

ABSTRACT
Strong viral enhancers in gammaretrovirus vectors have caused cellular proto-oncogene activation and leukemia, necessitating the use of cellular promoters in “enhancerless” self-inactivating integrating vectors. However, cellular promoters result in relatively low transgene expression, often leading to inadequate disease phenotype correction. Vectors derived from foamy virus, a nonpathogenic retrovirus, show higher preference for nongenic integrations than gammaretroviruses/lentiviruses and preferential integration near transcriptional start sites, like gammaretroviruses. We found that strong viral enhancers/promoters placed in foamy viral vectors caused extremely low immortalization of primary mouse hematopoietic stem/progenitor cells compared to analogous gammaretrovirus/lentivirus vectors carrying the same enhancers/promoters, an effect not explained solely by foamy virus' modest insertional site preference for nongenic regions compared to gammaretrovirus/lentivirus vectors. Using CRISPR/Cas9-mediated targeted insertion of analogous proviral sequences into the LMO2 gene and then measuring LMO2 expression, we demonstrate a sequence-specific effect of foamy virus, independent of insertional bias, contributing to reduced genotoxicity. We show that this effect is mediated by a 36-bp insulator located in the foamy virus long terminal repeat (LTR) that has high-affinity binding to the CCCTC-binding factor. Using our LMO2 activation assay, LMO2 expression was significantly increased when this insulator was removed from foamy virus and significantly reduced when the insulator was inserted into the lentiviral LTR. Our results elucidate a mechanism underlying the low genotoxicity of foamy virus, identify a novel insulator, and support the use of foamy virus as a vector for gene therapy, especially when strong enhancers/promoters are required.
IMPORTANCE Understanding the genotoxic potential of viral vectors is important in designing safe and efficacious vectors for gene therapy. Self-inactivating vectors devoid of viral long-terminal-repeat enhancers have proven safe; however, transgene expression from cellular promoters is often insufficient for full phenotypic correction. Foamy virus is an attractive vector for gene therapy. We found foamy virus vectors to be remarkably less genotoxic, well below what was expected from their integration site preferences. We demonstrate that the foamy virus long terminal repeats contain an insulator element that binds CCCTC-binding factor and reduces its insertional genotoxicity. Our study elucidates a mechanism behind the low genotoxic potential of foamy virus, identifies a unique insulator, and supports the use of foamy virus as a vector for gene therapy.
KEYWORDS: foamy virus, CCCTC-binding factor (CTCF), gene insulator, CRISPR/Cas, genotoxicity, gene therapy
INTRODUCTION
The severe adverse events due to insertional activation of proto-oncogenes by gammaretroviral (GV) vectors in several successful gene therapy trials, specifically, X-linked severe combined immunodeficiency (X-SCID) (1–3), chronic granulomatous disease (CGD) (4, 5), and Wiskott-Aldrich syndrome (WAS) (6), highlight the genotoxic risks of GV vectors. Here, strong enhancers in the U3 region of the GV vector long terminal repeat (LTR) caused proto-oncogene activation as a consequence of nonrandom integrations near transcriptional start sites of proto-oncogenes (7). Self-inactivating (SIN) GV and lentivirus (LV) vectors with a 3′ LTR U3 enhancer/promoter deletion and internal, weaker cellular/endogenous gene promoters driving transgene expression have been used to circumvent the risk of insertional oncogenesis by viral enhancers.
These SIN “LTR-less” or “enhancerless” vectors show reduced genotoxicity compared to LTR-intact GV vectors in experimental systems both in vitro and in vivo (8–10). However, expression of the transgene is often not robust, and complete correction of the disease phenotype is largely dependent on introduction of high numbers of transduction/vector copy numbers (VCN) per cell, except in diseases where modest levels of transgene expression are sufficient for correction.
Indeed, cellular promoters were reported to be inadequate for correcting the defect in canine leukocyte adhesion deficiency (LAD), where a strong viral enhancer/promoter from the murine stem cell virus (MSCV) LTR was required for phenotypic correction (11, 12). Similar results have been reported in CGD (13, 14) and WAS (15, 16), where partial or inadequate correction of the disease phenotype occurred with internal cellular/endogenous gene promoters in SIN vectors. Hence, in diseases requiring higher transgene expression per cell for complete phenotypic correction, the use of internal promoters requires enhancers, and enhancer-mediated genotoxicity will need to be addressed before clinical trials are initiated.
Foamy virus (FV) vectors have several favorable characteristics. FVs are the largest of the retroviruses with the largest packaging capacity (17, 18), they are nonpathogenic, and they have broad cellular tropism (19, 20). FV vectors efficiently transduce canine (21, 22), murine (23), and human (24–26) hematopoietic stem and progenitor cells (HSPCs) and have been used in preclinical animal studies correcting canine LAD (21), WAS (27), and β-thalassemia (28). Previous studies revealed that FV vectors show nearly half the propensity of LV vectors to integrate into genes, while they have a higher tendency to integrate near transcriptional start sites (TSS), similar to GV (21, 22, 29, 30).
A plasmid-based transient transfection assay with FV vectors revealed that the vector backbone prevents read-through transcription compared to LV- and GV-based proviruses, which may reduce their propensity to promote transcription of neighboring genes (31). Furthermore, despite their preference for integration near transcriptional start sites, FV vectors have shown less generation of clonal dominance in a SCID repopulating model than LV vectors (30). We sought to uncover the unexplained dichotomy between FV and LV vectors, with FV vectors causing less clonal dominance than LV vectors but having greater propensity to integrate near TSS than LV.
One mechanism the genome uses to shield a gene from the enhancer/promoter elements of neighboring genes is the use of insulators. Insulators shield genes from inappropriate cis-regulatory signals (32), which in the case of viral vectors are the enhancer elements. CCCTC-binding factor (CTCF) is the main insulator-binding protein in vertebrates (33) and also provides chromatin barrier functions (34). CTCF binds to different DNA sequences via various combinations of 11 zinc fingers (32). In addition to its insulator and barrier functions, CTCF binding sites have previously been shown to reduce the genotoxicity of viral vectors without diminishing viral titers (35).
In this study, we first directly compared the genotoxic potential of FV vectors carrying strong viral enhancers, compared to those of GV and LV vectors. We used the in vitro immortalization frequency of transduced primary mouse HSPCs (36) and assessed their genotoxic potential independently of site of integration by gene targeting of all three analogous proviral sequences into the same location in the LMO2 proto-oncogene. We found insulator activity in the FV backbone, which was mapped to the FV LTR and strongly bound CTCF. Furthermore, when this region was removed, insulator activity was abrogated. Our study showed that FV vectors have significantly reduced genotoxicity even when carrying strong viral enhancers, largely due to enhancer-blocking properties of the FV vector backbone derived from a 36-bp enhancer-blocking sequence in the foamy virus LTR.
RESULTS
Vector design for comparative genotoxicity.
The spleen focus-forming virus (SFFV) vector, a GV, was previously reported to generate a high frequency of immortalized clones in the in vitro immortalization (IVIM) assay (8, 9), which was correlated with the occurrence of leukemia in mice (9) and with a high incidence (80 to 100%) of myelodysplastic syndrome and leukemia in the CGD and WAS gene therapy trials. We constructed analogous enhanced green fluorescent protein (eGFP)-encoding FV and LV vectors carrying the internal enhancers/promoters from the SFFV LTR or FV and LV vectors carrying internal enhancers/promoters from the murine stem cell virus (MSCV) LTR (also a GV): (i) ΔΦSF.eGFP (SFFV-FV) carries an internal SFFV enhancer/promoter; (ii) ΔΦMSCV.eGFP (MSCV-FV) carries an internal MSCV enhancer/promoter. These vectors were compared to two analogous LV vectors, RRL.ppt.SF.eGFP.pre (SFFV-LV), which carries an internal SFFV enhancer/promoter, and RRL.ppt.MSCV.eGFP.pre (MSCV-LV), which carries an internal MSCV enhancer/promoter, and to two analogous GV vectors, SF91-eGFP.pre (SFFV-GV), which is driven by the SFFV LTR, and MSCV.eGFP.pre, which is driven by the MSCV LTR, as positive controls with known high genotoxic potential (Fig. 1A). A promoterless (Pr-less) FV vector, ΔΦ.eGFP, and mock transductions were included as negative controls.
FIG 1.

The immortalization frequency and replating efficiency of FV are significantly lower than those of LV and GV vectors. (A) Schematic representation of the proviral forms of the vectors. An SFFV LTR-driven GV vector, RSF91.eGFP.pre (SFFV-GV), and an MSCV LTR-driven GV, MSCV.eGFP.pre, are shown. The U3 LTR region of the SFFV-GV contains the enhancer/promoter elements of the SFFV and drives expression of eGFP cDNA. SIN lentiviral vectors, RRL.ppt.SFFV.eGFP.pre (SFFV-LV), have a 400-bp LTR deletion and are driven by the enhancer-promoter elements from the SFFV U3 LTR region placed internally, upstream of eGFP; similarly, RRL.ppt.MSCV.eGFP.pre (MSCV-LV) is driven by an internal MSCV enhancer/promoter element from the MSCV U3 LTR region. The ΔΦ series of vectors represent FV vectors with a 582-bp LTR deletion and are driven by the internal enhancer-promoter elements derived from SFFV and MSCV. All the vectors carry the eGFP cDNA. Pr-less is a promoterless vector (ΔΦ.eGFP). The wPRE is present downstream of eGFP for all vectors except ΔΦMSCV.eGFP. Δ represents an LTR with a U3 deletion. The vectors are not drawn to scale. (B and C) The replating frequencies of immortalized clones were assessed at 2 and 5 weeks. The x axis represents replating frequencies at 2 weeks (left) and 5 weeks (right) normalized to VCN. The y axis represents vectors tested using the IVIM assay. The immortalization potentials of SFFV-FV (B) and MSCV-FV (C) were compared to that of SFFV-GV. The replating frequencies of SFFV-LV and MSCV-LV carrying internal SFFV or MSCV enhancer/promoter elements were also compared to that of SFFV-GV. Mock transductions were done without addition of virus and were negative controls for each experiment. A promoterless FV was also included as a negative control. Each symbol represents the replating frequency normalized for VCN from one independent transduction experiment using the specified vector. The data points on the left of the vertical line represent independent transductions with no replating clones. The replating frequency was calculated based on Poisson statistics using L-Calc software and was normalized to the mean VCN of the Lin− bulk culture population prior to replating. Fold reductions in the frequencies of immortalized mutants are indicated in the graphs. Statistical significance between vector backbones is indicated by P values; ***, P < 0.001.
FV vectors showed significantly less immortalization of primary mouse HSPCs than GV and LV vectors.
To compare the genotoxic potentials, the above-mentioned viral vectors were used in the IVIM assay. This assay is widely used as a preclinical screening tool and is particularly sensitive for relative quantitative detection of myeloid lineage-related genotoxicity (37).
Lineage-negative (Lin−) cells from bone marrow of wild-type (WT) (C57BL/6J) mice were transduced with the SFFV- and MSCV-GV, -LV, and -FV vectors using optimized transduction protocols for each vector (5, 21, 38). The cells were expanded for 2 weeks and then cloned, as previously described (39). By 2 weeks, untransduced/mock-transduced Lin− cells terminally differentiated and died. If vector insertion conferred a proliferative potential, clonal outgrowth occurred, creating immortalized clones. The replating frequencies (immortalization frequencies) of cells transduced with GV, LV, and FV vectors were assessed at 2 weeks and at 5 weeks. All wells with immortalized cells were picked for expansion. Cells transduced with SFFV-driven GV or LV vectors expanded robustly, even at 5 weeks. Transduction efficiencies (measured by GFP marking) of Lin− cells in SFFV-GV-, MSCV-GV-, SFFV-LV-, MSCV-LV-, SFFV-FV-, and MSCV-FV-transduced Lin− cells were 91%, 70%, 89%, 99%, 93%, and 75%, respectively. The average VCN in Lin− cells transduced with the SFFV-GV, MSCV-GV, SFFV-LV, MSCV-LV, SFFV-FV, and MSCV-FV vectors were 8 ± 2, 9 ± 0.3, 10 ± 2, 27 ± 2, 7 ± 0.8, and 8.5 ± 0.7 (mean ± standard error of the mean [SEM]), respectively (Table 1).
TABLE 1.
Frequencies of IVIM assay mutants
| Vector | Independent transduction no. | Type of cells plateda | No. of IVIM wells per 96-well plate (at 2 wks)b | 2-wk replating frequencyc | VCNd | 5-wk replating frequencye | 5-wk replating frequency/VCNf |
|---|---|---|---|---|---|---|---|
| SFFV-GV | 1 | US | 15 | 0.00200 | 1.0 | 0.00200 | 0.002000 |
| S | 58 | 0.01000 | 5.7 | 0.00900 | 0.001579 | ||
| 2 | US | 17 | 0.00200 | 3.0 | 0.00200 | 0.000667 | |
| S | 60 | 0.01000 | 5.4 | 0.01000 | 0.001852 | ||
| 3 | S | 96 | 0.04500 | 12 | 0.04500 | 0.003750 | |
| 4 | US | 93 | 0.03400 | 14 | 0.03400 | 0.002429 | |
| 5 | US | 92 | 0.03200 | 15 | 0.03200 | 0.002133 | |
| 6 | US | 70 | 0.01300 | 8.0 | 0.01300 | 0.001625 | |
| 7 | S | 82 | 0.01800 | 7.3 | 0.01800 | 0.002466 | |
| Avg | 65 | 0.01844 (1 in 54) | 7.9 | 0.01844 (1 in 54) | 0.002311 (1 in 452) | ||
| SFFV-LV | 1 | US | 13 | 0.00100 | 11.5 | 0.00100 | 0.000087 |
| 2 | US | 11 | 0.00120 | 15.0 | 0.00120 | 0.000080 | |
| S | 20 | 0.00110 | 8.6 | 0.00110 | 0.000128 | ||
| 3 | US | 7 | 0.00080 | 6.0 | 0.00080 | 0.000133 | |
| 4 | US | 25 | 0.00300 | 14.0 | 0.00300 | 0.000214 | |
| 5 | US | 27 | 0.00300 | 10.0 | 0.00300 | 0.000300 | |
| 6 | US | 21 | 0.00260 | 10.5 | 0.00260 | 0.000248 | |
| Avg | 18 | 0.00181 (1 in 552) | 10.8 | 0.00181 (1 in 552) | 0.000168 (1 in 5,952) | ||
| SFFV-FV | 1 | US | 1 | 0.00003 | 3.7 | 0.00003 | 0.000008 |
| S | 2 | 0.00020 | 8.6 | 0.00020 | 0.000023 | ||
| 2 | US | 1 | 0.00002 | 6.8 | 0.00002 | 0.000003 | |
| S | 2 | 0.00020 | 8.3 | 0.00010 | 0.000012 | ||
| 3 | S | 5 | 0.00026 | 9 | 0.00020 | 0.000022 | |
| 4 | US | 4 | 0.00008 | 6 | 0.00003 | 0.000005 | |
| S | 3 | 0.00030 | 9.3 | 0.00003 | 0.000003 | ||
| Avg | 3 | 0.00015 (1 in 6667) | 7.4 | 0.00009 (1 in 11,111) | 0.000012 (1 in 83,333) | ||
| Promoterless FV | 1 | US | 0 | 0.00005 | 3.8 | 0.00000 | 0.000000 |
| 2 | US | 1 | 0.00010 | 16.8 | 0.00001 | 0.000001 | |
| 3 | US | 0 | 0.00005 | 6 | 0.00000 | 0.000000 | |
| 4 | US | 1 | 0.00010 | 12 | 0.00001 | 0.000001 | |
| Avg | 0.5 | 0.00008 (1 in 12,500) | 9.6 | 0.00001 (1 in 100,000) | 0.000001 (1 in 1,000,000) | ||
| MSCV-GV | 1 | US | 48 | 0.00680 | 9.6 | 0.00680 | 0.000708 |
| S | 36 | 0.00469 | 9.2 | 0.00469 | 0.000510 | ||
| 2 | US | 28 | 0.00344 | 8.2 | 0.00344 | 0.000420 | |
| S | 65 | 0.0110 | 9.6 | 0.0110 | 0.001146 | ||
| Avg | 44 | 0.00648 (1 in 154) | 9.2 | 0.00648 (1 in 154) | 0.000708 (1 in 1,412) | ||
| MSCV-LV | 1 | US | 40 | 0.00530 | 25 | 0.0053 | 0.000212 |
| 2 | US | 23 | 0.00270 | 29 | 0.0027 | 0.000093 | |
| Avg | 32 | 0.00400 (1 in 250) | 27 | 0.00400 (1 in 250) | 0.000148 (1 in 6,757) | ||
| MSCV-FV | 1 | S | 8 | 0.00087 | 9 | 0.00032 | 0.000036 |
| 2 | S | 9 | 0.00098 | 9 | 0.00021 | 0.000023 | |
| 3 | US | 0 | 0.00000 | 6.4 | 0.00000 | 0.000000 | |
| S | 3 | 0.00015 | 10 | 0.00005 | 0.000005 | ||
| 4 | US | 2 | 0.00010 | 6.5 | 0.00005 | 0.000008 | |
| S | 3 | 0.00015 | 10 | 0.00005 | 0.000005 | ||
| Avg | 4 | 0.00038 (1 in 2,631) | 8.5 | 0.00011 (1 in 9,091) | 0.000013 (1 in 76,923) | ||
| Mock | NAg | 0 | 0 | 0 | 0 | 0 | |
| NA | 0 | 0 | 0 | 0 | 0 | ||
| NA | 0 | 0 | 0 | 0 | 0 | ||
| NA | 0 | 0 | 0 | 0 | 0 | ||
| NA | 0 | 0 | 0 | 0 | 0 | ||
| NA | 0 | 0 | 0 | 0 | 0 | ||
| Avg | 0 | 0 | 0 | 0 | 0 |
Transduction pools that were sorted for eGFP+ cells by FACS. US, unsorted; S, sorted.
