

Abstract
Key points
Calcium homeostasis modulator 1 (CALHM1), a new voltage‐gated ATP‐ and Ca2+‐permeable channel, plays important physiological roles in taste perception and memory formation.
Regulatory mechanisms of CALHM1 remain unexplored, although the biophysical disparity between CALHM1 gating in vivo and in vitro suggests that there are undiscovered regulatory mechanisms.
Here we report that CALHM1 gating and association with lipid microdomains are post‐translationally regulated through the process of protein S‐palmitoylation, a reversible attachment of palmitate to cysteine residues.
Our data also establish cysteine residues and enzymes responsible for CALHM1 palmitoylation.
CALHM1 regulation by palmitoylation provides new mechanistic insights into fine‐tuning of CALHM1 gating in vivo and suggests a potential layer of regulation in taste and memory.
Abstract
Emerging roles of CALHM1, a recently discovered voltage‐gated ion channel, include purinergic neurotransmission of tastes in taste buds and memory formation in the brain, highlighting its physiological importance. However, the regulatory mechanisms of the CALHM1 channel remain entirely unexplored, hindering full understanding of its contribution in vivo. The different gating properties of CALHM1 in vivo and in vitro suggest undiscovered regulatory mechanisms. Here, in searching for post‐translational regulatory mechanisms, we discovered the regulation of CALHM1 gating and association with lipid microdomains via protein S‐palmitoylation, the only reversible lipid modification of proteins on cysteine residues. CALHM1 is palmitoylated at two intracellular cysteines located in the juxtamembrane regions of the third and fourth transmembrane domains. Enzymes that catalyse CALHM1 palmitoylation were identified by screening 23 members of the DHHC protein acyltransferase family. Epitope tagging of endogenous CALHM1 proteins in mice revealed that CALHM1 is basally palmitoylated in taste buds in vivo. Functionally, palmitoylation downregulates CALHM1 without effects on its synthesis, degradation and cell surface expression. Mutation of the palmitoylation sites has a profound impact on CALHM1 gating, shifting the conductance–voltage relationship to more negative voltages and accelerating the activation kinetics. The same mutation also reduces CALHM1 association with detergent‐resistant membranes. Our results comprehensively uncover a post‐translational regulation of the voltage‐dependent gating of CALHM1 by palmitoylation.
Keywords: ion channel, palmitoylation, taste
Key points
Calcium homeostasis modulator 1 (CALHM1), a new voltage‐gated ATP‐ and Ca2+‐permeable channel, plays important physiological roles in taste perception and memory formation.
Regulatory mechanisms of CALHM1 remain unexplored, although the biophysical disparity between CALHM1 gating in vivo and in vitro suggests that there are undiscovered regulatory mechanisms.
Here we report that CALHM1 gating and association with lipid microdomains are post‐translationally regulated through the process of protein S‐palmitoylation, a reversible attachment of palmitate to cysteine residues.
Our data also establish cysteine residues and enzymes responsible for CALHM1 palmitoylation.
CALHM1 regulation by palmitoylation provides new mechanistic insights into fine‐tuning of CALHM1 gating in vivo and suggests a potential layer of regulation in taste and memory.
Abbreviations
- 2BP
2‐bromopalmitate
- ABE
acyl–biotin exchange
- CALHM1
calcium homeostasis modulator 1
- CS
Cys‐to‐Ser
- DAPI
4′,6‐diamidino‐2‐phenylindole
- DRM
detergent‐resistant membrane
- DS
detergent soluble
- ECL
extracellular loop
- KI
knock‐in
- PAT
protein acyltransferase
- PTM
post‐translational modification
- TBST
Tris‐buffered saline containing 0.1% Tween 20
- TM
transmembrane
- WT
wild‐type
- 17‐ODYA
17‐octadecynoic acid
Introduction
Voltage‐gated ion channels play central roles in many physiological activities. They endow excitable cells, such as neurons, muscle cells and endocrine cells, with the ability to fire, shape and conduct action potentials, and convert membrane voltage changes into chemical reactions responsible for the functions of these cells. To meet changing physiological demands, the activity of voltage‐gated ion channels is finely regulated by various mechanisms, including interactions with auxiliary subunits, extracellular/intracellular ions, or ligands, and diverse post‐translational modifications (PTMs). Calcium homeostasis modulator 1 (CALHM1) is a voltage‐gated ion channel identified recently (Dreses‐Werringloer et al. 2008; Ma et al. 2016). In vitro experiments in heterologous expression systems have demonstrated that CALHM1 monomers homo‐hexamerize to form a plasma membrane voltage‐gated non‐selective ion channel with a large ion‐conducting pore that is permeable to both cations and anions, including Ca2+ and ATP (Ma et al. 2012; Siebert et al. 2013; Taruno et al. 2013b). Recent studies have demonstrated that CALHM1 is expressed in excitable cells in different species and plays important physiological and pathophysiological roles. Its expression has been identified in hippocampal and cortical neurons in human and mouse brains (Dreses‐Werringloer et al. 2008; Ma et al. 2012; Dreses‐Werringloer et al. 2013), and in sweet‐, bitter‐ and umami‐sensing taste cells in taste buds of human, macaque and mouse tongues (Moyer et al. 2009; Taruno et al. 2013b). A Caenorhabditis elegans homologue of CALHM1, CLHM‐1, is expressed in sensory neurons and body wall muscle cells (Tanis et al. 2013). In the brain, CALHM1 activity influences the onset of Alzheimer's disease by controlling the accumulation of the amyloid‐β (Dreses‐Werringloer et al. 2008), and controls memory flexibility by modulating long‐term synaptic potentiation through protein kinase A‐mediated phosphorylation of NMDA and AMPA receptors (Dreses‐Werringloer et al. 2013; Vingtdeux et al. 2016). In the tongue, CALHM1 activation in taste cells following a taste‐evoked action potential burst mediates purinergic neurotransmission of sweet, bitter and umami tastes, in which ATP released from CALHM1 in taste cells serves as the primary neurotransmitter to the afferent gustatory nerve terminals expressing P2X2/3 receptors (Taruno et al. 2013b). CLHM‐1 in body wall muscles regulates locomotion of C. elegans (Tanis et al. 2013). Although CALHM1 gating is known to be regulated by membrane voltage and extracellular Ca2+ (), no other regulatory mechanisms of the channel are currently known (Ma et al. 2016). In fact, voltage‐dependent gating properties are different between CALHM1 currents recorded in vivo and in vitro (Romanov et al. 2008; Ma et al. 2012), warranting further exploration into CALHM1 channel regulation to reveal novel mechanisms in regulating memory formation and taste perception.
Among proteinogenic amino acids, the side chain of a cysteine residue (Cys, C) contains a thiol group. Due to its susceptibility to oxidation and high nucleophilicity, it often undergoes diverse chemical modifications (i.e. PTMs) such as disulfide formation, S‐sulfenation, S‐nitrosylation, S‐glutathionylation, S‐prenylation, S‐acylation and ADP ribosylation (Bischoff & Schluter, 2012), all of which have been shown to influence function and/or localization of proteins. S‐Palmitoylation (hereafter referred to as palmitoylation) is the most common form of S‐acylation, and involves a reversible covalent attachment of 16‐carbon saturated fatty acid, palmitate, to specific intracellular Cys via a thioester linkage (Linder & Deschenes, 2007; Fukata et al. 2016). Because of the reversible nature of this modification (Rocks et al. 2005), palmitoylation dynamically regulates activity and localization of both soluble and membrane proteins. Palmitoylation and depalmitoylation are enzymatically driven by a family of DHHC protein acyltransferases (PATs) (Lobo et al. 2002; Fukata et al. 2004) and depalmitoylases (Lin & Conibear, 2015; Yokoi et al. 2016), respectively. An emerging body of evidence has demonstrated a role for palmitoylation of ion channel subunits in controlling every stage of their life cycle (Shipston, 2014), i.e. synthesis (Schmidt & Catterall, 1987; Zhang et al. 2007), forward trafficking (Takimoto et al. 2002; Hayashi et al. 2005; Hayashi et al. 2009; Tian et al. 2012), internalization (Hayashi et al. 2005; Lin et al. 2009) and degradation (Zhang et al. 2007; Jindal et al. 2008). Only a few studies have demonstrated palmitoylation‐mediated regulation of intrinsic channel gating properties (Gubitosi‐Klug et al. 2005; Mueller et al. 2010; Bosmans et al. 2011; Mukherjee et al. 2014; Pei et al. 2016).
CALHM1 is emerging as an important regulator of taste perception and memory formation, yet its regulatory mechanisms remain entirely unexplored. Here we explored roles of intracellular and transmembrane (TM) Cys in CALHM1 function and cell surface expression and discovered the different contributions of these residues, suggesting potential PTMs for these residues. Our results uncover palmitoylation of CALHM1 at two intracellular Cys close to the third and fourth TM domains. Three DHHC PAT members are identified as palmitoylation enzymes for CALHM1 by screening. Remarkably, Calhm1V5‐ires‐Cre knock‐in (KI) mice generated in this study overcame the difficulty in detecting CALHM1 proteins in vivo and revealed that CALHM1 is palmitoylated in taste cells. Palmitoylation has profound influences on CALHM1 function. Disruption of the palmitoylation sites promotes voltage‐dependent gating (voltage sensitivity and activation kinetics), and decreases the association with flotillin‐1‐enriched detergent resistant membrane (DRM) domains. Our results provide the first demonstration of post‐translational regulatory mechanism of the CALHM1 channel and comprehensively describe palmitoylation‐mediated CALHM1 regulation.
Methods
Ethics
Mice and African clawed frogs (Xenopus laevis) were used in this study. All procedures for the care and treatment of animals were carried out according to the Japanese Act on the Welfare and Management of Animals and the Guidelines for the Proper Conduct of Animal Experiments issued by the Science Council of Japan. All animal experiments were conducted in accordance with protocols approved by Institutional Animal Care and Use Committee of Kyoto Prefectural University of Medicine and Osaka medical college. All efforts were made to minimize suffering and to reduce the number of animals used in this study. This work complies with the ethical principles under which The Journal of Physiology operates (Grundy, 2015).
Cell culture
Neuro2a (N2a) and HeLa cells (no. CCL‐131 and CCL‐2, American Type Culture Collection, Rockville, MD, USA) were grown in plastic flasks at 37°C in a humidified incubator with 5% CO2‐in‐air in culture medium containing 90% (v/v) minimum essential medium (MEM) (for N2a cells) or Dulbecco's modified Eagle's medium (for HeLa cells), 10% (v/v) fetal bovine serum, and 1 × antibiotic–antimycotic (Thermo Fisher Scientific, Waltham, MA, USA).
Plasmid vectors
Mouse CALHM1 cDNA (Accession No. NM_001081271) was a kind gift from Dr J. Kevin Foskett (University of Pennsylvania, USA). The full coding sequence of CALHM1 was amplified by PCR (forward primer, 5′‐TTGAGAATTCCACCATGGATAAGTTTCGGATGATCTTC‐3′; reverse primer, 5′‐CGATAATCTTTCACACTTTGCTGAAGTAGGTGGC‐3′) and cloned into EcoRI and HindIII sites of pcDNA3.1(−) (Thermo Fisher Scientific). Single‐ and multiple‐point mutations were then made using site‐directed mutagenesis. We created a mammalian expression vector for fusing a 3×FLAG epitope tag at the carboxyl terminus of a protein (p3×FLAG) by replacing the EGFP sequence with a 3×FLAG sequence at KpnI and NotI sites in pEGFP‐N3 (Clontech, Mountain View, CA, USA). To express carboxyl‐terminally 3×FLAG‐tagged CALHM1 (CALHM1‐FLAG), CALHM1 sequences without stop codon were amplified by PCR (forward primer, 5′‐TTGACTCGAGATGGATAAGTTTCGGATGATCTTC‐3′; reverse primer, 5′‐TTGAGAATTCAACACTTTGCTGAAGTAGGTGGCCAC‐3′) and cloned in frame into XhoI and EcoRI sites of p3×FLAG. Wild‐type (WT) and C100/207S CALHM1 sequences were also subcloned into XhoI and EcoRI sites of pIRES2.AcGFP1 (Clontech), by PCR (forward primer, 5′‐TTGACTCGAGATGGATAAGTTTCGGATGATCTTC‐3′; reverse primer, 5′‐TCTGAATTCTTACACTTTGCTGAAGTAGGTGGC‐3′). All plasmid sequences were validated by Sanger sequencing using BigDye Terminator v3.1 (Applied Biosystems, Foster City, CA, USA). Plasmid DNA was transfected into cells using Lipofectamine 3000 (Thermo Fisher Scientific) according to the manufacturer's instruction. Unless otherwise stated, the CALHM1 cDNA clones in pcDNA3.1(−) and p3×FLAG were respectively transfected for functional analyses (ATP release assay and cell viability assay) and biochemical/immunocytochemical analyses, and the transfected cells were analysed 20–30 h after transfection. cDNAs encoding amino‐terminally 3×HA tagged mouse DHHCs and glutathione S‐transferase (GST) (DHHC‐HA and GST‐HA) cloned into pEF‐Bos‐HA were generous gifts from Dr Masaki Fukata (National Institute for Physiological Sciences, Japan) (Fukata et al. 2004). For cRNA production, CALHM1 constructs were subcloned into the pGEMHE expression vector. cRNAs were synthesized with mMESSAGE mMACHINE T7 kit (Thermo Fisher Scientific) from linearized plasmid DNAs.
Bioluminescence ATP release assay
Extracellular ATP concentration ([ATP]o) was continuously monitored by the luciferin–luciferase reaction as previously reported (Taruno et al. 2013b). HeLa cells were seeded onto 96‐well microplates (Corning Costar, Corning, NY, USA) at 2.0 × 104 cells/well. The next day, cells in each well were transfected with 0.1 μg pcDNA3.1(−) or p3×FLAG encoding WT or mutant CALHM1 cDNA or the empty vector. In experiments shown in Fig. 7 J–L, cells were transfected with 0.1 μg of CALHM1‐FLAG and/or DHHC3‐HA cDNAs, where total amount of DNA (0.2 μg) was adjusted with appropriate control vectors (empty p3×FLAG or pEF‐Bos‐HA encoding GST‐HA). Cells were then incubated for 20–30 h before being analysed, except for experiments shown in Fig. 7 F–H and Fig. 7 J–L, in which cells were analysed at 6 and 9 h post‐transfection, respectively. Media were replaced with 100 μl of the normal bath solution and cells were incubated for 1 h at 37°C. Immediately after, 75 μl of the bath solution was replaced with an equal volume of the Ca2+‐free bath solution containing 5 mm EGTA to remove extracellular free Ca2+ (final extracellular Ca2+ concentration ([Ca2+]o) ∼17 nm), 10 μl of ATP assay solution (FL‐AAM and FL‐AAB, Sigma‐Aldrich, St Louis, MO, USA) was dispensed into each well and the plate was placed in a microplate luminometer (Centro LB960, Berthold Technologies, Bad Wildbad, Germany). Luminescence was measured every 2 min at 25°C. [ATP]o was calculated from a standard curve created in each plate. The normal bath solution contained (in mm): 150 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 Hepes and 10 glucose, pH 7.4 adjusted with NaOH. The Ca2+‐free bath solution contained (in mm): 150 NaCl, 5 KCl, 5 EGTA, 1 MgCl2, 10 Hepes and 10 glucose, pH 7.4 adjusted with NaOH. To measure cellular ATP content, media were replaced with 100 μl of the normal bath solution containing 0.5% Triton X‐100, before 10 μl of ATP assay solution was added and the amount of ATP liberated into the extracellular milieu was measured.
