

Summary
Transcription and translation are two main pillars of gene expression. Due to the different timings, spots of action, and mechanisms of regulation, these processes are mainly regarded as distinct and generally uncoupled, despite serving a common purpose. Here, we sought for a possible connection between transcription and translation. Employing an unbiased screen of multiple human promoters, we identified a positive effect of TATA box on translation and a general coupling between mRNA expression and translational efficiency. Using a CRISPR-Cas9-mediated approach, genome-wide analyses, and in vitro experiments, we show that the rate of transcription regulates the efficiency of translation. Furthermore, we demonstrate that m6A modification of mRNAs is co-transcriptional and depends upon the dynamics of the transcribing RNAPII. Suboptimal transcription rates lead to elevated m6A content, which may result in reduced translation. This study uncovers a general and widespread link between transcription and translation that is governed by epigenetic modification of mRNAs.
Keywords: transcription, translation efficiency, N6-adenosine methylation, m6A, TATA, RNAPII, gene regulation
Graphical Abstract
Highlights
-
•
Transcription rates of mRNAs positively correlate with rates of their translation
-
•
Dynamics of RNA polymerase II impact the deposition of m6A on mRNAs
-
•
Suboptimal transcription enhances m6A modification of mRNAs
-
•
Excessive m6A modification is detrimental for the translation process
Slowly transcribed mRNAs show increased levels of N6-adenosine methylation and reduced translation efficiency, setting up a nuclear control on protein abundance.
Introduction
Transcription of genome-encoded information into mRNA and translation of mRNA into a functional protein are the main layers of gene expression. Due to the existential need to adjust gene expression to both intracellular requirements and extracellular stimuli, both processes are subject to regulation at multiple levels. Transcription is a highly controlled process that is extensively regulated at the levels of initiation, elongation, and termination. Recent studies in eukaryotes have linked transcription to other levels of mRNA regulation, such as alternative splicing (Dujardin et al., 2014), polyadenylation (Oktaba et al., 2015), localization and translation (Zid and O’Shea, 2014), and degradation (Dori-Bachash et al., 2012). Although splicing and polyadenylation are thought to be co-transcriptional, and therefore could be directly affected by the RNAPII dynamics, the effect on translation and degradation, which have distinct spatial and temporal dynamics, is more complicated to perceive. A recently formulated model explains the imprinting role of transcription by co-transcriptional recruitment of coordinator proteins (Haimovich et al., 2013), which accompany the synthesized transcript and are capable of regulating its future fate.
Translation of mRNAs is controlled mainly via initiation (Sonenberg and Hinnebusch, 2009) and elongation (Richter and Coller, 2015). Although several recent studies have suggested certain levels of dependency between transcription and translation (Elfakess and Dikstein, 2008, Harel-Sharvit et al., 2010, Tamarkin-Ben-Harush et al., 2014, Zid and O’Shea, 2014), it is not clear whether these are limited to certain subgroups of mRNAs or represent a general link. In general, transcription and translation are still regarded as mutually independent processes, characterized by different timings, cellular locations, functional complexes, and mechanisms of action.
N6-methyladenosine (m6A) is considered to be one of the most abundant RNA modifications, detected in thousands of human transcripts (Dominissini et al., 2012, Meyer et al., 2012). Several recent studies have connected m6A to the regulation of splicing (Xiao et al., 2016), translation (Meyer et al., 2015, Wang et al., 2015), and degradation (Wang et al., 2014). Overall, a growing body of evidence suggests that m6A plays an important role in multiple levels of mRNA regulation.
In this study, we tested a hypothesis suggesting a direct flow of information from transcription to translation. Combining an unbiased screen for examination of the effect of human promoters on mRNA translation and genome-wide analyses, we identified a positive correlation between mRNA expression and translation efficiency (TE) and found that rate of transcription positively affects TE. Moreover, we observed that transcriptional dynamics are reflected in the relative deposition of m6A on mRNAs that affects translation. This study establishes a general and robust link between transcription and translation of mRNAs and provides a mechanistic insight regarding the way transcription epigenetically imprints mRNA molecules.
Results
A Reporter Vector System to Examine the Transcription-Translation Relationship
To examine the relationship between transcription and translation, we set out to determine the effect of different human promoters on the translation of a reporter gene (Renilla luciferase [Rluc]). For this purpose, we defined promoters as 0.5- to 2.5-Kb-long regions characterized by high H3K4Me3 and low H3K4Me1 epigenetic marks upstream of transcriptional start sites (TSSs), supported by RNA sequencing (RNA-seq) data of MCF7 cells (Loayza-Puch et al., 2013). To make our screen versatile and diverse, we cloned promoters from genes connected to stress response, autophagy, ER metabolism, and metastasis, as well as multiple transcription factors. While cloning the promoter sequences, we avoided regions extending downstream the respective TSSs, as these might result in the inclusion of additional 5′ UTRs into the reporter transcripts, potentially complicating the interpretation of results. Because each of the cloned promoters drives the expression of the same reporter gene, we affiliated every promoter with a unique 10 nt barcode (cloned in the 3′ UTR of Rluc) in order to follow its expression in a pool of Rluc mRNAs (Figure S1A). Following these guidelines, we cloned 135 human promoters (Table S1) in order to create a library named Pro-Lib, where most of the promoters were associated with two or more different barcodes to provide higher experimental confidence. As expected, cloned promoter regions substantially induced Rluc expression (Figure S1B). For normalization of expression, Pro-Lib included an additional reporter gene, Firefly luciferase (Fluc), used as an inner control (Figure S1A). Last, we employed an Flp-FRT recombination system and used competent Flp-In MCF7 cells to stably integrate a single copy of a Pro-Lib vector per cell in the identical genetic locus in order to avoid any possible influence of different chromatin neighborhoods on the reporter gene expression.