Number of wells with immortalized cells after 100,000 Lin− cells were transduced, expanded, and plated in a limiting dilution at 100 cells/well in 96-well plates.
Frequency of wells with immortalized cells (replating frequency) at 2 weeks calculated by Poisson statistics using L-Calc software. The average replating frequencies from individual transductions for each vector are also shown. The data in parentheses are another way of showing the frequency of Lin− cells immortalized by vector insertion, e.g., a replating frequency of 0.001 means 1 in 1,000 Lin− cells are immortalized by that vector. For vector-transduced wells negative for replating clones, calculations are based on the assumption that a replating clone would be detected if 97 wells were plated instead of 96 (9).
VCN of transduced pools prior to plating in 96-well plates.
Replating frequency at 5 weeks. Since numerous immortalized clones were present with the SFFV-GV and SFFV-LV groups, based upon our previously reported equal fitness of these clones at 2 and 5 weeks, only a subset of 2-week-immortalized clones were expanded for 5 weeks with these vectors. However, all the immortalized clones from the foamy virus vector group were expanded for 5 weeks further analysis.
Immortalization frequency was normalized for vector copy number (with the assumption that one immortalization event is caused by a one-vector insertion and immortalization frequency correlates linearly with the vector copy number).
NA, not applicable.
The fitness of immortalized clones (i.e., the ability to be replated and expanded) after transduction with GV or LV vectors with SFFV/MSCV enhancers at 5 weeks was similar to that at 2 weeks. The number of immortalized clones with SFFV-GV and SFFV-LV was consistent with previously reported studies (40). Notably, however, the immortalization frequencies of SFFV-FV and MSCV-FV were remarkably lower, by more than 2 orders of magnitude, than those of the analogous SFFV-GV and MSCV-GV (P < 0.01) (Fig. 1B and C and Table 1). The analogous SFFV-LV and MSCV-LV showed a 10- to 14-fold reduction in immortalization frequency compared to SFFV-GV, consistent with prior reports (40). In addition, the immortalized clones derived from FV transduction were not as fit as those derived from LV or GV transduction, as they had a lower expansion potential than clones with SFFV-GV and SFFV-LV insertions and therefore lower 5-week replating frequencies. Mean Sca-1 and c-Kit expression trended lower for SFFV-FV and MSCV-FV clones (69.6 and 57.5%, respectively) than for SFFV-GV, SFFV-LV, and MSCV-LV clones (89.0, 76.6, and 70.5%, respectively), but the differences were not statistically significant (Mann-Whitney U test, one tailed). The percentage of unique insertions from FV and LV vectors with respect to gene transcriptional units and nongenic/repeat sequences demonstrated a unique integration profile for FV compared to LV insertions. Frequencies of integration sites for FV within genes, in nongenic/repeat sequences, and nonassignable were 27, 62, and 11%, respectively; for LV, they were 60, 6, and 34%, respectively. The numbers of unique insertions for SFFV-FV and MSCV-FV were 121 and 65, respectively. The number of unique insertions identified for MSCV-LV was 270.
To exclude differences in immortalization frequency due to VCN, the immortalization frequency of each vector was normalized to the VCN (Fig. 1B and C), allowing comparative analysis of the relative genotoxicity. When normalized for VCN, the MSCV-GV vector had a 3-fold-lower immortalization potential than the SFFV-GV vector. Importantly, at 2 weeks, the SFFV-FV and MSCV-FV vectors showed a 110-fold- and 156-fold-lower immortalization potential, respectively, than the SFFV-GV vector. By 5 weeks, the immortalization potentials of FV vectors declined even further, resulting in 155-fold- and 414-fold-lower immortalization potentials of SFFV-FV and MSCV-FV vectors, respectively, than of the SFFV-GV vector. The SFFV-LV and MSCV-LV vectors showed 12- and 14-fold reductions in immortalization frequency compared to the SFFV-GV vector, consistent with prior reports (9, 40). Supporting the notion that immortalization in this assay occurred secondarily to vector integration, (i) there were no detectable immortalized clones in the mock-transduced progenitor cultures and (ii) promoterless FV had live wells at 2 weeks, but they contained fewer cells that were lost by 5 weeks, suggesting that they were not truly immortalized.
CRISPR/Cas9-based targeted insertional genotoxicity.
The SFFV enhancer has been shown in the IVIM assay, in mice, and in human trials to be one of the most genotoxic enhancers (5, 10, 40). The remarkably reduced genotoxicity (150- to 400-fold less) in the IVIM assay from the SFFV/MSCV enhancers in an FV vector, as shown here, could not be fully explained by the reported 2-fold-higher propensity of FV to integrate in nongenic regions, especially when FV tends to integrate near TSS, like GV. We hypothesized that the FV backbone may have an enhancer-blocking/insulator effect. To assess the potential enhancer-blocking functionality of the vector backbone without the confounding effects of the enhancer/promoter, transgene, or integration site, we targeted proviral forms of SFFV-GV, SFFV-LV, and SFFV-FV into the LMO2 gene at the retroviral integration site (RIS), known to cause multiple cases of secondary leukemia (1–3). In order to isolate the genotoxic effects of viral vector backbone sequences from integration site effects, a CRISPR (clustered regularly interspaced short palindromic repeat)/Cas9-based assay was devised that allowed integration of the proviral sequences of GV, LV, and FV, all carrying an eGFP transgene driven by the SFFV enhancer/promoter, at precisely the same locus within LMO2 and in the same direction (Fig. 2A). The insertion site for the viral-vector sequences was based on a previous report of secondary leukemia in a patient following gene therapy for SCID-X1 using a GV vector (41).
FIG 2.

CRISPR/Cas9 facilitated insertion of GV, LV, and FV proviral sequences into a known locus previously shown to increase expression of LMO2. (A) General outline of CRISPR/Cas9 insertion of the proviral sequences into the LMO2 gene. The gRNA/Cas9 ribonucleoprotein complex creates a double-strand break (DSB) near the insertion site. The DSB is generally repaired by nonhomologous end joining (NHEJ) or by homologous recombination (HR) if a donor DNA, encoding the designed genetic modification flanked by homology arms (HA), is provided. (B) gRNA target sequences (purple hatched arrows) were chosen that target the LMO2 locus near the insertion site clinically found to be associated with insertional LMO2 transactivation and leukemogenesis. Insertion of proviral sequences occurred at the location indicated by the red arrow. (C) Donor templates for HR were plasmids constructed to insert vector sequences for GV, LV, and FV into the LMO2 gene at this locus. Viral sequences are flanked by HA corresponding to the region on either side of the insertion site. Each HA is ∼600 bp in length. All three vectors utilize the SFFV enhancer/promoter and carry an eGFP cDNA. Δ represents an LTR with a U3 deletion.
Five potential guide RNA (gRNA) target sequences, each with low predicted off-target activity and in close proximity to the insertion site, were identified (Fig. 2B). LMO2 gRNA 5 was the most efficient, with an indel generation efficiency of 24.1% (efficiencies for gRNAs 1 to 4 were 0, 13.3, 0, and 1.5%, respectively), and was used for subsequent experiments. Donor plasmids containing proviral cassettes for GV, LV, or FV were cloned (Fig. 2C). Sequences were constructed in reverse orientation to match the directionality of the insertion described previously (41). HeLa cells, which have very low LMO2 mRNA expression and lack LMO2 protein (42), were simultaneously transfected with the gRNA/Cas9 plasmid and one of the donor (provirus-containing) plasmids. Approximately 2 weeks after transfection, 9.7%, 28.2%, and 12.1% of GV-, LV-, and FV-transfected pools showed GFP transgene expression when green fluorescent protein-positive (GFP+) cells were sorted into single cells (Fig. 3A). Clones were then harvested and screened by PCR for homology-directed insertion of the proviruses (Fig. 3B to D).
FIG 3.
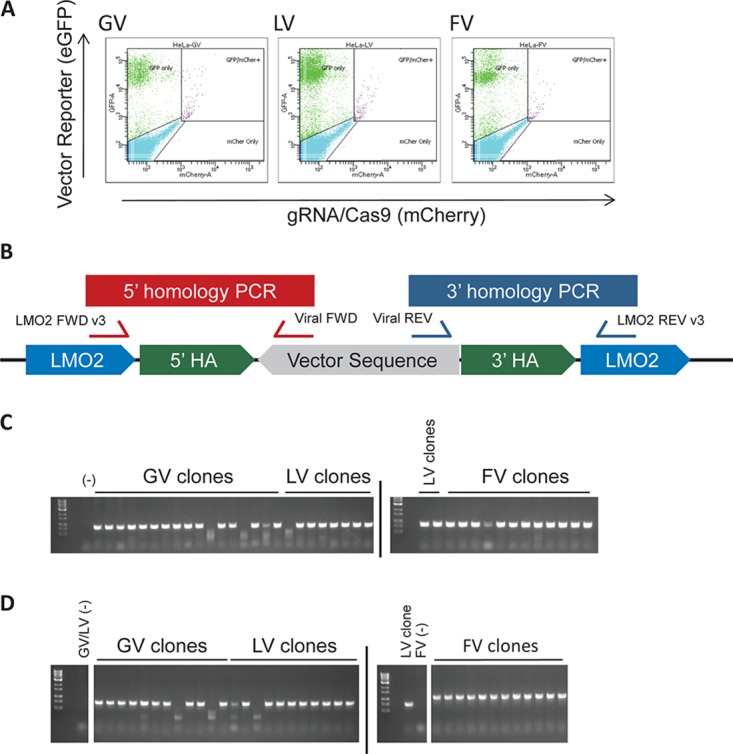
Establishing HeLa LMO2 clones. Following transfection and successful insertion of the proviral sequence via CRISPR/Cas9-mediated HR, cells express GFP. (A) Editing efficiency of HeLa cells assessed by GFP expression at 2 weeks posttransfection. GFP-positive cells were sorted into single cells to establish clones. (B) Schema for the two PCRs performed on each clone to detect homology-directed repair. (C) PCR 1 amplicon, which bridges the 5′ HA. (D) PCR 2 amplicon, which bridges the 3′ HA. PCR 2 used a different FWD primer for FV. Only correctly integrated sequences produced amplicons.
When editing with CRISPR/Cas9, it is possible to edit more than one allele in a given clone, especially in highly transfectable cell lines. In our case, this would either result in proviral sequences being integrated into multiple LMO2 alleles in a given HeLa cell (provirus-targeted alleles) or the double-strand break (DSB) would be repaired by nonhomologous end joining (NHEJ), creating a small indel (termed “edited nontargeted” alleles here). Since HeLa cells have very low LMO2 mRNA expression and do not express LMO2 protein and the proviruses are targeted to an intron of the LMO2 gene, LMO2 expression would be influenced by the virus enhancers only in the provirus-targeted alleles. Moreover, the reading frame of LMO2 (and its mRNA expression) would not be altered by an edited nontargeted allele. The proviral plasmid could also randomly integrate into the genome in HeLa cells, resulting in GFP expression, but would not affect LMO2 expression. Due to the potential for random integration, we could not directly determine whether the clones had more than one provirus-targeted LMO2 allele by quantitative PCR (qPCR) of the proviral sequences. We therefore took several steps to calculate the targeted-allele copy numbers indirectly (see Materials and Methods and Table 2). Briefly, fluorescence in situ hybridization (FISH) for the LMO2 locus on control HeLa cells showed four LMO2 alleles. Next, copy number analysis was used to detect unedited alleles and alleles containing small indels. PCR across the target site with a larger amplicon, followed by gel electrophoresis and sequencing, was used to detect larger indels that would not be detected by copy number analysis. One LMO2 allele with a 261-bp deletion was found in three of the LV clones that initially showed only one nontargeted/WT allele on copy number analysis. The number of targeted alleles for a given clone equals the number of LMO2 loci with the number of nontargeted alleles detected by copy number analysis and PCR subtracted (Table 2). Overall, comparable numbers of LMO2 alleles were targeted (2 or 3 targeted alleles in most clones) with FV, LV, or GV.
TABLE 2.
Numbers of WT or indel alleles in each clone
| Clone | No. of WT or indel alleles | Calculated no. of provirus-targeted allelesa | Used for Western blotting |
|---|---|---|---|
| GV A7 | 2 | 2 | Yes |
| GV B1 | 2 | 2 | Yes |
| GV B2 | 2 | 2 | Yes |
| GV B3 | 2 | 2 | |
| GV B4 | 2 | 2 | |
| GV B12 | 3 | 1 | |
| LV A2b | 2 | 2 | |
| LV A7c,e | 1 | 3 | Yes |
| LV A9 | 1 | 3 | Yes |
| LV A11 | 1 | 3 | Yes |
| LV B1 | 2 | 2 | |
| LV B3 | 1 | 3 | |
| LV B8 | 2 | 2 | |
| LV B11b | 2 | 2 | |
| LV C2b | 2 | 2 | |
| FV A2 | 2 | 2 | Yes |
| FV A3 | 2 | 2 | Yes |
| FV A7 | 2 | 2 | Yes |
| FV A8c,e | 2 | 2 | |
| FV A10 | 1 | 3 | |
| FV A11 | 2 | 2 | |
| FV B1c,e | 1 | 3 | |
| FV B8d,e | 2 | 2 | |
| FV C1 | 2 | 2 | |
| FV C2 | 2 | 2 | |
| FV C4c,e | 1 | 3 |
Assumes that all clones have 4 LMO2 alleles, like the parental HeLa cells from which they were derived.
Clone contains an allele with a 261-bp intronic deletion that would not be detected by copy number analysis.
Clone had no PCR amplicon when assessing for large deletions by PCR.