Figure 7. Palmitoylation downregulates CALHM1 function.
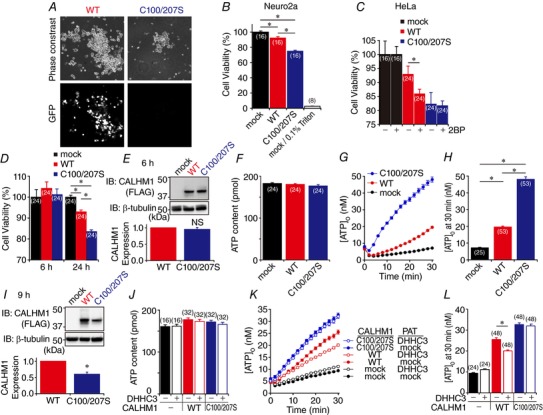
A, phase‐contrast (top) and GFP fluorescence (bottom) images of live N2a cells transfected with WT (left) or C100/207S (right) CALHM1 cloned in pIRES2.AcGFP1. B, cell viabilities of WT and C100/207S CALHM1‐transfected N2a cells 24 h after transfection normalized with respect to mock. Treatment with 0.1% Triton X‐100 was used as control. * P < 0.05 (Tukey's HSD test). C, effect of 2BP on cytotoxicity of WT and C100/207S CALHM1. Cell viabilities of HeLa cells 24 h after transfection with WT and C100/207S CALHM1 normalized to mock were measured with or without 2BP treatment. * P < 0.05 (Student's t test). D, cell viabilities of HeLa cells compared to mock 6 and 24 h after transfection with WT and C100/207S CALHM1‐FLAG. * P < 0.05 (Tukey's HSD test). E, a representative Western blot for analysis of WT and C100/207S CALHM1 expression 6 h after transfection. HeLa cells transfected with WT or C100/207S CALHM1‐FLAG were harvested at 6 h post‐transfection. β‐tubulin signal was used as a loading control. Shown below is quantitative summary of WT and C100/207S CALHM1 expression. WT and C100/207S CALHM1 levels after normalization to the respective β‐tubulin levels were further normalized with respect to WT. n = 7. NS, not significant (Student's t test). F and G, total cellular ATP content (F) and time courses of [ATP]o following removal of (G) in HeLa cells 6 h after transfection with mock, WT or C100/207S CALHM1‐FLAG. H, summary of [ATP]o at 30 min in G. * P < 0.05 (Tukey's HSD test). I, a representative Western blot for analysis of WT and C100/207S CALHM1 expression 9 h after transfection. Quantitative summary is shown below. n = 6. * P < 0.05 (Student's t test). J and K, total cellular ATP content (J) and time courses of [ATP]o following removal (K) in HeLa cells 9 h after transfection with mock, WT or C100/207S CALHM1‐FLAG, and/or DHHC3‐HA as indicated. L, summary of [ATP]o at 30 min in K. * P < 0.05 (Student's t test).
Surface biotinylation
N2a cells were seeded onto 12‐well plate (Corning) at 4.0 × 105 cells/well the day before transfection. Cells were transfected with 0.8 μg of WT or mutant CALHM1‐FLAG cDNA or the empty vector (p3×FLAG). Twenty‐four hours later, cells were washed twice with ice‐cold phosphate‐buffered saline (PBS) containing 2 mm CaCl2 and 1 mm MgCl2 (PBS‐2Ca) and incubated with 0.25 mg ml−1 EZ‐link Sulfo‐NHS‐SS‐biotin (Thermo Fisher Scientific) in 0.5 ml of PBS‐2Ca containing 100 μm GdCl3 for 30 min at 4°C. GdCl3, an inhibitor of the CALHM1 channel, was added to avoid permeation of the biotin reagents through the pore of the CALHM1 channel. The biotinylation reaction was stopped with the addition of 50 μl of 2 m glycine in PBS, followed by sequential washes with PBS containing 100 mm glycine (twice) and PBS (twice). After the final wash, cells were collected, lysed in 100 μl of the lysis buffer (PBS containing 1% Triton X‐100, 1 mm phenylmethylsulfonyl fluoride, and 1 × protease inhibitor cocktail (P8340, Sigma‐Aldrich)), and centrifuged at 20,000 g at 4°C for 10 min. The supernatants were collected as the whole cell lysates. Biotinylated proteins in 130 μg of the whole cell lysates were pulled down by 30 μl of Pierce NeutrAvidin agarose resin (Thermo Fisher Scientific) and eluted by incubating the resin in 30 μl of Laemmli sample buffer at 95°C for 5 min. To enable quantitative analysis, the amount of NeutrAvidin beads was determined so that no biotinylated proteins were detected in the flowthrough samples. The whole cell lysates (15 μg) denatured in Laemmli sample buffer (Total) and 30 μl of the eluate samples (Surface) were subjected to SDS‐PAGE/Western blotting analysis.
Acyl–biotin exchange method
The acyl–biotin exchange (ABE) method uses the conversion of thioester‐linked acyl modifications of proteins (S‐acylation), most commonly palmitoylation, to disulfide‐linked biotin, which involves blocking of unmodified free Cys thiols with N‐ethylmaleimide, cleavage of thioester bonds with hydroxylamine (NH2OH), and labelling of the newly generated free thiols with sulfhydryl‐reactive biotin reagents (biotin‐HPDP), which allows purification of formerly S‐acylated proteins on the streptavidin agarose resin (Wan et al. 2007; Suzuki et al. 2015).
One day after being plated on 12‐well plates at 4.0 × 105 cells/well, N2a cells were transfected with cDNAs encoding WT or mutant CALHM1‐FLAG, WT CALHM2‐FLAG, or WT CALHM3‐FLAG. In experiments shown in Fig. 2 E, 100 μm 2‐bromopalmitate (2BP) or vehicle (0.1% ethanol) was added to the media at 8 h post‐transfection. In experiments shown in Fig. 4 A–C, cells were transfected with CALHM1‐FLAG cDNA along with one of 23 DHHC‐HA cDNAs. GST‐HA cDNA was used as a negative control for DHHCs. In experiments shown in Fig. 6 E and F, cells transfected with WT or C100/207S CALHM1‐FLAG were treated with 20 μm MG‐132 or vehicle (0.1% water) for 8 h before being harvested. Twenty‐four hours after transfection, cells were washed three times with PBS and lysed in 400 μl of 4% SDS‐lysis buffer (PBS containing 4% SDS, 5 mm EDTA, and 1 mm phenylmethylsulfonyl fluoride). After centrifugation at 20,000 g for 10 min at 25°C, the supernatants were collected as the whole cell lysates. Eight microlitres of Bond‐Breaker TCEP (Thermo Fisher Scientific) was added to the lysates at a final concentration of 10 mm followed by incubation at room temperature for 30 min. To block free Cys thiol groups, 8 μl of 2 m N‐ethylmaleimide was added and the samples were incubated at 60°C for 150 min. Proteins were precipitated by the standard methanol–chloroform method to remove unbound N‐ethylmaleimide and resuspended in 100 μl of 4% SDS‐lysis buffer. Half of the sample (50 μl) was mixed with 150 μl NH2OH(+) buffer (1.33 m hydroxylamine, 1 mm biotin‐HPDP, 150 mm NaCl, 0.2% Triton X‐100, pH 7.2) to exchange thiol‐bound fatty acids for biotin, while the other half was mixed with NH2OH(−) buffer (1.33 m Tris–HCl, 1 mm biotin‐HPDP, 150 mm NaCl, 0.2% Triton X‐100, pH 7.2) to serve as a control. After incubation at 37°C for 1 h, proteins were precipitated to remove unbound biotin and resuspended in 1 ml of 0.2% SDS–0.2% Triton‐lysis buffer (PBS containing 0.2% SDS, 0.2% Triton X‐100, 5 mm EDTA and 1 mm phenylmethylsulfonyl fluoride). While 1 μg of the samples were separated as Input samples, biotinylated proteins in 60 μg of the samples were pulled down by 30 μl of Pierce NeutrAvidin agarose resin (Thermo Fisher Scientific) and eluted by incubating the resin in 30 μl of Laemmli sample buffer at 95°C for 5 min. The Input samples were denatured in Laemmli sample buffer with 16% β‐mercaptoethanol at 95°C for 5 min for the detection of CALHM1 or in Laemmli sample buffer with 10 mm DTT at 90°C for 2 min for the detection of DHHC proteins. The denatured input samples and 30 μl of the eluate samples (biotin) were subjected to SDS‐PAGE/Western blotting analysis.
Figure 2. CALHM1 is post‐translationally modified by protein S‐palmitoylation.
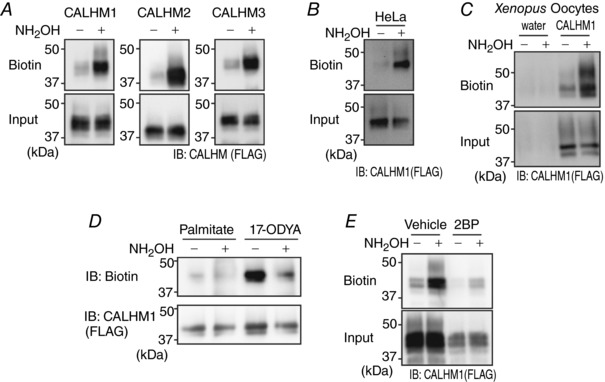
A, detection of S‐acylation on CALHM1, CALHM2 or CALHM3 by the ABE assay in N2a cells. The difference in the amount of biotinylated proteins (Biotin) between Tris‐treated (NH2OH ‘−’) and hydroxylamine‐treated (NH2OH ‘+’) samples (Input) quantitatively indicates the existence of S‐acylation. FLAG‐tagged CALHM proteins were expressed and detected by anti‐FLAG antibody. B, detection of CALHM1 S‐acylation in HeLa cells by the ABE assay. C, detection of CALHM1 S‐acylation in Xenopus oocytes by the ABE assay. Oocytes injected with water or CALHM1‐FLAG cRNA were analysed. D, detection of incorporation of 17‐ODYA into CALHM1 through a hydroxylamine‐cleavable linkage by metabolic labelling. E, effects of 2BP (100 μm, 16 h) on CALHM1 palmitoylation demonstrated by the ABE assay.
Figure 4. Screening for CALHM1 palmitoylating enzymes.
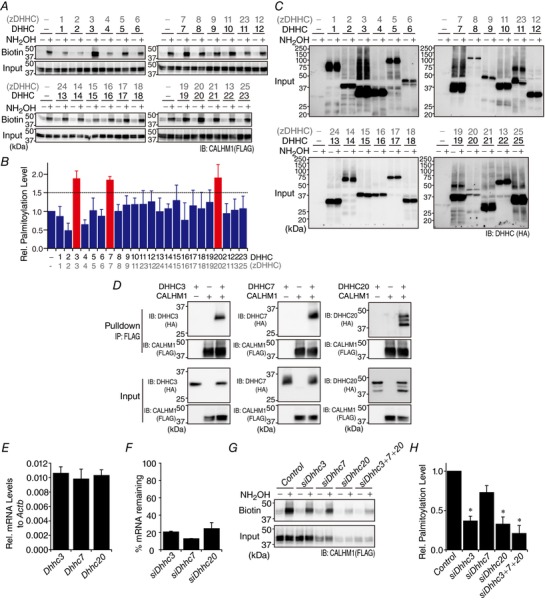
A, screening of 23 DHHC enzymes for their ability to enhance CALHM1 palmitoylation. Shown are representative Western blots of the ABE assay for CALHM1. CALHM1‐FLAG cDNA was co‐transfected with either one of 23 DHHC‐HA cDNAs. GST‐HA cDNA was used as mock transfection in place of DHHCs. DHHC clone numbers are shown in both the old (DHHC) and new (zDHHC) nomenclatures. B, quantitative summary of levels of CALHM1 palmitoylation in A. n = 3. C, expression of DHHC proteins in samples analysed in A. HA‐tagged DHHC proteins were detected by anti‐HA antibody. D, interaction between CALHM1 and DHHC3, 7 and 20, demonstrated by co‐immunoprecipitation. CALHM1‐FLAG and DHHC‐HA were expressed as indicated and respectively detected with anti‐FLAG and ‐HA antibodies in whole cell lysates (Input) and eluates after pulldown with anti‐FLAG antibody (Pulldown). ‘+’, cDNA transfected; ‘−’, mock transfection. E, mRNA expression of Dhhc3, 7 and 20 in N2a cells. The mRNA level of each gene quantified by qRT‐PCR is expressed as a fraction of that of Actb. n = 4. F, efficiency of siRNA‐mediated knockdown of Dhhc3, 7 and 20. For each Dhhc, the mRNA expression remaining after its knockdown is expressed as a percentage of its mRNA level in control cells. n = 3. G, effect of knockdown of Dhhc3, 7 and 20 on CALHM1 palmitoylation in N2a cells demonstrated by the ABE assay. H, quantitative summary of palmitoylation levels of CALHM1 in G. n = 3. * P < 0.05 (Tukey's HSD test).
Figure 6. Palmitoylation does not control CALHM1 protein degradation.