Figure S1.

Setup of the Promoter Library (Pro-Lib) and Barcoded Polysomal Profiling (BPP), Related to Figure 1
(A) Schematics of the Pro-Lib vector. The reporter Rluc gene is flanked upstream by sequences encoding for putative promoters of interest (magenta colored) and downstream by unique 10-nt barcodes (also magenta colored). Transcription of the control Fluc gene is driven by HSV-TK promoter in all vectors.
(B) Several Pro-Lib vectors with the indicated regions cloned upstream of Rluc gene were integrated into MCF7/FRT cells, grown separately and subjected to dual Rluc/Fluc assay. Note the uniform expression of the tested constructs that is substantially higher than the background signal (assessed by expression of vectors with no promoter or with random intron sequence cloned instead of a promoter) and milder than the expression of the strong SV40 promoter. Data are represented as mean ± SEM of n = 3.
(C) Total RNA isolated from fractions of a typical BPP experiment were separated on 1.5% agarose gels and documented. Note the characteristic segregation of RNA as well as the relative enrichment of ribosomal RNAs in the polysomal fractions.
(D) Total RNA concentrations in the different fractions were measured following lysis of cells in the presence or absence of EDTA and polysomal profiling procedure. Note the dramatic drop of the RNA content in the fractions 9-13 upon presence of EDTA in the lysis buffer.
(E) BPP experiment identifies two Rluc transcripts (driven by SV40 and CD44 promoters) exhibiting super-sensitivity to the inhibition of mTORC1. Note the multiple barcodes for each promoter showing identical behavior.
(F) The transcripts mentioned in (E) contain putative 5′TOP sequences (marked in blue). The TSS of the SV40 promoter was reported previously (Byrne et al., 1983), while the TSS of the CD44-driven construct was determined by 5′-RACE analysis in this study.
After establishing the library in a stable population, we performed a polysomal profiling experiment by using sucrose gradients, a classical method to separate mRNAs according to the amount of bound ribosomes. Since we examined only the barcoded Rluc mRNAs, their relative segregation in the gradient indicates ribosome density and TE estimation. We named the whole procedure barcoded polysomal profiling, or BPP (Figure 1A). Relative enrichment of Pro-Lib barcodes in the various fractions of the gradient showed that most Rluc mRNAs are localized in the initial polysomal fractions (i.e., 9–12; Figure 1B). Control total RNA segregation showed a characteristic pattern of polysomes and EDTA sensitivity, in line with the known dependence of polysomes on the availability of Mg2+ ions (Figures S1C and S1D).
Figure 1.

A Screen for Examination of the Relationship between Transcription and Translation
(A) Schematics of the barcoded polysomal profiling (BPP) approach.
(B) A typical segregation of Rluc mRNAs.
(C) Control Rluc transcript with 5′ TOP sequence exhibits rapid shift to non-translating fractions upon inhibition of mTORC1.
(D) Control Rluc transcript shifts to denser fractions following splicing. The bar diagram below represents normalized relative Rluc protein expression assessed by luciferase assay in two separate clones.
See also Figure S1.
Next, we tested whether BPP can detect changes in TE. For this purpose, we employed Rluc mRNA containing 5′-terminal oligopyrimidine tracts (5′ TOP) derived from RPL37a. Translation of mRNAs possessing 5′ TOP is highly dependent on mTOR activity, resulting in a rapid translational arrest after mTOR inhibition, as compared to other mRNAs (Thoreen et al., 2012). Indeed, inhibition of mTORC1 resulted in a global moderate shift of the Rluc mRNAs to fractions 9–11 (e.g., SPTBN1 promoter), whereas a 5′ TOP-containing transcript was depleted from the translated fractions of the gradient (Figure 1C). Interestingly, we also identified several additional Rluc transcripts exhibiting similar hypersensitivity toward the inhibition of mTORC1 (Figure S1E). Further analysis of the sequences adjacent to their TSSs (Figure S1F) identified stretches of pyrimidines that could serve as 5′ TOP signals, providing a probable explanation for their dramatic response. To further test detection capabilities of BPP, we examined the effect of splicing on TE. Indeed, we observed that spliced Rluc mRNA shifts to heavier polysomal fractions and yields more protein (Figure 1D), as expected from the known positive effect of splicing on translation (Nott et al., 2004). Altogether, our control experiments demonstrate that BPP is capable of examining multiple Rluc mRNAs in bulk and detecting changes in TE of individual transcripts.