Clone FV B8 was not assayed for large deletions due to lack of sample material.
The number of provirus-targeted alleles was calculated solely by copy number analysis.
The advantage of using the HeLa cell line without significant endogenous LMO2 expression is that editing events that abrogate gene expression does not significantly affect the overall increase in LMO2 expression due to directed proviral insertion events. We determined LMO2 mRNA expression in the generated HeLa clones by quantitative real-time (qRT) PCR with two probe and primer sets. The primers were selected to detect all spliced transcript variants expressed from the LMO2 promoter from both modified and WT alleles. Data from both primer sets using two different loading controls were very similar (Fig. 4A to D). Overall, we found that the SFFV enhancer in GV demonstrated the greatest fold increase in LMO2 mRNA expression (median increase, 280- ± 23-fold over unmodified HeLa cells), followed by the SFFV enhancer in LV (median increase, 200- ± 27-fold). It should be noted that SFFV-GV provirus had two copies of the enhancer at either LTR, while SFFV-LV (and SFFV-FV) had only one copy of the SFFV enhancer. However, the same SFFV enhancer in FV showed a remarkably lower (median, 45- ± 7-fold) increase in LMO2 mRNA expression, 4- and 6-fold lower expression than was seen with the SFFV enhancer in LV and GV, respectively. We then performed a Western blot analysis to detect LMO2 protein expression from three representative clones among the ones used for qRT-PCR (Fig. 5 and Table 2). We could not detect any LMO2 expression in SFFV-FV clones, which was similar to baseline in mock-transduced (nonedited) HeLa cells. However, significantly more LMO2 protein was detectable in GV and LV clones. Taken together, the qRT-PCR and Western blot analyses confirmed that the FV backbone/cis elements have a strong enhancer-blocking or insulator effect, which likely contributes to the reduced ability of the SFFV enhancer to upregulate the expression of LMO2.
FIG 4.

FV induces LMO2 mRNA expression to a lesser extent than either GV or LV. cDNA was generated from GV, LV, and FV clones, and LMO2 mRNA expression was determined using RT-PCR. Depicted are data from experiments using two different primer-probe sets and two different endogenous controls. (A) Hs001534473_m1 primer-probe set and GAPDH endogenous control. n = 8 GV, 9 LV, and 11 FV clones. (B) Hs001534473_m1 primer-probe set and PPIA endogenous control. n = 7 GV, 8 LV, and 8 FV clones. (C) Hs00277106_m1 primer-probe set and GAPDH endogenous control. n = 8 GV, 9 LV, and 11 FV clones. (D) Hs00277106_m1 primer-probe set and PPIA endogenous control. n = 7 GV, 8 LV, and 8 FV clones. The error bars indicate SEM.
FIG 5.
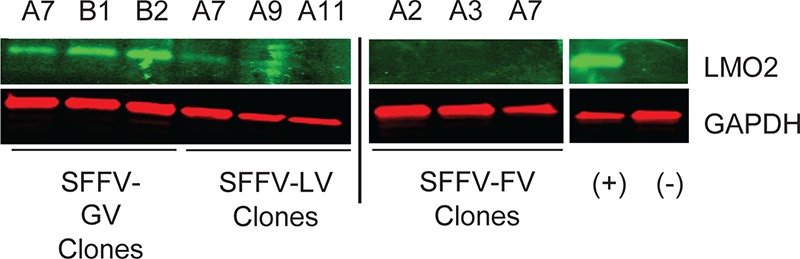
FV induces LMO2 protein expression to a lesser extent than either GV or LV. Western blot analysis for LMO2 expression (top) was performed on SFFV-GV, SFFV-LV, and SFFV-FV clones. The clones used are indicated by the letter and number designations above each row. Untransduced HeLa cells (−) and K562 cells (+) served as negative and positive controls, respectively. Endogenous GAPDH expression (bottom) was used as a loading control.
In silico insulator analysis.
A likely mechanism for the observed enhancer-blocking effect of the FV backbone is that it contains one or more insulator elements. To test our hypothesis, we performed an in silico analysis for CTCF binding sites, the main insulator in vertebrates. The proviral sequences of GV, LV, and FV vectors (excluding the SFFV enhancer/promoter, eGFP, and woodchuck hepatitis virus posttranscriptional regulatory element [wPRE] sequences) were analyzed for predicted CTCF binding sites, or consensus sequences, using the CTCFBSDB 2.0 database (http://insulatordb.uthsc.edu/) (32) to identify core motifs for CTCF binding, represented as position weight matrices (PWMs). The algorithm searches for identified core motifs for CTCF binding sites and represents the motifs as PWMs. PWM scores correspond to the log odds of the observed sequence being generated by the motif versus being generated by the background. A PWM score of >3.0 is suggestive of a significant match. A limitation of the prediction tool is that it returns only the best match for a given PWM in a sequence. Therefore, other putative CTCF consensus sequences for a given motif within the same analyzed sequence are not revealed. To partially account for this, sequences were divided into several fragments, and each fragment was analyzed separately.
Analysis of the vector backbone sequences identified totals of 4, 6, and 26 motifs with PWM scores of >3 for GV, LV, and FV, respectively. The locations of the motifs in the vector backbone are depicted in Fig. 6A and Table 3 and their PWM scores in Fig. 6B. Besides the number of significant PWMs, the PWM scores for GV were lower in general, ranging from 3.3 to 3.4, while the PWM scores for LV ranged from 5.1 to 9.9 and those for FV ranged from 3.4 to 12.6. Motifs for FV were dispersed both in the LTR and the cis sequences compared to those for GV. Overall, the in silico analysis suggested that FV had a greater number of predicted CTCF binding motifs and motifs with higher PWM scores than GV and LV.
FIG 6.

High number of CTCF insulator binding sites in the sequence of FV. (A) Proviral sequences (except SFFV, eGFP, and wPRE sequences) of GV, LV, and FV were analyzed in silico for potential CTCF binding motifs. The approximate locations of predicted binding motifs are indicated by asterisks above the construct. Multiple asterisks indicate multiple overlapping predicted binding motifs. (B) PWM scores for the predicted CTCF binding motifs.
TABLE 3.
Predicted CTCF binding motifs
| Vector | Motif PWMa | Motif sequence | Score | Location from 3′ HA (bp)b | Motif orientationc | Associated feature |
|---|---|---|---|---|---|---|
| GV | MIT_LM23 | AAACCTACAGGTGGGGTCTT | 3.40278 | 88 | − | 5′ LTR |
| EMBL_M1 | CCCCACCTGTAGGT | 3.2515 | 92 | + | 5′ LTR | |
| MIT_LM23 | AAACCTACAGGTGGGGTCTT | 3.40278 | 2105 | − | 3′ LTR | |
| EMBL_M1 | CCCCACCTGTAGGT | 3.2515 | 2109 | + | 3′ LTR | |
| LV | EMBL_M1 | TTCCCCCTGGCCTT | 5.30958 | 427 | − | After Psi |
| EMBL_M2 | GGAAGAGCA | 8.68716 | 679 | + | After Psi | |
| EMBL_M1 | CTCCTCCTCCAGGT | 7.14542 | 734 | − | After Psi | |
| MIT_LM7 | TCCCCAGGAGCTGTTGATCC | 5.08224 | 1091 | − | After Psi | |
| EMBL_M2 | GGCACAGCA | 9.92719 | 1145 | − | After Psi | |
| EMBL_M2 | GGTACAGCT | 8.25192 | 3579 | − | Before 3′ LTR | |
| FV | EMBL_M2 | AGCATTGCA | 9.5685 | 142 | − | 5′ LTR |
| MIT_LM23 | ATATCACTAGATGTCTCCCT | 4.14861 | 273 | − | 5′ LTR | |
| MIT_LM7 | ATATCACTAGATGTCTCCCT | 8.73461 | 273 | − | 5′ LTR | |
| MIT_LM2 | ATATCACTAGATGTCTCCC | 8.02923 | 274 | − | 5′ LTR | |
| EMBL_M1 | AGACATCTAGTGAT | 5.89526 | 277 | + | 5′ LTR | |
| EMBL_M2 | AGCATAGCG | 3.42649 | 429 | + | before gag | |
| EMBL_M2 | GGCATTGCC | 9.56461 | 1304 | − | Pro-Pol | |
| EMBL_M2 | GGAATTGCA | 12.3512 | 1882 | − | Pro-Pol/integrase | |
| REN_20 | TGGTCCAGGAGAGGGTGGCT | 9.14351 | 2319 | + | Pro-Pol/integrase | |
| MIT_LM23 | GGTCCAGGAGAGGGTGGCTA | 3.48392 | 2320 | + | Pro-Pol/integrase | |
| MIT_LM2 | GGTCCAGGAGAGGGTGGCT | 5.54859 | 2320 | + | Pro-Pol/integrase | |
| MIT_LM7 | GGTCCAGGAGAGGGTGGCTA | 9.1009 | 2320 | + | Pro-Pol/integrase | |
| EMBL_M1 | CACCCTCTCCTGGA | 12.5569 | 2322 | − | Pro-Pol/integrase | |
| MIT_LM2 | TGAACAGCAGAAGGAACAA | 4.26098 | 2570 | + | ENV | |
| MIT_LM23 | TGAACAGCAGAAGGAACAAA | 4.51999 | 2570 | + | ENV | |
| MIT_LM7 | TGAACAGCAGAAGGAACAAA | 6.26218 | 2570 | + | ENV | |
| EMBL_M2 | TGAACAGCA | 7.76981 | 2570 | + | ENV | |
| EMBL_M1 | TTCCTTCTGCTGTT | 9.13542 | 2572 | − | ENV | |
| MIT_LM7 | TAACGAGGAGAGGGTGTGGT | 4.82626 | 4898 | + | BEL3 | |
| EMBL_M1 | CACCCTCTCCTCGT | 5.68719 | 4900 | − | BEL3 | |
| EMBL_M2 | GGCATTCCA | 5.79638 | 4917 | − | BEL3 | |
| EMBL_M2 | AGCATTGCA | 9.5685 | 5234 | − | 3′ LTR | |
| MIT_LM23 | ATATCACTAGATGTCTCCCT | 4.14861 | 5365 | − | 3′ LTR | |
| MIT_LM7 | ATATCACTAGATGTCTCCCT | 8.73461 | 5365 | − | 3′ LTR | |
| MIT_LM2 | ATATCACTAGATGTCTCCC | 8.02923 | 5366 | − | 3′ LTR | |
| EMBL_M1 | AGACATCTAGTGAT | 5.89526 | 5369 | + | 3′ LTR |
Identified core motifs for CTCF binding sites represented as PWM.
Locations were determined by the distance from the end of the LMO2 3′ homology arm (HA).
The motif orientation is given in relation to the proviral sequence. +, positive; −, negative.
Mapping the enhancer-blocking element in the FV backbone.
To assess the binding of CTCF to the proviral sequences within HeLa cells, chromatin immunoprecipitation (ChIP) purification of CTCF-bound DNA was performed, followed by qualitative PCR for predicted binding sites within the FV proviral sequence (Fig. 7A). Using HeLa control cells, analysis of the ChIP input material showed amplification of only the H19-Igf2 locus, a known CTCF binding site. However, both the input material and the ChIP-purified DNA from one of the FV clones (FVA2) showed amplification of five tested sites with a high level of predicted binding. While ChIP analysis showed the presence of CTCF binding, the close proximity of the assayed regions limited the resolution between sites by this ChIP-PCR assay. Regardless, the ChIP-PCR assay demonstrated in-cell binding of CTCF to the FV proviral sequence.
FIG 7.

CTCF binding of FV proviral sequence. (A) CTCF-ChIP of the FV A2 clone, followed by qualitative PCR, was performed to interrogate in-cell binding of CTCF to the predicted binding sites. PCR was performed on the ChIP input for HeLa control cells and the FV A2 clone and on the ChIP product for the FV A2 clone. The amplicons for PCRs of H19, FV1, FV2, FV5, FV6, and FV7 were 165, 157, 188, 110, 115, and 155 bp, respectively. (B) Fluorescently labeled probes corresponding to predicted CTCF binding sites in FV and LV proviral sequences were allowed to bind recombinant CTCF protein and were resolved by EMSA, demonstrating binding between the FV2 probe and CTCF. (C) Competitive binding assay between the FV2 probe and unlabeled H19 probe. H19 was provided at the indicated molar excess. (D) Sequence of the FV2 probe (top) with predicted CTCF binding sites indicated in red and blue. Mutant FV2 probes (1 to 6) are listed, with mutated regions underlined in green. (E) EMSA utilizing mutant probes. The original FV2 probe was used as a positive control.
An electrophoretic mobility shift assay (EMSA) was then used to map the predicted CTCF binding sites within LV and FV. Eighty- to 90-bp DNA fragments corresponding to predicted CTCF binding sites by in silico analysis, labeled at both 5′ ends with fluorescent dye, were used as the EMSA probe. Five of the six predicted binding sites in LV were probed. All predicted binding sites in FV were probed. Probes LV1, LV2, and FV8 contained two predicted CTCF binding motif sequences. EMSA was conducted using recombinant human CTCF. An H19 oligonucleotide containing a consensus sequence known to bind CTCF with high affinity was the positive control.
None of the LV probes demonstrated any binding to CTCF. However, probe FV2, corresponding to the sequence ATATCACTAGATGTCTCCCT (located in the LTR and containing four motifs with PWM scores of 4.1, 8.7, 8.0, and 5.9) demonstrated a significant band shift (Fig. 7B). Additionally, the labeled probe could be competed off with unlabeled H19 probe (Fig. 7C). The sequence of the FV2 probe was analyzed in silico for predicted CTCF binding sites. In addition to the previously predicted site, a second site was identified, 5′-TGTAGTTCA-3′, with a score of 6.8. The central region of the FV2 probe was divided into four regions (1 to 4). Region 1 contained the newly identified predicted binding site. Regions 2 and 3 contained the original predicted binding site. Six mutant probes were designed that replaced one or more of the four regions with a scrambled DNA sequence (Fig. 7D). The sequences were analyzed to ensure that no new predicted CTCF binding sites were created. Mutating region 1, 2, or 3 reduced CTCF binding (EMSA band intensities are shown in Fig. 7E). Mutating region 4 appeared to have no effect. Mutating regions 1 and 3 together or 2 and 3 together further reduced CTCF binding. Therefore, we found that CTCF binds the 36-bp sequence defined by regions 1 to 3 of the FV2 probe. A BLAST search (https://blast.ncbi.nlm.nih.gov/) using the defined sequence did not reveal matches to any sequences other than those of foamy virus.
Verifying insulator function.
To verify the insulator function of the defined CTCF binding sequence, (i) the 36-bp sequence was precisely excised from the proviral SFFV-FV sequence, leaving the rest of the sequence intact, and (ii) the proposed insulator was inserted into the LTR of the SFFV-LV proviral sequence. The modified proviral sequences were then inserted into the LMO2 gene using our CRISPR/Cas9-based targeted insertional genotoxicity assay, as before. Expression of LMO2 relative to control HeLa cells and LV and FV clones used previously was determined by qPCR using the Hs001534473_m1 primer-probe set and PPIA endogenous control (Fig. 8). Removing the insulator from the FV LTR resulted in a >5-fold increase in relative LMO2 expression (9.4 to 47.4; P = 0.001). Inserting the sequence into the LV LTR resulted in a >13-fold drop in relative LMO2 expression (21.0 to 1.6; P = 0.0002). Interestingly, placing the insulator into the LTR of SFFV-LV resulted in LMO2 expression that was only 1.6-fold higher than that of control HeLa cells and significantly less than that of the original SFFV-FV containing the insulator (P = 0.002). The overall lower relative expression seen in this assay compared to the prior assay using GV, LV, and FV was due to higher observed expression of LMO2 in the control HeLa cells. Based on our prior assay, the number of inserted proviral sequences did not correlate well with LMO2 expression. The copy number, determined by qPCR only, predicted that all of the new FV clones without the insulator had three correctly placed proviral sequences. Of the new LV clones containing the insulator, 6, 1, and 2 clones contained 1, 2, and 3 copies of the proviral sequence, respectively. Again, the number of inserted proviral sequences did not seem to correlate with expression levels.