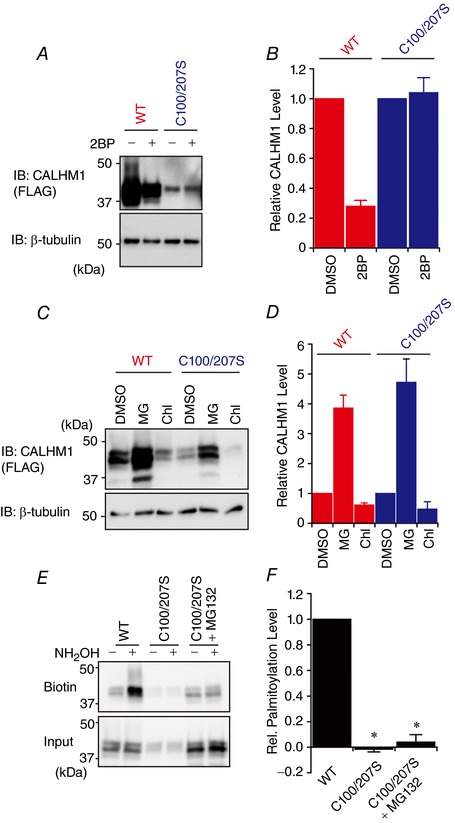
A, Western blot analysis of WT and C100/207S CALHM1. N2a cells were transfected with WT or C100/207S CALHM1‐FLAG and treated with 100 μm 2BP or vehicle (0.1% DMSO) for 8 h before being harvested at 24 h post‐transfection. β‐Tubulin signal was used as a loading control. B, quantitative summary of CALHM1 expression levels in A. For each of WT and C100/207S CALHM1, CALHM1 levels after normalization to the β‐tubulin levels were further normalized with respect to vehicle‐treated samples to highlight the effects of 2BP. n = 3. C, effects of 20 μm MG‐132 (MG) and 50 μm chloroquine (Chl) on expression levels of WT and C100/207S CALHM1. N2a cells transfected with WT or C100/207S CALHM1‐FLAG were treated with either one of the drugs for 8 h before being harvested at 24 h post‐transfection. D, quantitative summary of WT and C100/207S CALHM1 expression levels in C calculated as in B. n = 3. E, representative Western blots of the ABE assay for WT and C100/207S CALHM1, where N2a cells transfected with WT or C100/207S CALHM1‐FLAG were treated with 20 μm MG‐132 or vehicle for 8 h before being harvested at 24 h post‐transfection. F, quantitative summary of CALHM1 palmitoylation levels in E. n = 3. * P < 0.05 (Tukey's HSD test).
For the detection of CALHM1 palmitoylation in Xenopus oocytes, oocytes were obtained as described in ‘Electrophysiological recordings’ and 20 ng of CALHM1‐FLAG cRNA was injected into each oocyte. Water‐injected oocytes were prepared as a negative control. Oocytes were incubated for 2 days at 17°C in frog Ringer solution containing (in mm): 88 NaCl, 1 KCl, 2.4 NaHCO3, 0.3 Ca(NO3)2, 0.41 CaCl2 and 0.82 MgSO4, pH7.6 with 0.1% penicillin–streptomycin solution (Sigma‐Aldrich). They were then processed to obtain total membrane fractions as described previously (Leduc‐Nadeau et al. 2007). Forty oocytes were rinsed in PBS and homogenized in 750 μl of PBS containing 1 mm phenylmethylsulfonyl fluoride and 1 × protease inhibitor cocktail. Homogenates were centrifuged at 300 g for 10 min at 4°C to discard cell debris, and the supernatant was centrifuged at 20,000 g for 20 min at 4°C to pellet total membranes. Pellets were resuspended in 400 μl of 4% SDS‐lysis buffer. After centrifugation at 20,000 g for 10 min at 25°C, the supernatants were subjected to the ABE assay as described above.
For the detection of CALHM1 palmitoylation in taste cells in vivo, Calhm1V5‐ires‐Cre/+ KI mice were killed by CO2 inhalation and cervical dislocation. The tongues were dissected out from 10 mice and lingual epithelia were enzymatically peeled as described previously (Taruno et al. 2013b). Epithelial sheets containing circumvallate and foliate papillae (i.e. many taste buds) were cut out, collected and subjected to the ABE assay as described above. The whole cell lysate and the eluate samples (biotin) were analysed by SDS‐PAGE/Western blotting.
On‐bead click chemistry
N2a cells plated on 10 cm dishes (10 × 106 cells) were transfected with WT CALHM1‐FLAG cDNA (10 μg). Twenty‐four hours later, cells were washed twice with pre‐warmed PBS (37°C) and incubated in MEM containing 10% dialysed fetal bovine serum (Thermo Fisher Scientific) in a humidified incubator with 5% CO2‐in‐air at 37°C for 30 min. Media were then replaced with the labelling medium (MEM containing 10% dialysed fetal bovine serum and 25 μm 17‐octadecynoic acid (17‐ODYA) or palmitate) and the flasks were returned to the incubator for another 2 h before cells were washed twice in ice‐cold PBS, lysed in 600 μl of the lysis buffer (PBS containing 1% Triton X‐100, 1 mm phenylmethylsulfonyl fluoride, and 1 × protease inhibitor cocktail), and centrifuged at 20,000 g at 4°C for 10 min. The supernatants were collected as the whole cell lysates. To pull down CALHM1‐FLAG proteins, 2 mg of the whole cell lysates were incubated with 60 μl of mouse anti‐FLAG antibody‐bound rProtein G agarose resin (Thermo Fisher Scientific) in 1 ml of the IP buffer (PBS containing 1% Triton X‐100 and 1 mm phenylmethylsulfonyl fluoride) with head‐over‐tail rotation at 4°C overnight. The next day, the resin was washed three times with the IP buffer and incubated in 100 μl of Tris buffer (0.5 m Tris–HCl, 150 mm NaCl, 0.1% Triton X‐100, pH 7.2) or hydroxylamine buffer (0.5 m hydroxylamine, 150 mm NaCl, 0.1% Triton X‐100, pH 7.2) at room temperature for 1 h. Thereafter, the resin was washed twice with the IP buffer and twice with PBS and incubated in the click reaction buffer (PBS containing 1 mm CuSO4, 1 mm tris(2‐carboxyethyl)phosphine, 100 μm tris[(1‐benzyl‐1H‐1, 2, 3‐triazol‐4‐yl)methyl]amine and 100 μm biotin‐azide (Thermo Fisher Scientific)) at room temperature for 1 h with head‐over‐tail rotation. Finally, the resin was washed three times with the IP buffer and incubated in 30 μl of 1 × Laemmli sample buffer at 95°C for 5 min to elute CALHM1‐FLAG. The eluate samples were subjected to SDS‐PAGE/Western blotting analysis, in which 90 and 10% of the samples were analysed with anti‐biotin and FLAG antibodies, respectively.
Co‐immunoprecipitation
N2a cells (1.0 × 106 cells) were plated on 6‐well plates and, on the following day, transfected with CALHM1‐FLAG and/or DHHC‐HA cDNAs (1 μg each), where the total amount of DNA (2 μg) was adjusted with appropriate control vectors (empty p3×FLAG or pEF‐Bos‐HA encoding GST‐HA). Twenty‐four hours later, cells were washed twice with ice‐cold PBS and harvested in 100 μl of the lysis buffer (PBS containing 1% Triton X‐100, 1 mm phenylmethylsulfonyl fluoride, and 1 × protease inhibitor cocktail). After centrifugation at 20,000 g for 10 min at 4°C, the supernatants were collected as the whole cell lysates. The whole cell lysates (500 μg) were incubated with 40 μl of rProtein G agarose resin coupled to mouse anti‐FLAG antibody in 1 ml of the IP buffer (PBS containing 1% Triton X‐100 and 1 mm phenylmethylsulfonyl fluoride) with head‐over‐tail rotation at 4°C overnight. The next day, the resins were washed three times with the ice‐cold IP buffer and incubated in 30 μl of Laemmli sample buffer at 95°C for 5 min to elute proteins from the resins. The whole cell lysates (20 μg) (Input) and the eluate samples (Pulldown) were subjected to SDS‐PAGE/Western blotting analysis.
Lipid raft assay
N2a cells (6.0 × 106 cells) were plated on 10 cm dishes and, on the following day, transfected with WT or C100/207S CALHM1‐FLAG cDNA (12 μg). Twenty‐four hours later, cells were harvested in 0.5 ml of the initial lysis buffer (PBS containing 0.05% Triton X‐100, 1 mm phenylmethylsulfonyl fluoride and 1 × protease inhibitor cocktail), mixed by vortex, and incubated on ice for 10 min. Samples were then centrifuged at 20,000 g for 10 min at 4°C and the supernatants were kept as detergent‐soluble (DS) fractions. To wash detergent‐resistant (insoluble) pellets with the initial lysis buffer, pellets were resuspended in 1 ml of the initial lysis buffer, rigorously vortexed, and centrifuged at 20,000 g for 10 min at 4°C and the supernatants were discarded. This washing step was repeated twice. To solubilize proteins in DRMs, detergent‐resistant pellets were resuspended in 50 μl of B buffer from UltraRIPA kit for Lipid Raft (BioDynamics Laboratory Inc., Tokyo, Japan), rigorously vortexed, incubated at room temperature for 5 min, and centrifuged at 20,000 g for 10 min at 25°C. Finally, the supernatants were collected as DRM fractions. Protein concentrations in the DR and DRM fractions were measured by the Non‐Interfering protein assay (G‐Biosciences, St Louis, MO, USA) and equal protein amounts (35 μg) were subjected to SDS‐PAGE/Western blotting analysis. Flotillin1, one of palmitoylated substrates (Morrow et al. 2002), is broadly accepted as a lipid raft‐associated marker (Morrow & Parton, 2005), verifying the isolation of lipid raft‐associated proteins in the DRM fraction.
Western blotting
Proteins were separated in 10% polyacrylamide gels by SDS‐PAGE and transferred to nitrocellulose membranes as described previously (Sun et al. 2014). The membranes were blocked by 5% (w/v) non‐fat dry milk in Tris‐buffered saline containing 0.1% Tween 20 (5% milk‐TBST) at room temperature for 1 h and immunoblotted with anti‐FLAG, anti‐biotin, anti‐glyceraldehyde‐3‐phosphate dehydrogenase, anti‐β‐tublin, anti‐flotillin‐1, anti‐V5, or anti‐HA antibody in 5% milk‐TBST at room temperature for 1 h or at 4°C overnight. The membranes were then washed three times with TBST for 5 min each and incubated with horseradish peroxidase‐conjugated anti‐rabbit or mouse IgG antibody in 5% milk–TBST at room temperature for 1 h. After three washes with TBST, immunoreactive bands were detected using Amersham ECL chemiluminescent reagent (GE Healthcare Bio‐Sciences, Piscataway, NJ, USA) and ChemiDoc XRS+ system with Image Lab software (Bio‐Rad, Hercules, CA, USA). Band intensities were quantified using ImageJ software (https://imagej.nih.gov/ij/) after background correction.
RNA interference
Pre‐designed siRNAs targeted to mouse Dhhcs and negative control siRNA were purchased from Qiagen (Venlo, Netherlands). siRNAs (20 nm of each siRNA) were transfected into N2a cells using Lipofectamine RNAiMAX (Thermo Fisher Scientific) following the manufacturer's instruction. For qRT‐PCR, total RNA extraction was carried out 48 h after siRNA transfection. To test effects of knockdown of Dhhcs on CALHM1 palmitoylation, cells were transfected with CALHM1‐FLAG cDNA 48 h after siRNA transfection and harvested for the ABE assay 24 h later (total 72 h post siRNA transfection).
qRT‐PCR
Total RNA from N2a cells was extracted using an RNeasy Mini Kit (Qiagen) and treated with RNase‐free DNase I to avoid genomic DNA contamination. Three micrograms of RNA was reverse‐transcribed into cDNA in a final volume of 20 μl using the SuperScript III First‐Strand Synthesis System for RT‐PCR with random hexamers according to the manufacturer's instruction (Thermo Fisher Scientific). SYBR green dye gene expression assay was performed with THUNDERBIRD SYBR qPCR Mix (Toyobo, Osaka, Japan) on a StepOnePlus Real‐Time PCR system (Applied Biosystems) with 0.2 μl cDNA (30 ng total RNA equivalent) (see Table 1 for primer sequences). The PCR cycling condition was: 95°C for 60 s; 40 cycles of 95°C for 15 s, 60°C for 60 s. Each assay was run in triplicate. Expression levels after siRNA‐induced mRNA knockdown were normalized to control levels after calculation, with Actb as the endogenous control.
Table 1.
Primer sequences for RT‐PCR and qRT‐PCR
| Gene | Primer | Sequence (5′–3′) | Amplicon size (bp) |
|---|---|---|---|
| Calhm1‐V5 | Cal1‐F | TGCCCTGAGATCTATGATGG | 589 |
| V5‐R | GAGGGTTAGGGATAGGCTTACC | ||
| Calhm1 | Cal1‐F | TGCCCTGAGATCTATGATGG | 283 |
| Cal1‐R | TCATGGCTTCAAAGAACTGC | ||
| Calhm2 | Cal2‐F | CAGGCTCAAGTATGAGTCTC | 184 |
| Cal2‐R | TCGAATGCACCTCTGCTGTG | ||
| Calhm3 | Cal3‐F | CTTCAGCAACTCTGTTGACC | 189 |
| Cal3‐R | CAGCAAGGTTATACTCCAGC | ||
| Dhhc3 | Dhhc3‐F | TGGTGGGATTCCACTTCCTGCA | 106 |
| Dhhc3‐R | GCCTCAAAGCACAGCAGGATGA | ||
| Dhhc7 | Dhhc7‐F | GCTCTGTCTTCGGTTCATGCTC | 132 |
| Dhhc7‐R | CTCAAGGCACAGGAAGACCAAC | ||
| Dhhc20 | Dhhc20‐F | GCAAACCAGAGTGACTACGTCAG | 121 |
| Dhhc20‐R | CAGCTCCATTCTCTAGCCACTG | ||
| Actb | Actb‐F | TCCTGTGGCATCCATGAAAC | 213 |
| Actb‐R | GATCCACACAGAGTACTTGC |
Cholesterol depletion and filipin staining of cholesterol
Methyl‐β‐cyclodextrin (Sigma‐Aldrich) was used to sequester cholesterol from cultured cells. HeLa cells were treated with 2 mm methyl‐β‐cyclodextrin in the normal bath solution or the complete culture medium for 2 or 16 h. Cellular cholesterol was visualized by filipin fluorescence staining using a cholesterol assay kit (ab133116, Abcam, Cambridge, UK) and imaged with the 4′,6‐diamidino‐2‐phenylindole (DAPI) filter set on an inverted microscope (Olympus, Tokyo, Japan) equipped with a ×20/0.70 NA objective lens, a scientific CMOS camera (ORCA‐Flash 4.0 V2, Hamamatsu Photonics, Shizuoka, Japan) and an image acquisition software (HCImage, Hamamatsu).
