

Abstract
The programmed self-destruction of infected cells is a powerful antimicrobial strategy in metazoans. For decades, apoptosis represented the dominant mechanism by which the virus-infected cell was thought to undergo programmed cell death. More recently, however, new mechanisms of cell death have been described that are also key to host defense. One such mechanism in vertebrates is programmed necrosis, or ‘necroptosis’, driven by receptor-interacting protein kinase 3 (RIPK3). Once activated by innate-immune stimuli, including virus infections, RIPK3 phosphorylates the mixed lineage kinase domain-like protein (MLKL), which then disrupts cellular membranes to effect necroptosis. Emerging evidence demonstrates that RIPK3 can also mediate apoptosis and regulate inflammasomes. Here, we review studies on the mechanisms by which viruses activate RIPK3 and the pathways engaged by RIPK3 that drive cell death.
Keywords: RIPK1, RIPK3, necrosis, necroptosis, apoptosis, viruses
Introduction.
The cytolytic clearance of infected or damaged cells is a frontline strategy for the eradication of pathogens and the maintenance of health in metazoans. The earliest described mechanism of programmed cellular self-destruction is, of course, apoptosis. For decades, it has been known that, in many settings, apoptosis mediates the elimination of virus- and microbe-infected cells (1). During apoptosis, multiple upstream signaling cascades can activate caspases, which then function as executioners of the apoptotic program and systematically dismantle the infected cell, collapsing it from the inside into discrete packages for eventual recycling (2). The key contribution of apoptosis to virus clearance is underscored by the number of virus-encoded effector proteins that block this form of cell death. These include bona fide caspase inhibitors, (e.g., cowpox virus CrmA), inhibitors of membrane proximal signaling events by death receptors of the tumor necrosis factor-α (TNF-α) superfamily (e.g., adenovirus E3 proteins), and orthologs of the Bcl-2 class of mitochondrial apoptosis blockers (e.g., adenovirus E1B-19K) (3–5). Indeed, the importance of apoptosis to clearance of virus-infected cells was elegantly revealed by Hardwick and colleagues, who demonstrated that blockade of apoptosis by simple overexpression of Bcl-2 could change a lytic virus infection into a persistent one (6). In other words, prevention of cell death converted the host cell into a ‘factory’ for progeny virion production, underscoring the importance of auto-destruction of the infected cell as an altruistic host defense strategy to limit virus replication and spread.
Apoptosis, by definition, relies on the activity of caspases for its execution; thus, inhibition of caspases, whether by cellular- or virus-encoded inhibitory proteins, or by pharmacological means, are effective at nullifying cell death in many contexts (7). Rather paradoxically then, it was observed by several groups that caspase blockade in certain settings did not prevent cell death; rather, caspase inhibition greatly sensitized a subset of cell lines to cell death following stimulation by death receptors, or upon exposure to certain other innate-immune activators, including synthetic double-stranded (ds) RNA (a virus mimetic) and the cytokine interferon-γ (IFN-γ) (8–14). Notably, death induced by TNF-α, dsRNA, or IFN-γ was necrotic in morphology and very likely the ‘programmed’ end-result of a dedicated signaling cascade. For example, ablation of signaling intermediates in tumor necrosis factor receptor 1 (TNFR1) and Fas pathways abrogated not only apoptosis induced by these receptors, but also caspase-independent necrotic death as well (12). Somewhat oddly, but as will become clear later, the phenomenon of ‘programmed necrosis’ was restricted to a few cell types, including murine embryo fibroblasts (MEFs), the L929 fibrosacroma cell line, and the Jurkat T cell line (8, 10, 12, 15). In the vast majority of commonly-employed cell lines, however, caspase blockade expectedly prevents cell death activated by TNF-α and other innate-immune stimuli. Likely for this reason, programmed necrosis was considered a ‘niche phenomenon’ and remained underexplored for years.
Early molecular insight into programmed necrosis came from the work of Tschopp and colleagues, who, in 2000, identified the kinase RIPK1 as essential for caspase-independent cell death triggred by Fas (16). In 2008, Yuan and colleagues identified a class of small-molecule inhibitors of necrotic death, called necrostatins, and pinpointed RIPK1 as the molecular target of one of these inhibitors, necrostatin-1 (Nec-1) (17). This group also coined the term ‘necroptosis’ to describe the form of programmed necrosis mediated by RIPK1 and blocked by Nec-1 (18). Perhaps the most significant breakthrough in our understanding of the molecular sequelae of necroptosis came from the simultaneous discovery in 2009 by three independent groups that the kinase RIPK3 was essential for the execution of programmed necrosis (19–21), published over two decades after the phenomenon was first seen in TNF-α-treated cells (22). Quickly thereafter, the pseudokinase MLKL was identified as a direct target of RIPK3 (23, 24). In a few short years, a reasonably clear outline of the pathway leading to necroptotic death downstream of the TNF-α receptor has emerged. Following ligation of TNFR1 by TNF-α, and under circumstances when caspases are inhibited, RIPK1 and RIPK3 assemble into a cytosolic complex called the ‘necrosome’ (19, 25) (Fig. 1). From within the necrosome, RIPK3 phosphorylates MLKL on key serines, triggering MLKL oligomerization (24, 26, 27). Oligomerized MLKL acquires lipid binding capacity, with an affinity for phosphatidylinositol lipids; this newly-acquired property draws MLKL to cellular membranes, including the plasma membrane. MLKL oligomers then, either directly or indirectly, disrupt membrane integrity and perturb cytosolic osmotic balance, causing the cell to swell and eventually burst (28–31). Most adherent cell lines commonly used in cell culture for cell death and virological studies have lost expression of RIPK3 and/or other effectors of necroptosis (20, 32, 33), providing a straightforward explanation for why this pathway went undiscovered for as long as it did.
Figure 1. RIPK3-driven cell death during virus infections.

Multiple viruses activate RIPK3 by different upstream mechanisms during their life cycles, leading to phosphorylation of MLKL and necroptosis, as well as recruitment of FADD and caspase-8-mediated apoptosis. The survival advantage to the virus of blocking RIPK3 signaling is highlighted by the growing number of virus proteins that target activation of these pathways during species-specific co-evolution of viruses with their natural hosts. Activation or inhibition of RIPK3 signaling is often mediated by RHIM-based homotypic interactions; the RHIM in proteins containing this motif is shown as a red rectangle. (HSV, herpes simplex virus; MCMV, murine cytomegalovirus; IAV, influenza A virus; VV, vaccinia virus.)
In contrast to the orderly collapse and eventual resorption of cellular materials via apoptosis, necrotic cellular demise releases cellular debris into the extracellular space. As the presence of many intracellular components, including DNA itself, in the extracellular milieu are indicative of infection or injury, and thus potently inflammatory, necroptosis has been regarded as a powerfully immunogenic form of cell death (34–38). Support for this idea came from a whole-genome RNAi screen performed by Yuan and colleagues, which identified several innate immune pathway nodes as essential for effective execution of necroptosis, strongly suggesting that this form of programmed cell death was activated during innate-immune responses to pathogens (39).
Since the initial observations that caspase inhibition sensitized cells to dsRNA-induced necrotic death (10, 13), it has become clear that necroptosis and other RIPK3-driven cell death/inflammatory mechanisms are essential for host defense against a growing number of DNA and RNA viruses. In this review, we summarize current knowledge on the contributions of RIPK3-dependent cell death in protection against viruses for which necroptosis has emerged as an important antiviral mechanism: vaccinia virus (VV; Poxviridae), cytomegaloviruses (CMV; Herpesviridae), herpes simplex viruses (HSV; Herpesviridae), and influenza A viruses (IAV; Orthomyxoviridae).
Vaccinia virus.
VV is the prototypic member of Poxviridae, a large family of enveloped viruses with double-stranded (ds)DNA genomes, and well-known as the active component of the vaccine used in the eradication of smallpox. Unusual among DNA viruses, poxviruses replicate in the cytoplasm of host cells. The VV genome encodes ~250 proteins, many of which are dedicated to disabling innate host defense mechanisms and delaying cell death (40, 41).
In 2000, Li and Beg reported a paradoxical observation when they set out to test the effect of vaccinia virus infection on subsequent exposure to TNF-α in MEFs (10). As VV encodes an inhibitor of caspases [B13R, also called SPI-2, with strong similarity to the cowpox virus protein CrmA (40)], they expected wild type VV, but not VV mutants lacking the caspase inhibitor, to protect cells from TNF-α-induced apoptosis. Bizarrely, they instead found that prior infection of MEFs with VV greatly sensitized these cells to killing by TNF-α, and that this effect was attenuated when VV lacking the B13R gene was used (10). These results are, of course, the opposite of what was expected if TNF-α induced caspase-dependent apoptosis. Subsequent experiments revealed that TNF-α-treated cells instead died by necrosis, and that death was accompanied by elevated production of reactive oxygen species. Necrosis was effectively prevented by treating cells with antioxidants, demonstrating that, at least in this cell type, death was reliant on mitochondrial reactive oxygen species (ROS) (10). Li and Beg extended these observations to demonstrate that the caspase inhibitor carbobenzoxy-valyl-alanyl-aspartyl-[O-methyl]-fluoromethylketone (zVAD) could substitute for VV in sensitizing MEFs to TNF-α necrosis (10). In a prescient set of observations, they also demonstrated that zVAD sensitized MEFs to necrotic death upon exposure to the synthetic dsRNA and virus mimetic polyinosinic:polycytidylic acid (polyI:C), as well as to the cytokine IFN-γ, foreshadowing a broader role for programmed necrosis in innate immune responses (10).