FIG 8.
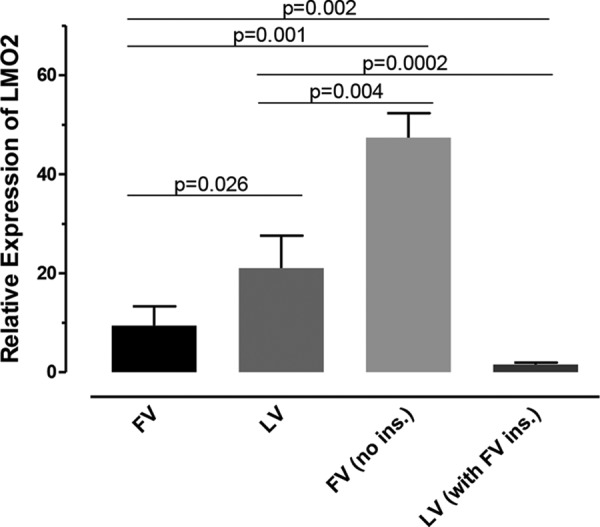
Induction of LMO2 mRNA expression by FV is increased when the insulator is removed, and induction of LMO2 mRNA expression by LV is decreased when the insulator is added to the LV LTR. cDNA was generated from LMO2-modified clones containing FV, LV, FV with no insulator (ins.), and LV with the FV insulator placed in the LTR. LMO2 mRNA expression was determined using qRT-PCR. The Hs001534473_m1 primer-probe set and PPIA endogenous control were used to acquire the data. n = 5, 6, 17, and 9 clones, respectively. The error bars indicate SEM.
Taking the data together, we show that FV LTRs contain a strong 36-bp CTCF binding motif with potent CTCF binding that produces an enhancer-blocking effect and serves to protect nearby genes from the enhancer activity of a delivered transgene. These data provide novel insight into the remarkably low immortalization potential of SFFV (and MSCV) in FV vectors, demonstrating a previously unreported and significant mechanism contributing to the lower genotoxicity of FV carrying strong viral enhancers.
DISCUSSION
Vector-driven genotoxicity is primarily caused by the use of strong enhancers (in the LTR) (7–9, 43) and by the integration site preference of the vector (10, 44). GV LTR enhancers ubiquitously and strongly enhance expression of the transgene, leading to a therapeutic correction, but also enhance the expression of genes flanking the transgene insertion site, which can lead to leukemia. In addition, both GV and LV integrases target the provirus to gene-rich regions nearly 60 to 70% of the time, while GV vectors tend to integrate near TSS and LV vectors have a strong preference for integrating within introns of active transcriptional units and thus have a lower propensity to activate transcription of cellular genes (7–9, 43). Overall, LV vectors carrying GV LTR enhancers tend to have an approximately 10- to 20-fold lower genotoxic potential in in vitro and in vivo experimental systems than GV vectors carrying the same enhancers (8, 10).
Prior studies have demonstrated via RIS analysis that FV vector integrations resemble those of GV vectors with insertions near TSS, albeit at 50% lower frequency, and show approximately 30 to 35% of integrations into transcribed regions of the genome compared to 60 to 70% by LV (29). Therefore, we were surprised at the remarkably low (150- to 400-fold lower) immortalizing frequency of primary hematopoietic progenitor cells by FV vectors carrying the strong viral LTR enhancer from the SFFV-GV vector, otherwise known to be highly genotoxic in GV vectors that also target TSS. There was a 10- to 15-fold reduction in immortalization frequency when the same enhancers were placed in LV vectors, likely due in part to their tendency to integrate farther from TSS than with GV vectors.
Our data confirmed a prior report that FV insertions were predominantly in nongenic regions (29). However, given the remarkable magnitude of the reduction in genotoxicity, we wanted to assess genotoxic potential when we controlled for integration site preference. Hence, a CRISPR/Cas9 method was devised to insert the proviral sequences of GV, LV, and FV vectors carrying the same eGFP transgene into the LMO2 locus, in which insertional oncogenesis occurred after viral integration in clinical trials of GV vectors.
Ryu and colleagues (45) previously used a two-step system to target insertions of proviruses into the LMO2 locus. An AAV vector was used to integrate a loxP site-flanked LTR-eGFP construct into Jurkat cells at the LMO2 locus, and a clone was derived. This allowed insertion of GV and LV vectors by Cre recombination-mediated provirus cassette exchange. The CRISPR/Cas9-based method we developed is a flexible, extremely efficient one-step process in which a Cas9/gRNA plasmid and a donor proviral plasmid are transfected together, with derivation of clones within 2 to 3 weeks following transfection, and is easily implementable in a variety of cell types. Additionally, it can serve as a technology platform for testing any vector near any other proto-oncogene, only requiring cloning of a donor plasmid with the provirus sequence flanked by relevant homology arms and changing the gRNA sequence.
LMO2-targeted GV, LV, and FV clones assayed by qRT-PCR and Western blotting for LMO2 expression mirrored the immortalization assay results: a 6-fold-reduced SFFV enhancer effect was seen with FV compared to GV. This may be partly explained by the fact that the GV vector has two SFFV enhancers while the FV vector has only one. However, FV also had a 4-fold reduction in enhancer effect compared to LV, which also has only a single SFFV enhancer placed internally, similar to FV. Similarly, Western blot analysis of LMO2 protein expression demonstrated a 3-fold reduction in LMO2 expression in LV clones compared to GV clones. LMO2 expression was undetectable for FV clones, showing very potent SFFV enhancer blocking by the FV vector sequences.
This remarkable reduction in the effect of a strong enhancer such as SFFV led us to look for an insulator in the FV vector cis sequences. In silico analysis for binding sites for CTCF, the primary vertebrate insulator protein, showed CTCF PWMs for GV in the beginning of the U3 region of each LTR just prior to the enhancer/promoter, but all with low PWM scores (below 3.5) (Table 3 lists the locations of the motifs). LV had 6 unique CTCF PWMs with scores ranging from 5.3 to 9.9. FV had 26 CTCF PWMs with scores ranging from 3.5 to 12.6 that were present in the SIN LTR and the portions of gag, pro-pol, env, and bel cis sequences retained in the FV vector, of which 11 were nonoverlapping CTCF PWMs and 2 were within the R regions of the LTRs. If a more stringent cutoff for the PWM score was used, e.g., 3.5, GV lost all of its CTCF binding sites, while none of the LV and only 2 of the FV CTCF binding sites were lost. However, we did not find any CTCF binding to the tested in silico-predicted LV CTCF binding sites.
Binding of CTCF to one of the predicted binding sites within the FV LTR sequences was demonstrated using EMSA, and the CTCF binding was mapped to a 36-bp region. The location within the LTR is notable, as it provides insulation at both ends of the proviral sequence. Recently, CTCF binding sites experimentally inserted into the LV or FV LTR have been shown to reduce genotoxicity (35, 46, 47). Our studies show that inherent CTCF binding sites already exist within the FV LTR. By removing the CTCF binding site from the FV proviral sequence, the reduction in genotoxicity seen in our LMO2 activation assay was abrogated. Conversely, inserting this site into the LV LTR significantly reduced genotoxicity in the same assay to levels only slightly above those of the control.
Overall, it appears that multiple factors combine to provide FV vectors with reduced genotoxicity compared to GV and LV vectors. Besides higher integration into noncoding regions, its backbone exerts enhancer-blocking activity upon integration into the genome. Given the prominent role that CTCF plays in gene insulation and enhancer blocking (48), these data show that CTCF binding sites within the FV vector play a role in insulating nearby genes from genotoxic effects of an enhancer placed within the vector. CTCF has been shown to be important in the life cycles of numerous viruses. In human papillomavirus (HPV) infection, CTCF regulates viral oncogene expression by controlling the activity of the viral promoter and regulating splicing of viral transcripts (49). Knockdown of CTCF during HPV infection leads to blockage of genome amplification (50). CTCF contributes to establishing restricted latency in Epstein-Barr virus infection (51). In herpes simplex virus 1 (HSV-1) infection, CTCF knockdown reduces viral transcription, viral genome copy numbers, and virus yield during lytic infection (52). Additionally, CTCF binding clusters within HSV-1 have been shown to serve as classical insulators capable of acting as blockers of the latency-associated transcript enhancer (53). In the case of human T cell leukemia virus type 1 (HTLV-1) (a retrovirus, unlike the DNA viruses listed above), CTCF has been shown to bind HTLV-1 and to act as an enhancer blocker, to regulate mRNA splicing, and to form long-distance interactions with flanking host chromatin (54). However, it remains to be seen what role it plays in the life cycle of FV. Future studies using mutational analysis may provide further insight into the role of CTCF in the FV life cycle.
Importantly, these studies led to identification of a novel insulator sequence with strong enhancer-blocking activity, making FV an attractive vector for expressing transgenes requiring strong enhancers. Additionally, our data suggest that the sequence may be transportable into other vectors, with the possibility of reducing genotoxicity. The risk of insertional immortalization has been reported to be greatly reduced in SIN GV and LV vectors expressing transgenes via the elongation factor EF-1 (EFS) and phosphoglycerate kinase (PGK) promoters due to their weak/negligible enhancer activity (9). However, a major limitation of the use of cellular promoters is the problem of obtaining and maintaining a consistently high level of transgene expression. In diseases like LAD (11), CGD (13, 14), and WAS (15, 16), where a high level of transgene expression is required to achieve a therapeutic benefit, cellular promoters were insufficient to mediate a therapeutic effect unless high VCN were present and strong enhancers were used. In fact, we have recently reported that expression of perforin from cellular or endogenous promoters in LV only partially corrects the hemophagocytic lymphohistiocytosis (HLH) phenotype, and strong viral enhancers are necessary for complete disease phenotype correction (55).
This study shows that strong viral LTR enhancers within FV vectors may be applicable, as the combined tendency to integrate in nongenic regions combined with an inherent insulator effect can remarkably reduce genotoxicity. Browning et al. have recently incorporated known insulator sequences into FV, resulting in a reduction in integration near hot spots (46). The same group has also designed retargeted FV vectors that integrate less frequently near proto-oncogenes (56).
In summary, direct comparison of the genotoxic potentials induced by analogous GV, LV, and FV vectors displays remarkably reduced immortalization potential of HSPCs from insertions of FV vectors carrying some of the most genotoxic viral enhancers. Interrogating the sole effect of vector backbones on genotoxicity using CRISPR/Cas9-mediated targeted integration of GV, LV, and FV sequences at a specific LMO2 locus revealed that the mechanism of this reduced genotoxicity is in large part due to an enhancer-blocking insulator effect in the FV LTRs that strongly bind CTCF. Our study provides valuable insights into the genotoxicity of FV vectors and has relevance to clinical vector design. The identified small (36-bp) insulator sequence could be of potential use in a wide variety of vectors, especially when addition of strong enhancers is critical for disease phenotype correction.
MATERIALS AND METHODS
Vector design and production.
The vectors SFFV-GV (RSF91.eGFP.pre) and SFFV-LV (RRL.ppt.SF.eGFP.pre) used in this study have been described previously (8, 57). MSCV-GV (MSCV.eGFP.pre) has also been described previously (58). MSCV-LV (RRL.ppt MSCV.eGFP.pre) was generated in the laboratory of Dennis Hickstein (National Cancer Institute, Bethesda, MD). A 388-bp region of the MSCV promoter (identical in sequence to the MSCV LTR enhancer/promoter in ΔΦMSCV CD18 [21]) was obtained through PCR amplification with artificial XhoI and AgeI ends and then cloned into XhoI/AgeI restriction enzyme-digested pRRLSIN.cPPT.PGK.eGFP.WPRE (Addgene, Cambridge, MA) to create pRRL.ppt.MSCV.eGFP.pre (M. J. Hunter and D. D. Hickstein, unpublished results). All FVs were in the ΔΦ backbone. The FV ΔΦMSCV.eGFP has been described previously (59, 60). The SFFV promoter replaced the MSCV promoter in the ΔΦMSCV.eGFP FV vector to create ΔΦSF.eGFP. All the vectors carry eGFP cDNA. The promoterless FV was derived from the ΔΦMSCV.eGFP vector by removing the MSCV enhancer/promoter and religation.
Ecotropic GV supernatants were produced in 293T cells by transient transfection, as described previously, and titers were determined on NIH 3T3 fibroblasts (American Type Culture Collection [ATCC]) (39). Virus titers were in the range of 106 to 107 infectious units (IU)/ml. The LV vectors SFFV-LV and MSCV-LV were produced by transient cotransfection of 293T cells (ATCC) as described previously (38). SFFV-FV, MSCV-FV, and Pr-less FV were produced by four-plasmid (pCiES [Env], pCiGSΔΨ [Gag], pCiPs [Pol], and vector [pΔΦ]) transient transfection, as described previously (59). pCiGSΔΨ is the Gag expression cassette (D. W. Russell, unpublished data) with a more complete deletion in the packaging signal. pΔΦ is a deleted FV backbone with a polylinker to insert the transgene cassette.
FVs were resuspended in Stemspan (Stem Cell Technologies, Vancouver, BC, Canada) containing 2% heat-inactivated fetal bovine serum (FBS) (HyClone, Logan, UT) and 5% dimethyl sulfoxide (DMSO) (Sigma, St. Louis, MO) and were stored frozen in 5% DMSO until use. Titers of Pr-less FV were determined by measuring the genome copy number of transduced HT1080 cells by quantitative real-time PCR using primers that recognize the wPRE, while titers of the other vectors were determined by quantifying GFP expression by fluorescence-activated cell sorter (FACS). The titers of FV were in the range of 3 × 107 IU/ml to 1 × 108 IU/ml.
Isolation of Lin− cells.
Bone marrow from C57BL/6J mice was used for the isolation of Lin− cells using biotinylated lineage-specific antibodies (lineage cell depletion kit; BD Biosciences, San Jose, CA) as described previously (39, 40). The biotin-labeled Lin− cells were incubated with anti-biotin microbeads (Miltenyi Biotech), followed by magnetic sorting of unlabeled Lin− cells. The isolated Lin− cells were prestimulated for viral transductions in Stemspan medium (Stem Cell Technologies) containing 1% penicillin-streptomycin, 50 ng/ml mouse stem cell factor (mSCF), 100 ng/ml human interleukin 11 (hIL-11), and 10 ng/ml mIL-3.
In vitro immortalization assay.
Lin− cells were prestimulated overnight in Stemspan medium containing 1% penicillin-streptomycin, 50 ng/ml mSCF, 100 ng/ml hIL-11, and 10 ng/ml mIL-3. On day 2, 100,000 Lin− cells were used for each LV vector transduction. The Lin− cells were transduced at a multiplicity of infection (MOI) of 20 twice at 8-h intervals using concentrated LV vector supernatants. For GV transduction, lineage-negative cells were prestimulated for 2 days in Stemspan-cytokine cocktail. GV transductions were performed on day 3 and day 4 on RetroNectin recombinant human fibronectin fragment (TaKaRa Bio Inc., Kusatsu, Shiga, Japan)-coated 24-well dishes preloaded with retroviral vectors SFFV-GV and MSCV-GV at an MOI of 20. After the final transductions, the transduced Lin− cells were washed and expanded as bulk cultures in Stemspan-cytokine cocktail for 19 days.