Live cell imaging
N2a cells on poly‐l‐lysine‐coated glass coverslips placed in 24‐well plates were transfected with 0.8 μg of WT or C100/207S CALHM1 cDNA cloned in pIRES2.AcGFP1. Twenty‐four hours after transfection, phase contrast and fluorescence images of cells bathed in the normal bath solution were captured using an inverted microscope equipped with a ×20/0.70 NA objective lens, a scientific CMOS camera (ORCA‐Flash 4.0 V2) and HCImage software.
Immunocytochemistry
N2a cells on poly‐l‐lysine‐coated glass coverslips placed in 24‐well plates were transfected with WT or C100/207S CALHM1‐FLAG cDNA. The medium was replaced by fresh medium containing 100 μg ml−1 cycloheximide or vehicle (0.1% water) 6.5 h after transfection. After incubation for another 1.5 h (i.e. 8 h after transfection), cells were washed twice with ice‐cold PBS, fixed in PBS containing 4% paraformaldehyde at room temperature for 20 min, washed in PBS for 3 × 3 min, blocked for 1 h at room temperature with the blocking buffer (PBS containing 3% normal goat serum and 0.1% Triton X‐100), and incubated at 4°C overnight with mouse anti‐FLAG antibody (Sigma‐Aldrich) diluted in the blocking buffer (16 μg ml−1). The next day, cells were washed with PBS at room temperature for 3 × 10 min, incubated in the dark at room temperature for 1 h with Alexa Fluor 488 goat anti‐mouse IgG (Thermo Fisher Scientific) diluted in the blocking buffer (8 μg ml−1), and washed with PBS in the dark at room temperature for 3 × 10 min before coverslips were mounted on slides in VectaShield with DAPI (H‐1500, Vector Laboratories, Burlingame, CA, USA). Images of stained cells were captured with an LSM510 confocal scanning microscope (Carl Zeiss, Oberkochen, Germany) using an EC Plan‐Neofluar ×40/1.30 NA oil objective. Images show single optical sections, collected with the pinhole set to 1 Airy unit for the green channel. DAPI (blue channel) was excited with two photons using a MaiTai titanium–sapphire laser tuned to 780 nm (Spectra‐Physics, Santa Clara, CA, USA).
Cell viability assay
Cytotoxicity of WT and C100/207S CALHM1 expression and treatment with 2BP was evaluated by quantifying cell viability using Cell Counting Kit 8 (Dojindo, Kumamoto, Japan). HeLa and N2a cells were plated and transfected the same way as in the bioluminescence ATP release assay. After 6 or 24 h, 10 μl of Cell Counting Kit 8 solution was added to each well and cells were incubated for 2 h at 37°C before absorbance at 450 nm was measured using a SpectraMax M2 microplate reader (Molecular Devices, Sunnyvale, CA, USA). The cell viability relative to mock‐transfected cells (control) was calculated by the following formula:
where A is absorbance at 450 nm after background signal subtraction and the denominator is the average absorbance in mock‐transfected cells. In experiments shown in Fig. 7 B, 0.1% Triton X‐100 was added to some wells 30 min before adding Cell Counting Kit 8 solution. In experiments shown in Fig. 7 C, either 100 μm 2BP or vehicle (0.1% ethanol) was added to each well at 8 h post‐transfection.
Electrophysiological recordings
Frogs (Xenopus laevis) were obtained from a commercial supplier (Hamamatsu Seibutsu Kyozai, Hamamatsu, Japan), kept in a water tank at 18°C, and fed three times a week. A total of 15 female frogs were used in this study. They were killed by decapitation after the final collection of oocytes. Oocytes were collected from frogs anesthetized in water containing 0.15% tricaine and treated with collagenase (2 mg ml−1; type 1; Sigma‐Aldrich) for 5 h to completely remove the follicular cell layer. One nanogram of CALHM1 cRNA was injected into oocytes with 80 ng of Xenopus connexin‐38 antisense oligonucleotide to inhibit endogenous Cx38 currents (Ma et al. 2012), and oocytes were incubated at 17°C in frog Ringer solution. One to three days after cRNA injection, CALHM1 currents were recorded under two‐electrode voltage clamp using an amplifier (OC725C; Warner Instruments, Hamden, CT, USA) and pCLAMP10 software (Molecular Devices). Oocytes were injected with a 23 nl of 50 mm BAPTA to minimize endogenous Ca2+‐activated Cl− currents at least 30 min before the recording (Ma et al. 2012). The microelectrodes were drawn from borosilicate glass capillaries (World Precision Instruments, Sarasota, FL, USA) to a resistance of 0.2–0.5 MΩ when filled with 3 m KCl. The bath solution contained (in mm): 96 NaCl, 2 KCl, 1.8 CaCl2, 1 MgCl2 and 5 Hepes, pH 7.2. Oocytes were held at −80 mV, stepped up to various voltages for 5 s every 30 s. Data from the amplifier were digitized at 10 kHz with a Digidata 1440 (Molecular Devices) and filtered at 1 kHz by pCLAMP10. All experiments were performed at room temperature (22 ± 2°C). Oocytes with a holding current larger than −0.2 μA at −80 mV were excluded from the analysis to avoid unhealthy oocytes. Conductance–voltage (G–V) relationships were taken from tail current amplitude at −80 mV and were fitted using pCLAMP10 software to a two‐state Boltzmann equation: G = G min + (G max − G min)/(1+e− zF ( V−V 1/2 )/ RT), where G max and G min are the maximum and minimum tail current amplitudes, z is the effective charge, V 1/2 is the half‐activation voltage, T is the temperature in kelvins, F is Faraday's constant, and R is the gas constant. G/G max, which represents the normalized tail current amplitude, was plotted against membrane voltage for presentation of the G–V relationships.
Generation of Calhm1V5‐ires‐Cre knock‐in mice
To fuse CALHM1 with a V5 epitope tag (CALHM1‐V5) and express Cre recombinase under the control of Calhm1 promoter in mice, a V5‐stop codon–IRES–NLS‐Cre–poly(A) sequence was inserted immediately before the stop codon in the Calhm1 locus on chromosome 19 by the CRISPR/Cas9 genome editing technology (Yang et al. 2013) (Fig. 5 A). Here, the V5‐stop codon sequence is 5′‐ GGTAAGCCTATCCCTAACCCTCTCCTCGGTCTCGATTCTACGTGA‐3′. IRES, NLS‐Cre, and poly(A) refer to the encephalomyocarditis virus internal ribosomal entry site and the bacteriophage P1 Cre recombinase containing a nuclear localization signal, and the simian virus 40 late polyadenylation signal, respectively. Gene targeting in one‐cell‐stage embryos and generation of mutant mice on the C57BL/6 background were performed at the Laboratory Animal Resource Centre, University of Tsukuba. The resulting Calhm1V5‐ires‐Cre KI allele was validated by sequencing genomic DNA purified from tails (Wizard Genomic DNA Purification kit, Promega) using BigDye Terminator v3.1 (Applied Biosystems) (Fig. 5 B). Calhm1V5‐ires‐Cre/+ KI mice are viable and fertile. They were backcrossed at least three times with WT C57BL/6 mice obtained from Shimizu Laboratory Supplies (Shizuoka, Japan). Breeding of mice carrying the Calhm1V5‐ires‐Cre allele resulted in expected frequencies of genotypes. All mice were maintained on a 12 h–12 h light–dark cycle with lights off at 19.00 h and had ad libitum access to tap water and standard rodent chow. A total of 66 heterozygous Calhm1V5‐ires‐Cre/+ KI mice and their WT littermates (Calhm1+/+) (adult males and females, 10–20 weeks of age) were killed in this study: 3 KI and 3 WT in Fig. 5 C and D (1 animal from each group per experiment × 3 experiments); 12 KI and 12 WT in Fig. 5 E (4 animals from each group per experiment × 3 experiments); 30 KI in Fig. 5F (10 animals per experiment × 3 experiments).
Figure 5. CALHM1 is palmitoylated in taste cells in vivo .
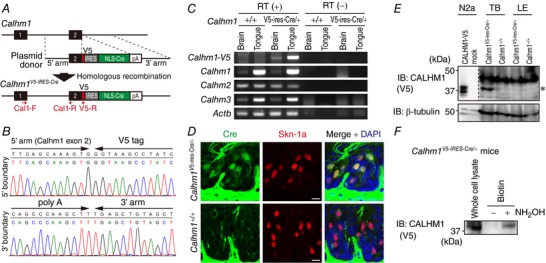
A, strategy for the generation of a Calhm1V5‐ires‐Cre KI allele. Black boxes, exons 1 and 2 of Calhm1; 5′ arm and 3′ arm, homologous arms of the plasmid donor; arrows, PCR primers (Cal1‐F, Cal1‐R and V5‐R) for RT‐PCR. B, genome sequencing across the 5′ and 3′ boundaries of the targeting region confirmed correct fusion of a V5 tag to the last codon of Calhm1 and insertion of the full length transgene sequence. C, RT‐PCR of mRNA of CALHM1‐V5 (mutant transcript), CALHM1, CALHM2, CALHM3 and Actb from the brain and the tongue in wild type (Calhm1+/+) and heterozygous Calhm1 KI (Calhm1V5‐ires‐Cre/+) mice. RT(−), no reverse transcriptase control. Primer sequences and PCR conditions are shown in Table 1. D, immunohistochemical detection of Cre in taste buds of Calhm1V5‐ires‐Cre/+ mice. Cre (green) and Skn‐1a (red) were fluorescently double‐labelled in 10 μm‐thick tissue sections containing the circumvallate papillae of Calhm1+/+ and Calhm1V5‐ires‐Cre/+ mice. Sections were counterstained with DAPI. Scale bars, 10 μm. E, Western blot detection of CALHM1‐V5 in taste buds of Calhm1V5‐ires‐Cre/+ mice. Lysates (95 μg) from lingual epithelial sheets bearing taste buds (TB) and non‐gustatory lingual epithelial sheets devoid of taste buds (LE) collected from Calhm1+/+ and Calhm1V5‐ires‐Cre/+ mice (4 mice/sample) were analysed by Western blotting with lysates (1 μg) of N2a cells expressing CALHM1‐V5 and control N2a cells as positive and negative controls, respectively. A band for V5 at the size corresponding to CALHM1‐V5 (*) was observed only in TB of Calhm1V5‐ires‐Cre/+ mice. F, palmitoylation of CALHM1 in taste buds in vivo demonstrated by the ABE assay. Lingual epithelial sheets containing circumvallate and foliate papillae were collected from 10 Calhm1+/V5‐ires‐Cre KI mice and subjected to the ABE assay. Replacing hydroxylamine (NH2OH) with a Tris‐buffer (NH2OH ‘−’) served as a negative control to prove efficient initial blockade of free Cys thiols.
RT‐PCR
Mice were killed with an overdose of sodium pentobarbital (> 300 mg kg−1, i.p.) and transcardially perfused with ice‐cold PBS. The middle one‐third portion of the whole brain in the rostrocaudal axis containing the hippocampus, the cerebral cortex and the thalamus, and a small region of the tongue containing circumvallate papilla were dissected out and kept in RNAlater Stabilization Solution (Thermo Fisher Scientific). Total RNA from the brain and tongue tissues was extracted using an RNeasy Mini Kit (Qiagen, Venlo, Netherlands) and treated with RNase‐free DNase I. Five micrograms of RNA was reverse‐transcribed into cDNA in a final volume of 20 μl using the SuperScript III First‐Strand Synthesis System for RT‐PCR with random hexamers according to the manufacturer's instructions (Thermo Fisher Scientific). PCRs were performed with 2 μl cDNA (500 ng total RNA equivalent), 0.5 μm of each primer (see Table 1 for PCR primer sequences), 0.2 mm dNTPs, 1 × Takara Ex Taq buffer and 0.625 U Takara Ex Taq polymerase (Takara Bio, Shiga, Japan). PCR cycling conditions were: 94°C for 60 s; 45 cycles of 94°C for 30 s, 54°C for 30 s, 72°C for 60 s; 72°C for 60 s; 4°C (hold).
Immunohistochemistry
Mice were used for immunofluorescence staining of taste buds. After mice were killed with an overdose of sodium pentobarbital (> 300 mg kg−1, i.p.), tissue preparation and staining were performed as previously described (Taruno et al. 2013b). Briefly, tongue tissues containing circumvallate papilla fixed with 4% paraformaldehyde were cryosectioned at 10 μm thickness and mounted on glass slides. Slides were blocked, and incubated at room temperature overnight with primary antibodies: mouse anti‐Cre antibody (EMD Millipore, Billerica, MA, USA) and rabbit anti‐Skn‐1a antibody (Santa Cruz Biotechnology, Dallas, TX, USA). The next day, the slides were washed and incubated at room temperature for 1 h with Alexa Fluor‐conjugated secondary antibodies: Alexa Fluor 488 goat anti‐mouse IgG and Alexa Fluor 546 goat anti‐rabbit IgG (Thermo Fisher Scientific). Finally, slides were washed and mounted in VectaShield with DAPI (Vector Laboratories). Confocal images of stained sections were captured as described in ‘Immunocytochemistry’.
Statistical analysis
All data are presented in graphical form using Kaleida Graph (Synergy Software, Reading, PA, USA) and Igor Pro (WaveMatrics, Portland, OR, USA). One‐way analysis of variance (ANOVA) with Tukey's HSD post hoc test and Student's two‐tailed unpaired t test were used for multiple comparisons (three or more groups) and comparison of two groups, respectively. A P value of <0.05 was considered as statistically significant. All data are presented as means ± SEM. n refers to the number of experiments throughout the text.
Results
Single cysteine residues control CALHM1 function and cell surface expression
Membrane voltage and extracellular divalent cations are currently the only known mechanisms of CALHM1 regulation (Ma et al. 2012). To explore other modes of CALHM1 regulation, we examined the roles for Cys residues in the intracellular and TM domains on function and cell surface expression of the channel. According to the membrane topology of a CALHM1 monomer that was predicted by the TMpred algorithm (http://www.ch.embnet.org/software/TMPRED_form.html) and has been experimentally supported (Siebert et al. 2013), mouse CALHM1 possesses eight intracellular and TM Cys: C23 in TM1, C100 in the intracellular loop, C123 and C126 in TM3, and C207, C234, C245 and C312 in the carboxyl terminus (Fig. 1 A). To elucidate the contribution of these Cys to CALHM1 function, we quantified cellular ATP release mediated by WT CALHM1 and eight single Cys‐to‐Ser (CS) point mutants in which a Cys is replaced with a serine (Ser, S) by monitoring [ATP]o after removal of (Fig. 1 C and D). Lowered [Ca2+]o evoked a robust increase in [ATP]o in WT CALHM1‐expressing cells but not in control cells, indicating ATP release into the extracellular milieu through CALHM1 as reported previously (Taruno et al. 2013b). Single CS mutations caused changes in the amount of ATP release without affecting the total cellular ATP content (Fig. 1 B). Compared to WT, C23S mutation potentiated, C123S and C126S abolished, C234S, C245S and C312S partially reduced, and C100S and C207S only slightly altered ATP release, suggesting that these Cys differentially contribute to the channel function. Next, cell surface expression of WT and CS mutants were examined by the cell surface biotinylation assay (Fig. 1 E). Single CS mutants exhibited various levels of cell surface targeting (Fig. 1 F). Compared to WT, C23S and C245S mutations increased, C123S and C126S diminished, and the others did not alter trafficking of the channel to the cell surface. Note that effects of mutations on C23, C123 and C126 on cell surface targeting agree with those on ATP release, implying that these Cys regulate CALHM1 function mainly by controlling trafficking. In contrast, the contributions of C234, C245 and C312 to ATP release cannot be explained by those to cell surface localization, suggesting that these residues regulate CALHM1 function predominantly by controlling individual channel activity. These results demonstrate that intracellular and TM Cys are differentially involved in the regulation of the activity and cell surface expression of CALHM1, indicating that potential PTMs on these Cys are crucial for CALHM1 regulation.