TATA Box Confers Higher TE
Inspecting the segregation of the barcoded Rluc mRNAs on sucrose gradients, we identified 12 promoters that caused a shift of the reporter transcript toward higher ribosomal occupancy fractions in at least two independent experiments. Intriguingly, four of these promoters contained a TATA-box element (Figure 2A) and four others had TA-rich sequences that could potentially serve as non-canonical TATA boxes (Figure S2A), indicating that this promoter element could positively influence translation. To test this possibility, we supplied several TATA-less promoters with an artificial TATA element (consisting of the TATA sequence followed by the short downstream sequence derived from human ACTB). In all cases, this manipulation resulted in Rluc transcripts occupying denser fractions of the gradient (i.e., 11–13; Figures 2B and S2B), supporting the previous observations. Taking the ASNSD1 promoter as a model, we observed a positive effect of TATA addition on TE under various conditions (Figure S2C), thus indicating a robust phenomenon.
Figure 2.

Presence of the TATA Element in Promoters Enhances TE
(A) Pro-Lib vectors encoding for TATA-containing promoters result in Rluc mRNAs shifted to the denser fractions of the gradient compared to other transcripts (e.g., under SPTBN1 promoter).
(B) Pro-Lib vectors supplied with an artificial TATA yield Rluc mRNAs that are shifted to the denser fractions of the gradient compared to the parental TATA-less promoters.
(C) Northern blot analysis of Rluc mRNAs; quantification reflects relative Rluc/Fluc ratio.
(D) Relative levels of Rluc mRNAs and proteins produced by promoters with or without TATA element; note the super-induction of the protein expression. Data are represented as mean ± SEM, n = 4.
(E) Relative levels of Rluc mRNAs and proteins produced by SV40 promoter upon mutagenesis of TATA; data are represented as mean ± SEM, n = 3.
(F) Mutagenesis of SV40 promoter-derived TATA results in less efficient translation of Rluc mRNA as detected by BPP; data are represented as mean ± SEM of a representative gradient, n = 2.
(G) Schematics of the CRISPR-Cas9-mediated mutagenesis of the endogenous TATA (in pink square) of c-Myc. Below: characterization of the different c-Myc alleles of one of the isolated clones, including relative abundances.
(H) Mutagenesis of the c-Myc TATA results in lowered mRNA levels (left chart) and further reduced c-Myc activity (right chart); data are represented as mean ± SEM of n = 3.
(I) Western blot analysis of c-Myc protein in the TATA-mutated clone and WT MCF7 cells; see Figure S2I for the uncropped blot.
(J) Polysomal profilings of c-Myc mRNAs isolated from the clone with mutated TATA and WT MCF7 cells; data are represented as mean ± SEM of a characteristic gradient; n = 3.
See also Figures S2G–S2K for the characterization of an additional clone.
Figure S2.

Effect of TATA Box on TE, Related to Figure 2
(A) Pro-Lib vectors encoding for promoters with TA-rich regions at their 3′ ends result in Rluc mRNAs shifted to the denser fractions of the gradient, compared to other transcripts (e.g., under SPTBN1 promoter).
(B) Average standardized profiles of polysome fractions for the TATA+/TATA- paired constructs in Pro-Lib.
(C) Promoter region of ASNSD1 gene yields better translated Rluc mRNAs when supplemented with an artificial TATA box at its 3′end. Cells expressing Rluc mRNAs from either native ASNSD1 promoter or with artificial TATA box were subjected to BPP and plotted for a direct comparison of TE (left panel). To test the robustness of the positive effect of TATA on TE, cells were subjected to various stress conditions, prior to BPP (right panels). Note that in all listed conditions the presence of TATA box enhances TE.
(D) Point mutagenesis of the artificial TATA box reduces mRNA levels and prevents super-induction of the protein production. SZT2 and ASNSD1 promoters supplemented with an artificial TATA box were subjected to site-directed mutagenesis of the TATA box sequence (muTATA). Levels of the Rluc mRNAs and protein compared to the native promoters (lacking TATA) are shown; data are represented as mean ± SEM of n = 3.
(E) Rluc mRNAs resulting from either native ASNSD1 promoter, one with the artificial TATA box (+TATA) or with a point-mutated TATA sequence (muTATA) were subjected to the BPP procedure. Note the shift toward denser fractions caused by the artificial addition of TATA box as well as the shift to the opposite direction upon mutation of the TATA sequence.
(F) Screenshot of the DBTSS database (release 9.0) showing the distribution of TSS of c-Myc gene in MCF7 cells. Note that most of TSSs stem from a single TATA box-containing promoter (see zoom-in window).
(G) Genotyping of the c-Myc proximal promoter region of the second clone bearing modified c-Myc TATA box. Numbers on the left represent relative abundances of the different alleles.
(H) Lysates of the clone described in (G) were subjected to polysomal profiling and plotted against wild-type MCF7 cells. SEM represents three measurements of a characteristic gradient; n = 2. ∗∗∗p < 0.005, ∗∗∗∗p < 0.001.
(I) Lysates of the two clones bearing modified c-Myc TATA boxes together with wild-type MCF7 cells (WT TATA) and cells transfected with vector encoding for c-Myc (Myc OE), were resolved on SDS-PAGE and probed for c-Myc and GAPDH proteins. Replicates on the blot represent biological repeats.
(J) Relative quantification of the c-Myc bands presented in (I).
(K) TE of a control gene (ALG8) was assessed by probing polysomal fractions described in Figure 2J with primers detecting ALG8 mRNA. SEM represents three measurements of a characteristic gradient; n = 3.