Lenardo and colleagues later showed that VV-induced necrosis in Jurkat T cells required TNFR2 signaling and the kinase activity of RIPK1 (42). They reported higher virus burden and defective adaptive immune responses in TNFR2-deficient animals, consistent with the idea that TNFR2 facilitates programmed necrosis of VV-infected tissues and triggers an inflammatory reaction that is crucial for the subsequent initiation of adaptive immunity against this virus (42). TNFR signaling activates multiple pathways (43) and RIPK1 deficiency results in perinatal lethality (44), so the in vivo role of necroptosis in host defense against VV (and other viruses) required a better mouse model.
When Chan and colleagues later identified RIPK3 as critical for necroptosis, they drew on these earlier observations (10, 42) to demonstrate that RIPK3 was required for TNF-α-induced cell death following infection of MEFs with the VV B13R mutant (19). Employing RIPK3 gene knockout mice produced by Dixit and colleagues (45), they showed that RIPK3 was essential for controlling VV replication in vivo (19). In wild type mice, VV inoculated in the foot pad typically causes a localized inflammatory reaction, following which it travels to the liver and spleen, where it replicates and from where it is eventually cleared by host immune mechanisms. In agreement, VV-infected RIPK3-containing mice displayed inflammation in their visceral foot pads, which, notably, surrounded areas of fat cell necrosis (19). The livers of these mice also displayed large areas of necrosis, with accompanying infiltration of immune cells. In remarkable contrast, necrosis and inflammation were conspicuously absent in tissues from RIPK3-deficient mice, correlating with dramatic increases in virus titers in organs from these mice. Consequently, RIPK3-deficient mice succumbed to VV-induced lethality while wild type mice mounted effective immune responses and cleared the virus. Mechanistically, Chan and colleagues provided evidence that VV-induced TNF-α production drives RIPK3 activation duting infection: TNF-α was readily detected in virus-infected tissues and, echoing earlier findings (42), TNFR2-deficient mice were protected from VV-induced inflammation and necrosis. Taken together, these results supply compelling evidence that VV triggers production of TNF-α, which then activates necroptosis to not only destroy the infected cell, but also to alert and activate an antiviral immune response (Fig. 1). It will be interesting to learn if VV itself can also directly activate RIPK3-mediated death signaling independently of TNF-α, perhaps via a host protein that senses VV and triggers RIPK3 (as MCMV and IAV can do), or by means of a virus-endoded protein that binds and stimulates this kinase (as HSV-1 does in murine cells).
Cytomegaloviruses.
Cytomegaloviruses (CMVs) are members of the Herpesviridae, another family of enveloped viruses with large, linear dsDNA genomes. Like poxviruses, herpesviruses encode a large number of gene products (70–170, depending on the virus) with many devoted to innate immune evasion and cell death suppression, although unlike poxviruses (which are cytoplasmic), all herpesviruses replicate their genome in the nucleus of infected cells. CMVs, the prototypic betaherpesviruses, have life cycles characterized by both lytic and latent phases, are highly species-specific, and persist for the lifetime of the host. Like herpesviruses in general, CMVs have received significant attention for the battery of apoptosis inhibitors they employ to prolong cell survival and promote lytic replication (46). In addition to a viral inhibitor of caspase-8 activation (vICA), CMVs encode additional inhibitors of pro-apoptotic Bcl-2 family members, and deletion or mutation of any one of these cell death suppressors derepresses the corresponding pathway of programmed cell death (46).
In 2001, Brune and colleagues reported results from a genetic screen designed to identify novel determinants of cell tropism encoded by murine CMV (MCMV) (47). From this screen, they isolated a cohort of mutant viruses that induced premature cell death in endothelial cells and macrophages, both important targets of MCMV in vivo, and mapped the defect to mutations in the MCMV M45 gene. MCMV M45 is annotated as encoding the large subunit of ribonucleotide reductase (RR1), and is conserved among all herpesvirus families (48) (Fig. 2). However, while this enzyme is crucial to a subset of herpesviruses for maintaining ribonucleotide pools in infected non-cycling cells, the MCMV M45 gene product itself lacks enzymatic activity (49).
Figure 2. RHIM containing proteins.
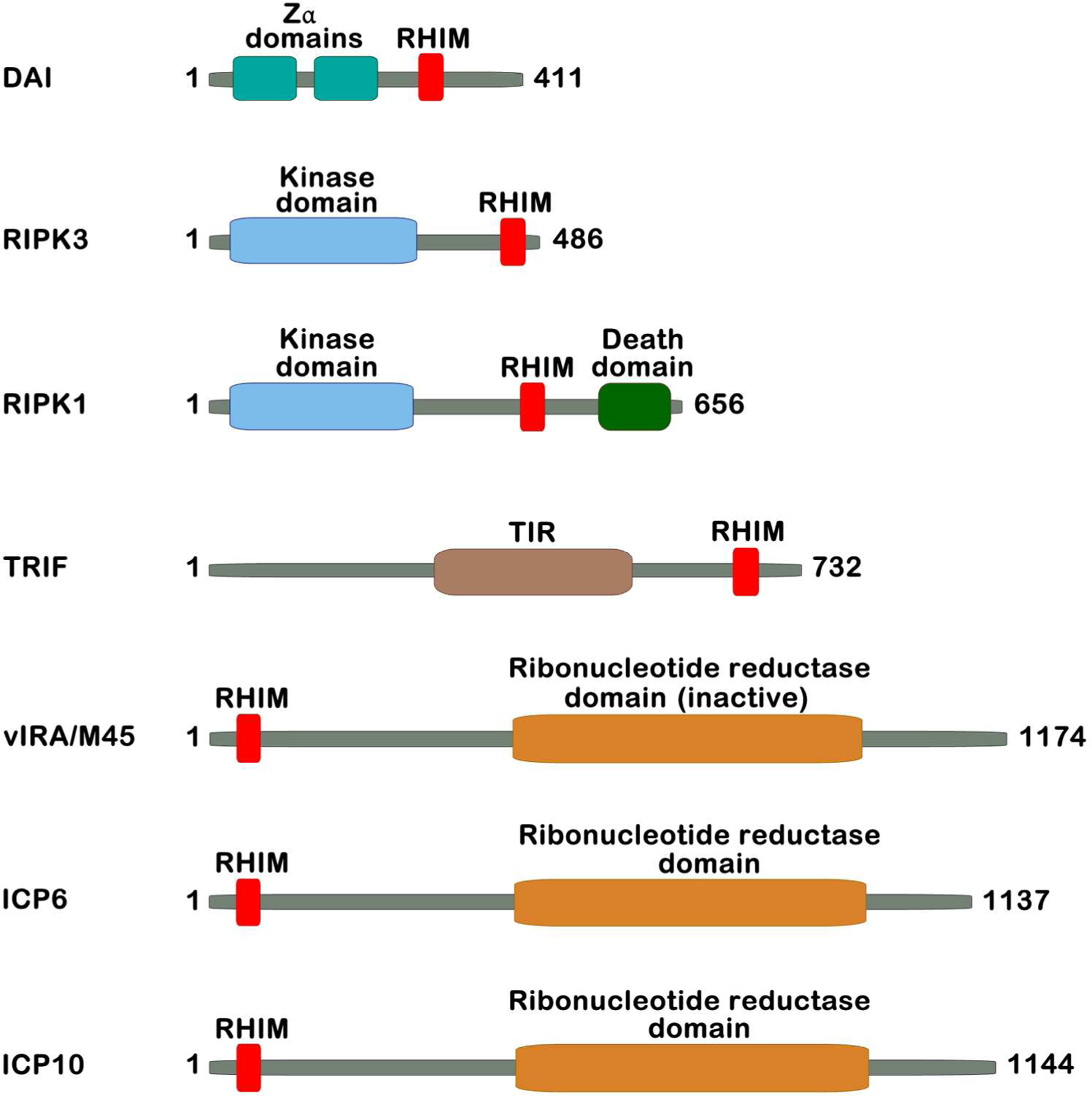
Four vertebrate proteins (DAI, RIPK1, RIPK3, and TRIF) and three virus proteins (MCMV vIRA, HSV-1 ICP6, and HSV-2 ICP10) have known RHIMs. Amino acid lengths are for murine orthologs of each protein, where relevant.