FV stocks were rapidly thawed by adding warm prestimulation medium, and the Lin− cells were transduced once with FV in 48-well plates precoated with RetroNectin recombinant human fibronectin fragment at a concentration of 8 μg/cm2 at an MOI of 50. A higher MOI was chosen for transduction, as foamy virus transductions were done only once compared to two transductions with lentiviral vectors. After 16 h, the cells were washed and expanded in the Stemspan-cytokine cocktail. We observed toxicity in Lin− cells (50 to 60% viability) following foamy virus vector transductions even at ≤1% DMSO (final concentration) during transduction. At day 4 after the final transduction, transgene expression from the transduced bulk cultures was analyzed using flow cytometry. In bulk cultures with lower gene transfer efficiency, GFP+ cells were sorted using a BD FACS Aria II (BD Biosciences) and expanded until plated. Bulk cultures with higher gene transfer efficiency were also sorted for GFP+ cells, and the replating frequencies of the sorted pools and unsorted transduced pools were compared. During expansion, the transduced bulk cultures were maintained at a concentration of 2 × 105 to 5 × 105 cells/ml. After expansion, the cells were plated in 96-well plates at a density of 100 cells/well. After 2 weeks of plating, the 96-well plates were examined and scored for the presence of wells with proliferating cell populations. Under these conditions, the mock-transduced cells/untransduced cells barely survived. The positive wells were further expanded for molecular analysis. At 5 weeks, some of the clones expanded at 2 weeks had terminally differentiated and died by 5 weeks. The replating frequency of each vector tested at 2 weeks and 5 weeks was calculated based on Poisson statistics using L-Calc software (Stem Cell Technologies).
Immortalized clones derived from SFFV-GV could be replated at the same frequency at 2 weeks and 5 weeks, allowing comparison of the genotoxicities of our vectors relative to the highly genotoxic SFFV-GV vector. In contrast, clones derived from vectors with low genotoxic potential showed initial growth and replating potential at 2 weeks but terminally differentiated thereafter and lost their replating frequency by 5 weeks. To be able to compare immortalization frequencies/VCN of sorted and unsorted populations, a portion of the transduced bulk cultures from SFFV-GV-, MSCV-GV-, SFFV-LV-, MSCV-LV-, SFFV-FV-, and MSCV-FV-transduced Lin− cells were sorted for GFP expression and showed proportional immortalization before and after sorting, validating this modification to give a similar immortalization readout. Immortalization frequencies/VCN before sorting of SFFV-GV-, SFFV-LV-, and SFFV-FV-transduced cells were 0.001755, 0.000176, and 0.000006, respectively. After sorting, the frequencies were 0.002016, 0.000128, and 0.000023, respectively. For the vector-transduced group negative for replating clones, calculations were based on the assumption that a replating clone would be detected if 97 wells were plated instead of 96 wells (9).
Phenotypic analysis of immortalized clones.
Immortalized clones were labeled with antibodies that recognize the cell surface markers Sca-1 phycoerythrin (PE) (clone D7; catalog number 553108) and c-Kit allophycocyanin (APC) (clone 2B8; catalog number 553356) from BD Biosciences and analyzed using a FACS Canto (BD Biosciences).
Vector copy number analysis.
Quantitative real-time PCR was performed to assess the gene transfer efficiencies of GV, LV, and FV vector-transduced bulk cultures. For GV vectors, LV vectors, and FV vectors, primers that recognize the wPRE region were used to measure the VCN in bulk cultures. Genomic DNA from a single-copy NIH 3T3 cell clone carrying a single copy of the MM13 vector was used as a standard for copy number analysis. The MM13 plasmid has been described previously (61). Primers in the FV backbone were used to measure the copy number, as well (FV backbone forward primer, 5′-AATCCTTTACATGGAGAAGTTATAGGTCTT-3′, and reverse primer, 5′-TGGCCAAATCCATAGCCTTAGA-3′). PCR was carried out with TaqMan probe (5′-ATCTGAAATCTCTCAATTTGTCCCCACCA-3′) with tetramethyl-6-carboxyrhodamine dye as a quencher. The FV- or wPRE-specific signal was normalized to mouse ApoB in each sample. Genomic DNA (50 ng) from a single-copy murine erythroleukemia (MEL) cell clone transduced with FV was diluted with untransduced MEL DNA to generate copy number standards. Quantitative PCR was performed with an Applied Biosystems 7900HT real-time PCR system (Thermo Fisher, Grand Island, NY) using a thermocycler protocol for 96-well plates, according to the manufacturer's instructions.
LAM PCR to determine insertion sites in immortalized clones.
For ligation amplification-mediated (LAM) PCR, the junction sequences between the viral LTR and the mouse genome were linearly amplified twice with 100 ng of genomic DNA from FV immortalized clones using 0.25 pmol of the FV-specific 5′-end-biotinylated primer (5′-GAACCTTGTGTCTCTCATCCC-3′) and 2.5 units of Taq polymerase (Qiagen, Hilden, Germany), with cycling conditions as follows: initial denaturation at 95°C for 3 min, 50 cycles of amplification (95°C for 30 s, 55°C for 30 s, and 72°C for 1 min), and a final extension at 72°C for 3 min. After DNA enrichment of the biotinylated DNA, hexanucleotide primer extension was carried out using Klenow enzyme (Promega, Madison, WI), and the primer-extended product was digested with TasI (New England BioLabs [NEB], Ipswich, MA). Following TasI digestion, the DNA was ligated to TasI-specific double-stranded linkers (5′-GACCCGGGAGATCTGAATTCAGTGGCACAGCAGTTAGG-3′/5′-AATTCCTAACTGCTGTGCCACTGAATTCAGATC-3′). The first exponential amplification of the linked products was performed using 12.5 pmol each of FV-specific primer (5′-GTCTATGAGGAGCAGGAGTA-3′) and the linker cassette-specific primer (5′-GACCCGGGAGATCTGAATTC-3′). Eight percent of the first exponential PCR was then used as the template for a second exponential nested-PCR amplification using 12.5 pmol each of nested FV-specific primer (5′-CCTCCTTCCCTGTAATACTC-3′) and nested linker cassette-specific primer (5′-AGTGGCACAGCAGTTAGG-3′) under the same conditions as the first PCR. To detect the insertion sites from the MSCV-GV-immortalized clones, 100 ng of genomic DNA was linearly amplified using MSCV LTR-specific 5′-end-biotinylated primer LTR1 (5′-CTGGGGACCATCTGTTCTTGGCCCT-3′), enriched with Dynabeads M-280 streptavidin (Thermo Fisher), digested with Tsp5091 (NEB), and linked to an asymmetric linker cassette (5′-AATTCTCTAGTATGCTACTCGCACCGATTATCTCCGCTGTCAGT-3′ and 5′-ACTGACAGCGGAGATAATCGGTGCGAGTAGCATACTAGAG-3′). The ligation products were then amplified with LTR- and linker-specific primer LTR2 (5′-GACTTGTGGTCTCGCTGTTCCTTGG-3′) and a linker cassette primer, LC1 (5′-ACTGACAGCGGAGATAATCG-3′) (first exponential PCR). The second exponential PCR was carried out with primers LTR3 (5′-GGTCTCCTCTGAGTGATTGACTACC-3′) and LC2 (5′-GTGCGAGTAGCATACTAGAG-3′) (62).
Next-generation sequencing of LAM PCR products.
The products from the second exponential PCR were processed for next-generation DNA sequencing at the Cincinnati Children's Hospital Medical Center (CCHMC) DNA Sequencing Core. LAM second exponential PCR products were purified using a QIAquick PCR purification kit (Qiagen) and then rendered blunt ended by end repair with T4 DNA polymerase, Klenow enzyme, and T4 PNK (Promega) in the presence of 10 mM deoxynucleoside triphosphates (dNTPs) (Thermo Fisher). The blunt-ended products were randomly concatenated by treatment with T4 Quick Ligase (NEB) at room temperature for 15 min. Next-generation sequencing libraries compatible with the Illumina system were prepared using a Nextera in vitro transposition kit (Epicentre, Madison, WI) according to the manufacturer's recommendations and amplified using a different molecular barcode for each sample. After another round of PCR purification, all 10 libraries were quality checked on an Agilent Bioanalyzer (Agilent, Santa Clara, CA) and then mixed in equal amounts in a single pool. Sequencing was conducted on an Illumina HiSeq2000 (Illumina, San Diego, CA) in single-read mode with indexing, producing 100-base-long sequences.
After demultiplexing of all the sequences in the pool and assignment to their respective samples, reads were processed and aligned to the mm9 mouse reference assembly using the CASAVA 1.8 package. The results were generated in the QSEQ SORTED file format so that alignments could be visualized using the ChIP sequencing (ChIP-seq) module of Illumina's Genome Studio software. Insertion sites detected by LAM PCR are characterized by an LTR sequence upstream of the insertion and an adapter sequence downstream. While the aligner was configured to position reads that contained only mouse genome sequence, reads that contained some LTR or some adapter sequence along with a majority of mouse sequence were also positioned. By zooming in to the base level display in Genome Studio (Illumina), it was possible to determine the edge of the covered regions and sides that matched the adapter sequence and the sides that matched the LTR sequence, allowing the determination of the insertion point and the direction in which the provirus integrated. All identified insertions were compared to the National Center for Biotechnology Information (NCBI) mouse build 37 genome database (http://www.ncbi.nlm.nih.gov).
CRISPR/Cas9 insertion of proviral sequences. (i) gRNA development.
The reference sequence used for the initial description (41) of the LMO2 integration site (Homo sapiens chromosome 11 [chr11] clone RP1-22J9 map of p12-14.1; GenBank accession number AL135799.8) was obtained from NCBI. It corresponds to GRCh38.p2 chr11:33890271. Genomic DNA was isolated from Jurkat cells, and the region around the insertion site was PCR amplified using Q5 polymerase (NEB) and sequenced by the CCHMC DNA Sequencing and Genotyping Core. The PCR primers were LMO2 FWD PCR (5′-TTTAGGTTGCCCTGAAAAGGTG-3′) and LMO2 REV PCR (5′-GCCAAACACTCCTAGGCTCTTG-3′). The sequencing primers were LMO2 FWD PCR, LMO2 REV PCR, and LMO2 seq1 (5′-GTCTCTCGCAGCCACATGGG-3′). The region around the insertion site was analyzed for potential gRNA target sites using the CRISPR design program (Benchling, Inc., San Francisco, CA). Five gRNAs were chosen on the basis of proximity to the planned insertion site and low predicted off-target effects. A plasmid containing both a gRNA and a Cas9-T2A-eGFP expression cassette (pX458m) was a kind gift from Yueh-Chiang Hu, Transgenic Animal and Genome Editing Core at CCHMC. eGFP cDNA was first replaced with an mCherry reporter (pX458m-mCherry). Site-directed mutagenesis was performed using a QuikChange II XL site-directed mutagenesis kit (Agilent) to remove a BbsI site within the mCherry sequence (primers 5′-CCCGTAATGCAGAAGAAAACCATGGGCTGGGAGGC-3′ and 5′-GCCTCCCAGCCCATGGTTTTCTTCTGCATTACGGG-3′). DNA oligonucleotides for cloning the target sequences into the pX458m-mCherry vector were designed and obtained from Integrated DNA Technologies (IDT) (Coralville, IA). The oligonucleotides used to generate gRNA 1 with targeting sequence GATACCAATAGATATCAATC were LMO2 gRNA 1 FWD (5′-CACCGGGATACCAATAGATATCAATC-3′) and LMO2 gRNA 1 REV (5′-AAACGATTGATATCTATTGGTATCCC-3′). The oligonucleotides used to generate gRNA 2 with targeting sequence ATCACCAGATTGATATCTAT were LMO2 gRNA 2 FWD (5′-CACCGGGATCACCAGATTGATATCTAT-3′) and LMO2 gRNA 2 REV (5′-AAACATAGATATCAATCTGGTGATCCC-3′). The oligonucleotides used to generate gRNA 3 with targeting sequence AATTGCATAGTCGTGAAGTC were LMO2 gRNA 3 FWD (5′-CACCGGGAATTGCATAGTCGTGAAGTC-3′) and LMO2 gRNA 3 REV (5′-AAACGACTTCACGACTATGCAATTCCC-3′). The oligonucleotides used to generate gRNA 4 with targeting sequence ATTGCATAGTCGTGAAGTCA were LMO2 gRNA 4 FWD (5′-CACCGGGATTGCATAGTCGTGAAGTCA-3′) and LMO2 gRNA 4 REV (5′-AAACTGACTTCACGACTATGCAATCCC-3′). The oligonucleotides used to generate gRNA 5 with targeting sequence TCGTGAAGTCAGGGCTTCTA were LMO2 gRNA 5 FWD (5′-CACCGGGTCGTGAAGTCAGGGCTTCTA-3′) and LMO2 gRNA 5 REV (5′-AAACTAGAAGCCCTGACTTCACGACCC-3′).
pX458m-mCherry was digested with FastDigest BbsI (Thermo Fisher) and simultaneously dephosphorylated with FastAP (Thermo Fisher). The digested product was then gel purified. Oligonucleotide pairs were phosphorylated and annealed in a reaction mixture of 100 μM each oligonucleotide and T4 polynucleotide kinase (NEB), placed in a Veriti 96-well fast thermal cycler (Thermo Fisher) at 37°C for 30 min and 95°C for 5 min, and then ramped down to 25°C at 5°C/min. The annealed oligonucleotides were then ligated into the cleaved pX458m plasmid and transformed into PX5-α competent cells (Protein Express, Cincinnati, OH). A plasmid was subsequently prepared using an EndoFree Plasmid Maxi kit (Qiagen); 2 μg of each gRNA/Cas9 plasmid was transfected into 2.4 × 105 Jurkat cells in a 24-well plate using Lipofectamine 3000 (Thermo Fisher) according to the manufacturers' protocol. At day 7, cells were harvested and genomic DNA was purified. The area around the target site was amplified using Q5 polymerase and sequenced (PCR primers, LMO2 FWD v3, 5′-GCTTGGGTTTTACACGTCTTC-3′, and LMO2 REV v3, 5′-TCAGCTAGAAAACAAGTACTTGC-3′; sequencing primer, LMO2 seq1, 5′-GTCTCTCGCAGCCACATGGG-3′). The gRNA efficiency was determined using the tracking of indels by decomposition (TIDE) assay (63).
(ii) Donor vector templates for homology-directed repair (HDR).
After sequencing the LMO2 region in Jurkat cells, ∼600-bp homology arms were designed with a multiple-cloning site region at the chosen insertion site. The homology vector was ordered as a plasmid in the pUC57 backbone from GenScript. The foamy virus pΔΦ.SF.eGFP.PRE was cut at the LTRs with XbaI and EcoNI and inserted between NheI and EcoNI sites. The lentivirus pRRL.PPT.SF.eGFP.PRE was cut at the LTRs with BsaI and PsiI and inserted between BbsI and NaeI sites. Retrovirus pRSF91.eGFP.PRE was cut at the LTRs between XhoI and HindIII and inserted between BsmFI and XhoI sites. The resulting clones were checked by restriction digestion and sequenced for verification. For GV, the entire LTR sequences, which contain SFFV enhancers/promoters, were contained in the cloned sequence. To facilitate cloning of LV and FV, sequences from the R region of the 5′ LTR through the entire 3′ LTR were cloned from viral production plasmids. For the LV vector, the 40 bp of the 5′ ΔU3 region (left after deletion of the U3 enhancer/promoter in the LTR) were not part of the cloned “proviral” construct. This small region does not have enhancer/promoter activity and therefore was inconsequential for the purpose of studying genotoxicity. Similarly, in the case of the FV vector, the omitted 5′ U3 region contains a 582-bp deletion that removes the U3 TATA box and transcriptional enhancer sites of the LTR, leading to silencing of the LTR (64). Here, we use the term “provirus” for these LV and FV constructs.
