-time trajectories with r0 extracts the microscopic residence times of a single regulator at any specific binding site (Figure 5c). Analysis of the distribution of microscopic residence times using a kinetic model gave the apparent unbinding rate constant ( ) of the regulator from its chromosomal specific binding sites. Strikingly, but as predicted from the in vitro work in Section 2.2, for both apo- and holo-forms of CueR and ZntR, their get larger with increasing regulator concentrations in the cell (Figure 5d), demonstrating concentration-enhanced unbinding in vivo, which likely results from assisted-dissociation and direction-substitution pathways that we delineated in vitro for CueR (Figure 2d).
Figure 5.

(a) Single-molecule tracking (left) using stroboscopic imaging scheme together with super-resolution analysis generates the moving trajectories (right) and displacement (r) trajectory (panel c) in living cells. (b) Histogram of displacement r and the corresponding resolved diffusion states. The black solid line is the overall probability-density-function. Red, green, and blue lines represent the populations of the regulator at the specifically-bound (SB), non-specifically bound (NB), and freely-diffusing (FD) states, respectively. The vertical red dashed line denotes the displacement threshold, r0, to select out microscopic residence time of CueR molecules bound to specific binding sites. (c) Displacement-per-time-lapse-versus-time trajectory for the tracked CueR. τ1 and τ2 are two microscopic residence times (two grey shades) thresholded by r0 (horizontal red dashed line). (d) Dependences of on free CueR concentration in cells. (e) Proposed transcription regulation processes in live bacteria using the concentration-dependent unbinding mechanism. Panels b–d reproduced with permission from Reference20. Copyright 2015 Nature Publishing Group.
The facilitated unbinding of apo- and holo-CueR (and ZntR) from specific binding sites could provide a new mechanism for the cell to switch facilely between transcription activation and repression of the respective regulons. For example, after intracellular metal stresses have been relieved through transcription activation of metal resistance genes, a bacterial cell can promptly turn off the transcription of the metal resistance genes using the intracellular free apo-repressor forms of the regulator. This can be achieved via the assisted-dissociation pathway followed by the binding of an apo-repressor, or via the direct-substitution pathway (Figure 5e, left). On the other hand, bacteria can also facilely turn on the transcription when challenged with excess metals, which are likely scavenged initially by intracellular free apo-regulator proteins, which are present in many copies, rather than by the promoter-bound one. Instead of waiting for the metal to find the promoter-bound apo-repressor through a competition process (i.e., against many other apo-repressors in the cell), a free holo-regulator can facilitate the unbinding of the incumbent apo-repressor and may directly substitute it to activate the transcription of metal resistance genes (Figure 5e, right).
4. Concluding remarks
Unbinding of protein from DNA is a fundamental process in protein-DNA interactions. Here we have reviewed the few identified proteins whose unbinding can be facilitated by their concentrations. Mechanistically, concentration-dependent protein unbinding from DNA likely only needs multivalent protein-DNA contacts. This multivalency can occur via multiple DNA-binding sites or a single-binding site that could be partially vacated, making possible for multiple-protein binding to the same DNA (Figure 6). Formation of a ternary complex involves an original bound protein to partially unbind and expose a (partial) binding site for a competitor to bind, leading to the concentration dependence on the competitor, which could be protein itself. The broad functional range of these proteins studied so far highlight the generality and importance of the protein unbinding on cellular functions. In fact, facilitated unbinding (from other proteins instead of DNA) was also observed in the bacterial flagellar-switch-protein FliM, in which the turnover of FliM molecules was abolished in the CheY knockout strain.47 These discoveries also corroborate the thought that protein unbinding from DNA is perhaps as much regulated as binding in the cell.
Figure 6.