Because artificial introduction of TATA may alter the TSS, possibly impacting translational capacity (Rojas-Duran and Gilbert, 2012), we investigated in detail the 5′ UTRs produced from the TATA-containing and TATA-less promoter pairs. By northern blotting, we observed that most of the tested promoters resulted in reporter transcripts of a similar length, with a noticeable enhancement of mRNA levels in TATA-containing promoters (Figure 2C). To establish precisely the 5′ ends of the transcripts, we performed 5′ rapid amplification of cDNA ends (RACE) analyses, which showed a very narrow peak ∼25 nt downstream of the inserted TATA element (Figures S3A and S3B). To test whether these alterations in the 5′ UTRs could explain the observed changes in translation, we in vitro synthesized Rluc transcripts bearing the different 5′ UTRs and examined their relative TE (Figure S3C). Given that the 5′ UTR generated after the insertion of TATA sequence did not confer higher TE, we conclude that the observed positive effect of TATA on translation is unlikely to stem from the differences in the 5′ UTRs of Rluc mRNAs.
Figure S3.

Analysis of Rluc mRNAs, Related to Figures 2, 3, 5, and 6
(A and B) Rluc mRNAs transcribed by ASNSD1 (A) or SZT2 (B) promoters either lacking or bearing an artificial TATA box were subjected to 5′RACE analysis in order to determine their TSSs. Reads were plotted in a quantitative manner using IGV software. Note the first ATG of the Rluc ORF and the precise focusing of the TSS by the TATA box.
(C) Evaluation of the effect of the different 5′UTRs on TE. Rluc transcripts bearing the different 5′UTRs identified in A,B (see the numbered arrows) were transcribed in vitro using T7 RNA polymerase, polyadenylated and transfected into MCF7 cells in fold-wise amounts. Renilla luciferase activity was measured 24 hr later and plotted in a relative manner to draw the trend lines that represent the respective ratios of translation. The rightmost panel shows the absolute Rluc signals; n = 4.
(D) Rluc mRNAs expressed from either induced or non-induced TRex-Rluc gene were subjected to 5′RACE analysis. Reads (numbers of sequenced colonies) were plotted in a quantitative manner on a scale ranging from TATA box (−243) to ATG (+1).
(E) Rluc transcripts produced in vitro using HeLa extract in optimal conditions (1.5mM Mg++), upon high MgCl2 (3mM Mg++) or from promoter with mutated TATA element (muTATA) were subjected to 5′- and 3′-RACE analyses. The reads from both assays were analyzed and plotted in a quantitative manner using IGV software.
(F) Transcripts described in (E) were subjected to analysis of the length of polyA-tails.
(G) The TRex-Rluc cassette was induced for 24 hr in cells transfected with the indicated siRNAs. After isolation of total RNA, the 5′- and 3′ ends of the Rluc mRNAs were determined as detailed in (E).
So far, we have inferred TE from ribosome occupancy measurements reflected by migration within sucrose gradients. Next, we tested TE changes by measuring separately the levels of the reporter mRNA and protein in the promoter pairs. As expected, we observed significantly more mRNA produced from TATA-containing promoters (7- to 9-fold, Figure 2D), consistent with the levels measured by northern blotting (Figure 2C). In contrast, the increase in the level of protein activity was significantly higher (>20-fold), supporting the connection between the presence of TATA in promoter and enhanced protein production. To further test the role of the TATA element in translation, we mutagenized it within the SV40 promoter. Remarkably, loss of TATA reduced the reporter mRNA expression by ∼14-fold and protein activity by ∼80-fold (Figure 2E), suggesting reduced TE. Indeed, mRNA produced by a TATA-mutated promoter was enriched in the lighter polysomal fractions (Figure 2F), further supporting this indication. We observed similar results upon mutagenesis of TATA in other promoters (Figures S2D and S2E), suggesting that this effect is not restricted to any particular promoter.
Next, we tested whether the positive effect of TATA on TE also applies to the endogenous mammalian gene expression. For this purpose, we chose c-Myc, a ubiquitously expressed gene with a single active TATA-positive promoter in MCF7 cells (Figure S2F) and used the CRISPR-Cas9 system to alter its TATA sequence (Figure 2G). This strategy yielded isolated cell clones with disrupted c-Myc TATA on most of their alleles (Figure 2G and Figure S2G). Examination of c-Myc expression in these clones revealed a reduction of ∼25% in c-Myc mRNA and ∼50% in both protein activity and expression (Figures 2H and 2I and S2I and S2J). Moreover, polysomal profiling of c-Myc transcripts showed reduced ribosomal occupancy upon mutation of the TATA element (Figures 2J and S2H), whereas a control transcript displayed very similar profiles in both clones (Figure S2K), indicating a c-Myc-specific effect. Thus, we conclude that the presence of the TATA element in a promoter enhances the efficiency of mRNA translation and note the validity of this observation to multiple promoters and genes.