After identifying the 36-bp insulator region in the FV LTR, the LMO2 donor containing pΔΦ.SF.eGFP.PRE was modified at the 5′ LTR and 3′ LTR to remove the identified CTCF binding sites. The initial sequence of this region was AGT AAA AGG ATT TGT ATA TTA GCC TTG CTA AGG GAG ACA TCT AGT GAT ATA AGT GTG AAC TAC ACT TAT CTT AAA TGA TG to AGT AAA AGG ATT TGT ATA TTA GCC TTG CTA AGC ACA TTC GAT AGT GAT ATA AGA GGC TTT ATA TCT TAT CTT AAA TGA TG (the insulator sequence is underlined). For the 3′ LTR of the proviral sequence, a gene block containing the modified insulator sequence was ordered from IDT to replace the ∼800-bp region between EcoNI and MluI sites. For the 5′ LTR, a gene synthesis product was ordered from GenScript to replace the ∼550-bp region between PacI and AvrII sites. The resulting plasmid was confirmed by sequencing.
To add the 36-bp insulator sequence to the lentiviral LTRs, the following sequence was added ahead of the R regions of the LTRs in the LMO2 donor containing pRRL.PPT.SF.eGFP.PRE: AAG GGA GAC ATC TAG TGA TAT AAG TGT GAA CTA CAC. Two gene blocks were ordered from IDT to replace the ∼1-kb region between BsiWI and MluI sites encompassing the 3′ LTR and the ∼900-bp region between BspEI and MfeI sites encompassing the 5′ LTR. The resulting plasmid was confirmed by sequencing.
(iii) Generation of HeLa clones.
On day −1, 5 × 104 cells were seeded into a 24-well plate. The cells were transfected with the LMO2 gRNA 5 plasmid, as well as with one of the three (GV, LV, or FV) LMO2 donor plasmids. Five hundred nanograms of total DNA was transfected, divided at an approximate molar ratio of 1:2 (LMO2 gRNA 5 plasmid to donor plasmid). Transfection was performed using 1.5 μl of Lipofectamine 3000 (Thermo Fisher) according to the manufacturer's recommendations. Successful transfection was verified on day 2 by analyzing a portion of the cells for expression of both eGFP (donor plasmid) and mCherry (LMO2 gRNA 5 plasmid) using a FACS Canto (BD Biosciences). At 2 weeks, the cells were reanalyzed for eGFP and mCherry by FACS. GFP-positive and mCherry-negative cells were sorted as single cells into a 96-well plate by the Research Flow Cytometry Core at CCHMC using a BD FACS Aria II.
After reaching at least 80% confluence, a portion of the cells were harvested. DNA was purified by resuspending the cell pellet in 20 μl of QuickExtract DNA extraction solution (Epicentre) and incubated at 65°C for 15 min, at 68°C for 15 min, and at 98°C for 10 min. The purified DNA was then screened for correct integration of the donor sequence by PCR using primer sets flanking the homology arms. The first PCR for ensuring correct 5′ homology used primers LMO2 FWD v3 and Viral FWD (5′-CGAGCGTTGGTAAGAGAAGC-3′). The second PCR for ensuring correct 3′ homology used primers LMO2 REV v3 and either Viral REV1 (5′-GAGATCTGTCCCGCTAGCA-3′) for GV and LV or Viral REV2 (5′-GGATAATTTACAAATAAACCCGACTTATATTCG-3′) for FV. Clones that had correct bands for both PCRs were considered to have correctly inserted viral sequences by HDR. The steps to estimate targeted-allele copy numbers were as follows. (i) HeLa cells have been reported to contain 3 or 4 copies of chromosome 11p, on which the LMO2 gene resides. We first confirmed by fluorescent in situ hybridization that our unedited/WT HeLa cells had 4 LMO2 alleles (Fig. 9A). (ii) We determined the number of edited nontargeted or WT LMO2 alleles, using primers in the LMO2 gene that flank the Cas9 DSB/proviral insertion site, so that only WT/edited nontargeted LMO2 would be detected and LMO2 loci containing a provirus sequence insertion would not be amplified. WT/nontargeted LMO2 copy number analysis showed that all evaluable clones had 1 or 2 WT/nontargeted LMO2 copies (Fig. 9B). (iii) Clones were further interrogated by PCR amplifying across the gRNA target site, followed by sequencing (the PCR/sequencing primers were the same as those used for the TIDE assay) to assess for the presence of large deletions and indels in each clone (Fig. 9C).
FIG 9.

LMO2 copy number analysis. (A) FISH was performed for a region overlapping the targeted LMO2 loci, revealing 4 LMO2 alleles in HeLa control cells. FISH was performed using an RP11-1006P23 FISH probe (Empire Genomics, Buffalo, NY) recognizing chr11 (33,736,494 to 33,907,488). (B) Copy number analysis was performed across the insertion site of LMO2 (intron 1). The numbers of nontargeted/WT LMO2 alleles were calculated relative to unedited HeLa cells. There were relatively similar nontargeted/WT LMO2 copy numbers between clones. n = 3 for each clone. The HeLa control sample is represented by the white bar. (C) PCR amplification of the region bridging the gRNA target site. Three LV clones (indicated by asterisks [panel B] and arrows [panel C]) were found to have a 261-bp deletion upon sequencing of the PCR product. One LV clone and three FV clones did not amplify.
LMO2 expression analysis by qRT-PCR.
RNA was prepared by lysing cells in RNA Stat-60 (AMS Biotechnology, Abingdon, United Kingdom) and passing them over a QIAshredder column (Qiagen). RNA was isolated by chloroform phase separation. The aqueous layer was precipitated with isopropanol, and the resulting pellet was washed with 75% ethanol. The RNA pellets were resuspended in nuclease-free molecular-grade water and dissolved by incubation at 55°C. The RNA was quantified using a NanoDrop 1000 spectrophotometer (Thermo Fisher). cDNA was prepared with purified RNA using a high-capacity cDNA reverse transcription kit (Thermo Fisher). We determined LMO2 mRNA expression in HeLa clones by qRT-PCR with two probe and primer sets, LMO2 TaqMan genomic assays Hs00277106 and Hs00153473 (Thermo Fisher), corresponding to two different regions in LMO2 cDNA, and using two different validated loading controls for HeLa cells, human PPIA and GAPDH (glyceraldehyde-3-phosphate dehydrogenase) (Thermo Fisher) (4). The LMO2 regions amplified are found in all reported transcript variants. Both probe-primer sets bridge exons, so only mRNA is amplified. For LMO2 transcript variant 1 (NM_005574.3), the probe-primer sets bridge exons 4 and 5 and exons 5 and 6, respectively. qRT-PCR mixtures were prepared with iTaq Universal Probes Supermix (Bio-Rad, Hercules, CA). The ABI 7900HT real-time PCR system was used to run the qRT-PCR. Data from the RT-PCR was analyzed by relative quantification by the 2−ΔΔCT method (65) using human GAPDH/PPIA to normalize the results and to determine fold induction. Unedited HeLa samples were used as calibrators.
WT LMO2 copy number analysis.
Genomic DNA was isolated from HeLa clones. Reactions were prepared with iTaq Universal Probes Supermix (Bio-Rad). The human ApoB gene was used as an endogenous control gene. The CFX Connect real-time PCR detection system (Bio-Rad) was used to run the qRT-PCR. Data from the RT-PCR were analyzed by relative quantification by the 2−ΔΔCT method (65) using a K562 cell line and unedited HeLa cells to normalize the results and to determine copy numbers. The primer and probe sets were as follows: LMO2-CN-FW (5′-TGGGGAACAAGTACAATTTTGTG-3′), LMO2-CN-RV2 (5′-CAATGTGGTGATATCAATCTGGTG-3′), LMO2-CN-Probe (5′-ACAAGCGTAAATTGCATAGTCGTGA-3′), hApoB-CN-FW (5′-CTTGGTTTATGAATCTGGCTC-3′), hApoB-CN-RV (5′-GCCTTTAGCAGTTAGAACAC-3′), and hApoB-CN-Probe (5′-ACATGCTGGGAATCGACTTGTGAT-3′).
Western blot analysis of LMO2 protein expression.
Cells were lysed in RIPA lysis buffer (Santa Cruz, Dallas, TX), and 20 μl of protein lysate (containing between 30 and 87 μg protein per sample) was separated on a 4 to 15% Mini-Protean TGX precast protein gel (Bio-Rad) and transferred onto a nitrocellulose polyvinylidene difluoride (PVDF) membrane (Bio-Rad). The membrane was blocked with Odyssey blocking buffer (Li-Cor Biosciences, Lincoln, NE) and probed with a primary human LMO2 antibody and secondary anti-goat IRDye 800CW antibody (Li-Cor Biosciences). The membrane was then reprobed with a primary human GAPDH antibody (Fitzgerald, Acton, MA) and secondary anti-mouse IRDye 680LT (Li-Cor Biosciences). Signals were visualized using an Odyssey 9120 infrared imager (Li-Cor Biosciences).
Electrophoretic mobility shift assay.
Oligonucleotides corresponding to predicted CTCF binding sites in the LV and FV proviral sequences were designed. Oligonucleotides labeled with IRDye 700 at the 5′ end were ordered from IDT. LV1 probe, made by annealing oligonucleotides 5′-ACAAGATAGAGGAAGAGCAAAACAAAAGTAAGACCACCGCACAGCAAGCGGCCGCTGATCTTCAGACCTGGAGGAGGAGATATGAGGGA-3′ and 5′-TCCCTCATATCTCCTCCTCCAGGTCTGAAGATCAGCGGCCGCTTGCTGTGCGGTGGTCTTACTTTTGTTTTGCTCTTCCTCTATCTTGT-3′, contained the GGAAGAGCA and CTCCTCCTCCAGGT sequence motifs. LV2 probe, made by annealing oligonucleotides 5′-GATACCTAAAGGATCAACAGCTCCTGGGGATTTGGGGTTGCTCTGGAAAACTCATTTGCACCACTGCTGTGCCTTGGAATGCT-3′ and 5′-AGCATTCCAAGGCACAGCAGTGGTGCAAATGAGTTTTCCAGAGCAACCCCAAATCCCCAGGAGCTGTTGATCCTTTAGGTATC-3′, contained the TCCCCAGGAGCTGTTGATCC and GGCACAGCA sequence motifs. LV3 probe, made by annealing oligonucleotides 5′-GTCGGGGAAGCTGACGTCCTTTCGAATTCGATATCAAGCTGTACCTTTAAGACCAATGACTTACAAGGCAGCTGTAGATC-3′ and 5′-GATCTACAGCTGCCTTGTAAGTCATTGGTCTTAAAGGTACAGCTTGATATCGAATTCGAAAGGACGTCAGCTTCCCCGAC-3′, contained the GGTACAGCT sequence motif. FV1 probe, made by annealing oligonucleotides 5′-TCCATTAACACTCTGCTTATAGATTGTAAGGGTGATTGCAATGCTTTCTGCATAAAACTTTGGTTTTCTTGTTAATCAAT-3′ and 5′-ATTGATTAACAAGAAAACCAAAGTTTTATGCAGAAAGCATTGCAATCACCCTTACAATCTATAAGCAGAGTGTTAATGGA-3′, contained the AGCATTGCA sequence motif. FV2 probe, made by annealing oligonucleotides 5′-AGTAAAAGGATTTGTATATTAGCCTTGCTAAGGGAGACATCTAGTGATATAAGTGTGAACTACACTTATCTTAAATGATG-3′ and 5′-CATCATTTAAGATAAGTGTAGTTCACACTTATATCACTAGATGTCTCCCTTAGCAAGGCTAATATACAAATCCTTTTACT-3′, contained the ATATCACTAGATGTCTCCCT and overlapping sequence motifs. FV3 probe, made by annealing oligonucleotides 5′-TCGGGTTTATTTGTAAATTATCCCTAGGGACCTCCGAGCATAGCGGGAGGCATATAAAAGCCAATAGACAATGGCTAGCA-3′ and 5′-TGCTAGCCATTGTCTATTGGCTTTTATATGCCTCCCGCTATGCTCGGAGGTCCCTAGGGATAATTTACAAATAAACCCGA-3′, contained the AGCATAGCG sequence motif. FV4 probe, made by annealing oligonucleotides 5′-GGCATCAGCCTACAAATACCAGTATTCATACTGAAGGCAATGCCCTAGCAGATAAGCTTGCCACCCAAGGAAGTTATGTA-3′ and 5′-TACATAACTTCCTTGGGTGGCAAGCTTATCTGCTAGGGCATTGCCTTCAGTATGAATACTGGTATTTGTAGGCTGATGCC-3′, contained the GGCATTGCC sequence motif. FV5 probe, made by annealing oligonucleotides 5′-CGCAACTGTTAAATCTCTCAATGTACTCACTAGTATTGCAATTCCAAAGGTGATTCACTCTGATCAAGGTGCAGCATTCA-3′ and 5′-TGAATGCTGCACCTTGATCAGAGTGAATCACCTTTGGAATTGCAATACTAGTGAGTACATTGAGAGATTTAACAGTTGCG-3′, contained the GGAATTGCA sequence motif. FV6 probe, made by annealing oligonucleotides 5′-CTCGTTCCTGGTCTCCTGTTGTTGGCCAATTGGTCCAGGAGAGGGTGGCTAGGCCTGCTTCTTTGAGACCTCGTTGGCAT-3′ and 5′-ATGCCAACGAGGTCTCAAAGAAGCAGGCCTAGCCACCCTCTCCTGGACCAATTGGCCAACAACAGGAGACCAGGAACGAG-3′, contained the TGGTCCAGGAGAGGGTGGCT and overlapping sequence motifs. FV7 probe, made by annealing oligonucleotides 5′-ATGAGGCACTTCAGAATACAACAACTGTGACTGAACAGCAGAAGGAACAAATTATACTGGACATTCAAAATGAAGAAGTA-3′ and 5′-TACTTCTTCATTTTGAATGTCCAGTATAATTTGTTCCTTCTGCTGTTCAGTCACAGTTGTTGTATTCTGAAGTGCCTCAT-3′, contained the TGAACAGCAGAAGGAACAAA and overlapping sequence motifs. FV8 probe, made by annealing oligonucleotides 5′-TATGGAAGCTTATGGACCTCAGAGAGGAAGTAACGAGGAGAGGGTGTGGTGGAATGCCACTAGAAACCAGGGAAAACAAG-3′ and 5′-CTTGTTTTCCCTGGTTTCTAGTGGCATTCCACCACACCCTCTCCTCGTTACTTCCTCTCTGAGGTCCATAAGCTTCCATA-3′, contained TAACGAGGAGAGGGTGTGGT, GGCATTCCA, and overlapping sequence motifs. Similarly, FV2 mutant 1 probe was made by annealing oligonucleotides 5′- AGTAAAAGGATTTGTATATTAGCCTTGCTAAGGGAGACATCTAGTGATATAAGaggctttatatcTTATCTTAAATGATG-3′ and 5′- CATCATTTAAGATAAgatataaagcctCTTATATCACTAGATGTCTCCCTTAGCAAGGCTAATATACAAATCCTTTTACT-3′. FV2 mutant 2 probe was made by annealing oligonucleotides 5′- AGTAAAAGGATTTGTATATTAGCCTTGCTAAGGGAGACATCaggctttatatcTGTGAACTACACTTATCTTAAATGATG-3′ and 5′- CATCATTTAAGATAAGTGTAGTTCACAgatataaagcctGATGTCTCCCTTAGCAAGGCTAATATACAAATCCTTTTACT-3′. FV2 mutant 3 probe was made by annealing oligonucleotides 5′- AGTAAAAGGATTTGTATATTAGCCTTGCTaggctttatatcTAGTGATATAAGTGTGAACTACACTTATCTTAAATGATG-3′ and 5′- CATCATTTAAGATAAGTGTAGTTCACACTTATATCACTAgatataaagcctAGCAAGGCTAATATACAAATCCTTTTACT-3′. FV2 mutant 4 probe was made by annealing oligonucleotides 5′- AGTAAAAGGATTTGTATaggctttatatcAAGGGAGACATCTAGTGATATAAGTGTGAACTACACTTATCTTAAATGATG-3′ and 5′- CATCATTTAAGATAAGTGTAGTTCACACTTATATCACTAGATGTCTCCCTTgatataaagcctATACAAATCCTTTTACT-3′. FV2 mutant 5 probe was made by annealing oligonucleotides 5′- AGTAAAAGGATTTGTATATTAGCCTTGCTaagcacattcgaTAGTGATATAAGaggctttatatcTTATCTTAAATGATG-3′ and 5′- CATCATTTAAGATAAgatataaagcctCTTATATCACTAtcgaatgtgcttAGCAAGGCTAATATACAAATCCTTTTACT-3′. FV2 mutant 6 probe was made by annealing oligonucleotides 5′- AGTAAAAGGATTTGTATATTAGCCTTGCTaagcacattcgaaggctttatatcTGTGAACTACACTTATCTTAAATGATG-3′ and 5′- CATCATTTAAGATAAGTGTAGTTCACAgatataaagccttcgaatgtgcttAGCAAGGCTAATATACAAATCCTTTTACT-3′. Lowercase letters represent sequences divergent from the original FV2 probe.