The mechanism of cell death induced by M45-mutant viruses was initially characterized as apoptosis (47). But several years later, in the course of investigating the role of MCMV M45 in endothelial cell tropism, we made the observation that a short sequence towards the amino-terminal region of M45 possessed significant similarity to a sequence in RIPK1 termed the ‘RIP homotypic interaction motif’ (RHIM) (50) (Fig. 2). The RHIM is a small motif, comprising a highly-conserved core of hydrophobic amino acid residues, that mediates homotypic interactions between RHIM-containing proteins. To date, four vertebrate proteins have been found to possess RHIMs: RIPK1, RIPK3, Toll/interleukin-1 receptor domain-containing adaptor-inducing interferon-β (TRIF, an adaptor for Toll-like receptor 3 [TLR3] and TLR4 ), and the nucleic acid sensor protein DNA-dependent activator of IFN-regulatory factors (DAI, also called Z-DNA binding protein-1 [ZBP-1]) (25) (Fig. 2). As the RHIM in RIPK1 mediates its interaction with RIPK3 (51), identification of a RHIM in MCMV M45 immediately suggested the attractive possibility that this virus-encoded protein was modulating RIP kinase activity to suppress cell death in infected cells. We demonstrated that MCMV M45 interacted with both RIPK1 and RIPK3, and that its RHIM was critical for these interactions (50). As these results provided the first molecular insights into the mechanism of action of the M45 gene product in modulating cell death responses, we renamed the M45 gene product ‘viral inhibitor of RIP activation’ (vIRA).
We next sought to formally investigate the role of the vIRA RHIM in the pathogenesis of MCMV. A recombinant mutant MCMV was constructed, in which the core ‘IQIG’ sequence in the vIRA RHIM was mutated to render it non-functional. In cell culture systems, this mutant virus, MCMV-M45mutRHIM, behaved similarly to the M45-deficient MCMV mutants characterized by Brune and colleagues years earlier (47). The new mutant virus did not exhibit any significant growth defect in the NIH3T3 fibroblast cell line, but was severely attenuated in endothelial and macrophage cell lines. In both endothelial cells and macrophages, MCMV-M45mutRHIM triggered rampant and premature cell death, greatly dampening progeny virion yield (52). These results indicated that the major function of MCMV vIRA was to prevent cell death, via RHIM-based interactions. As endothelial cells infected with the MCMV-M45mutRHIM remained sensitive to cell death even in the presence of a pan-caspase inhibitor, and as caspase activity was largely undetectable in infected cells, the mode of death induced by this mutant virus was not via canonical caspase-dependent apoptosis. These observations stood in remarkable contrast to results obtained with an MCMV mutant lacking a bona fide apoptosis inhibitor, in which cell death was reliant on caspase activity (53). As Brune and colleagues also found that an M45-deficient MCMV mutant induced caspase-independent cell death (54), we considered the possibility that vIRA was inhibiting RIPK1-dependent necroptosis.
Surprisingly, our efforts to implicate RIPK1 as the critical mediator of MCMV-induced necroptosis fell short of their intended goals. Neither RIPK1 kinase inhibition (by means of pharmacological agents) nor genetic ablation of RIPK1 reversed MCMV-M45mutRHIM induced cell death (52). Instead, expression levels of RIPK3 correlated with susceptibility to cell death, and knock-down or genetic ablation of RIPK3 abolished death (52). These results pointed to RIPK3 as a central player in MCMV-induced necroptosis, and suggested that vIRA was targeting the RHIM of RIPK3, not RIPK1, to suppress cell death.
The MCMV-M45mutRHIM virus is severely attenuated in wild type (C57BL/6) mice (52). Hypothesizing that this attenuation was due to the premature activation of RIPK3-dependent necroptosis in infected cells, we predicted that genetic deletion of host RIPK3 would reverse the attenuated phenotype of the mutant virus by preventing necroptosis. A similar strategy was used to demonstrate that the host kinase PKR was the direct target of the herpes simplex virus (HSV)-1 protein ICP34.5: replication and pathogenesis of an ICP34.5 mutant of HSV-1, normally attenuated in wild-type mice, was ‘restored’ by concurrent deletion of PKR in the host animal (55). In agreement with our hypothesis, we found that deletion of RIPK3 normalized replication and dissemination of the MCMV-M45mutRHIM virus to wild-type levels, clearly demonstrating that MCMV directly targets RIPK3 to suppress necroptosis and facilitate pathogenesis, and that RIPK3 is a bona fide target of vIRA.
While these studies were in progress, RIPK3 was identified as critical for TNF-α-induced programmed necrosis (19–21). Although RIPK3 was essential for both TNF-α- and MCMV-induced necroptosis, the upstream mechanisms by which these stimuli activated RIPK3 appeared to be significantly different. For example, while necroptosis induced by TNF-α relied on the kinase activity of RIPK1 (17, 19), our findings showed that RIPK1 activity was dispensible for MCMV-induced death. We also ruled out a role for the RHIM containing TLR adaptor TRIF as driver of virus-induced necroptosis (52), suggesting that MCMV activated RIPK3 by a new upstream mechanism, distinct from either TNF-α- or TLR signaling. In searching for this mechanism, we considered the possibility that DAI, the fourth known RHIM-containing vertebrate protein (in addition to RIPK1, RIPK3, and TRIF) might function to link MCMV to RIPK3.
DAI was first implicated in antiviral host defense as a sensor of cytosolic DNA in the pathway leading to production of type I IFNs (56), but later studies showed that DAI was dispensible for the host type I IFN response to most classes of DNA viruses (57, 58). Instead, we and others recognized that the central portion of DAI contains a functional RHIM, and that DAI can interact with both RIPK1 and RIPK3 via this motif (59, 60). In addition, DAI possesses two tandem Zα domains towards its N-terminus, which we call Zα1 and Zα2 (Fig. 2). In previous work, Zα2 is sometimes referred to as ‘Zβ’, but Zα2 is a more-accurate descriptor, distinguishing this domain from the functionally-distinct Zβ domain of adenosine deaminase acting on RNA-1 (ADAR-1) (61). The Zα domain was first identified by (and named for) its capacity to bind Z-form (i.e., left-handed) DNA double-helices (62). Subsequent work has since shown that Zα can also bind B-DNA (56), as well as Z-RNA (63, 64). As DAI can bind nucleic acids via its Zα domains (56), and as many innate-immune pathways are initiated by the sensing of virus nucleic acid by host proteins (65), we hypothesized that DAI functioned as a sensor of MCMV infection that triggered RIPK3-dependent necroptosis (when such signaling was not inhibited by vIRA). We found that DAI was readily detectable in cells sensitive to MCMV-induced necroptosis, and that boosting DAI levels in resistant cells containing low levels of RIPK3 was sufficient to sensitize them to death induced by MCMV-M45mutRHIM (66). Expectedly, mutation of the RHIM completely abolished the ability of DAI to elicit death upon infection (66), suggesting that vIRA inhibits RIPK3 activation by preventing its association with DAI. In agreement with this model, we found that a DAI-RIPK3 necrosome complex formed in cells infected with M45mutRHIM, but not with wild-type MCMV (66). As with deletion of RIPK3 (52), deletion of DAI restored replication and dissemination of MCMV-M45mutRHIM virus in vivo (66).
Together, our results provide compelling evidence that a DAI-RIPK3 axis mediates necroptotic clearance of MCMV-infected cells, and that this pathway is efficiently antagonized by the MCMV protein vIRA (Fig. 1). These results also raise a number of questions, many centered on where and how DAI senses MCMV. As MCMV is a DNA virus, and as DAI can bind DNA in vitro (56), the simplest model predicts that DAI recognizes MCMV genomes to trigger necroptosis. However, neither incoming viral DNA nor newly synthesized DNA are sufficient to trigger DAI-dependent cell death (47) (JU, unpublished observations). Moreover, the timing and execution of MCMV-induced necroptosis coincides with viral gene expression and our preliminary findings indicate that transcription of viral RNAs may be necessary for activation of DAI, raising the intriguing possibility that DAI may sense a putative RNA-containing replication intermediate to trigger necroptosis (JU, unpublished observations). Also currently unknown is where in the cell DAI senses MCMV. Although primarily considered cytosolic proteins, both DAI and RIPK3 can shuttle between the cytosol and nucleus (67–69), and recent work from the Wallach lab suggests that TNF-α-induced necroptosis is preceded by nuclear translocation of RIPK3, RIPK1 and MLKL (70). Whether DAI senses MCMV replication intermediates in the nucleus, and whether subsequent activation of RIPK3 also requires a nuclear step, remain to be determined. Finally, it remains to be established if caspase-8 suppression (via the vICA protein) is necessary for activation of necroptosis by MCMV. The generation of a vIRA/vICA double-mutant virus may help answer this question: if blockade of caspase-8 is needed for RIPK3-driven necroptosis, then MLKL will not be activated in cells infected with the double mutant; on the other hand, if MLKL and caspase-8 activities are concurrently detected, then perhaps caspase-8 suppression is not an obligate requirement for activation of necroptosis. This latter scenario (i.e., activation of MLKL without need for suppression of capase-8) is what plays out upon activation of DAI by influenza A virus, as will be described later. Demonstrating during MCMV infection that the DAI-RIPK3 axis can activate MLKL even when caspase-8 is not blocked should help experimentally address the prevalent idea that necroptosis is a ‘back-up’ form of cell death that is triggered only when capases are disabled.