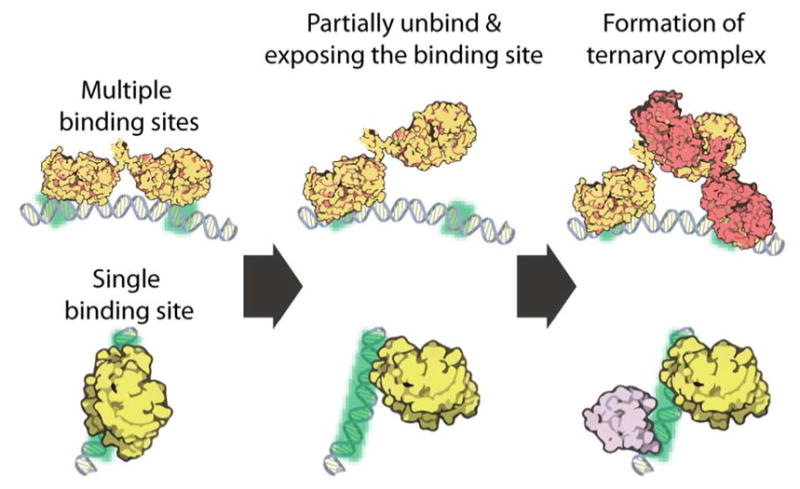
General mechanism of protein-concentration-dependent unbinding from DNA.
Other cellular cues, aside from protein concentration, could play important roles as well in affecting unbinding kinetics, for example, those perturbing the stability of protein-DNA complexes. Recent studies showed salt concentrations,11,27 mechanical force,48 and chromosomal organization20 could also alter protein-unbinding kinetics, even in living cells.20 Moreover, post-translational modifications such as acetylation and phosphorylation were found to alter proteins’ DNA-binding affinity,49 which may serve another pathway to alter protein unbinding. It is clear that both protein-binding and unbinding pathways are crucial for normal cellular functions, and single-molecule-level mechanistic studies could provide molecular insights into these cellular processes.
Acknowledgments
T.-Y.C. acknowledges the Department of Chemistry and the Division of Research at the University of Houston for startup funds. P.C. acknowledges support from the National Institute of Health (GM109993, AI117295, and GM106420) and Army Research Office (W911NF-15-1-0268).
Biographies
Tai-Yen Chen is an assistant professor of Chemistry at the University of Houston. He obtained his Ph.D. with Prof. Dong Hee Son in physical chemistry from Texas A&M University in 2010. He then did postdoctoral research in single-molecule biophysics with Prof. Peng Chen at Cornell University before starting at the University of Houston in 2016. His current research focuses on single-molecule metallo-neurobiology.
Yu-Shan Cheng obtained her Ph.D. in biochemistry from Texas A&M University in 2016, and has been a postdoctoral fellow in the Department of Chemistry at the University of Houston in Prof. Tai-Yen Chen’s lab since 2016.
Pei-San Huang obtained her Ph.D. in Veterinary Integrative Bioscience from Texas A&M University in 2012, and has been a postdoctoral fellow in the Department of Chemistry at the University of Houston in Prof. Tai-Yen Chen’s lab since2016.
Peng Chen is the Peter J. W. Debye Professor of Chemistry at Cornell University. He did his Ph.D. with Edward Solomon in bioinorganic/physical inorganic chemistry at Stanford University and postdoctoral research with Sunney Xie in single-molecule biophysics at Harvard University, before starting at Cornell in 2005. His current research focuses on single-molecule imaging of catalysis and bioinorganic/biophysical chemistry.
Footnotes
Author Contribution
The manuscript was written through contributions of all authors, who also have approved the final version of the manuscript.
The authors declare no competing financial interest.