Of note, one LV predicted binding site was not interrogated. A probe corresponding to the H19–Igf2 locus (H19), previously shown to bind CTCF, was used as a positive control (66). Unlabeled H19 probe was used for competition assays. Oligonucleotides were annealed in duplex buffer (IDT). Purified full-length human recombinant CTCF protein (Abnova, Taipei City, Taiwan) and labeled oligonucleotides were incubated at room temperature for 30 min in 20 mM HEPES (pH 7.5), 50 mM KCl, 5 mM MgCl2, 3.3 μM ZnSO4, 1 mM dithiothreitol, 0.3 mg/ml bovine serum albumin (BSA), 0.5 μg poly(dI-dC), 5% glycerol, and 0.5% Triton X-100 (67). Binding reaction mixtures were then resolved on 6% Novex Tris-borate-EDTA (TBE) gels (Thermo Fisher) using 0.5× TBE running buffer (Thermo Fisher). The gels were imaged using an Odyssey 9120 infrared imager.
Chromatin immunoprecipitation.
Briefly, HeLa cell clones (1 × 107 to 2 × 107 cells) from transfected FV and LV and untransfected control HeLa cells were treated with formaldehyde (1% final concentration) and incubated at 37°C for 10 min to cross-link histones to DNA. The formaldehyde was neutralized with 2.5 M glycine (final concentration, 0.25 M) for 5 to 10 min at room temperature and centrifuged for 5 min at 2,000 rpm. The cells were pelleted and stored at −80°C. For ChIP, cells were thawed and the pellet was resuspended in 200 μl of SDS lysis buffer (Millipore, Billerica, MA) and incubated on ice for 10 min, and protease inhibitors (Pierce protease inhibitor; Thermo Fisher) were added (1 mM phenylmethylsulfonyl fluoride [PMSF], 1 μg/ml aprotinin, and 1 μg/ml pepstatin A) to the cell lysate. The lysate was sheared under optimized conditions (in a Covaris TM S220 [Covaris, Woburn, MA] for 70 s at 4°C [peak power, 105; duty factor, 10; and cycles/burst, 200]) to generate cross-linked DNA fragments 200 to 1,000 bp in length. The sonicated samples were centrifuged for 10 min at 13,000 rpm at 4°C. The supernatant was diluted 10-fold with ChIP dilution buffer (Millipore), and protease inhibitors were added as described above. A portion (1%) of the sample was retained as the input sample. The diluted samples were then precleared with 75 μl of salmon sperm DNA-protein A agarose, 50% slurry (Millipore) for 30 min at 4°C with agitation. The agarose was pelleted by brief centrifugation, and 2 μl anti-CTCF antibody (Millipore) was added to 2 ml of precleared supernatant and incubated overnight at 4°C with constant rotation. The next day, 60 μl of salmon sperm DNA-protein A agarose, 50% slurry was added for 1 h at 4°C with rotation to collect the antibody-histone complex. The agarose was pelleted by gentle centrifugation at 1,000 × g for 1 min, and the supernatant with unbound chromatin was discarded. The protein A agarose-antibody-chromatin complex was washed sequentially with 1 ml each of low-salt immune complex (Millipore), high-salt immune complex (Millipore), and LiCl immune complex (Millipore) and twice with Tris-EDTA (TE) buffer (Millipore) for 5 min at 4°C with rotation. The TE wash buffer was removed, and the protein A agarose-antibody-chromatin complex was resuspended in 250 μl of fresh elution buffer (1% SDS, 0.1 M NaHCO3) and incubated at room temperature for 15 min with rotation. The agarose beads were spun down. The process was repeated twice, and the eluates were combined. The cross-links were reversed by adding 20 μl of 5 M NaCl (Millipore) to the combined eluates and heating at 65°C for 4 h, followed by the addition of 10 μl of 0.5 M EDTA (Millipore), 20 μl of 1 M Tris-HCl, pH 6.5 (Millipore), and 2 μl of 10-mg/ml proteinase K to the eluates. This mixture was incubated for 1 h at 45°C. DNA was recovered from the eluate by using a PCR clean-up kit (Qiagen). Following ChIP purification, the eluted products were analyzed by qualitative PCR. The following primers, corresponding to predicted CTCF binding sites, were utilized: FV1, 5′-CGAGACTCTCCAGGTTTGGTAA-3′ and 5′-GGTTCTCGAATCAAGTCGGTTT-3′; FV2, 5′-AACCGACTTGATTCGAGAACCT-3′ and 5′-GTTGGGCGCCAATTGTCAT-3′; FV5, 5′-ACTAAGGCTCCTTCTACTAGCG-3′ and 5′-GTTGAAGAAGTGAATGCTGCAC-3′; FV6, 5′-TTATACCATCCATCCACCCCTC-3′ and 5′-GTTTATGCCAACGAGGTCTCAA-3′; and FV7, 5′-GCATGAGGCACTTCAGAATACA-3′ and 5′-AGGCCAATACTCTTGAGCTAGT-3′. The H19 locus was again used as a positive control, using primers 5′-CCCATCTTGCTGACCTCAC-3′ and 5′-AGACCTGGGACGTTTCTGTG-3′. The sizes of the amplicons for H19, FV1, FV2, FV5, FV6, and FV7 were 165, 157, 188, 110, 115, and 155 base pairs, respectively. ChIP input for HeLa control cells and a HeLa FV clone (FVA2), as well as the ChIP product for the HeLa FV clone, were assayed.
Statistical analysis.
Two-tailed Student unpaired t tests, using GraphPad software (GraphPad Software, Inc., La Jolla, CA), were used to calculate the statistical differences between groups in the immortalization assay. The immortalization frequencies of the SFFV and MSCV series of FV and LV vectors were compared to the replating frequency of SFFV-GV. Data are presented as means ± standard errors of the mean (SEM), and differences with a P value of <0.05 were considered statistically significant. RT-PCR data were analyzed using a one-tailed Mann-Whitney U test.
ACKNOWLEDGMENTS
We acknowledge the assistance of the Research Flow Cytometry Core in the Division of Rheumatology at CCCHMC. Sanger sequencing was performed at the CCHMC DNA Sequencing and Genotyping Core Facility. We acknowledge the technical assistance of the Transgenic Animal and Genome Editing Core at CCHMC, operated under the direction of Yueh-Chiang Hu; the Mouse Cytogenetics Core at CCHMC for performing karyotype analysis; and the Viral Vector Core at CCHMC for assistance in production of vectors used in this study.
This work was supported by a Doris Duke Clinical Scientist Award to P.M. and a Pediatric Scientist Development Program Award (K12-HD000850) from the Eunice Kennedy Shriver National Institute of Child Health and Human Development to M.A.G. The Research Flow Cytometry Core in the Division of Rheumatology at CCCHMC is supported in part by NIH AR-47363, NIH DK78392, and NIH DK90971.
REFERENCES
- 1.Hacein-Bey-Abina S, Hauer J, Lim A, Picard C, Wang GP, Berry CC, Martinache C, Rieux-Laucat F, Latour S, Belohradsky BH, Leiva L, Sorensen R, Debre M, Casanova JL, Blanche S, Durandy A, Bushman FD, Fischer A, Cavazzana-Calvo M. 2010. Efficacy of gene therapy for X-linked severe combined immunodeficiency. N Engl J Med 363:355–364. doi: 10.1056/NEJMoa1000164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hacein-Bey-Abina S, Garrigue A, Wang GP, Soulier J, Lim A, Morillon E, Clappier E, Caccavelli L, Delabesse E, Beldjord K, Asnafi V, MacIntyre E, Dal Cortivo L, Radford I, Brousse N, Sigaux F, Moshous D, Hauer J, Borkhardt A, Belohradsky BH, Wintergerst U, Velez MC, Leiva L, Sorensen R, Wulffraat N, Blanche S, Bushman FD, Fischer A, Cavazzana-Calvo M. 2008. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J Clin Invest 118:3132–3142. doi: 10.1172/JCI35700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Howe SJ, Mansour MR, Schwarzwaelder K, Bartholomae C, Hubank M, Kempski H, Brugman MH, Pike-Overzet K, Chatters SJ, de Ridder D, Gilmour KC, Adams S, Thornhill SI, Parsley KL, Staal FJ, Gale RE, Linch DC, Bayford J, Brown L, Quaye M, Kinnon C, Ancliff P, Webb DK, Schmidt M, von Kalle C, Gaspar HB, Thrasher AJ. 2008. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J Clin Invest 118:3143–3150. doi: 10.1172/JCI35798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stein S, Ott MG, Schultze-Strasser S, Jauch A, Burwinkel B, Kinner A, Schmidt M, Kramer A, Schwable J, Glimm H, Koehl U, Preiss C, Ball C, Martin H, Gohring G, Schwarzwaelder K, Hofmann WK, Karakaya K, Tchatchou S, Yang R, Reinecke P, Kuhlcke K, Schlegelberger B, Thrasher AJ, Hoelzer D, Seger R, von Kalle C, Grez M. 2010. Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease. Nat Med 16:198–204. doi: 10.1038/nm.2088. [DOI] [PubMed] [Google Scholar]
- 5.Ott MG, Schmidt M, Schwarzwaelder K, Stein S, Siler U, Koehl U, Glimm H, Kuhlcke K, Schilz A, Kunkel H, Naundorf S, Brinkmann A, Deichmann A, Fischer M, Ball C, Pilz I, Dunbar C, Du Y, Jenkins NA, Copeland NG, Luthi U, Hassan M, Thrasher AJ, Hoelzer D, von Kalle C, Seger R, Grez M. 2006. Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1. Nat Med 12:401–409. doi: 10.1038/nm1393. [DOI] [PubMed] [Google Scholar]
- 6.Boztug K, Schmidt M, Schwarzer A, Banerjee PP, Diez IA, Dewey RA, Bohm M, Nowrouzi A, Ball CR, Glimm H, Naundorf S, Kuhlcke K, Blasczyk R, Kondratenko I, Marodi L, Orange JS, von Kalle C, Klein C. 2010. Stem-cell gene therapy for the Wiskott-Aldrich syndrome. N Engl J Med 363:1918–1927. doi: 10.1056/NEJMoa1003548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maruggi G, Porcellini S, Facchini G, Perna SK, Cattoglio C, Sartori D, Ambrosi A, Schambach A, Baum C, Bonini C, Bovolenta C, Mavilio F, Recchia A. 2009. Transcriptional enhancers induce insertional gene deregulation independently from the vector type and design. Mol Ther 17:851–856. doi: 10.1038/mt.2009.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Modlich U, Bohne J, Schmidt M, von Kalle C, Knoss S, Schambach A, Baum C. 2006. Cell-culture assays reveal the importance of retroviral vector design for insertional genotoxicity. Blood 108:2545–2553. doi: 10.1182/blood-2005-08-024976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zychlinski D, Schambach A, Modlich U, Maetzig T, Meyer J, Grassman E, Mishra A, Baum C. 2008. Physiological promoters reduce the genotoxic risk of integrating gene vectors. Mol Ther 16:718–725. doi: 10.1038/mt.2008.5. [DOI] [PubMed] [Google Scholar]
- 10.Montini E, Cesana D, Schmidt M, Sanvito F, Bartholomae CC, Ranzani M, Benedicenti F, Sergi LS, Ambrosi A, Ponzoni M, Doglioni C, Di Serio C, von Kalle C, Naldini L. 2009. The genotoxic potential of retroviral vectors is strongly modulated by vector design and integration site selection in a mouse model of HSC gene therapy. J Clin Invest 119:964–975. doi: 10.1172/JCI37630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hunter MJ, Zhao H, Tuschong LM, Bauer TR Jr, Burkholder TH, Persons DA, Hickstein DD. 2011. Gene therapy for canine leukocyte adhesion deficiency with lentiviral vectors using the murine stem cell virus and human phosphoglycerate kinase promoters. Hum Gene Ther 22:689–696. doi: 10.1089/hum.2010.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bauer TR, Olson EM, Huo Y, Tuschong LM, Allen JM, Li Y, Burkholder TH, Russell DW. 2011. Treatment of canine leukocyte adhesion deficiency by foamy virus vectors expressing CD18 from a PGK promoter. Gene Ther 18:553–559. doi: 10.1038/gt.2010.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barde I, Laurenti E, Verp S, Wiznerowicz M, Offner S, Viornery A, Galy A, Trumpp A, Trono D. 2011. Lineage- and stage-restricted lentiviral vectors for the gene therapy of chronic granulomatous disease. Gene Ther 18:1087–1097. doi: 10.1038/gt.2011.65. [DOI] [PubMed] [Google Scholar]
- 14.Chiriaco M, Farinelli G, Capo V, Zonari E, Scaramuzza S, Di Matteo G, Sergi LS, Migliavacca M, Hernandez RJ, Bombelli F, Giorda E, Kajaste-Rudnitski A, Trono D, Grez M, Rossi P, Finocchi A, Naldini L, Gentner B, Aiuti A. 2014. Dual-regulated lentiviral vector for gene therapy of X-linked chronic granulomatosis. Mol Ther 22:1472–1483. doi: 10.1038/mt.2014.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aiuti A, Biasco L, Scaramuzza S, Ferrua F, Cicalese MP, Baricordi C, Dionisio F, Calabria A, Giannelli S, Castiello MC, Bosticardo M, Evangelio C, Assanelli A, Casiraghi M, Di Nunzio S, Callegaro L, Benati C, Rizzardi P, Pellin D, Di Serio C, Schmidt M, Von Kalle C, Gardner J, Mehta N, Neduva V, Dow DJ, Galy A, Miniero R, Finocchi A, Metin A, Banerjee PP, Orange JS, Galimberti S, Valsecchi MG, Biffi A, Montini E, Villa A, Ciceri F, Roncarolo MG, Naldini L. 2013. Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott-Aldrich syndrome. Science 341:1233151. doi: 10.1126/science.1233151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hacein-Bey Abina S, Gaspar HB, Blondeau J, Caccavelli L, Charrier S, Buckland K, Picard C, Six E, Himoudi N, Gilmour K, McNicol AM, Hara H, Xu-Bayford J, Rivat C, Touzot F, Mavilio F, Lim A, Treluyer JM, Heritier S, Lefrere F, Magalon J, Pengue-Koyi I, Honnet G, Blanche S, Sherman EA, Male F, Berry C, Malani N, Bushman FD, Fischer A, Thrasher AJ, Galy A, Cavazzana M. 2015. Outcomes following gene therapy in patients with severe Wiskott-Aldrich syndrome. JAMA 313:1550–1563. doi: 10.1001/jama.2015.3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rethwilm A. 1995. Regulation of foamy virus gene expression. Curr Top Microbiol Immunol 193:1–24. [DOI] [PubMed] [Google Scholar]
- 18.Falcone V, Schweizer M, Neumann-Haefelin D. 2003. Replication of primate foamy viruses in natural and experimental hosts. Curr Top Microbiol Immunol 277:161–180. [DOI] [PubMed] [Google Scholar]
- 19.Russell DW, Miller AD. 1996. Foamy virus vectors. J Virol 70:217–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mergia A, Heinkelein M. 2003. Foamy virus vectors. Curr Top Microbiol Immunol 277:131–159. [DOI] [PubMed] [Google Scholar]