Altough MCMV M45 is indispensible for suppressing necroptosis and allowing virus replication in its murine host, the orthologous gene in human CMV (HCMV), UL45, does not encode an obvious RHIM-containing gene product. As no other RHIM-containing effectors appear to be encoded by the HCMV genome, it was unclear if suppressing necroptosis in humans represented an important, or even relevant, survival strategy for HCMV. Mocarski and colleagues first addressed this question by establishing RIPK3-expressing human fibroblasts and demonstrated their ability to undergo necroptosis upon exposure to TNF-α, as well as upon infection with the MCMV M45mutRHIM virus (71). To determine if HCMV was capable of inhibiting necroptosis, they first infected the RIPK3-expressing fibroblasts with HCMV, before stimulating necroptosis in these cells. Cells pre-infected with HCMV were protected from death induced by either TNF-α or MCMV-M45mutRHIM, indicating that HCMV suppresses necroptosis. In these cells, phosphorylation of MLKL proceeded normally although cell death itself was prevented, implying that HCMV, unlike MCMV, inhibited necroptosis at a step subsequent to necrosome assembly (71). While the precise mechanism by which HCMV inhibits necroptosis is not known, Mocarski and colleagues implicated a requirement for an early, IE1-regulated viral gene product(s) in preventing cell death at a step that follows MLKL phosphorylation, but precedes membrane leakage (71) (Fig. 1). Whether DAI plays a role in sensing HCMV infection to induce necroptosis is unclear, although human cells are capable of dying in response to M45mutRHIM MCMV infection (71), indicating the DAI-RIPK3 pathway is conserved between humans and mice.
Herpes simplex viruses.
HSV-1 and HSV-2, are alphaherpesviruses of the family Herpesviridae. Like CMVs and other herpesviruses, HSV-1 and HSV-2 are enveloped viruses with large dsDNA genomes. HSV-1 causes oral herpes, and is responsible for most cases of ‘cold sores’ in humans, while HSV-2 typically causes genital herpes. Both viruses infect epithelial cells and neurons, establishing life-long latent infections in the latter cell type.
Like MCMV, the genomes of both HSV-1 and HSV-2 each carry a gene (U39) that encodes a RHIM-containing ribonucleotide reductase large subunit (RR1), called infected cell polypeptide 6 (ICP6) for HSV-1 and ICP10 for HSV-2 (Fig. 2). Unlike MCMV, the HSV RR1 orthologs possess ribonucleotide reductase activity and are required for virus growth in non-dividing cells (72). They also have been ascribed a number of additional novel functions, including roles in protein folding, translation, and kinase signaling (73–75).
Recent studies from the Han, He, and Mocarski laboratories have not only begun to define the roles of ICP6 and ICP10 in modulating necroptosis during HSV infection, but have provided intriguing evolutionary insight into a possible role for RIPK3-driven cell death in restricting HSVs to humans (76–78). Although both HSVs are human pathogens, these viruses can infect mice, but replication of most strains of HSV-1 is less efficient in murine cells that it is in human cells (79). The Han and He groups both showed that RIPK3-dependent necroptosis limits HSV-1 infection in cultured murine cells and in mice. While HSV-1 triggered robust cell death in wild-type murine fibroblasts, it failed to do so in fibroblasts lacking RIPK3, resulting in significantly higher levels of HSV-1 progeny output from these latter cells (77, 78). In vivo, RIPK3-deficient mice were unable to efficiently control HSV-1 replication, resulting in increased mortality and high virus loads in the serum, brain, liver, spleen, and trigeminal ganglia of infected animals (77, 78). Interestingly, the mechanism by which HSV-1 activated RIPK3 in murine cells was independent of TNFR1/2, TLR3, cyclic GMP-AMP Synthase (cGAS), retinoic acid-inducible gene-I (RIG-I), or DAI signaling (77, 78). Reasoning that HSV-1 activated necroptosis by means distinct from previously reported mechanisms, the Han and He groups each employed mass spectrometry to identify proteins that interacted with RIPK3 during an active infection. In both cases, ICP6 was identified as a major RIPK3-associated protein, and this interaction was shown to rely on the ICP6 RHIM (77, 78). Recombinant viruses lacking ICP6, unlike their wild type counterparts, did not trigger cell death in cultured cells, and replicated to higher levels in vivo (77, 78). Surprisingly, both groups found that simple ectopic expression of ICP6 in murine cells was sufficient to drive RIPK3-dependent necroptosis, without need for virus (77, 78). This result was in striking contrast to our findings with MCMV vIRA, which protects cells from necroptosis when expressed on its own in murine cells (52). In an elegant domain-swapping experiment, Han and colleagues demonstrated that the RHIMs of MCMV vIRA and ICP6 had opposing effects on cell fate: replacing the RHIM in ICP6 with that of vIRA abolished the capacity of ICP6 to induce necroptosis, while inserting the ICP6 RHIM into vIRA now conferred on vIRA the ability to trigger cell death (77). The same group then provided mechanistic insight into how ICP6 triggers necroptosis by demonstrating that ICP6, likely as it accumulates during an active infection, first dimerizes and then nucleates a RIPK3 necrosome via RHIM-RHIM interactions (Fig. 1). Why the ICP6 RHIM promotes, rather than prevents, a RIPK3 necrosome in murine cells is still not clear, but interestingly, the core tetrapeptide sequence in the ICP6 and ICP10 RHIMs both contain a cysteine in the third position (VQCG), while ‘canonical’ RHIMs (including the one from vIRA) have a V,L, or I at this position. The ICP6 and ICP10 RHIMs also have a second cysteine three amino acids C-terminal to this sequence, whereas all other known RHIMs have an asparagine at this position. Possibly, these amino acid differences contribute to the dramatically different effects of HSV- versus MCMV-encoded RHIMs in murine settings.
The effects of ICP6 and ICP10 in human cells is quite different from those in murine cells. In human cell lines, Mocarski and colleagues found that ICP6 and ICP10 also bind to RIPK1 and RIPK3 in a RHIM-dependent manner, but do so to potently inhibit necroptosis (76) (Fig.1). Infection of human cells with wild-type HSV-1, but not ICP6-deficient virus, protected cells from TNF-α-induced necroptosis (76). Protection against necroptosis was, expectedly, dependent on the presence of a functional RHIM in ICP6 (76). Necroptosis also required the C-terminal half of ICP6, a segment that contains not only most of its ribonucleotide reductase domain, but also its caspase-8 inhibitory activity, supporting the idea that caspase-8 suppression by ICP6 is needed to unleash necroptosis (76). Nonetheless, additional roles for the C-terminus of ICP6 in promoting necroptosis cannot be ruled out, particularly as Han and colleagues identified this region as required for ICP6 self-association (77). Mutagenesis studies on ICP6 that abolish caspase-8 inhibitory activity but preserve its capacity to dimerize or function as an enzyme will help in this regard. It will also be interesting to interrogate a potential role for DAI in cell death and antiviral responses during HSV infection of permissive human target cell types.
In agreement with the findings of Mocarski and colleagues, the Han and He groups also found that HSVs failed to activate necroptosis in human cells as they did in murine cells (77, 80), outcomes attributable to the observations that ICP6 and ICP10 disrupted necrosome assembly in human cells but promoted necrosome formation in murine cells. Taken together, these studies demonstrate how HSV has co4evolved with its natural host – the human – to maintain a persistent infection by counteracting RIPK3-driven cell death in an exquisitely species-specific manner.
Influenza A virus.
Influenza A viruses (IAV) are members of the Orthomyxoviridae family of enveloped RNA viruses with segmented negative-sense genomes. IAV subtypes infect certain avaian and mammalian species, including humans, in whom they can cause a highly-contagious respiratory disease that is often severe and is responsible for significant worldwide morbidity and mortality each year. Uniquely for an RNA virus, all viral RNA synthesis takes places in the nucleus of the host cell. Virus replication is usually accompanied by the lysis of the infected cell, both in cell culture and in vivo (1, 81). For decades, apoptosis was thought to be the dominant mechanism underlying such cell death, although histological studies revealed evidence of both necrosis and apoptosis in infected human lung tissue (82, 83). In retrospect, it now appears that IAV-activated necrosis went unnoticed in cell culture studies largely because most commonly-employed cell lines used in IAV research (e.g., A549 lung adenocarcinoma cells) are deficient in necroptosis signaling (20, 32, 33).
The first indication that RIPK3 was involved in IAV-induced cell death came from the work of Saleh and colleagues, who reported in 2014 that mice deficient in the E3 ubiquitin ligase cellular inhibitor of apoptosis 2 (cIAP2) were susceptible to lethal IAV infection (84). They found that IAV-induced lethality in the absence of cIAP2 was not due to impaired adaptive immunity or increased virus replication in the infected lung, but rather due to uncontrolled RIPK3-driven necroptosis of airway epithelial cells, which resulted in severe, and ultimately lethal, degradation of the bronchiolar epithelium (84). Notably, co-deletion of RIPK3 or blockade of RIPK1 kinase activity ameliorated disease in cIAP2-deficient animals, as did deletion in hematopoetic cells of the necroptosis-inducing TNF-α family members Fas Ligand and TNF-related apoptosis-inducing ligand (TRAIL) (84). Collectively, these findings demonstated that IAV could activate RIPK3, in this case indirectly via Fas and TRAIL, and that aberrant RIPK3 activity had lethal consequences to the host when not held in check by cIAP2. Mechanistically, cIAP2 deficiency lowered the threshold for assembly of the RIPK3 necrosome in infected lungs, skewing cell death towards necroptosis (84). Saleh and colleagues speculated that airway epithelial cell necroptosis in infected cIAP2-deficient lungs was likely the result of bystander activity of Fas Ligand/TRAIL on surrounding, uninfected cells, rather than the direct consequence of virus replication in infected cells (84). The question of whether IAV could directly induce RIPK3-driven cell death in the infected cell, and the consequences of such cell death to host defense, was thus still unresolved.