References
- 1.Mueller F, Stasevich TJ, Mazza D, McNally JG. Quantifying transcription factor kinetics: at work or at play? Crit Rev Biochem Mol Biol. 2013;48:492–514. doi: 10.3109/10409238.2013.833891. [DOI] [PubMed] [Google Scholar]
- 2.Elf J, Li GW, Xie XS. Probing transcription factor dynamics at the single-molecule level in a living cell. Science. 2007;316:1191–1194. doi: 10.1126/science.1141967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beshnova DA, Cherstvy AG, Vainshtein Y, Teif VB. Regulation of the nucleosome repeat length in vivo by the DNA sequence, protein concentrations and long-range interactions. PLOS Comput Biol. 2014;10:e1003698. doi: 10.1371/journal.pcbi.1003698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Doucleff M, Clore GM. Global jumping and domain-specific intersegment transfer between DNA cognate sites of the multidomain transcription factor Oct-1. Proc Natl Acad Sci USA. 2008;105:13871–13876. doi: 10.1073/pnas.0805050105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Iwahara J, Zweckstetter M, Clore GM. NMR structural and kinetic characterization of a homeodomain diffusing and hopping on nonspecific DNA. Proc Natl Acad Sci USA. 2006;103:15062–15067. doi: 10.1073/pnas.0605868103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kozlov AG, Lohman TM. Kinetic mechanism of direct transfer of Escherichia coli SSB tetramers between single-stranded DNA molecules. Biochemistry. 2002;41:11611–11627. doi: 10.1021/bi020361m. [DOI] [PubMed] [Google Scholar]
- 7.Lieberman BA, Nordeen SK. DNA intersegment transfer, how steroid receptors search for a target site. J Biol Chem. 1997;272:1061–1068. doi: 10.1074/jbc.272.2.1061. [DOI] [PubMed] [Google Scholar]
- 8.Ruusala T, Crothers DM. Sliding and intermolecular transfer of the lac repressor: kinetic perturbation of a reaction intermediate by a distant DNA sequence. Proc Natl Acad Sci USA. 1992;89:4903–4907. doi: 10.1073/pnas.89.11.4903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fried MG, Crothers DM. Kinetics and mechanism in the reaction of gene regulatory proteins with DNA. J Mol Biol. 1984;172:263–282. doi: 10.1016/s0022-2836(84)80026-1. [DOI] [PubMed] [Google Scholar]
- 10.Graham JS, Johnson RC, Marko JF. Concentration-dependent exchange accelerates turnover of proteins bound to double-stranded DNA. Nucleic Acids Res. 2011;39:2249–2259. doi: 10.1093/nar/gkq1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hadizadeh N, Johnson RC, Marko JF. Facilitated Dissociation of a Nucleoid Protein from the Bacterial Chromosome. J Bacteriol. 2016;198:1735–1742. doi: 10.1128/JB.00225-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Loparo JJ, Kulczyk AW, Richardson CC, van Oijen AM. Simultaneous single-molecule measurements of phage T7 replisome composition and function reveal the mechanism of polymerase exchange. Proc Natl Acad Sci USA. 2011;108:3584–3589. doi: 10.1073/pnas.1018824108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kath JE, Jergic S, Heltzel JM, Jacob DT, Dixon NE, Sutton MD, Walker GC, Loparo JJ. Polymerase exchange on single DNA molecules reveals processivity clamp control of translesion synthesis. Proc Natl Acad Sci USA. 2014;111:7647–7652. doi: 10.1073/pnas.1321076111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kath JE, Chang S, Scotland MK, Wilbertz JH, Jergic S, Dixon NE, Sutton MD, Loparo JJ. Exchange between Escherichia coli polymerases II and III on a processivity clamp. Nucleic Acids Res. 2016;44:1681–1690. doi: 10.1093/nar/gkv1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Geertsema HJ, Kulczyk AW, Richardson CC, van Oijen AM. Single-molecule studies of polymerase dynamics and stoichiometry at the bacteriophage T7 replication machinery. Proc Natl Acad Sci USA. 2014;111:4073–4078. doi: 10.1073/pnas.1402010111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lewis JS, Spenkelink LM, Jergic S, Wood EA, Monachino E, Horan NP, Duderstadt KE, Cox MM, Robinson A, Dixon NE. Single-molecule visualization of fast polymerase turnover in the bacterial replisome. eLife. 