Figure 1. Intracellular and transmembrane Cys regulate function and cell surface expression of CALHM1.
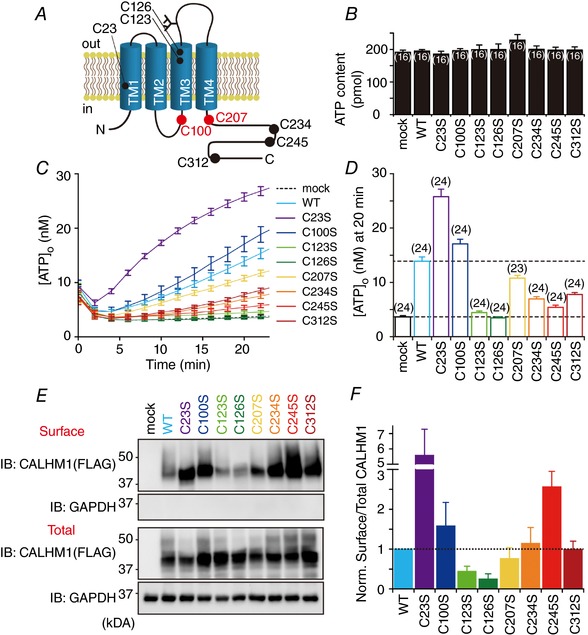
A, schematic diagram of the putative membrane topology of a CALHM1 monomer, identifying Cys in the intracellular and TM domains. The palmitoylated Cys are indicated in red. TM, transmembrane domain; C, Cys. B, total cellular ATP content of HeLa cells transfected with the empty vector (mock), WT CALHM1, or single CS mutant. C, time courses of [ATP]o due to release from HeLa cells expressing WT or single CS mutant CALHM1 as well as mock‐transfected cells following exposure to zero (∼17 nm) [Ca2+]o. Untagged CALHM1 constructs were expressed in B and C. D, summary of [ATP]o at 20 min in C. Number of experiments in parentheses. E, representative Western blots of the surface biotinylation analysis of WT and single CS mutant CALHM1 in N2a cells. Biotinylated proteins (Surface) were avidin affinity‐purified from the whole cell lysates (Total) and analysed by Western blotting. Carboxyl‐terminally FLAG tagged CALHM1 (CALHM1‐FLAG) constructs were expressed and detected by anti‐FLAG antibody for biochemistry throughout and glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) was detected as a loading control. F, summary of the ratios of surface‐to‐total CALHM1 levels normalized to WT. n = 3.
CALHM1 is a substrate for S‐palmitoylation
Among PTMs on the Cys thiol, growing evidence has drawn attention to the roles of palmitoylation, a major form of S‐acylation, on ion channel regulation (Shipston, 2014). To examine whether CALHM1 and its homologues, CALHM2 and CALHM3, are palmitoylated, the ABE assay was performed in N2a cells to purify S‐acylated proteins from whole cell lysates. As seen in Fig. 2 A, the amount of avidin‐purified biotinylated proteins of all the CALHM homologues was increased by hydroxylamine cleavage, the essential process in the ABE assay, indicating that CALHM1, CALHM2 and CALHM3 are all S‐acylated, most likely palmitoylated, by endogenous PATs. CALHM1 S‐acylation was also detected in HeLa cells and Xenopus laevis oocytes (Fig. 2 B and C), indicating that this PTM occurs on CALHM1 in a cell type‐independent manner. Although palmitate is commonly thought to be the major species of lipid in most S‐acylated proteins, the ABE assay does not define the identity of the bound lipid. To confirm whether palmitate is incorporated into CALHM1 proteins, we carried out metabolic labelling of CALHM1‐expressing cells with palmitate or 17‐ODYA, an alkyne‐containing palmitate analogue, immunoprecipitation of CALHM1, treatment with Tris or hydroxylamine, and a following copper‐catalysed click chemistry reaction to biotin azide for Western blot detection of 17‐ODYA‐labelled CALHM1 proteins. Hydroxylamine‐sensitive immunosignals for biotin on CALHM1 were detected in cells treated with 17‐ODYA but not in palmitate‐treated cells (Fig. 2 D). Furthermore, treatment of CALHM1‐expressing cells with 2BP, an inhibitor of palmitoylation (Resh, 2006; Zheng et al. 2013), abolished CALHM1 S‐acylation detected by the ABE assay (Fig. 2 E). Taken together, three lines of biochemical and pharmacological evidence demonstrate that CALHM1 is a substrate for palmitoylation.
CALHM1 is S‐palmitoylated at C100 and C207
We then set out to identify Cys that are involved in palmitoylation. Since DHHC PATs are multipass membrane proteins and possess the shared catalytic DHHC (Asp–His–His–Cys) Cys‐rich domain in their intracellular regions, palmitoylation of membrane proteins occurs on intracellular Cys. Therefore, C100 in the intracellular loop and C207, C234, C245 and C312 in the carboxyl terminus are potential palmitoylation sites on CALHM1 (Fig. 1 A). Systematic introduction of CS mutations into these Cys and evaluation of palmitoylation of the resultant mutants with the ABE assay were performed. First, to determine the contribution of these intracellular domains to palmitoylation, we compared palmitoylation of WT, C100S, C207/234/245/312S and C100/207/234/245/312S CALHM1 (Fig. 3 B). All the four Cys in the carboxyl terminus are replaced with Ser in C207/234/245/312S and all the five intracellular Cys residues are mutated in C100/207/234/245/312S. C100S and C207/234/245/312S mutants retained palmitoylation, whereas the palmitoyl modification was abolished in C100/207/234/245/312S, indicating that palmitoylation occurs at multiple sites, C100 in the intracellular loop and at least one Cys in the carboxyl terminus. Second, to identify the residues that are palmitoylated, we analysed palmitoylation of WT, C100/207/234/245/312S, C207/234/245/312S, C100/234/245/312S, C100/207/245/312S, C100/207/234/312S and C100/207/234/245S CALHM1 (Fig. 3 C). C207/234/245/312S and C100/234/245/312S remained palmitoylated, whereas C100/207/234/245/312S, C100/207/245/312S, C100/207/234/312S and C100/207/234/245S showed no palmitoylation, identifying C100 and C207 as two strong candidates (Fig. 3 A). Third, we further quantified palmitoylation levels of WT, C100S, C207S, C100/207S and C234/245/312S CALHM1 (Fig. 3 D and E). Palmitoylation was absent only in the double CS mutant, C100/207S, while the other mutants retained palmitoylation. Quantitatively, C100S mutation partially decreased the level of palmitoylation but C207S did not affect it. The insensitivity of CALHM1 palmitoylation to C234/245/312S mutation excluded the possibility of palmitoylation at these residues, on the condition that C100 and/or C207 are palmitoylated. Our data establish C100 and C207 residues as the sites for palmitoylation.
Figure 3. Identification of palmitoylation sites on CALHM1.
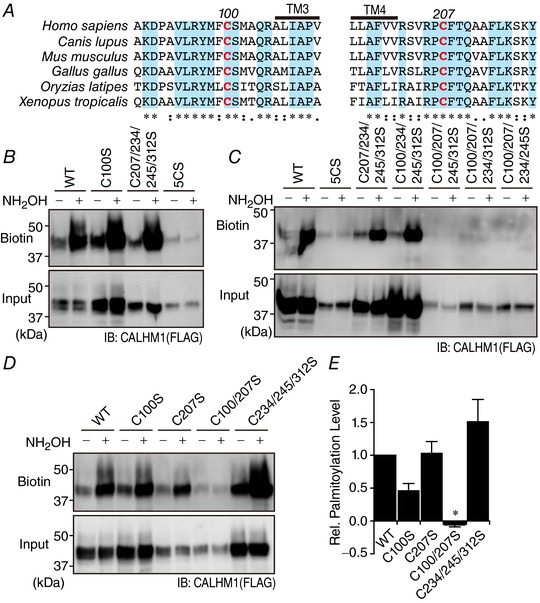
A, alignment of primary sequences surrounding C100 and C207 for different species. *represents a fully conserved residue, and ‘:’ and ‘.’ indicate strong and weak similarities, respectively. The putative TM3 and TM4 are indicated. Amino acid numbering is based on the murine CALHM1. B–D, representative Western blots of the ABE assay for WT, C100S, C207/234/245/312S and C100/207/234/245/312S (5CS) CALHM1 (B), for WT, 5CS, C207/234/245/312S, C100/234/245/312S, C100/207/245/312S, C100/207/234/312S and C100/207/234/245S CALHM1 (C), and for WT, C100S, C207S, C100/207S and C234/245/312S CALHM1 (D). E, quantitative summary of palmitoylation levels of WT and mutant CALHM1 in D. Levels of palmitoylation were calculated by comparison of biotinylated CALHM1 levels in corresponding samples with and without NH2OH treatment after normalization to the input levels and were normalized with respect to WT. n = 3. * P < 0.05 (Tukey's HSD test).
DHHC palmitoyl transferases for CALHM1
There are 23 members in the mammalian DHHC PAT family. To identify PATs that catalyse CALHM1 palmitoylation, we screened all the 23 DHHCs for their ability to enhance palmitoylation level of CALHM1. As displayed in Fig. 4 A and summarized in Fig. 4 B, noticeable (> 1.5‐fold) increases in the palmitoylation level detected by the ABE assay were observed by co‐expression of DHHC3, 7 or 20, identifying these three DHHC proteins as potential candidates of PATs for CALHM1 in vivo. Despite different expression levels of recombinant DHHC proteins (Fig. 4 C), their effects on CALHM1 palmitoylation do not appear to correlate with their relative expression levels. We also found that DHHC3, 7 and 20 were co‐immunoprecipitated with CALHM1 (Fig. 4 D), demonstrating that CALHM1 interacts with these DHHCs. In N2a cells, mRNAs of Dhhc3, 7 and 20 were expressed at similar levels (Fig. 4 E). We further examined effects of RNA interference‐mediated gene knockdown of Dhhc3, 7 and 20 on CALHM1 palmitoylation in N2a cells by the ABE assay. Transfection of siRNAs targeted to these Dhhcs decreased their mRNA levels by ∼80% (Fig. 4 F). Knockdown of Dhhc3 and 20 markedly reduced CALHM1 palmitoylation by ∼65% (Fig. 4 G and H). Dhhc7 knockdown showed a tendency to lower it but the reduction is not statistically significant (P = 0.17, Tukey's HSD test), suggesting that DHHC3 and 20 but not DHHC7 are responsible for endogenous CALHM1 palmitoylation in N2a cells. There is, however, still the possibility that the remaining DHHC7 after knockdown was sufficient for maintaining CALHM1 palmitoylation. Thus, these results together demonstrate that DHHC3 and 20, and potentially DHHC7, are PATs for CALHM1.
CALHM1 is palmitoylated in taste cells in vivo
Until now, endogenous CALHM1 proteins have never been detected in any native tissues or cell lines due to a lack of specific antibodies. After repeated failed attempts to generate anti‐CALHM1 antibodies which can detect CALHM1 in native tissues, we produced Calhm1V5‐ires‐Cre KI mice in which endogenous CALHM1 is carboxyl‐terminally tagged with a V5 epitope tag for highly sensitive biochemical analysis (Fig. 5 A and B). In these mice, the transgene mRNA expression was demonstrated by RT‐PCR in the brain and the tongue, which are the two organs where CALHM1 is endogenously expressed (Fig. 5 C). Expression of uncharacterized homologue genes Calhm2 and Calhm3, whose loci are located directly next to the Calhm1 locus, was unchanged by the transgene insertion (Fig. 5 C). Selective Cre recombinase expression in taste cells expressing Skn‐1a, a marker for sweet‐, bitter‐, and umami‐sensing taste cells, in Calhm1V5‐ires‐Cre/+ mice (Fig. 5 D) demonstrates that the transgene expression is confined to sweet‐, bitter‐, and umami‐sensing taste cells, where CALHM1 is endogenously expressed, but not in other taste cells, ruling out ectopic transgene expression (Matsumoto et al. 2011; Taruno et al. 2013b). Using these KI mice, we succeeded in detecting selective expression of CALHM1‐V5 proteins in the taste buds but not in the surrounding non‐sensory epithelium by Western blotting (Fig. 5 E). Finally, to examine whether CALHM1 is palmitoylated in native taste cells, tongue epithelial sheets which contain circumvallate and foliate papillae were dissected out from Calhm1V5‐ires‐Cre/+ mice and the ABE assay was performed. As shown in Fig. 5 F, we observed hydroxylamine cleavage‐dependent recovery of biotinylated CALHM1‐V5 proteins, demonstrating CALHM1 palmitoylation in taste cells in vivo.
Palmitoylation is not involved in CALHM1 degradation
Both pharmacological inhibition of cellular palmitoylation by 2BP (Fig. 2 E) and disruption of the palmitoylation sites (Fig. 3) caused sizable decreases in CALHM1 protein expression detected by Western blotting. It is likely that a defect in palmitoylation on CALHM1 caused a reduction in CALHM1 expression because the effects of 2BP and the palmitoylation‐deficient mutation, C100/207S, on CALHM1 expression were not additive (Fig. 6 A and B). Therefore, we utilized the C100/207S mutant in further experiments to analyse the effects of palmitoylation on CALHM1. We found that CALHM1 is mainly degraded by the ubiquitin‐proteasome pathway because MG‐132 (a proteasome inhibitor) but not chloroquine (a lysosomal inhibitor) robustly potentiated CALHM1 expression (Fig. 6 C). However, the potentiation by MG‐132 was not significantly different between WT and C100/207S (Fig. 6 D), demonstrating that the loss of palmitoylation reduces CALHM1 expression independently of its degradation process.