In 2016, we reported that IAV could directly activate RIPK3-dependent death in infected cells, and outlined twin signaling pathways downstream of RIPK3 that mediated such cell death (85) (Fig. 1). Motivated by earlier work that necroptosis likely represented a direct (i.e cell-intrinsic) innate-immune antiviral mechanism (10, 19, 39), we screened a panel of RNA viruses for their capacity to induce RIPK3-dependent death over a 24 hour-period in murine embryo fibroblasts (MEFs). From this screen, three phenotypes emerged: (1) viruses that did not induce much death (e.g., Sendai virus [Paramyxovirdae]); (2) viruses that induced significant cell death, but in a manner that did not require RIPK3 (e.g., vesicular stomatitis virus [Rhabdoviridae]); and (3) viruses that induced RIPK3-dependent cell death. In this third category were the orthomyxoviruses IAV and influenza B virus (IBV). IAV and IBV subtypes both induced readily observable cytopathic effect (CPE) and cell death in wild-type MEFs within 24 hours, but failed to do so in RIPK3-deficient MEFs in the same timeframe. Neither virus infectivity nor replication was impeded in the absence of RIPK3, arguing against a non-permissive environment (and consequent decreased virus proliferation) as reason for reduced cell death. Instead, we found that IAV directly activated RIPK3 and stimulated assembly of a necrosome containing the additional adaptors RIPK1, FADD and MLKL (85).
Downstream of RIPK3, we found, much to our surprise, that IAV activated not only necroptosis, but also apoptosis (Fig. 1). This result was unforeseen, as necropstosis was thought to unfold only when caspase activity was suppressed. Necroptosis, expectedly, was driven by MLKL. But loss of MLKL did nothing to diminish the magntitude or kinetics of IAV-induced cell death. Instead, the manner by which cells died switched from both necroptosis and apoptosis in wild-type cells to pure caspase-driven apoptosis in the absence of MLKL. We subsequently showed that this MLKL-independent pathway of apoptosis downstream of RIPK3 was initiated by RIPK1 and propelled by FADD and caspase-8. RIPK3 functioned as a kinase for the activation of necroptosis, but signaled as a kinase-independent adaptor for deployment of apoptosis. Only concurrent inhibition of both apoptosis and necroptosis pathways downstream of RIPK3, for example, via co-deletion of FADD and MLKL, or by co-blockade of caspase- and RIPK3 kinase activity, phenocopied loss of RIPK3 and protected against IAV-induced cell death. Singly eliminating either axis of death only switched the form of death to the other pathway, indicating that both apoptosis and necroptosis can be simultaneously deployed downstream of RIPK3 during an active IAV infection (85) (Fig. 1).
So what dictates whether RIPK3 triggers necroptosis versus apoptosis in infected cells, or are both pathways activated simultaneously to kill the infected cell? This decision may be a stochastic one, determined perhaps by local availability of necroptosis versus apoptosis effector proteins in the immediate vicinity of active RIPK3, at least in cell types (such as MEFs and airway epithelial cells) in which both pathways can be activated with equivalent kinetics. Alternatively, a more-active ‘molecular switch’ mechanism may exist that flips cell fate downstream of RIPK3 between necroptosis and apoptosis, just as ubiquitylation toggles RIPK1 between its roles as an initiator of NF-κB cell-survival signaling and an executor of cell death in the TNF-α pathway (86). RIPK3 is ubiquitylated by IAP family members upon TLR stimulation (87), and, as noted eariler, loss of cIAP2 exacerbates RIPK3-mediated necrosis of bronchiolar epithelial cells upon IAV infection (84), making it very likely that ubiquitylation of RIPK3 by the IAPs is an example of a molecular switch that regulates RIPK3-mediated cell fate outcomes.
It is noteworthy that IAV triggers necroptosis in cells without need for concurrent inhibition of caspase-8. In fact, caspase-8 is readily activated by IAV with kinetics that parallel phosphorylation of MLKL (85), representing, to our knowledge, the first example of a physiological activator of RIPK3 that stimulates in parallel both apoptosis and necroptosis downstream of this kinase. These observations support the ‘stochastic availability’ model of cell death, in which RIPK3 in the infected cell can activate either (or both) apoptosis or necroptosis with equivalent odds, but cell-intrinsic differences in abundance or availability of downstream effectors determine evenutual cell fate outcomes (88). Intriguingly, a subset of lung club cells were found by tenOever and colleagues to be resistant to IAV-induced cell death in vivo (89); in such cells, perhaps none of these pathways are activated. The “stochastic availability” and “molecular switch” models are not necessarily mutually exclusive in the context of an active infection in vivo; each may operate in settings that are dictated by temporal and cell-type-specific cues.
While these studies demonstrate that IAV could activate RIPK3 and initiate parallel, redundant downstream pathways leading to apoptotic and necroptotic cell death, the mechanisms by which the infected cell sensed the presence of IAV and initiated cell death signaling remained unknown. We found that activation of RIPK3 by IAV necrosome was independent of known RNA sensing pathways (85), arguing for a mechanism that was driven either by combinations of these known pathways, or by an as-yet undiscovered upstream sensor(s). We therefore carried out a focused screen to identify this sensor, systematically evaluating RIPK3 activity and cell death responses to IAV in MEFs from mice carrying gene-targeted deletions in known antiviral pathways. From this screen, we made the unanticipated discovery that the protein DAI, considered a sensor of DNA viruses, was essential for IAV induced RIPK3 activation and consequent cell death (90). DAI was required for nucleation of the RIPK3 necrosome and activation of both MLKL and caspase-8 downstream of RIPK3 (Fig. 1). DAI also drove a residual pathway of RIPK3-independent apoptosis, using the adaptor RIPK1 to recruit FADD and activate caspase-8 in IAV-infected RIPK3-deficient cells. Consequently, cells lacking DAI ware remarkably resistant to IAV-triggered cell death, even more so than cells lacking RIPK3 (90).
As DAI is capable of binding nucleic acids (56), and as our observations revealed an absolute requirement for this protein in RIPK3 activation and cell death responses upon IAV infection (90), we next examined if DAI represented the elusive sensor that linked replicating IAV to RIPK3-dependent (and, indeed, independent) cell death. We found that DAI could bind IAV genomic RNA, and that the interaction between IAV RNA and DAI required the second of its Zα domains (Zα2). Mutating or deleting Zα2 abrogated both association with RNA as well as cell death responses, strongly implicating DAI as the upstream sensor in the IAV-initiated pathway leading to RIPK3 activation and cell death (90). These and other findings allowed us to propose a straightforward ‘induced-proximity’ model for how DAI detects the presence of IAV and activates RIPK3. In this model, DAI recognizes nascent IAV genomic RNA species, likely following nuclear export. As previous crystallographic studies have shown that Zα domains associate with Z-form nucleic acids (including Z-RNA) as dimers (64, 91), we suggest that complexing with IAV RNA, at a minimum, dimerizes DAI and provides a platform for RHIM-based associations between DAI and RIPK3 to cluster this kinase and nucleate the necrosome, initating downstream death signaling. In the absence of RIPK3, similar interactions between multimerized DAI and RIPK1 may drive RIPK3-independent apoptosis (Fig. 1).
DAI appears to preferentially associate with shorter IAV genomic (i.e., negative polarity) RNAs, particularly those representing subgenomic internally-deleted versions of the longer polymerase gene segments. Such subgenomic viral (v) RNAs are formed when the IAV polymerase falls off its template RNAs but re-engages further downstream along the same RNA strands. Truncated vRNAs produced in this manner nonetheless retain the packaging signals found in the 3’ and 5’ termini of full-length IAV genome segments and can be sorted into nascent virus-like particles (92, 93). Such particles are, of course, not replication competent, and referred to as ‘defective interfering’ or DI particles (92, 93). Notably, DAI also associates with some of the shorter IAV full-length gene segments (e.g., NA, NP) across their entire span, so dsRNA complexes of these genomic RNAs may also serve as ligands for this sensor. As DAI is a Z-form nucleic acid binding protein, and as our in silico models suggest that it is unlikely to associate with A-form dsRNAs, it will be interesting to see if DAI-associated IAV vRNAs adopt the Z-confirmation, either because of tortional stress arising from negative supercoiling during active viral replication, or because these vRNAs undergo an A-Z transformation once associated with DAI. Previous studies have shown that Zα domains can promote the A-Z transition of dsRNAs under near-physiological conditions (63), so conversion of IAV dsRNAs from A-form to Z-form by DAI Zα domains is a strong possibility. It is noteworthy that the spectrum of IAV vRNAs associated with DAI bears striking similarity to those bound by the RNA sensor RIG-I during active replication (94), suggesting that these RNA species, especially those destined for packaging into DI particles, are likely improperly encapsidated and therefore perhaps more accessible to the host innate immune machinery.