- 21.Bauer TR Jr, Allen JM, Hai M, Tuschong LM, Khan IF, Olson EM, Adler RL, Burkholder TH, Gu YC, Russell DW, Hickstein DD. 2008. Successful treatment of canine leukocyte adhesion deficiency by foamy virus vectors. Nat Med 14:93–97. doi: 10.1038/nm1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Beard BC, Keyser KA, Trobridge GD, Peterson LJ, Miller DG, Jacobs M, Kaul R, Kiem HP. 2007. Unique integration profiles in a canine model of long-term repopulating cells transduced with gammaretrovirus, lentivirus, or foamy virus. Hum Gene Ther 18:423–434. doi: 10.1089/hum.2007.011. [DOI] [PubMed] [Google Scholar]
- 23.Vassilopoulos G, Trobridge G, Josephson NC, Russell DW. 2001. Gene transfer into murine hematopoietic stem cells with helper-free foamy virus vectors. Blood 98:604–609. doi: 10.1182/blood.V98.3.604. [DOI] [PubMed] [Google Scholar]
- 24.Josephson NC, Trobridge G, Russell DW. 2004. Transduction of long-term and mobilized peripheral blood-derived NOD/SCID repopulating cells by foamy virus vectors. Hum Gene Ther 15:87–92. doi: 10.1089/10430340460732481. [DOI] [PubMed] [Google Scholar]
- 25.Josephson NC, Vassilopoulos G, Trobridge GD, Priestley GV, Wood BL, Papayannopoulou T, Russell DW. 2002. Transduction of human NOD/SCID-repopulating cells with both lymphoid and myeloid potential by foamy virus vectors. Proc Natl Acad Sci U S A 99:8295–8300. doi: 10.1073/pnas.122131099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leurs C, Jansen M, Pollok KE, Heinkelein M, Schmidt M, Wissler M, Lindemann D, Von Kalle C, Rethwilm A, Williams DA, Hanenberg H. 2003. Comparison of three retroviral vector systems for transduction of nonobese diabetic/severe combined immunodeficiency mice repopulating human CD34+ cord blood cells. Hum Gene Ther 14:509–519. doi: 10.1089/104303403764539305. [DOI] [PubMed] [Google Scholar]
- 27.Uchiyama T, Adriani M, Jagadeesh GJ, Paine A, Candotti F. 2012. Foamy virus vector-mediated gene correction of a mouse model of Wiskott-Aldrich syndrome. Mol Ther 20:1270–1279. doi: 10.1038/mt.2011.282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morianos I, Siapati EK, Pongas G, Vassilopoulos G. 2012. Comparative analysis of FV vectors with human alpha- or beta-globin gene regulatory elements for the correction of beta-thalassemia. Gene Ther 19:303–311. doi: 10.1038/gt.2011.98. [DOI] [PubMed] [Google Scholar]
- 29.Trobridge GD, Miller DG, Jacobs MA, Allen JM, Kiem HP, Kaul R, Russell DW. 2006. Foamy virus vector integration sites in normal human cells. Proc Natl Acad Sci U S A 103:1498–1503. doi: 10.1073/pnas.0510046103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Everson EM, Olzsko ME, Leap DJ, Hocum JD, Trobridge GD. 2016. A comparison of foamy and lentiviral vector genotoxicity in SCID-repopulating cells shows foamy vectors are less prone to clonal dominance. Mol Ther Methods Clin Dev 3:16048. doi: 10.1038/mtm.2016.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hendrie PC, Huo Y, Stolitenko RB, Russell DW. 2008. A rapid and quantitative assay for measuring neighboring gene activation by vector proviruses. Mol Ther 16:534–540. doi: 10.1038/sj.mt.6300398. [DOI] [PubMed] [Google Scholar]
- 32.Ziebarth JD, Bhattacharya A, Cui Y. 2013. CTCFBSDB 2.0: a database for CTCF-binding sites and genome organization. Nucleic Acids Res 41:D188–D194. doi: 10.1093/nar/gks1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ong CT, Corces VG. 2014. CTCF: an architectural protein bridging genome topology and function. Nat Rev Genet 15:234–246. doi: 10.1038/nrg3663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cuddapah S, Jothi R, Schones DE, Roh TY, Cui K, Zhao K. 2009. Global analysis of the insulator binding protein CTCF in chromatin barrier regions reveals demarcation of active and repressive domains. Genome Res 19:24–32. doi: 10.1101/gr.082800.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu M, Maurano MT, Wang H, Qi H, Song CZ, Navas PA, Emery DW, Stamatoyannopoulos JA, Stamatoyannopoulos G. 2015. Genomic discovery of potent chromatin insulators for human gene therapy. Nat Biotechnol 33:198–203. doi: 10.1038/nbt.3062. [DOI] [PubMed] [Google Scholar]
- 36.Du Y, Spence SE, Jenkins NA, Copeland NG. 2005. Cooperating cancer-gene identification through oncogenic-retrovirus-induced insertional mutagenesis. Blood 106:2498–2505. doi: 10.1182/blood-2004-12-4840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kiem H-P, Baum C, Bushman FD, Byrne BJ, Carter BJ, Cavagnaro J, Malech HL, Mendell JR, Naldini LM, Sorrentino BP, Williams DA, Flotte TR. 2014. Charting a clear path: the ASGCT Standardized Pathways Conference. Mol Ther 22:1235–1238. doi: 10.1038/mt.2014.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Puthenveetil G, Scholes J, Carbonell D, Qureshi N, Xia P, Zeng L, Li S, Yu Y, Hiti AL, Yee JK, Malik P. 2004. Successful correction of the human beta-thalassemia major phenotype using a lentiviral vector. Blood 104:3445–3453. doi: 10.1182/blood-2004-04-1427. [DOI] [PubMed] [Google Scholar]
- 39.Arumugam PI, Higashimoto T, Urbinati F, Modlich U, Nestheide S, Xia P, Fox C, Corsinotti A, Baum C, Malik P. 2009. Genotoxic potential of lineage-specific lentivirus vectors carrying the beta-globin locus control region. Mol Ther 17:1929–1937. doi: 10.1038/mt.2009.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Modlich U, Navarro S, Zychlinski D, Maetzig T, Knoess S, Brugman MH, Schambach A, Charrier S, Galy A, Thrasher AJ, Bueren J, Baum C. 2009. Insertional transformation of hematopoietic cells by self-inactivating lentiviral and gammaretroviral vectors. Mol Ther 17:1919–1928. doi: 10.1038/mt.2009.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hacein-Bey-Abina S, Von Kalle C, Schmidt M, McCormack MP, Wulffraat N, Leboulch P, Lim A, Osborne CS, Pawliuk R, Morillon E, Sorensen R, Forster A, Fraser P, Cohen JI, de Saint Basile G, Alexander I, Wintergerst U, Frebourg T, Aurias A, Stoppa-Lyonnet D, Romana S, Radford-Weiss I, Gross F, Valensi F, Delabesse E, Macintyre E, Sigaux F, Soulier J, Leiva LE, Wissler M, Prinz C, Rabbitts TH, Le Deist F, Fischer A, Cavazzana-Calvo M. 2003. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science 302:415–419. doi: 10.1126/science.1088547. [DOI] [PubMed] [Google Scholar]
- 42.Natkunam Y, Zhao S, Mason DY, Chen J, Taidi B, Jones M, Hammer AS, Hamilton Dutoit S, Lossos IS, Levy R. 2007. The oncoprotein LMO2 is expressed in normal germinal-center B cells and in human B-cell lymphomas. Blood 109:1636–1642. doi: 10.1182/blood-2006-08-039024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kustikova O, Fehse B, Modlich U, Yang M, Dullmann J, Kamino K, von Neuhoff N, Schlegelberger B, Li Z, Baum C. 2005. Clonal dominance of hematopoietic stem cells triggered by retroviral gene marking. Science 308:1171–1174. doi: 10.1126/science.1105063. [DOI] [PubMed] [Google Scholar]
- 44.Montini E, Cesana D, Schmidt M, Sanvito F, Ponzoni M, Bartholomae C, Sergi Sergi L, Benedicenti F, Ambrosi A, Di Serio C, Doglioni C, von Kalle C, Naldini L. 2006. Hematopoietic stem cell gene transfer in a tumor-prone mouse model uncovers low genotoxicity of lentiviral vector integration. Nat Biotechnol 24:687–696. doi: 10.1038/nbt1216. [DOI] [PubMed] [Google Scholar]
- 45.Ryu BY, Evans-Galea MV, Gray JT, Bodine DM, Persons DA, Nienhuis AW. 2008. An experimental system for the evaluation of retroviral vector design to diminish the risk for proto-oncogene activation. Blood 111:1866–1875. doi: 10.1182/blood-2007-04-085506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Browning DL, Collins CP, Hocum JD, Leap DJ, Rae DT, Trobridge GD. 2015. Insulated foamy viral vectors. Hum Gene Ther 27:255–266. doi: 10.1089/hum.2015.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Browning DL, Everson EM, Leap DJ, Hocum JD, Wang H, Stamatoyannopoulos G, Trobridge GD. 2017. Evidence for the in vivo safety of insulated foamy viral vectors. Gene Ther 24:187–198. doi: 10.1038/gt.2016.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schmidt D, Schwalie PC, Wilson MD, Ballester B, Goncalves A, Kutter C, Brown GD, Marshall A, Flicek P, Odom DT. 2012. Waves of retrotransposon expansion remodel genome organization and CTCF binding in multiple mammalian lineages. Cell 148:335–348. doi: 10.1016/j.cell.2011.11.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Paris C, Pentland I, Groves I, Roberts DC, Powis SJ, Coleman N, Roberts S, Parish JL. 2015. CCCTC-binding factor recruitment to the early region of the human papillomavirus 18 genome regulates viral oncogene expression. J Virol 89:4770–4785. doi: 10.1128/JVI.00097-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mehta K, Gunasekharan V, Satsuka A, Laimins LA. 2015. Human papillomaviruses activate and recruit SMC1 cohesin proteins for the differentiation-dependent life cycle through association with CTCF insulators. PLoS Pathog 11:e1004763. doi: 10.1371/journal.ppat.1004763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hughes DJ, Marendy EM, Dickerson CA, Yetming KD, Sample CE, Sample JT. 2012. Contributions of CTCF and DNA methyltransferases DNMT1 and DNMT3B to Epstein-Barr virus restricted latency. J Virol 86:1034–1045. doi: 10.1128/JVI.05923-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lang F, Li X, Vladimirova O, Hu B, Chen G, Xiao Y, Singh V, Lu D, Li L, Han H, Wickramasinghe JM, Smith ST, Zheng C, Li Q, Lieberman PM, Fraser NW, Zhou J. 2017. CTCF interacts with the lytic HSV-1 genome to promote viral transcription. Sci Rep 7:39861. doi: 10.1038/srep39861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ertel MK, Cammarata AL, Hron RJ, Neumann DM. 2012. CTCF occupation of the herpes simplex virus 1 genome is disrupted at early times postreactivation in a transcription-dependent manner. J Virol 86:12741–12759. doi: 10.1128/JVI.01655-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Satou Y, Miyazato P, Ishihara K, Yaguchi H, Melamed A, Miura M, Fukuda A, Nosaka K, Watanabe T, Rowan AG, Nakao M, Bangham CR. 2016. The retrovirus HTLV-1 inserts an ectopic CTCF-binding site into the human genome. Proc Natl Acad Sci U S A 113:3054–3059. doi: 10.1073/pnas.1423199113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tiwari S, Hontz A, Terrell CE, Arumugam P, Carmo M, Risma K, Jordan M, Malik P. 2016. High level of perforin expression is required for effective correction of hemophagocytic lymphohistiocytosis. Hum Gene Ther 27:847–859. doi: 10.1089/hum.2016.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hocum JD, Linde I, Rae DT, Collins CP, Matern LK, Trobridge GD. 2016. Retargeted foamy virus vectors integrate less frequently near proto-oncogenes. Sci Rep 6:36610. doi: 10.1038/srep36610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schambach A, Bohne J, Chandra S, Will E, Margison GP, Williams DA, Baum C. 2006. Equal potency of gammaretroviral and lentiviral SIN vectors for expression of O6-methylguanine-DNA methyltransferase in hematopoietic cells. Mol Ther 13:391–400. doi: 10.1016/j.ymthe.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 58.Bauer TR Jr, Hai M, Tuschong LM, Burkholder TH, Gu YC, Sokolic RA, Ferguson C, Dunbar CE, Hickstein DD. 2006. Correction of the disease phenotype in canine leukocyte adhesion deficiency using ex vivo hematopoietic stem cell gene therapy. Blood 108:3313–3320. doi: 10.1182/blood-2006-03-006908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Trobridge G, Vassilopoulos G, Josephson N, Russell DW. 2002. Gene transfer with foamy virus vectors. Methods Enzymol 346:628–648. doi: 10.1016/S0076-6879(02)46082-X. [DOI] [PubMed] [Google Scholar]
- 60.Kiem H-P, Allen J, Trobridge G, Olson E, Keyser K, Peterson L, Russell DW. 2007. Foamy virus-mediated gene transfer to canine repopulating cells. Blood 109:65–70. doi: 10.1182/blood-2006-04-016741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Will E, Speidel D, Wang Z, Ghiaur G, Rimek A, Schiedlmeier B, Williams DA, Baum C, Ostertag W, Klump H. 2006. HOXB4 inhibits cell growth in a dose-dependent manner and sensitizes cells towards extrinsic cues. Cell Cycle 5:14–22. doi: 10.4161/cc.5.1.2304. [DOI] [PubMed] [Google Scholar]
- 62.Shou Y, Ma Z, Lu T, Sorrentino BP. 2006. Unique risk factors for insertional mutagenesis in a mouse model of XSCID gene therapy. Proc Natl Acad Sci U S A 103:11730–11735. doi: 10.1073/pnas.0603635103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Brinkman EK, Chen T, Amendola M, van Steensel B. 2014. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res 42:e168. doi: 10.1093/nar/gku936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Trobridge G, Josephson N, Vassilopoulos G, Mac J, Russell DW. 2002. Improved foamy virus vectors with minimal viral sequences. Mol Ther 6:321–328. doi: 10.1006/mthe.2002.0672. [DOI] [PubMed] [Google Scholar]
- 65.Schmittgen TD, Livak KJ. 2008. Analyzing real-time PCR data by the comparative CT method. Nat Protoc 3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 66.Hark AT, Schoenherr CJ, Katz DJ, Ingram RS, Levorse JM, Tilghman SM. 2000. CTCF mediates methylation-sensitive enhancer-blocking activity at the H19/Igf2 locus. Nature 405:486–489. doi: 10.1038/35013106. [DOI] [PubMed] [Google Scholar]
- 67.Spencer RJ, del Rosario BC, Pinter SF, Lessing D, Sadreyev RI, Lee JT. 2011. A boundary element between Tsix and Xist binds the chromatin insulator Ctcf and contributes to initiation of X-chromosome inactivation. Genetics 189:441–454. doi: 10.1534/genetics.111.132662. [DOI] [PMC free article] [PubMed] [Google Scholar]