2017;6:e23932. doi: 10.7554/eLife.23932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Joshi CP, Panda D, Martell DJ, Andoy NM, Chen TY, Gaballa A, Helmann JD, Chen P. Direct substitution and assisted dissociation pathways for turning off transcription by a MerR-family metalloregulator. Proc Natl Acad Sci USA. 2012;109:15121–15126. doi: 10.1073/pnas.1208508109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gibb B, Ye LF, Gergoudis SC, Kwon Y, Niu H, Sung P, Greene EC. Concentration-dependent exchange of replication protein A on single-stranded DNA revealed by single-molecule imaging. PLoS One. 2014;9:e87922. doi: 10.1371/journal.pone.0087922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ma CJ, Gibb B, Kwon Y, Sung P, Greene EC. Protein dynamics of human RPA and RAD51 on ssDNA during assembly and disassembly of the RAD51 filament. Nucleic Acids Res. 2017;45:749–761. doi: 10.1093/nar/gkw1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen TY, Santiago AG, Jung W, Krzeminski L, Yang F, Martell DJ, Helmann JD, Chen P. Concentration- and chromosome-organization-dependent regulator unbinding from DNA for transcription regulation in living cells. Nat Commun. 2015;6:7445. doi: 10.1038/ncomms8445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tsai MY, Zhang B, Zheng W, Wolynes PG. Molecular Mechanism of Facilitated Dissociation of Fis Protein from DNA. J Am Chem Soc. 2016;138:13497–13500. doi: 10.1021/jacs.6b08416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Paramanathan T, Reeves D, Friedman LJ, Kondev J, Gelles J. A general mechanism for competitor-induced dissociation of molecular complexes. Nat Commun. 2014;5:5207. doi: 10.1038/ncomms6207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dahlke K, Sing CE. Facilitated Dissociation Kinetics of Dimeric Nucleoid-Associated Proteins Follow a Universal Curve. Biophys J. 2017;112:543–551. doi: 10.1016/j.bpj.2016.11.3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen C, Bundschuh R. Quantitative models for accelerated protein dissociation from nucleosomal DNA. Nucleic Acids Res. 2014;42:9753–9760. doi: 10.1093/nar/gku719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Andoy NM, Sarkar SK, Wang Q, Panda D, Benítez JJ, Kalininskiy A, Chen P. Single-Molecule Study of Metalloregulator CueR-DNA Interactions Using Engineered Holliday Junctions. Biophys J. 2009;97:844–852. doi: 10.1016/j.bpj.2009.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Swinger KK, Rice PA. Structure-based analysis of HU-DNA binding. J Mol Biol. 2007;365:1005–1016. doi: 10.1016/j.jmb.2006.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Giuntoli RD, Linzer NB, Banigan EJ, Sing CE, de la Cruz MO, Graham JS, Johnson RC, Marko JF. DNA-Segment-Facilitated Dissociation of Fis and NHP6A from DNA Detected via Single-Molecule Mechanical Response. J Mol Biol. 2015;427:3123–3136. doi: 10.1016/j.jmb.2015.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hadizadeh N, Johnson RC, Marko JF. Facilitated dissociation of a nucleoid protein from the bacterial chromosome. J Bacteriol. 2016;198:1735–1742. doi: 10.1128/JB.00225-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sing CE, Olvera de la Cruz M, Marko JF. Multiple-binding-site mechanism explains concentration-dependent unbinding rates of DNA-binding proteins. Nucleic Acids Res. 2014;42:3783–3791. doi: 10.1093/nar/gkt1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kamar RI, Banigan EJ, Erbas A, Giuntoli RD, Olvera de la Cruz M, Johnson RC, Marko JF. Facilitated dissociation of transcription factors from single DNA binding sites. Proc Natl Acad Sci USA. 2017;114:E3251–E3257. doi: 10.1073/pnas.1701884114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.O’Halloran TV, Frantz B, Shin MK, Ralston DM, Wright JG. The MerR heavy metal receptor mediates positive activation in a topologically novel transcription complex. Cell. 1989;56:119–129. doi: 10.1016/0092-8674(89)90990-2. [DOI] [PubMed] [Google Scholar]