In vivo, loss of either DAI or RIPK3, or combined loss of apoptosis and necroptosis pathways downstream of RIPK3 (85), render mice incapable of controlling IAV replication in lungs, resulting in lethal infection. Loss of only MLKL does not appreciably increase susceptibility to IAV, demonstrating the redundant nature of necroptosis and revealing an important role for RIPK3-activated apoptosis in host defense against this virus. Whether MLKL can similarly compensate for an absence of apoptosis signaling remains to be seen: germline deficiency in any of the apoptosis effectors downstream of RIPK3 (i.e., RIPK1, FADD, and caspase-8) results in embryonic or perinatal lethality (44, 95, 96), precluding their use in such studies.
In 2016, Kanneganti and colleagues also identified DAI as an innate sensor of IAV in the pathway leading to activation of RIPK3 (97). Taking a different approach from ours, they began with the observation that bone-marrow derived murine macrophages (BMDMs) defective in IFN signaling were resistant to IAV-induced cell death. They next enriched microarray datasets from IAV-infected wild-type and IFN signaling-deficient BMDMs, looking for gene-expression differences in inflammatory- and nucleic acid sensing pathways that might explain why BMDMs lacking IFN signaling were resistant to IAV-induced cell death despite being permissive to this virus. From these analyses, they identified zbp1 (encoding DAI), as among the most under-expressed genes in infected IFN-deficient BMDMs, compared to controls (97). In agreement with our findings, Kanneganti and colleagues also observed that DAI was required for the execution of not only necroptosis but also apoptosis upon IAV infection, and that this requirement was unique, at least among RNA viruses tested, to IAV (97). They further determined that DAI was required for IAV-triggered activation of pyroptosis, placing this sensor at the apex of the host cell death response to IAV (98) (Fig. 1). Interestingly, Kanneganti and colleagues identified the IAV proteins NP and PB1 as interacting partners and putative ligands for DAI (97). It is possible that direct sensing of NP/PB1 by DAI functions in tandem with detection of vRNA to drive DAI signaling; alternatively, vRNAs that bind PB1 and NP indirectly bridge these proteins to DAI to facilitate death signaling, paralleling how incoming viral nucleocapsids are ligands for RIG-I (99). Although the structural features of the IAV genome and/or its protein components that trigger DAI remain unclear, what is clear is that DAI is an innate sensor essential for activation of all major forms of programmed cell death upon IAV infection.
Concluding remarks.
RIPK3-driven cell death mechanisms may contribute to antiviral host defense in at least two ways. First, they limit virus spread by preventing the infected cell from becoming a virus factory, and second, they stimulate the adaptive immune response by providing virus antigens for cross-presentation, as well as danger associated molecular patterns. The importance of RIPK3-dependent cell death to host immunity is underscored by examples from multiple paradigms of virus infection that blockade or loss of RIPK3 signaling increases virus proliferation and spread. At least two DNA viruses, HSV and MCMV, encode inhibitors of RIPK3 cell death signaling, and the genes encoding DAI, RIPK3, and MLKL are under strong positive selection in primates, indicative of evolutionary pressure on their gene products to evade inhibition by viruses (Harmit Malik, personal communication). It is thus likely that many viruses target RIPK3 signaling for inhibition, arguing for a crucial role of RIPK3-mediated cell death in host defense to viruses, and perhaps other microbes.
Given these observations, it is somewhat surprising that the necroptosis machinery, largely restricted to vertebrates in the first place, is poorly conserved across the classes of this subphylum (100). For example, both DAI and RIPK3 are found in amphibians, but not in birds. Even within mammals, DAI, RIPK3, and MLKL are absent in marsupials and, curiously, MLKL appears missing in carnivorous placentals (100). A suggested explanation for the poor conservation of necroptosis pathway genes is that RIPK3-mediated cell death may represent a redundant, back-up mechanism that has specifically evolved to clear cells in which caspases have been suppressed by virus inhibition and apoptosis fails to eliminate infected cells (100, 101). But we have found that the RNA virus IAV robustly activates necroptosis without need for concurrent caspase-8 blockade. Thus, RIPK3 is not always a back-up effector of cell death when apoptosis pathways are blocked.
An alternative explanation for the checkered conservation of necroptosis effectors through evolution lies in the way this signaling pathway is wired. Under homeostatic conditions, RIPK3 is kept inactive by the concerted and often cue-specific repressive activities of multiple checkpoints, including those mediated by an enzymatically active complex of the apoptosis pathway members FADD, cellular FLICE-like inhibitory protein (c-FLIP), and caspase-8, as well as by the anti-necroptotic effects of RIPK1, ubiquitylation, and certain NF-κB signaling components (36, 102). These negative regulators actively keep the RIPK3-MLKL axis from inopportune activation by innate-immune stimuli, and eliminating such repressive activity can unleash RIPK3-driven cell death not only during mammalian embryogenesis, but also during parturition in placental mammals, and even in multiple tissues in the adult. For example, germline deletion of FADD or caspase-8 results in embryonic lethality (95, 96), mice lacking RIPK1 fail to survive much past birth (44), and tissue-specific elimination of FADD in the skin or gut triggers severe inflammation in adult mice (103, 104), each of which are partially or wholly-driven by the activity of RIPK3 and MLKL (36, 102–104). In many of these scenarios, RIPK3-mediated morbidity and mortality is initiated by innate-immune signaling pathways (e.g., TLRs, IFNs, TNF-α) often upon exposure to commensal and environmental microbes and viruses (e.g., in the birth canal during parturition, and in the skin and gut of adult mice). Thus, the evolutionary benefits gained from necroptosis as a host defense mechanism are constrained by the deleterious, and often catastrophic, consequences of activating necroptosis inappropriately. In line with this argument, Vandenabeele and colleagues speculate that a microbe- and virus-rich diet of raw meat might have driven the loss of MLKL in carnivorous mammals (100). Similarly, the extended non-sterile ex vivo stage in the early development of marsupial young may have also selected for loss of necroptosis signaling in this mammalian infraclass. Of note, deleting necroptosis effectors RIPK3 or MLKL does not prompt activation of apoptosis, and mice lacking either RIPK3 or MLKL develop into adulthood without any overt developmental problems (45, 105).
We envision three major areas of future thrust in the field of virus-activated necroptosis. First, the molecular mechanisms that restrain RIPK3 and other effectors under homeostatic conditions remain incompletely understood, as are mechanisms that modulate the activity of this pathway during acute infections to ensure its appropriate magnitude and duration. Other innate pathways (e.g., RLR and TLR signaling) are tightly regulated at numerous steps by transcriptional induction of their effectors and regulators, as well as by post-translational modifications of these players. Necroptosis signaling during virus infections is likely similarly controlled; these mechanisms, many of which may be cell type- and tissue-specific, await discovery. Second, the viruses currently shown to activate necroptosis and described in this review almost certainly represent only a small fraction of the complete spectrum of virus families that can trigger necroptosis in the cells they infect. How these other viruses [e.g., human immunodeficiency virus-1 (106), reovirus (107)] activate RIPK3, and what strategies they may employ to subvert RIPK3-induced cell death, are fertile areas for future research. Finally, necroptosis may represent an effective mechanism of host species restriction, as was described elsewhere in this review for HSV-1. Whether necroptosis similarly contributes to the species distribution of other viruses among vertebrate hosts, is unexplored. Clearly, only the opening notes of this host-pathogen ‘La Danse Macabre’ have yet been heard.
Acknowledgements.
We thank Roshan Thapa and Glenn Rall for helpful comments. We are grateful to Holly Gillin for secretarial assitance. This work was supported by a Cancer Prevention & Research Institute of Texas (CPRIT) Scholar Award R1202 to J.W.U., and NIH grants CA168621 and CA190542 to S.B. Additional funds were provided by NIH Cancer Center Support Grant P30CA006927 and an appropriation from the Commonwealth of Pennsylvania to S.B.
References.