We exploited the effects of MG‐132 on CALHM1 protein expression to further confirm that C100 and C207 are the only residues that are palmitoylated, because the C100/207S mutation substantially lowered protein expression (Fig. 3 D) thus making interpretation of the ABE assays less reliable. As shown in Fig. 6 E and F, treatment with MG‐132 rescued the expression of the C100/207S mutant to a comparable level to that of WT, yet the mutant completely lacked palmitoylation, confirming the aforementioned conclusion.
Palmitoylation downregulates CALHM1 function
Next, we observed WT and C100/207S CALHM1‐expressing cells. N2a cells were transfected with WT or C100/207S CALHM1 cloned into pIRES‐AcGFP1, a bicistronic expression vector, to visualize transfected cells with AcGFP1 fluorescence. Unexpectedly, while many AcGFP1‐positive cells were observed in WT‐transfected cells, AcGFP1‐positive cells were rarely seen in C100/207S‐transfected cells (Fig. 7 A). Meanwhile, a significant decrease in the number of cells for the C100/207S‐transfected cells were noticed, implying that the C100/207S mutant is highly toxic to cells. Therefore, we compared the cell viability of N2a cells 24 h after transfection with WT or C100/207S CALHM1. As demonstrated previously (Tanis et al. 2013), cell viability was moderately compromised by WT CALHM1 expression (Fig. 7 B) possibly because of Ca2+ overload, loss of cellular nutrients, and/or membrane depolarization. Compared with WT, C100/207S showed greater cytotoxicity (Fig. 7 B). Here, we reasoned that C100/207S is a gain‐of‐function mutation and expression of the hyperactive mutant channel killed most of successfully transfected cells. As in N2a cells (Fig. 7 B), C100/207S exhibited significantly higher cytotoxicity than WT in HeLa cells (Fig. 7 C). Treatment with 2BP, which by itself was not cytotoxic, potentiated the cytotoxicity of WT but not C100/207S (Fig. 7 C). This observation supports the hypothesis that losing palmitoylation augments CALHM1 function and makes the channel more cytotoxic. This hypothesis also explains why C100/207S CALHM1 expression was apparently lower than WT in Western blotting.
We then moved on to directly address the effects of palmitoylation on CALHM1 function. To avoid the influence of cell damage, the ATP release assay was carried out at 6 h post‐transfection, when neither WT nor C100/207S yet showed cytotoxic activity compared with mock transfection (Fig. 7 D) and thereby both WT and the mutant channels are expressed at similar levels (Fig. 7 E) without affecting the total cellular ATP content (Fig. 7 F). A reduction in [Ca2+]o appreciably induced release of ATP from WT CALHM1 cells (Fig. 7 G), indicating that even at this early time point functional channel expression can fairly be achieved. Strikingly, the amount of ATP released from C100/207S CALHM1 cells was considerably larger than that from WT CALHM1 cells (Fig. 7 G and H). Conversely, DHHC3, a PAT for CALHM1 (Fig. 4), suppressed WT but not C100/207S CALHM1‐mediated ATP release (Fig. 7 K and L) measured at 9 h post‐transfection, when CALHM1 and DHHC3 expression DHHC3 showed no influences on the total cellular ATP content (Fig. 7 J) but a reduction in C100/207S expression was already apparent (Fig. 7 I), demonstrating that DHHC3 palmitoylates and downregulates CALHM1. Taken altogether, these results clearly demonstrate that palmitoylation has a negative impact on CALHM1 function.
Palmitoylation promotes CALHM1 association with DRMs
The amount of cellular ATP release mediated by CALHM1 depends on the number of channels in the plasma membrane and the activity of individual channels. Subcellular localization of CALHM1 visualized by immunocytochemistry was identical between WT and C100/207S with or without cycloheximide treatment, which enhances the visibility of cell surface localization of the channels by blocking protein synthesis (Fig. 8 A). Furthermore, the cell surface biotinylation assay showed that cell surface targeting was quantitatively comparable between WT and C100/207S (Fig. 8 B and C). These results suggest that palmitoylation does not control cell surface expression of the channel.
Figure 8. Palmitoylation downregulates CALHM1 association with DRMs.
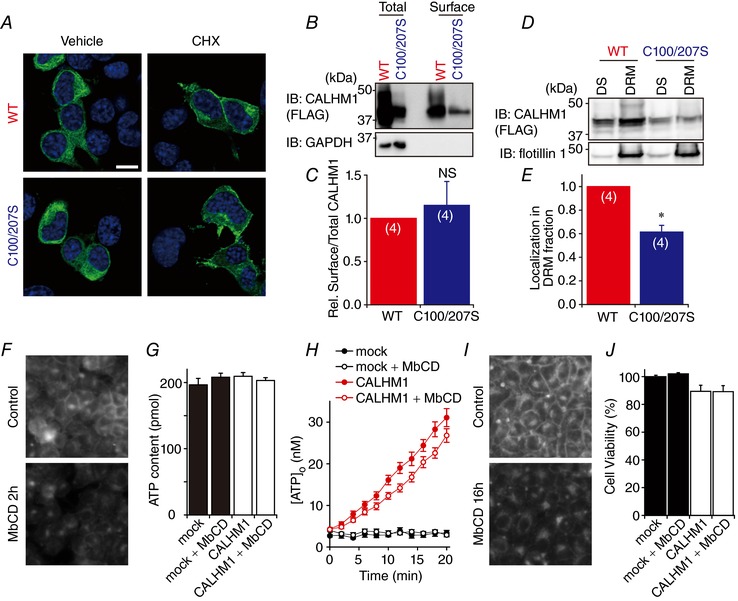
A, immunofluorescence images of N2a cells expressing WT (top) or C100/207S (bottom) CALHM1‐FLAG. Cells were treated with 100 μg mg−1 cycloheximide (CHX, right) or vehicle (left) for 90 min before being fixed at 8 h post‐transfection. CALHM1‐FLAG proteins were fluorescently labelled with anti‐FLAG antibody. Labelling of CALHM1‐FLAG and DAPI is green and blue, respectively. Confocal 2‐colour merged images taken at ×40 magnification are displayed. Scale bars, 10 μm. B, representative Western blots of the surface biotinylation analysis of WT and C100/207S CALHM1. Biotinylated proteins (Surface) were avidin affinity‐purified from the whole cell lysates (Total) and analysed by Western blotting. C, summary of the ratios of surface‐to‐total CALHM1 levels normalized to WT. n = 4. D, representative Western blots detecting WT and C100/207S CALHM1 in DS and DRM fractions. Flotillin 1 is a DRM marker protein. DS, detergent‐soluble fraction; DRM, detergent‐resistant membrane fraction. E, localization of WT and C100/207S CALHM1 in DRM fraction normalized with respect to WT. n = 4. NS, not significant; * P < 0.05 (Student's t test). F, filipin staining of cholesterol in HeLa cells. Cells were treated with or without 2 mm methyl‐β‐cyclodextrin (MbCD) in the normal bath solution for 2 h at 37°C followed by incubation without MbCD for 1 h at 37°C before being fixed and stained with filipin. G and H, total ATP content (G) and time courses of [ATP]o following removal of (H) in HeLa cells expressing WT CALHM1 after MbCD treatment as in F. I, filipin staining of cholesterol in HeLa cells. Cells were cultured in medium with or without 2 mm MbCD for 16 h before being subjected to filipin staining. Note that, after MbCD treatment, signals in the plasma membrane are markedly reduced whereas intracellular signals are relatively maintained. J, effect of MbCD on cytotoxicity of WT CALHM1. Cell viabilities of HeLa cells 24 h after transfection with mock or WT CALHM1 were measured with or without MbCD treatment (2 mm, 16 h) as in I. Viability of mock‐transfected cells without MbCD treatment were considered as 100%.
To further determine whether palmitoylation controls the affinity of CALHM1 for particular membrane domains as reported for other proteins (Levental et al. 2010a,b), DS and DRM fractions of N2a cells expressing WT or C100/207S CALHM1 were obtained and analysed by Western blotting. As is the case for P2X7 receptor (P2X7R) (Gonnord et al. 2009), the vast majority of CALHM1 was solubilized with 1% Triton X‐100 and appeared in the DS fraction (data not shown), whereas an appreciable amount of CALHM1 was recovered in the DRM fraction containing flotillin‐1, a DRM marker protein, when solubilized with 0.05% Triton X‐100 (Fig. 8 D). Although WT and C100/207S were detected in both the DS and DRM fractions, residence of the mutant channel in the flotillin‐1‐positive DRM fraction was ∼40% smaller than that of WT (Fig. 8 E). This observation suggests that palmitoylation increases affinity of CALHM1 to DRMs, also known as lipid rafts.
We further investigated the possible link between CALHM1 interaction with DRMs and channel function. To this end, we used methyl‐β‐cyclodextrin, a commonly used cholesterol sequestering reagent, to disrupt lipid microdomains (Christian et al. 1997) and measured low [Ca2+]o‐induced ATP release from WT CALHM1‐expressing HeLa cells after 2 h‐incubation with 2 mm methyl‐β‐cyclodextrin. The amount of ATP release was not significantly affected by methyl‐β‐cyclodextrin ([ATP]o at 20 min, control/CALHM1 (31.1 ± 2.2 nm) vs. methyl‐β‐cyclodextrin/CALHM1 (26.8 ± 2.2 nm), P = 0.105 by Student's t test) (Fig. 8 H). The cholesterol depletion by methyl‐β‐cyclodextrin was confirmed by filipin staining (Fig. 8 F) and total cellular ATP content was not significantly different between groups (Fig. 8 G). Thus, breaking up lipid microdomains did not alter CALHM1 function. Furthermore, cholesterol depletion by methyl‐β‐cyclodextrin (2 mm for 16 h, Fig. 8 I) had no effects on cytotoxicity of the channel (Fig. 8 J). All these data suggest that the submembrane distribution and function of CALHM1 are independently regulated by palmitoylation.
Palmitoylation regulates gating of CALHM1
Having ruled out the possibility of palmitoylation regulating CALHM1 channel number at the cell surface, it is likely that the channel activity itself is regulated by palmitoylation. Therefore, we compared electrophysiological properties of WT and C100/207S channels expressed in Xenopus oocytes. We used the oocyte system for two reasons: (1) the small membrane conductance generated by CALHM1 in mammalian expression systems is not sufficient for gating analysis (Ma et al. 2012), and (2) the C100/207S mutant is so cytotoxic that transfected cells could not be visually identified by a fluorescent marker protein (Fig. 7 A). We first demonstrated that, like in N2a cells, CALHM1 is palmitoylated at C100 and C207 when expressed in oocytes (Fig. 9 A), indicating that oocytes are equipped with a mechanism for CALHM1 palmitoylation similar to that in N2a cells. Two‐electrode voltage clamp recordings were made in Xenopus oocytes injected with WT or C100/207S CALHM1 cRNAs. Of note, C100/207S cRNA injection led to oocyte deterioration to a greater extent than WT, suggested by more depolarized resting membrane potentials and a decrease in the number of healthy‐looking oocytes. In the condition in which endogenous currents were inhibited (see Methods), the membrane voltage was held at −80 mV and stepped to various values from −40 to +70 mV with 10 mV increments for 5 s. Both WT and C100/207S generated voltage‐dependent membrane conductances as displayed in Fig. 9 B. Current–voltage relationships derived from the current amplitudes at the end of the voltage steps (Fig. 9 C) demonstrate an increase in CALHM1 channel conductance by the C100/207S mutation, in agreement with the augmented function of the mutant channel as observed above. Normalized G–V relationships were constructed from inward tail current amplitudes at −80 mV and fitted with a two‐state Boltzmann equation (Fig. 9 D). The G–V relationship for C100/207S was shifted to more hyperpolarized voltages than that for WT. The V 1/2 values for WT and C100/207S were 67 ± 3 and 48 ± 2 mV, respectively (P = 0.00004). The outward currents were not fitted well with exponential functions. The normalized outward currents at +70 mV are shown in Fig. 9 E and the time to half activation is plotted against the membrane voltage (Fig. 9 F). Although acceleration of the activation speed was seen for both WT and C100/207S channels with increases in the membrane voltage, the voltage‐dependent acceleration was more pronounced for C100/207S than for WT. Above +40 mV, the time to half‐activation for C100/207S was significantly shorter than WT. In contrast, the inward tail currents recorded at −80 mV were satisfactorily fitted with double‐exponential functions (Fig. 9 G) and the deactivation constants were not significantly different between WT and C100/207S (Fig. 9 H and I). Taken together, palmitoylation profoundly regulates the voltage‐dependent gating of the CALHM1 channel, shifting its voltage dependence to more depolarized potentials and slowing down its activation kinetics.
Figure 9. Palmitoylation controls voltage sensitivity and activation kinetics of CALHM1.
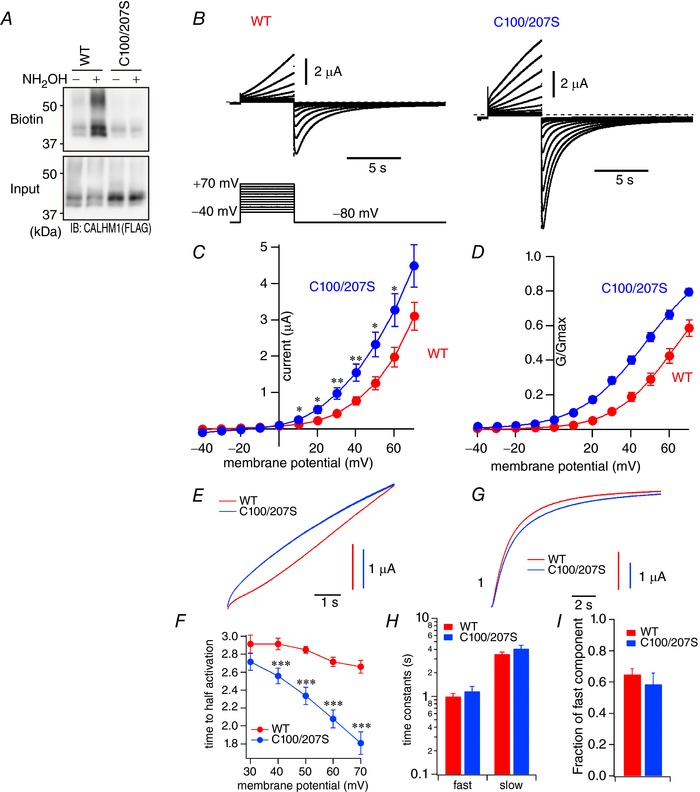
A, representative Western blots of the ABE assay showing the presence and absence of palmitoylation in WT and C100/207S CALHM1 expressed in Xenopus oocytes, respectively. B, macroscopic currents observed in oocytes expressing WT (left) or C100/207S (right) CALHM1 in the bath solution containing 1.8 mm Ca2+ and 1 mm Mg2+ in response to 5 s voltage pulses ranging from −40 to +70 mV with a holding potential of −80 mV. The pulse protocol is shown at the bottom. C, current–voltage relationships for WT and C100/207S CALHM1 derived from the current amplitudes measured at the end of the depolarizing pulses. D, normalized G–V relationships for WT and C100/207S CALHM1 derived from the maximum tail currents measured at −80 mV after a series of voltage pulses by fitting the current magnitude with a two‐state Boltzmann function (lines). WT, V 1/2 = 67 ± 3 mV, z = 1.7 ± 0.1; C100/207S, V 1/2 = 48 ± 2 mV, z = 1.5 ± 0.1. E, normalized traces showing the current activation phase for WT and C100/207S at +70 mV. F, the time required to reach half the current amplitude recorded at the end of depolarization pulses (5 s) plotted against pulse voltages. G, normalized traces showing the current deactivation phase for WT and C100/207S CALHM1 at −80 mV. H, time constants for fast and slow exponential components of the deactivation phase calculated at −80 mV. I, fraction of the fast component in the deactivation phase at −80 mV. n = 13 (WT) and 11 (C100/207S). *** P < 0.001 vs. WT (Student's t test).