General Association, but Not Causal Link, between mRNA Levels and TE
In all the cases we studied, presence of TATA in a promoter stimulated both mRNA expression levels and TE. To examine whether TE positively correlates with mRNA expression levels, we clustered the Pro-Lib BPP data to separate the transcripts of our library into two relative groups, one with lower TE (Figures S4A and S4B) and another with higher TE (Figures S4C and S4D), and compared the expression levels between these two groups. Indeed, we found that transcripts with higher TE tend to be more abundant (Figure 3A). To test whether this coupling is TATA dependent, we repeated this analysis while omitting the promoters with artificially added TATA elements. Notably, this analysis yielded similar albeit less significant results (Figure 3B), thus suggesting a positive correlation between the abundance of Rluc transcripts and their TE, with no dependency on TATA. To further test this observation, we employed cells expressing an inducible version of the barcoded reporter gene (TRex-Rluc), in which the levels of Rluc mRNA are stimulated ∼17-fold after induction (Figure 3C). Importantly, induction of this gene yielded a significantly greater enhancement of protein activity (∼60-fold, Figure 3C), suggesting a translational boost. Indeed, subsequent BPP analysis revealed a strong increase in the ribosome occupancy of the induced reporter mRNA (Figure 3D and Figure S5A), supporting the notion that increased expression results in higher TE. Notably, we performed 5′ RACE analysis of both induced and non-induced TRex-Rluc mRNAs and found the scattering of TSSs to be very similar (Figure S3D). Altogether, we conclude that the observed link between the expression levels of mRNAs and their TE is not restricted to the TATA element and might therefore represent a more general phenomenon.
Figure S4.

Global TE Assessment of the Pro-Lib mRNAs, Related to Figure 3
Barcoded Rluc mRNAs with sufficient read coverages (> 10.000 reads over 15 polysomal fractions) in three independent BPP experiments were divided into two main groups by hierarchical clustering. One arm includes barcodes that showed peak in fractions 9-11 (lower TE, A-B) and the other arm includes barcodes whose polysomal profile peaked in fraction 11-13 (higher TE, C-D). Plots to the right of the heatmaps show the mean profile of the vectors assigned to each group over the three repeats; error bars = SD.
Figure 3.

Levels of mRNAs Positively Correlate with TE but Do Not Dictate It
(A) Reporter mRNAs from the Pro-Lib screen were separated into groups with relatively high or low TE (derived from the polysomal profiles, see Figure S4) and compared with their expression levels (estimated from read counts observed for each vector over all fractions). p values calculated using Wilcoxon’s test.
(B) Same comparison as described in (A) was performed after exclusion of mRNAs transcribed from promoters with artificial TATA element.
(C) Levels of mRNA and protein resulting from the induced TRex-Rluc gene were measured and plotted relatively to the non-induced condition; data are represented as mean ± SEM of n = 3.
(D) BPP examination of either induced or non-induced TRex-Rluc mRNAs; data are represented as mean ± SEM of a characteristic gradient; n > 5, see also Figure S5A.
(E) Upper panels: paired RNA-seq and Ribo-seq datasets from different human cell lines were examined for relationship between mRNA expression level and TE (calculated as the (log2) ratio between densities of ribosome footprint and RNA-seq reads). Lower panels: comparisons between the 10% of genes with lowest and highest expression levels are presented; p values calculated using Wilcoxon’s test.
(F) MCF7 cells were transfected with fold-wise amounts of in vitro transcribed (using HeLa nuclear extract) Rluc mRNA followed by measurement of RLuc activity after 18 hr; data are represented as mean ± SEM of n = 3.
(G) Levels of Rluc mRNA and protein were compared between two populations of MCF7 cells expressing near single or multiple integrated copies of Lenti-Rluc unit. Data are represented as mean ± SEM of n = 3.
(H) BPP analysis of Rluc transcripts described in (G); data are represented as mean ± SEM of a characteristic gradient, n = 3.
Figure S5.

Transcription rates impacts translation, Related to Figures 3 and 4
(A) Segregation of the induced versus non-induced Rluc mRNAs on sucrose gradients. Cells expressing barcoded Rluc gene were treated for 21 hr to induce the expression of Rluc or left untreated, and subjected to BPP procedure.
(B) Upper panels: test for correlation between mRNA expression and TE, as described in Figure 3E, was performed here on data from BJ cells in quiescent, senescent, and transformed states. Lower panels: direct comparison between 10% of mRNAs with the lowest and 10% with the highest abundance.
(C) In MCF7 cells, a similar correlation was tested in p53-activated (by Nutlin3 treatment) versus the control cells. Both datasets are from (Loayza-Puch et al., 2013), representation as detailed for (B).
(D) Cells with induced Rluc were treated with CPT (or DMSO) for 5 hr and subjected to BPP analysis.
To test whether genome-wide data support a global relationship between mRNA level and TE, we analyzed pairs of RNA-seq and Ribo-seq (ribosomal footprinting sequencing) datasets from multiple human cell lines. Intriguingly, we observed a positive global correlation between mRNA expression level and TE, which was rather weak but consistent and statistically significant (Figure 3E). Moreover, this correlation between mRNA levels and TE was also apparent in various stress conditions (Figures S5B and S5C). These results indicate that there is a positive correlation between expression levels of mRNAs and their TE in mammalian genomes as well.