- 1.Yatim N, Albert ML. Dying to replicate: the orchestration of the viral life cycle, cell death pathways, and immunity. Immunity.2011;35:478–490. [DOI] [PubMed] [Google Scholar]
- 2.Green DR. Apoptotic pathways: the roads to ruin. Cell.1998;94:695–698. [DOI] [PubMed] [Google Scholar]
- 3.Galluzzi L, Brenner C, Morselli E, Touat Z, Kroemer G. Viral control of mitochondrial apoptosis. PLoS Pathog.2008;4:e1000018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Upton JW, Chan FK. Staying alive: cell death in antiviral immunity. Mol Cell.2014;54:273–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hardwick JM. Viral interference with apoptosis. Seminars in cell & developmental biology.1998;9:339–349. [DOI] [PubMed] [Google Scholar]
- 6.Levine B, Huang Q, Isaacs JT, Reed JC, Griffin DE, Hardwick JM. Conversion of lytic to persistent alphavirus infection by the bcl-2 cellular oncogene. Nature.1993;361:739–742. [DOI] [PubMed] [Google Scholar]
- 7.Salvesen GS, Dixit VM. Caspase activation: the induced-proximity model. Proc Natl Acad Sci U S A.1999;96:10964–10967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vercammen D, et al. Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J Exp Med.1998;187:1477–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vercammen D, et al. Dual signaling of the Fas receptor: initiation of both apoptotic and necrotic cell death pathways. J Exp Med.1998;188:919–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li M, Beg AA. Induction of necrotic-like cell death by tumor necrosis factor alpha and caspase inhibitors: novel mechanism for killing virus-infected cells. J Virol.2000;74:7470–7477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Khwaja A, Tatton L. Resistance to the cytotoxic effects of tumor necrosis factor alpha can be overcome by inhibition of a FADD/caspase-dependent signaling pathway. J Biol Chem.1999;274:36817–36823. [DOI] [PubMed] [Google Scholar]
- 12.Jaattela M, Tschopp J. Caspase-independent cell death in T lymphocytes. Nat Immunol.2003;4:416–423. [DOI] [PubMed] [Google Scholar]
- 13.Kalai M, et al. Tipping the balance between necrosis and apoptosis in human and murine cells treated with interferon and dsRNA. Cell Death Differ.2002;9:981–994. [DOI] [PubMed] [Google Scholar]
- 14.Hirsch T, et al. The apoptosis-necrosis paradox. Apoptogenic proteases activated after mitochondrial permeability transition determine the mode of cell death. Oncogene.1997;15:1573–1581. [DOI] [PubMed] [Google Scholar]
- 15.Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol.2010;11:700–714. [DOI] [PubMed] [Google Scholar]
- 16.Holler N, et al. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol.2000;1:489–495. [DOI] [PubMed] [Google Scholar]
- 17.Degterev A, et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol.2008;4:313–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Degterev A, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol.2005;1:112–119. [DOI] [PubMed] [Google Scholar]
- 19.Cho YS, et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell.2009;137:1112–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.He S, et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell.2009;137:1100–1111. [DOI] [PubMed] [Google Scholar]
- 21.Zhang DW, et al. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science.2009;325:332–336. [DOI] [PubMed] [Google Scholar]
- 22.Laster SM, Wood JG, Gooding LR. Tumor necrosis factor can induce both apoptic and necrotic forms of cell lysis. J Immunol.1988;141:2629–2634. [PubMed] [Google Scholar]
- 23.Zhao J, et al. Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc Natl Acad Sci U S A.2012;109:5322–5327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sun L, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell.2012;148:213–227. [DOI] [PubMed] [Google Scholar]
- 25.Vanden Berghe T, Hassannia B, Vandenabeele P. An outline of necrosome triggers. Cell Mol Life Sci.2016;73:2137–2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cai Z, et al. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol.2013;16:55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hildebrand JM, et al. Activation of the pseudokinase MLKL unleashes the four-helix bundle domain to induce membrane localization and necroptotic cell death. Proc Natl Acad Sci U S A.2014;111:15072–15077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang H, et al. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell.2014;54:133–146. [DOI] [PubMed] [Google Scholar]
- 29.Quarato G, et al. Sequential Engagement of Distinct MLKL Phosphatidylinositol-Binding Sites Executes Necroptosis. Mol Cell.2016;61:589–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dondelinger Y, et al. MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep.2014;7:971–981. [DOI] [PubMed] [Google Scholar]
- 31.Chen X, et al. Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res.2014;24:105–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morgan MJ, Kim YS. The serine threonine kinase RIP3: lost and found. BMB reports.2015;48:303–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koo GB, et al. Methylation-dependent loss of RIP3 expression in cancer represses programmed necrosis in response to chemotherapeutics. Cell Res.2015;25:707–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wallach D, Kang TB, Dillon CP, Green DR. Programmed necrosis in inflammation: Toward identification of the effector molecules. Science.2016;352:aaf2154. [DOI] [PubMed] [Google Scholar]
- 35.Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature.2015;517:311–320. [DOI] [PubMed] [Google Scholar]
- 36.Silke J, Rickard JA, Gerlic M. The diverse role of RIP kinases in necroptosis and inflammation. Nat Immunol.2015;16:689–697. [DOI] [PubMed] [Google Scholar]
- 37.Chan FK, Luz NF, Moriwaki K. Programmed necrosis in the cross talk of cell death and inflammation. Annu Rev Immunol.2015;33:79–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yatim N, et al. RIPK1 and NF-kappaB signaling in dying cells determines cross-priming of CD8(+) T cells. Science.2015;350:328–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hitomi J, et al. Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell.2008;135:1311–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shisler JL, Moss B. Immunology 102 at poxvirus U: avoiding apoptosis. Semin Immunol.2001;13:67–72. [DOI] [PubMed] [Google Scholar]
- 41.Moss B, Shisler JL. Immunology 101 at poxvirus U: immune evasion genes. Semin Immunol.2001;13:59–66. [DOI] [PubMed] [Google Scholar]
- 42.Chan FK, et al. A role for tumor necrosis factor receptor-2 and receptor-interacting protein in programmed necrosis and antiviral responses. J Biol Chem.2003;278:51613–51621. [DOI] [PubMed] [Google Scholar]
- 43.Dempsey PW, Doyle SE, He JQ, Cheng G. The signaling adaptors and pathways activated by TNF superfamily. Cytokine Growth Factor Rev.2003;14:193–209. [DOI] [PubMed] [Google Scholar]
- 44.Kelliher MA, Grimm S, Ishida Y, Kuo F, Stanger BZ, Leder P. The death domain kinase RIP mediates the TNF-induced NF-kappaB signal. Immunity.1998;8:297–303. [DOI] [PubMed] [Google Scholar]
- 45.Newton K, Sun X, Dixit VM. Kinase RIP3 is dispensable for normal NF-kappa Bs, signaling by the B-cell and T-cell receptors, tumor necrosis factor receptor 1, and Toll-like receptors 2 and 4. Mol Cell Biol.2004;24:1464–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fliss PM, Brune W. Prevention of cellular suicide by cytomegaloviruses. Viruses.2012;4:1928–1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brune W, Menard C, Heesemann J, Koszinowski UH. A ribonucleotide reductase homolog of cytomegalovirus and endothelial cell tropism. Science.2001;291:303–305. [DOI] [PubMed] [Google Scholar]
- 48.Lembo D, Brune W. Tinkering with a viral ribonucleotide reductase. Trends Biochem Sci.2009;34:25–32. [DOI] [PubMed] [Google Scholar]
- 49.Lembo D, et al. The ribonucleotide reductase R1 homolog of murine cytomegalovirus is not a functional enzyme subunit but is required for pathogenesis. J Virol.2004;78:4278–4288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Upton JW, Kaiser WJ, Mocarski ES. Cytomegalovirus M45 cell death suppression requires receptor-interacting protein (RIP) homotypic interaction motif (RHIM)-dependent interaction with RIP1. J Biol Chem.2008;283:16966–16970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sun X, Yin J, Starovasnik MA, Fairbrother WJ, Dixit VM. Identification of a novel homotypic interaction motif required for the phosphorylation of receptor-interacting protein (RIP) by RIP3. J Biol Chem.2002;277:9505–9511. [DOI] [PubMed] [Google Scholar]
- 52.Upton JW, Kaiser WJ, Mocarski ES. Virus inhibition of RIP3-dependent necrosis. Cell Host Microbe.2010;7:302–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cicin-Sain L, et al. Dominant-negative FADD rescues the in vivo fitness of a cytomegalovirus lacking an antiapoptotic viral gene. J Virol.2008;82:2056–2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mack C, Sickmann A, Lembo D, Brune W. Inhibition of proinflammatory and innate immune signaling pathways by a cytomegalovirus RIP1-interacting protein. Proc Natl Acad Sci U S A.2008;105:3094–3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Leib DA, Machalek MA, Williams BR, Silverman RH, Virgin HW. Specific phenotypic restoration of an attenuated virus by knockout of a host resistance gene. Proc Natl Acad Sci U S A.2000;97:6097–6101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Takaoka A, et al. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature.2007;448:501–505. [DOI] [PubMed] [Google Scholar]
- 57.Ishii KJ, et al. TANK-binding kinase-1 delineates innate and adaptive immune responses to DNA vaccines. Nature.2008;451:725–729. [DOI] [PubMed] [Google Scholar]