Discussion
Emerging roles of CALHM1 highlight its broad physiological importance, yet complete understanding of its contribution is hindered by lack of knowledge on its regulatory mechanisms (Ma et al. 2016). In this study, we aimed to uncover post‐translational regulatory mechanisms of CALHM1. Various lines of biochemical and functional evidence demonstrated that CALHM1 is post‐translationally regulated by palmitoylation. We identified two Cys residues and enzymes responsible for CALHM1 palmitoylation and analysed the effects of palmitoylation on various stages in the life cycle of the CALHM1 channel, including its gating, DRM association, synthesis, degradation and forward trafficking.
Post‐translational modification of CALHM1
Little is known about CALHM1 PTMs. Asparagine‐linked glycosylation in the second extracellular loop (ECL2) is the only known PTM on the channel (Dreses‐Werringloer et al. 2008). The initial experiments in this study (Fig. 1) revealed that each Cys differentially regulates the CALHM1 channel, although the possibility that the mutations analysed in these experiments affected the channel function and trafficking simply by changing its normal structure cannot be ruled out. In addition to the two palmitoylation sites, our results provide targets for future studies for elucidating post‐translational CALHM1 regulation.
S‐Palmitoylation of CALHM1
We defined two intracellular palmitoylation sites, C100 and C207, located near TM3 and TM4, respectively. The location of the palmitate addition in close proximity to TM domains is consistent with palmitoylation sites experimentally determined for many other membrane proteins (Shipston, 2014). The two palmitoylation sites on CALHM1 are well conserved among vertebrates including frog, fish, chicken, mouse, dog and human (Fig. 3 A). Although C. elegans CLHM‐1 does not align well with vertebrate CALHM1, CLHM‐1 also possesses intracellular Cys near its TM3 and TM4, suggesting that palmitoylation on these Cys may be an evolutionarily conserved, essential regulatory mechanism of CALHM1 channels. Palmitoylation and depalmitoylation respectively down‐ and up‐regulated the channel function. Notably, single mutations on either one of the two palmitoylation sites only subtly affected channel activity (Fig. 1), whereas the double mutation on these residues robustly increased it (Figs 7 G and H and 9). This indicates that palmitoylation on one residue can compensate for loss of the other. Compensation of total palmitoylation level is also seen in the C207S mutant (Fig. 3 D). Thus, having two compensatory sites ensures that the channel is kept under the control of palmitoylation. Fine tuning of CALHM1 activity by palmitoylation may be indispensable for maintaining cellular homeostasis as well as controlling its primary function, because CALHM1 activation is toxic to cells (Fig. 7 A–D) (Tanis et al. 2013).
Members in the DHHC family are bona fide PATs in mammals (Fukata et al. 2004; Chamberlain & Shipston, 2015), whereas depalmitoylation enzymes are currently being explored (Chamberlain & Shipston, 2015; Lin & Conibear, 2015; Yokoi et al. 2016) and are not fully understood yet. DHHC PAT members vary greatly in substrate specificity, subcellular localization and tissue expression. CALHM1 can be palmitoylated by three DHHCs, 3, 7 and 20 (Fig. 4). DHHC3 and DHHC7 localize to the Golgi apparatus and DHHC20 to the plasma membrane (Ohno et al. 2006; Korycka et al. 2012; He et al. 2014). Thus, we suggest that palmitoylation regulates CALHM1 in different stages of its life cycle. Of note, DHHC20 may exert dynamic control of CALHM1 function at the cell surface.
Palmitoylation and CALHM1 gating
The Cys palmitoylation has at least two obvious effects on CALHM1. First, the voltage‐dependent gating of CALHM1 is significantly altered by depalmitoylation with the double mutation on C100 and C207. A 20 mV hyperpolarizing shift of the G–V relationship by the mutation (Fig. 9 D) indicates that voltage sensing by CALHM1 is tightly regulated by palmitoylation. In addition, the same mutant displayed faster activation kinetics and more pronounced voltage‐dependent acceleration of the activation speed than WT (Fig. 9 E and F), whereas the deactivation kinetics remained relatively unaltered, although C100/207S showed a tendency to deactivate more slowly than WT (Fig. 9 G–I). These observations indicate that the channels are stabilized at closed states by palmitoylation. CALHM1 has been established as a voltage‐gated ion channel with an intrinsic voltage sensor and an extracellular Ca2+‐binding site (Ma et al. 2012). However, none of structural features that define the gate, Ca2+‐binding site and voltage sensor domain have been discovered. Given the reported structural and functional similarities between CALHM1 and connexins (Siebert et al. 2013), one could imagine that CALHM1 would have a structure similar to connexin 26. The resolved structure of connexin 26 (Maeda et al. 2009) revealed that its TM1 and TM2 face the interior, namely the permeation pathway, whereas TM3 and TM4 face the surrounding membrane environment. In CALHM1, palmitates are attached to two cytoplasmic Cys residues near TM3 and TM4, respectively (Fig. 1 A). Thus, palmitate groups may be located at the protein–membrane interface so that the presence of these hydrophobic moieties can affect the interplay between the voltage sensor and membrane lipids. The voltage sensor may, therefore, reside in or near TM3 and TM4 because it is likely that palmitoylation primarily affects the movement of TM3 and TM4. Alternatively, the altered movement of these TM domains may influence the movement of a remotely located voltage sensor. Thus, the structural determinants of the voltage sensor of CALHM1 still remain an open question.
Palmitoylation and CALHM1 synthesis, degradation, forward trafficking and DRM association
Second, the C100/207S mutant exhibited reduced association with DRMs. Biochemical separation of CALHM1‐expressing cells into DRM and non‐DRM fractions (Fig. 8 D and E) revealed the presence of CALHM1 in DRMs as well as non‐DRMs. Although our experiment does not determine whether CALHM1 is preferentially localized to DRMs, the results demonstrate that C100/207S mutation caused a reduction in the relative residence of the channels in DRMs by approximately 40%. Of note, no relationship between DRM association and function of CALHM1 was observed (Fig. 8 F–J). Other proteins including ion channels have also been shown to associate with DRMs in a palmitoylation‐dependent manner (Levental et al. 2010a,b). For example, palmitoylation of P2X7R, an ATP‐gated ion channel, is required for its association with DRMs (Gonnord et al. 2009). It is worth noting that the palmitoylation‐deficient mutant P2X7R exhibits increased retention in the ER and a shorter life time than WT because of increased protein degradation through proteasomes, suggesting that palmitoylation may also play a role in the correct folding of P2X7R. In contrast, CALHM1 degradation also occurs mainly in the proteasome but the palmitoylation deficiency does not enhance its degradation rate (Fig. 6 C and D), and forward trafficking of CALHM1 toward the cell surface is not influenced by palmitoylation (Fig. 8 A–C), suggesting that palmitoylation does not play a role in the macromolecular organization of CALHM1 channels.
CALHM1 palmitoylation in vivo
The discovery of CALHM1 was a major breakthrough in taste research because it identified the long‐sought molecular mechanism of neurotransmission linking taste bud cells to the nervous system (Taruno et al. 2013a,b; Liman et al. 2014). Since Calhm1 mRNA is specifically expressed in taste cells dedicated to sweet, bitter and umami (savoury taste) sensing, a taste stimulus evokes action potentials in those cells to activate CALHM1 channels leading to liberation of the neurotransmitter, ATP, toward the afferent gustatory nerves expressing P2X2/3 receptors, and genetic ablation of Calhm1 in mice makes the animals indifferent to those taste qualities (Taruno et al. 2013a,b). Nevertheless, the CALHM1‐associated voltage‐dependent current recorded in mouse taste cells activates at more negative membrane voltages than heterologous CALHM1 currents. V 1/2 for the native CALHM1 current is 31 mV (Romanov et al. 2008), whereas V 1/2 values for recombinant mouse CALHM1 (Fig. 9 D), human CALHM1 (Ma et al. 2012) and C. elegans CLHM‐1 (Tanis et al. 2013) are 67 ± 3, 82 ± 3 and 66 ± 0.4 mV, respectively. In addition, the activation kinetics of the native current is much faster than that of heterologous CALHM1 (Romanov et al. 2008; Ma et al. 2012; Tanis et al. 2013; Fig. 9 F): the times to half‐activation at 70 mV for the native current and mouse CALHM1 are ∼7 ms and 2.66 ± 0.07 s, respectively. These observations clearly suggest an involvement of a previously unidentified mechanism(s) in the regulation of CALHM1 gating in taste cells. This study demonstrated that palmitoylation is a strong regulator of the voltage sensitivity and activation kinetics of CALHM1 gating, suggesting that palmitoylation may be a part of the mechanisms which taste cells utilize to control CALHM1 gating in vivo. CALHM1 is indeed palmitoylated to some degree in taste cells in vivo as revealed by Calhm1V5‐ires‐Cre KI mice (Fig. 5 F). Although it remains unresolved whether different levels of palmitoylation on CALHM1 in vivo and in vitro account for the difference in channel gating, the gating properties of CALHM1 without palmitoylation are still appreciably different from those of CALHM1 in vivo: V 1/2 and the time to half‐activation at +70 mV for the taste cell CALHM1 current and C100/207S CALHM1 are, respectively, 31 and 48 ± 2 mV and ∼7 ms and 1.81 ± 0.12 s. These significant residual differences imply additional regulatory mechanism(s).
In the brain, we were unable to detect CALHM1 proteins by any method, including Calhm1V5‐ires‐Cre KI mice. This may reflect CALHM1 protein expression in specific microdomains in a small subset of brain cells and/or in a developmentally controlled manner. In fact, expression of CALHM1 transcripts in the brain is extremely low compared with in the tongue (Fig. 5 C). Meanwhile, CALHM1 mRNA has been reproducibly detected in the whole brain by RT‐PCR but not yet by in situ hybridization (Dreses‐Werringloer et al. 2008, 2013; Vingtdeux et al. 2016; Fig. 5 C). Therefore, brain regions and cells where CALHM1 is expressed remain unclear, although single cell RT‐PCR detected CALHM1 expression in primary cultured cortical pyramidal neurons (Ma et al. 2012). Nevertheless, previous studies indicated that proline86‐to‐leucine86 polymorphism downregulates CALHM1 function and is associated with the risk for late onset Alzheimer's disease in humans (Dreses‐Werringloer et al. 2008) and that gene knockout of Calhm1 impaired cortical neuron excitability (Ma et al. 2012) and memory flexibility (Vingtdeux et al. 2016) in mice, suggesting that CALHM1 proteins are indeed expressed and function in the brain including cortical neurons. It is conceivable that palmitoylation could occur on CALHM1 in neurons and be important in neuronal physiology and pathophysiology because all the DHHC PATs for CALHM1 identified in this study are expressed in the brain including neurons (Korycka et al. 2012). Future studies will need to resolve this question.
Summary
CALHM1 research is still in its fledgling stages. Our data have identified a novel post‐translational regulation of CALHM1 by protein S‐palmitoylation. The location and effects of palmitoylation provide insights into the gating mechanism of the channel. Modulation of the voltage‐dependent gating by palmitoylation may partially explain the biophysical discrepancy between CALHM1 gating in vivo and in vitro. Also, the demonstration of CALHM1 palmitoylation in taste cells in vivo unravelled a potential layer of regulation in taste. Cross‐talk of palmitoylation with other nearby PTMs is an emerging concept (Pickering et al. 1995; Abrami et al. 2006; Abrami et al. 2008; Tian et al. 2008; Hayashi et al. 2009; Lin et al. 2009; Zhou et al. 2012). Although palmitoylation may directly regulate CALHM1, there is a possibility that palmitoylation regulates other undefined PTMs which are crucial for CALHM1 function. Further studies are necessary in order to fully understand the gating and regulatory mechanisms of CALHM1. On the whole, palmitoylation is an important regulatory mechanism of CALHM1 channels and likely physiological and pathophysiological phenomena involving CALHM1.
Additional information
Competing interests
The authors declare no conflict of interest.
Author contributions
Conception and design of the research: A.T. and Y.M.; data collection: A.T., H.S., K.N., T.M. and Y.O.; analysis and interpretation of data: A.T., H.S., K.N., T.M., Y.O., M.A.K, F.O. and Y.M.; drafting the work or revising it for important intellectual content: A.T., H.S., K.N., T.M., Y.O., M.A.K, F.O. and Y.M. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was supported by Grants‐in‐Aid from Japan Society of the Promotion of Science (26713008 and 16K15181 to A.T., 16K18991 to H.S., 25670111 and 15K15034 to Y.M.); Salt Science (1235 to Y.M., 1429 and 1542 to A.T.); Society for Research on Umami Taste to A.T.; KITKPUM‐KPU‐KPhU Collaborative Research Grant (2013 and 2015) to Y.M.; Kyoto‐Funding for Innovation in Health‐related R&D Fields to Y.M.; Fuji Foundation for Protein Research to Y.M.; Cell Research Conference to Y.M.
Acknowledgements
We thank Dr Yoshihiro Kubo for critical reading of an earlier version of the manuscript and good advice, Dr J Kevin Foskett and Dr Masaki Fukata for providing us with CALHM and DHHC cDNAs, respectively, Dr Hiroyuki Hioki and Dr Akiyuki Nishimura for technical assistance, and Emi Mura for statistical analysis.