To examine whether mRNA levels could directly regulate TE, we transfected different amounts of in vitro transcribed and purified Rluc mRNA into MCF7 cells and monitored the activity of the produced Rluc protein. We anticipated that if levels of mRNA positively stimulated TE, transfecting increasing amounts of mRNA would result in an exponential increase of protein production. However, we observed a clear linear dependency between the two parameters (Figure 3F), suggesting indifference of the protein production rates to mRNA abundance. To test this conclusion further, we employed lentiviral-mediated stable integration of a transcriptional unit including a promoter and Rluc gene and generated two stable populations with either nearly single or multiple integrations of the same transcriptional unit per cell, resulting in an ∼9-fold difference in Rluc mRNA levels (Figure 3G). Also here, increase in protein activity was similar to the increase in mRNA levels, indicating comparable TEs. Supporting this conclusion, Rluc mRNAs derived from both cell populations exhibited similar segregation patterns in sucrose gradients (Figure 3H). Altogether, these results suggest that while mRNA expression levels positively correlate with TE in multiple cellular models, mRNA abundance does not directly regulate it.
Rate of Transcription Positively Affects TE
Next we searched for the causal origin of the observed correlation between mRNA levels and TE. We considered the rate of transcription to be the most likely candidate since it directly regulates the expression levels of mRNAs. To test this possibility, we correlated transcription rates, as estimated by global run-on sequencing (GRO-seq) (Core et al., 2008), and TE, as determined by Ribo-seq and RNA-seq data, in BJ and MCF7 cell lines. In both cell types, we observed a significant positive association between the rates of transcription and TE of genes that do not possess upstream ORFs (Figure 4A and Figures S6A and S6B). This correlation is notable given that this analysis integrates distinct datasets independently obtained from three genomic techniques in two different cellular systems.
Figure 4.

TE is Affected by the Rate of Transcription
(A) Upper panels: positive genome-wide correlations between translational efficacies and rates of transcription in BJ and MCF7 cells. Lower panels: direct comparisons between 10% of genes with lowest and highest transcription rates. GRO-seq data were from (Korkmaz et al., 2016, Léveillé et al., 2015); Ribo-seq and RNA-seq data were from (Loayza-Puch et al., 2013); p values were calculated using Wilcoxon’s test.
(B) Expression levels of Rluc mRNA and protein were examined following parallel induction of the TRex-Rluc gene and CPT treatment for 7 hr; data are represented as mean ± SEM of n = 3.
(C) Lysates of the two populations described in (B) were subjected to BPP procedure; data are represented as mean ± SEM of a characteristic gradient, see also Figure S5D; n = 3.
(D) Upper panels: genome-wide correlations between rates of transcription and TE changes after treatment with CPT. Lower panels: direct comparison of the effect on TE between the 10% of most highly and lowly transcribed genes; p values were calculated using Wilcoxon’s test.
(E) Polysomal profilings of mRNAs after CPT treatment (red) show reduced TE compared to untreated cells (green). Columns represent the relative mRNA levels as detected by qRT-PCR; ALG8 was used as a control gene. Data are presented as mean ± SEM of three technical measurements of a characteristic graident; n = 3. See also Figure S6C.
(F) Cells with barcoded TRex-IRES-Rluc cassette (schematics) were induced for 18 hr and subjected for examination of Rluc mRNA and protein levels relative to the non-induced cells; data are presented as mean ± SEM of n = 3.
(G) Same cells as in (F) were treated in a similar way and subjected to BPP procedure. Data are presented as mean ± SEM of three measurements of a characteristic gradient; n = 3.
(H) Pro-Lib vectors with SZT2 promoters, with or without the TATA element, were supplemented with IRES-encoding sequence as shown on the schematics. Cells expressing these constructs were subjected to quantification of mRNA and protein levels; data are presented as mean ± SEM of n = 3.
Figure S6.

Related to Figures 4 and 5 and 6
(A) Genes with high ribosomal occupancy in their 5′UTRs show overall reduced TE in both BJ and MCF7 cells; p values were calculated Wilcoxon’s test.
(B) These genes do not display any clear correlation between their transcription rate and TE (showing two independent replicates), and were removed from the genome-wide analysis.
(C) Treatment with CPT causes attenuation of translation. Transcripts showed in Figure 4E and additional mRNAs were detected in polysomal profiling experiments using primers against their 3′ UTRs.
(D) Rluc mRNAs produced in cells stably expressing Lenti-Rluc cassette integrated at either single- or multiple-copy numbers, were subjected to MeRIP procedure and analyzed by qRT-PCR. In each experiment, the level of recovery from cells with multi-copy integration was calculated relatively to the recovery of the single-copy population; n = 4.
(E) Rluc mRNAs transcribed from the promoter pairs with or without intact TATA box element were subjected to MeRIP procedure and analyzed by qRT-PCR; n = 2.
(F) Efficiency of METLL14 and YTHDF1 siRNA-mediated knockdown. MCF7 cells were transfected with siRNAs targeting METTL14 or YTHDF1 genes or control non-targeting siRNA. After 56 hr, the cells were harvested, subjected to RNA isolation and tested for the levels of the respective mRNAs using qRT-PCR.
(G) MCF7 cells transfected with the indicated siRNAs were subjected to polysomal profiling procedure and an average levels of RNA in the collected fractions were measured using NanoDrop and plotted; n = 2.