- 58.Dempsey A, Bowie AG. Innate immune recognition of DNA: A recent history. Virology.2015;4794480:146–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kaiser WJ, Upton JW, Mocarski ES. Receptor-interacting protein homotypic interaction motif-dependent control of NF-kappa B activation via the DNA-dependent activator of IFN regulatory factors. J Immunol.2008;181:6427–6434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rebsamen M, et al. DAI/ZBP1 recruits RIP1 and RIP3 through RIP homotypic interaction motifs to activate NF-kappaB. EMBO Rep.2009;10:916–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Athanasiadis A. Zalpha-domains: at the intersection between RNA editing and innate immunity. Seminars in cell & developmental biology.2012;23:275–280. [DOI] [PubMed] [Google Scholar]
- 62.Herbert A, Alfken J, Kim YG, Mian IS, Nishikura K, Rich A. A Z-DNA binding domain present in the human editing enzyme, double-stranded RNA adenosine deaminase. Proc Natl Acad Sci U S A.1997;94:8421–8426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Brown BA 2nd, Lowenhaupt K, Wilbert CM, Hanlon EB, Rich A. The zalpha domain of the editing enzyme dsRNA adenosine deaminase binds left-handed Z-RNA as well as Z-DNA. Proc Natl Acad Sci U S A.2000;97:13532–13536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Placido D, Brown BA 2nd, Lowenhaupt K, Rich A, Athanasiadis A. A left-handed RNA double helix bound by the Z alpha domain of the RNA-editing enzyme ADAR1. Structure.2007;15:395–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell.2006;124:783–801. [DOI] [PubMed] [Google Scholar]
- 66.Upton JW, Kaiser WJ, Mocarski ES. DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe.2012;11:290–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Deigendesch N, Koch-Nolte F, Rothenburg S. ZBP1 subcellular localization and association with stress granules is controlled by its Z-DNA binding domains. Nucleic Acids Res.2006;34:5007–5020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pham HT, Park MY, Kim KK, Kim YG, Ahn JH. Intracellular localization of human ZBP1: Differential regulation by the Z-DNA binding domain, Zalpha, in splice variants. Biochem Biophys Res Commun.2006;348:145–152. [DOI] [PubMed] [Google Scholar]
- 69.Lee YS, Dayma Y, Park MY, Kim KI, Yoo SE, Kim E. Daxx is a key downstream component of receptor interacting protein kinase 3 mediating retinal ischemic cell death. FEBS Lett.2013;587:266–271. [DOI] [PubMed] [Google Scholar]
- 70.Yoon S, Bogdanov K, Kovalenko A, Wallach D. Necroptosis is preceded by nuclear translocation of the signaling proteins that induce it. Cell Death Differ.2016;23:253–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Omoto S, Guo H, Talekar GR, Roback L, Kaiser WJ, Mocarski ES. Suppression of RIP3-dependent necroptosis by human cytomegalovirus. J Biol Chem.2015;290:11635–11648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Goldstein DJ, Weller SK. Herpes simplex virus type 1-induced ribonucleotide reductase activity is dispensable for virus growth and DNA synthesis: isolation and characterization of an ICP6 lacZ insertion mutant. J Virol.1988;62:196–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Smith CC. The herpes simplex virus type 2 protein ICP10PK: a master of versatility. Front Biosci.2005;10:2820–2831. [DOI] [PubMed] [Google Scholar]
- 74.Walsh D, Mohr I. Assembly of an active translation initiation factor complex by a viral protein. Genes Dev.2006;20:461–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chabaud S, et al. The R1 subunit of herpes simplex virus ribonucleotide reductase has chaperone-like activity similar to Hsp27. FEBS Lett.2003;545:213–218. [DOI] [PubMed] [Google Scholar]
- 76.Guo H, et al. Herpes simplex virus suppresses necroptosis in human cells. Cell Host Microbe.2015;17:243–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Huang Z, et al. RIP1/RIP3 binding to HSV-1 ICP6 initiates necroptosis to restrict virus propagation in mice. Cell Host Microbe.2015;17:229–242. [DOI] [PubMed] [Google Scholar]
- 78.Wang X, et al. Direct activation of RIP3/MLKL-dependent necrosis by herpes simplex virus 1 (HSV-1) protein ICP6 triggers host antiviral defense. Proc Natl Acad Sci U S A.2014;111:15438–15443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lopez C. Genetics of natural resistance to herpesvirus infections in mice. Nature.1975;258:152–153. [DOI] [PubMed] [Google Scholar]
- 80.Yu X, et al. Herpes Simplex Virus 1 (HSV-1) and HSV-2 Mediate Species-Specific Modulations of Programmed Necrosis through the Viral Ribonucleotide Reductase Large Subunit R1. J Virol.2015;90:1088–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sanders CJ, Doherty PC, Thomas PG. Respiratory epithelial cells in innate immunity to influenza virus infection. Cell Tissue Res.2011;343:13–21. [DOI] [PubMed] [Google Scholar]
- 82.Korteweg C, Gu J. Pathology, molecular biology, and pathogenesis of avian influenza A (H5N1) infection in humans. Am J Pathol.2008;172:1155–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mauad T, et al. Lung pathology in fatal novel human influenza A (H1N1) infection. Am J Respir Crit Care Med.2010;181:72–79. [DOI] [PubMed] [Google Scholar]
- 84.Rodrigue-Gervais IG, et al. Cellular inhibitor of apoptosis protein cIAP2 protects against pulmonary tissue necrosis during influenza virus infection to promote host survival. Cell Host Microbe.2014;15:23–35. [DOI] [PubMed] [Google Scholar]
- 85.Nogusa S, et al. RIPK3 Activates Parallel Pathways of MLKL-Driven Necroptosis and FADD-Mediated Apoptosis to Protect against Influenza A Virus. Cell Host Microbe.2016;20:13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Weinlich R, Green DR. The two faces of receptor interacting protein kinase-1. Mol Cell.2014;56:469–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lawlor KE, et al. RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat Commun.2015;6:6282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cook WD, et al. RIPK1- and RIPK3-induced cell death mode is determined by target availability. Cell Death Differ.2014;21:1600–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Heaton NS, Langlois RA, Sachs D, Lim JK, Palese P, tenOever BR. Long-term survival of influenza virus infected club cells drives immunopathology. J Exp Med.2014;211:1707–1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Thapa RJ, et al. DAI Senses Influenza A Virus Genomic RNA and Activates RIPK3-Dependent Cell Death. Cell Host Microbe.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ha SC, Kim D, Hwang HY, Rich A, Kim YG, Kim KK. The crystal structure of the second Z-DNA binding domain of human DAI (ZBP1) in complex with Z-DNA reveals an unusual binding mode to Z-DNA. Proc Natl Acad Sci U S A.2008;105:20671–20676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Nayak DP, Chambers TM, Akkina RK. Defective-interfering (DI) RNAs of influenza viruses: origin, structure, expression, and interference. Curr Top Microbiol Immunol.1985;114:103–151. [DOI] [PubMed] [Google Scholar]
- 93.Saira K, et al. Sequence analysis of in vivo defective interfering-like RNA of influenza A H1N1 pandemic virus. J Virol.2013;87:8064–8074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Baum A, Sachidanandam R, Garcia-Sastre A. Preference of RIG-I for short viral RNA molecules in infected cells revealed by next-generation sequencing. Proc Natl Acad Sci U S A.2010;107:16303–16308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yeh WC, et al. FADD: essential for embryo development and signaling from some, but not all, inducers of apoptosis. Science.1998;279:1954–1958. [DOI] [PubMed] [Google Scholar]
- 96.Varfolomeev EE, et al. Targeted disruption of the mouse Caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally. Immunity.1998;9:267–276. [DOI] [PubMed] [Google Scholar]
- 97.Kuriakose T, et al. ZBP1/DAI is an Innate Sensor of Influenza Virus Triggering the NLRP3 Inflammasome and Programmed Cell Death Pathways. Science Immunology.2016;1:aag2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Krug RM. Functions of the influenza A virus NS1 protein in antiviral defense. Current opinion in virology.2015;12:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Weber-Gerlach M, Weber F. Standing on three legs: antiviral activities of RIG-I against influenza viruses. Curr Opin Immunol.2016;42:71–75. [DOI] [PubMed] [Google Scholar]
- 100.Dondelinger Y, Hulpiau P, Saeys Y, Bertrand MJ, Vandenabeele P. An evolutionary perspective on the necroptotic pathway. Trends Cell Biol.2016;26:721–732. [DOI] [PubMed] [Google Scholar]
- 101.Mocarski ES, Kaiser WJ, Livingston-Rosanoff D, Upton JW, Daley-Bauer LP. True grit: programmed necrosis in antiviral host defense, inflammation, and immunogenicity. J Immunol.2014;192:2019–2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Dillon CP, Tummers B, Baran K, Green DR. Developmental checkpoints guarded by regulated necrosis. Cell Mol Life Sci.2016;73:2125–2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bonnet MC, et al. The adaptor protein FADD protects epidermal keratinocytes from necroptosis in vivo and prevents skin inflammation. Immunity.2011;35:572–582. [DOI] [PubMed] [Google Scholar]
- 104.Welz PS, et al. FADD prevents RIP3-mediated epithelial cell necrosis and chronic intestinal inflammation. Nature.2011;477:330–334. [DOI] [PubMed] [Google Scholar]
- 105.Murphy JM, et al. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity.2013;39:443–453. [DOI] [PubMed] [Google Scholar]
- 106.Pan T, et al. Necroptosis takes place in human immunodeficiency virus type-1 (HIV-1)-infected CD4+ T lymphocytes. PLoS One.2014;9:e93944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Berger AK, Danthi P. Reovirus activates a caspase-independent cell death pathway. mBio.2013;4:e00178–00113. [DOI] [PMC free article] [PubMed] [Google Scholar]