A. Taruno and H. Sun contributed equally to this work.
Contributor Information
Akiyuki Taruno, Email: taruno@koto.kpu-m.ac.jp.
Yoshinori Marunaka, Email: marunaka@koto.kpu-m.ac.jp.
References
- Abrami L, Kunz B, Iacovache I & van der Goot FG (2008). Palmitoylation and ubiquitination regulate exit of the Wnt signaling protein LRP6 from the endoplasmic reticulum. Proc Natl Acad Sci USA 105, 5384–5389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abrami L, Leppla SH & van der Goot FG (2006). Receptor palmitoylation and ubiquitination regulate anthrax toxin endocytosis. J Cell Biol 172, 309–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischoff R & Schluter H (2012). Amino acids: chemistry, functionality and selected non‐enzymatic post‐translational modifications. J Proteomics 75, 2275–2296. [DOI] [PubMed] [Google Scholar]
- Bosmans F, Milescu M & Swartz KJ (2011). Palmitoylation influences the function and pharmacology of sodium channels. Proc Natl Acad Sci USA 108, 20213–20218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain LH & Shipston MJ (2015). The physiology of protein S‐acylation. Physiol Rev 95, 341–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian AE, Haynes MP, Phillips MC & Rothblat GH (1997). Use of cyclodextrins for manipulating cellular cholesterol content. J Lipid Res 38, 2264–2272. [PubMed] [Google Scholar]
- Dreses‐Werringloer U, Lambert JC, Vingtdeux V, Zhao H, Vais H, Siebert A, Jain A, Koppel J, Rovelet‐Lecrux A, Hannequin D, Pasquier F, Galimberti D, Scarpini E, Mann D, Lendon C, Campion D, Amouyel P, Davies P, Foskett JK, Campagne F & Marambaud P (2008). A polymorphism in CALHM1 influences Ca2+ homeostasis, Aβ levels, and Alzheimer's disease risk. Cell 133, 1149–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreses‐Werringloer U, Vingtdeux V, Zhao H, Chandakkar P, Davies P & Marambaud P (2013). CALHM1 controls the Ca2+‐dependent MEK, ERK, RSK and MSK signaling cascade in neurons. J Cell Sci 126, 1199–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukata M, Fukata Y, Adesnik H, Nicoll RA & Bredt DS (2004). Identification of PSD‐95 palmitoylating enzymes. Neuron 44, 987–996. [DOI] [PubMed] [Google Scholar]
- Fukata Y, Murakami T, Yokoi N & Fukata M (2016). Local palmitoylation cycles and specialized membrane domain organization. Curr Top Membr 77, 97–141. [DOI] [PubMed] [Google Scholar]
- Gonnord P, Delarasse C, Auger R, Benihoud K, Prigent M, Cuif MH, Lamaze C & Kanellopoulos JM (2009). Palmitoylation of the P2X7 receptor, an ATP‐gated channel, controls its expression and association with lipid rafts. FASEB J 23, 795–805. [DOI] [PubMed] [Google Scholar]
- Grundy D (2015). Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology . J Physiol 593, 2547–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gubitosi‐Klug RA, Mancuso DJ & Gross RW (2005). The human Kv1.1 channel is palmitoylated, modulating voltage sensing: Identification of a palmitoylation consensus sequence. Proc Natl Acad Sci USA 102, 5964–5968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T, Rumbaugh G & Huganir RL (2005). Differential regulation of AMPA receptor subunit trafficking by palmitoylation of two distinct sites. Neuron 47, 709–723. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Thomas GM & Huganir RL (2009). Dual palmitoylation of NR2 subunits regulates NMDA receptor trafficking. Neuron 64, 213–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He M, Abdi KM & Bennett V (2014). Ankyrin‐G palmitoylation and βII‐spectrin binding to phosphoinositide lipids drive lateral membrane assembly. J Cell Biol 206, 273–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jindal HK, Folco EJ, Liu GX & Koren G (2008). Posttranslational modification of voltage‐dependent potassium channel Kv1.5: COOH‐terminal palmitoylation modulates its biological properties. Am J Physiol Heart Circ Physiol 294, H2012–H2021. [DOI] [PubMed] [Google Scholar]
- Korycka J, Lach A, Heger E, Boguslawska DM, Wolny M, Toporkiewicz M, Augoff K, Korzeniewski J & Sikorski AF (2012). Human DHHC proteins: a spotlight on the hidden player of palmitoylation. Eur J Cell Biol 91, 107–117. [DOI] [PubMed] [Google Scholar]
- Leduc‐Nadeau A, Lahjouji K, Bissonnette P, Lapointe JY & Bichet DG (2007). Elaboration of a novel technique for purification of plasma membranes from Xenopus laevis oocytes. Am J Physiol Cell Physiol 292, C1132–C1136. [DOI] [PubMed] [Google Scholar]
- Levental I, Grzybek M & Simons K (2010a). Greasing their way: lipid modifications determine protein association with membrane rafts. Biochemistry 49, 6305–6316. [DOI] [PubMed] [Google Scholar]
- Levental I, Lingwood D, Grzybek M, Coskun U & Simons K (2010b). Palmitoylation regulates raft affinity for the majority of integral raft proteins. Proc Natl Acad Sci USA 107, 22050–22054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liman ER, Zhang YV & Montell C (2014). Peripheral coding of taste. Neuron 81, 984–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin DT & Conibear E (2015). ABHD17 proteins are novel protein depalmitoylases that regulate N‐Ras palmitate turnover and subcellular localization. Elife 4, e11306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin DT, Makino Y, Sharma K, Hayashi T, Neve R, Takamiya K & Huganir RL (2009). Regulation of AMPA receptor extrasynaptic insertion by 4.1N, phosphorylation and palmitoylation. Nat Neurosci 12, 879–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linder ME & Deschenes RJ (2007). Palmitoylation: policing protein stability and traffic. Nat Rev Mol Cell Biol 8, 74–84. [DOI] [PubMed] [Google Scholar]
- Lobo S, Greentree WK, Linder ME & Deschenes RJ (2002). Identification of a Ras palmitoyltransferase in Saccharomyces cerevisiae . J Biol Chem 277, 41268–41273. [DOI] [PubMed] [Google Scholar]
- Ma Z, Siebert AP, Cheung KH, Lee RJ, Johnson B, Cohen AS, Vingtdeux V, Marambaud P & Foskett JK (2012). Calcium homeostasis modulator 1 (CALHM1) is the pore‐forming subunit of an ion channel that mediates extracellular Ca2+ regulation of neuronal excitability. Proc Natl Acad Sci USA 109, E1963–E1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Z, Tanis JE, Taruno A & Foskett JK (2016). Calcium homeostasis modulator (CALHM) ion channels. Pflugers Arch 468, 395–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda S, Nakagawa S, Suga M, Yamashita E, Oshima A, Fujiyoshi Y & Tsukihara T (2009). Structure of the connexin 26 gap junction channel at 3.5 A resolution. Nature 458, 597–602. [DOI] [PubMed] [Google Scholar]
- Matsumoto I, Ohmoto M, Narukawa M, Yoshihara Y & Abe K (2011). Skn‐1a (Pou2f3) specifies taste receptor cell lineage. Nat Neurosci 14, 685–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow IC & Parton RG (2005). Flotillins and the PHB domain protein family: rafts, worms and anaesthetics. Traffic 6, 725–740. [DOI] [PubMed] [Google Scholar]
- Morrow IC, Rea S, Martin S, Prior IA, Prohaska R, Hancock JF, James DE & Parton RG (2002). Flotillin‐1/reggie‐2 traffics to surface raft domains via a novel Golgi‐independent pathway. Identification of a novel membrane targeting domain and a role for palmitoylation. J Biol Chem 277, 48834–48841. [DOI] [PubMed] [Google Scholar]
- Moyer BD, Hevezi P, Gao N, Lu M, Kalabat D, Soto H, Echeverri F, Laita B, Yeh SA, Zoller M & Zlotnik A (2009). Expression of genes encoding multi‐transmembrane proteins in specific primate taste cell populations. PLoS One 4, e7682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller GM, Maarouf AB, Kinlough CL, Sheng N, Kashlan OB, Okumura S, Luthy S, Kleyman TR & Hughey RP (2010). Cys palmitoylation of the beta subunit modulates gating of the epithelial sodium channel. J Biol Chem 285, 30453–30462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee A, Mueller GM, Kinlough CL, Sheng N, Wang Z, Mustafa SA, Kashlan OB, Kleyman TR & Hughey RP (2014). Cysteine palmitoylation of the gamma subunit has a dominant role in modulating activity of the epithelial sodium channel. J Biol Chem 289, 14351–14359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno Y, Kihara A, Sano T & Igarashi Y (2006). Intracellular localization and tissue‐specific distribution of human and yeast DHHC cysteine‐rich domain‐containing proteins. Biochim Biophys Acta 1761, 474–483. [DOI] [PubMed] [Google Scholar]
- Pei Z, Xiao Y, Meng J, Hudmon A & Cummins TR (2016). Cardiac sodium channel palmitoylation regulates channel availability and myocyte excitability with implications for arrhythmia generation. Nat Commun 7, 12035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickering DS, Taverna FA, Salter MW & Hampson DR (1995). Palmitoylation of the GluR6 kainate receptor. Proc Natl Acad Sci USA 92, 12090–12094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resh MD (2006). Use of analogs and inhibitors to study the functional significance of protein palmitoylation. Methods 40, 191–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocks O, Peyker A, Kahms M, Verveer PJ, Koerner C, Lumbierres M, Kuhlmann J, Waldmann H, Wittinghofer A & Bastiaens PI (2005). An acylation cycle regulates localization and activity of palmitoylated Ras isoforms. Science 307, 1746–1752. [DOI] [PubMed] [Google Scholar]
- Romanov RA, Rogachevskaja OA, Khokhlov AA & Kolesnikov SS (2008). Voltage dependence of ATP secretion in mammalian taste cells. J Gen Physiol 132, 731–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt JW & Catterall WA (1987). Palmitylation, sulfation, and glycosylation of the alpha subunit of the sodium channel. Role of post‐translational modifications in channel assembly. J Biol Chem 262, 13713–13723. [PubMed] [Google Scholar]
- Shipston MJ (2014). Ion channel regulation by protein S‐acylation. J Gen Physiol 143, 659–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siebert AP, Ma Z, Grevet JD, Demuro A, Parker I & Foskett JK (2013). Structural and functional similarities of calcium homeostasis modulator 1 (CALHM1) ion channel with connexins, pannexins, and innexins. J Biol Chem 288, 6140–6153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H, Niisato N, Inui T & Marunaka Y (2014). Insulin is involved in transcriptional regulation of NKCC and the CFTR Cl− channel through PI3K activation and ERK inactivation in renal epithelial cells. J Physiol Sci 64, 433–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki M, Murakami T, Cheng J, Kano H, Fukata M & Fujimoto T (2015). ELMOD2 is anchored to lipid droplets by palmitoylation and regulates adipocyte triglyceride lipase recruitment. Mol Biol Cell 26, 2333–2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takimoto K, Yang EK & Conforti L (2002). Palmitoylation of KChIP splicing variants is required for efficient cell surface expression of Kv4.3 channels. J Biol Chem 277, 26904–26911. [DOI] [PubMed] [Google Scholar]
- Tanis JE, Ma Z, Krajacic P, He L, Foskett JK & Lamitina T (2013). CLHM‐1 is a functionally conserved and conditionally toxic Ca2+‐permeable ion channel in Caenorhabditis elegans . J Neurosci 33, 12275–12286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taruno A, Matsumoto I, Ma Z, Marambaud P & Foskett JK (2013a). How do taste cells lacking synapses mediate neurotransmission? CALHM1, a voltage‐gated ATP channel. Bioessays 35, 1111–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taruno A, Vingtdeux V, Ohmoto M, Ma Z, Dvoryanchikov G, Li A, Adrien L, Zhao H, Leung S, Abernethy M, Koppel J, Davies P, Civan MM, Chaudhari N, Matsumoto I, Hellekant G, Tordoff MG, Marambaud P & Foskett JK (2013b). CALHM1 ion channel mediates purinergic neurotransmission of sweet, bitter and umami tastes. Nature 495, 223–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian L, Jeffries O, McClafferty H, Molyvdas A, Rowe IC, Saleem F, Chen L, Greaves J, Chamberlain LH, Knaus HG, Ruth P & Shipston MJ (2008). Palmitoylation gates phosphorylation‐dependent regulation of BK potassium channels. Proc Natl Acad Sci USA 105, 21006–21011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian L, McClafferty H, Knaus HG, Ruth P & Shipston MJ (2012). Distinct acyl protein transferases and thioesterases control surface expression of calcium‐activated potassium channels. J Biol Chem 287, 14718–14725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vingtdeux V, Chang EH, Frattini SA, Zhao H, Chandakkar P, Adrien L, Strohl JJ, Gibson EL, Ohmoto M, Matsumoto I, Huerta PT & Marambaud P (2016). CALHM1 deficiency impairs cerebral neuron activity and memory flexibility in mice. Sci Rep 6, 24250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan J, Roth AF, Bailey AO & Davis NG (2007). Palmitoylated proteins: purification and identification. Nat Protoc 2, 1573–1584. [DOI] [PubMed] [Google Scholar]
- Yang H, Wang H, Shivalila CS, Cheng AW, Shi L & Jaenisch R (2013). One‐step generation of mice carrying reporter and conditional alleles by CRISPR/Cas‐mediated genome engineering. Cell 154, 1370–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoi N, Fukata Y, Sekiya A, Murakami T, Kobayashi K & Fukata M (2016). Identification of PSD‐95 depalmitoylating enzymes. J Neurosci 36, 6431–6444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Foster K, Li Q & Martens JR (2007). S‐acylation regulates Kv1.5 channel surface expression. Am J Physiol Cell Physiol 293, C152–C161. [DOI] [PubMed] [Google Scholar]
- Zheng B, DeRan M, Li X, Liao X, Fukata M & Wu X (2013). 2‐Bromopalmitate analogues as activity‐based probes to explore palmitoyl acyltransferases. J Am Chem Soc 135, 7082–7085. [DOI] [PubMed] [Google Scholar]
- Zhou X, Wulfsen I, Korth M, McClafferty H, Lukowski R, Shipston MJ, Ruth P, Dobrev D & Wieland T (2012). Palmitoylation and membrane association of the stress axis regulated insert (STREX) controls BK channel regulation by protein kinase C. J Biol Chem 287, 32161–32171. [DOI] [PMC free article] [PubMed] [Google Scholar]