(H) Cells expressing “normal speed” or “slow” mutants of RNAPII were subjected to polysomal profiling on sucrose gradients. Several mRNAs were detected using qRT-PCR to monitor changes in their TE; a characteristic profile is shown, n = 2.
To further investigate the relationship between rates of transcription and TE, we employed camptothecin (CPT), a chemical compound that inhibits topoisomerase I and, in mild concentrations, slows down the progression of transcribing RNA polymerase II (RNAPII) (Dujardin et al., 2014). Indeed, when CPT was applied in parallel to the induction of TRex-Rluc gene, we observed a reduction of ∼60% of Rluc mRNA, but a more prominent reduction of Rluc activity of ∼80% (Figure 4B). This difference could be explained by the reduced ribosome occupancy, as reflected by polysomal profiling analyses (Figure 4C and Figure S5D). Thus, impediment of RNAPII progression does not only reduce the transcriptional rate of TRex-Rluc but also attenuates its TE. To further test the role of RNAPII dynamics on translation, we assessed TE of multiple genes in CPT-treated cells. Overall, CPT treatment resulted in TE reduction of approximately 700 genes by more than 1.5-fold in two independent experiments (Table S2). Given that highly transcribed genes showed higher TE (Figure 4A), we speculated that CPT treatment would cause a more pronounced repressive effect on the translation of these genes. Indeed, intersection with GRO-seq data demonstrated that impediment of RNAPII caused a significant reduction in TE of genes with a relatively high transcription rate (Figure 4D). We further examined the polysomal segregation of several mRNAs, suggested by the genome-wide experiments, and validated that these mRNAs indeed displayed reduced TE coupled with reduced expression, as compared to ALG8 control mRNA (Figure 4E). To eliminate the possibility that the transcripts exhibiting translational shifts are truncated due to the CPT treatment, we re-examined these and other mRNAs by using primers annealing to 3′ UTRs only. Also in this case, the shift of mRNAs was apparent (Figure S6C). Taken together, we conclude that transcription rate is an important positive determinant of TE.
Transcription-Dependent Translation Regulation Requires Canonical Translation Initiation
To further examine the mechanism by which transcription affects TE, we introduced an internal ribosome entry site (IRES) sequence into the inducible TRex-Rluc cassette (TRex-IRES-Rluc, see schematics in Figure 4F) to bypass the canonical 5′ cap-mediated initiation of translation. Interestingly, induction of TRex-IRES-Rluc resulted in Rluc activity that mirrored the changes in the mRNA levels (i.e., ∼12- to 15-fold induction at both levels, Figure 4F), which contrasted the previously observed induction of the Rluc protein activity (>60-fold, Figure 3C). Moreover, BPP analyses revealed similar segregation of both induced and non-induced IRES-Rluc mRNAs (Figure 4G), further suggesting lack of translational induction. Furthermore, introduction of IRES into constructs containing the artificial TATA element (e.g., SZT2 promoter) abolished the previously observed enhancement of TE (Figure 4H, compare with Figure 2D) and resembled the effect of TATA mutagenesis (Figure S2D). Thus, we conclude that canonical translation initiation is required for transcription-dependent enhancement of translation.
m6A Modification Mediates Transcription-Responsive Translation
Next, we looked for a molecular event that could explain the link between transcription and translation. Since changes in transcription affected translation of multiple genes (Table S2), we reasoned that this molecular event should be robust and widespread. m6A is an abundant nuclear dynamic mRNA modification identified in thousands of human transcripts (Dominissini et al., 2012, Meyer et al., 2012), recently implicated in translational regulation (Wang et al., 2015). Therefore, we asked whether m6A could bridge transcription and translation.
First, we examined whether m6A levels could be affected by different rates of transcription. We therefore performed immunoprecipitations of m6A-modified RNAs (MeRIP) from cells with either induced or non-induced TRex-Rluc and compared the relative recoveries of the different Rluc transcripts. Strikingly, we observed very significant differences between the two Rluc populations; the non-induced mRNA was isolated much more efficiently in all tested mRNA regions (Figure 5A). Importantly, a control mRNA (AHNAK) showed similar recovery in both samples, suggesting a specific enrichment of the non-induced Rluc transcripts by MeRIP. We also assessed the methylation levels of Rluc mRNAs of the two populations expressing different copy numbers of Lenti-Rluc (Figure 3G) and found them to be very similar (Figure S6D), suggesting that the differences observed in MeRIP are unlikely to stem from unequal mRNA expression levels. Furthermore, we observed differences in the post-MeRIP recovery of Rluc mRNAs transcribed from either TATA-containing or TATA-less promoters (Figure S6E), suggesting that promoters with intact TATA elements yield mRNAs with relatively lower m6A content. Next, we in vitro transcribed Rluc in m6A-compatible nuclear HeLa extract (Shimba et al., 1995), using conditions that reduce rate of transcription (e.g., elevated MgCl2 [Wildeman et al., 1984] or a template with a mutated TATA box), and tested the different transcripts for m6A content. Indeed, both changes resulted in the relatively lower levels of Rluc mRNA, which were associated with higher deposition of m6A (Figure 5B). Importantly, both the UTRs and polyA tails of the different transcripts were similar (Figures S3E and S3F). Furthermore, treatment with CPT reduced mRNA levels in parallel to the relative enhancement of their m6A content (Figure 5C), suggesting a negative correlation between transcription elongation and m6A deposition. Additionally, we performed MeRIP analysis of genes that exhibited reduction of both mRNA levels and TE after CPT treatment (Figure 4E). These genes indeed displayed lower levels of mRNA coupled with enhanced m6A content (Figure 5D). To further address the link between the rate of transcription and m6A deposition in an additional independent system, we interrogated α-amanitin resistant RNAPII mutants exhibiting either normal (N792D) or slow (C4/R749H) rates of elongation (Fong et al., 2014). Similarly to the previous observations, we found that cells expressing the slow RNAPII mutant contained relatively lower levels of mRNA, associated with elevated methylation content (Figure 5E). Thus, various experimental approaches indicate that a suboptimal rate of transcription results in higher m6A deposition on mRNAs.