- 32.Chen P, Keller AM, Joshi CP, Martell DJ, Andoy NM, Benítez JJ, Chen TY, Santiago AG, Yang F. Single-Molecule Dynamics and Mechanisms of Metalloregulators and Metallochaperones. Biochemistry. 2013:52. doi: 10.1021/bi400597v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bastin-Shanower SA, Brill SJ. Functional analysis of the four DNA binding domains of Replication Protein A: the role of RPA2 in ssDNA binding*. The Journal of biological chemistry. 2001;276:36446–36453. doi: 10.1074/jbc.M104386200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gasior SL, Olivares H, Ear U, Hari DM, Weichselbaum R, Bishop DK. Assembly of RecA-like recombinases: distinct roles for mediator proteins in mitosis and meiosis. Proc Natl Acad Sci USA. 2001;98:8411–8418. doi: 10.1073/pnas.121046198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim C, Snyder RO, Wold MS. Binding properties of replication protein A from human and yeast cells. Mol Cell Biol. 1992;12:3050–3059. doi: 10.1128/mcb.12.7.3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee JY, Finkelstein IJ, Crozat E, Sherratt DJ, Greene EC. Single-molecule imaging of DNA curtains reveals mechanisms of KOPS sequence targeting by the DNA translocase FtsK. Proc Natl Acad Sci USA. 2012;109:6531–6536. doi: 10.1073/pnas.1201613109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Visnapuu ML, Fazio T, Wind S, Greene EC. Parallel arrays of geometric nanowells for assembling curtains of DNA with controlled lateral dispersion. Langmuir. 2008;24:11293–11299. doi: 10.1021/la8017634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gibb B, Silverstein TD, Finkelstein IJ, Greene EC. Single-stranded DNA curtains for real-time single-molecule visualization of protein-nucleic acid interactions. Anal Chem. 2012;84:7607–7612. doi: 10.1021/ac302117z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Johnson DE, Takahashi M, Hamdan SM, Lee SJ, Richardson CC. Exchange of DNA polymerases at the replication fork of bacteriophage T7. Proc Natl Acad Sci USA. 2007;104:5312–5317. doi: 10.1073/pnas.0701062104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Furukohri A, Goodman MF, Maki H. A dynamic polymerase exchange with Escherichia coli DNA polymerase IV replacing DNA polymerase III on the sliding clamp. J Biol Chem. 2008;283:11260–11269. doi: 10.1074/jbc.M709689200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Duderstadt KE, Geertsema HJ, Stratmann SA, Punter CM, Kulczyk AW, Richardson CC, van Oijen AM. Simultaneous Real-Time Imaging of Leading and Lagging Strand Synthesis Reveals the Coordination Dynamics of Single Replisomes. Mol Cell. 2016;64:1035–1047. doi: 10.1016/j.molcel.2016.10.028. [DOI] [PubMed] [Google Scholar]
- 42.Aberg C, Duderstadt KE, van Oijen AM. Stability versus exchange: a paradox in DNA replication. Nucleic Acids Res. 2016;44:4846–4854. doi: 10.1093/nar/gkw296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang J, Zhuang Z, Roccasecca RM, Trakselis MA, Benkovic SJ. The dynamic processivity of the T4 DNA polymerase during replication. Proc Natl Acad Sci USA. 2004;101:8289–8294. doi: 10.1073/pnas.0402625101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Beattie TR, Kapadia N, Nicolas E, Uphoff S, Wollman AJ, Leake MC, Reyes-Lamothe R. Frequent exchange of the DNA polymerase during bacterial chromosome replication. eLife. 2017;6:e21763. doi: 10.7554/eLife.21763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.English BP, Hauryliuk V, Sanamrad A, Tankov S, Dekker NH, Elf J. Single-molecule investigations of the stringent response machinery in living bacterial cells. Proc Natl Acad Sci USA. 2011;108:E365–373. doi: 10.1073/pnas.1102255108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Javer A, Long Z, Nugent E, Grisi M, Siriwatwetchakul K, Dorfman KD, Cicuta P, Cosentino Lagomarsino M. Short-time movement of E. coli chromosomal loci depends on coordinate and subcellular localization. Nat Commun. 2013;4:3003. doi: 10.1038/ncomms3003. [DOI] [PubMed] [Google Scholar]
- 47.Delalez NJ, Wadhams GH, Rosser G, Xue Q, Brown MT, Dobbie IM, Berry RM, Leake MC, Armitage JP. Signal-dependent turnover of the bacterial flagellar switch protein FliM. Proc Natl Acad Sci USA. 2010;107:11347–11351. doi: 10.1073/pnas.1000284107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xiao B, Zhang H, Johnson RC, Marko JF. Force-driven unbinding of proteins HU and Fis from DNA quantified using a thermodynamic Maxwell relation. Nucleic Acids Res. 2011;39:5568–5577. doi: 10.1093/nar/gkr141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brent MM, Anand R, Marmorstein R. Structural basis for DNA recognition by FoxO1 and its regulation by posttranslational modification. Structure. 2008;16:1407–1416. doi: 10.1016/j.str.2008.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]