Figure 5.

Enhancement of m6A Modification of mRNAs upon Attenuated Transcription
(A) Cells bearing the TRex-Rluc gene were induced for 18 hr and subjected to the MeRIP procedure, together with untreated cells. Recovery efficiencies of regions spanning the Rluc mRNA were examined, normalized to the input levels and compared; AHNAK was used as a control methylated mRNA. Data are represented as mean ± SEM of n = 3.
(B) Rluc mRNAs were transcribed in vitro using HeLa extract as detailed, resolved on agarose gels and quantified (left chart, n = 3) and further subjected to the relative comparison of their m6A contents (right chart, n = 2).
(C) Total RNA isolated from MCF7 cells treated with CPT for 5 hr was enriched for polyA+ population and quantified (left chart, n = 3) and further subjected to the relative comparison of their m6A contents (right chart, n = 3).
(D) mRNAs presented in Figure 4E were tested for recovery after MeRIP procedure (striped columns) relatively to their RNA levels (plain columns) using qRT-PCR. Data are represented as mean ± SEM of n = 2. See also Figures S6D and S6E.
(E) Relative levels of polyA+ RNA populations (plain columns) in cells expressing different RNAPII mutants, and their relative m6A levels (striped columns) were examined; n = 2.
(F) MCF7 cells were treated with CPT for 5 hr and subjected to immuno-precipitation of RNAPII, followed by detection of METTL3 protein using western blot. A characteristic blot is presented, n = 4. In the input panel, GAPDH was probed on a separate blot.
This conclusion raised the possibility that m6A modification of mRNAs occurs co-transcriptionally, and therefore we anticipated a physical interaction between RNAPII and the methyltransferase complex (MTC). To test this, we immunoprecipitated RNAPII from either normally growing or CPT-treated cells and probed the eluate for METTL3, a catalytic core of MTC (Wang et al., 2016). Remarkably, whereas in untreated conditions we observed no visible interaction, upon CPT treatment, METTL3 was efficiently co-precipitated (Figure 5F), suggesting co-transcriptional m6A modification and enhancement of the interaction between RNAPII and MTC upon impediment of RNAPII elongation dynamics.
Next, we tested whether elevated levels of m6A could negatively affect translation. We first examined the translation capacity of the in vitro transcribed Rluc mRNAs in the conditions of reduced transcription and enhanced m6A (as presented in Figure 5B). In line with our hypothesis, conditions associated with attenuated transcription resulted in lower efficiency of protein production compared with the normal transcription conditions (Figure 6A). Next, we examined the global effects of CPT treatment and expression of the slow RNAPII mutant on polysomes. Figures 6B and 6C show that these two treatments considerably reduced the RNA content of the polysomal fractions, suggesting a general reduction of translation and providing further evidence for the positive feedback between transcription and translation.
Figure 6.

Effect of the m6A Modifications on Translation
(A) Rluc mRNAs were transcribed in vitro using HeLa extract in the indicated conditions, and transfected into MCF7 cells in fold-wise amounts; RLuc enzymatic activity was measured after 24 hr. The trend lines are represented by the dashed lines; n = 3.
(B) MCF7 cells were treated with CPT for 5 hr and subjected to polysomal profiling with subsequent measurement of the average levels of total RNA in the collected fractions. Data are presented as mean ± SEM, n = 3.
(C) Polysomal segregation of total RNA from cells expressing “normal speed” or “slow” RNAPII; data are presented as mean ± SEM of n = 3. See also Figure S6H.
(D) TRex-Rluc gene was induced for 18 hr in MCF7 cells transfected with the detailed siRNAs, or left uninduced. Expression levels of RLuc protein were assessed; data are represented as mean ± SEM of n = 3.
(E) Cells bearing the TRex-Rluc gene were transfected with the indicated siRNAs, grown for 56 hr and subjected to BPP. Upper panel: non-induced Rluc; lower panel: Rluc expression was induced for 24 hr prior to harvesting; data are represented as mean ± SEM of three measurements of a characteristic gradient; n = 3.
(F) Results of the MeRIP experiment presented in Figure 5A were normalized to calculate the total relative recovery of Rluc mRNAs by the different regions. Error bars represent mean ± SEM of n = 3.
(G) Rluc mRNAs were in vitro transcribed using T7 polymerase in the presence of the indicated proportions of m6A nucleoside and in vitro translated using indicated amounts of mRNA as input. Data are represented as mean ± SEM of n = 4.
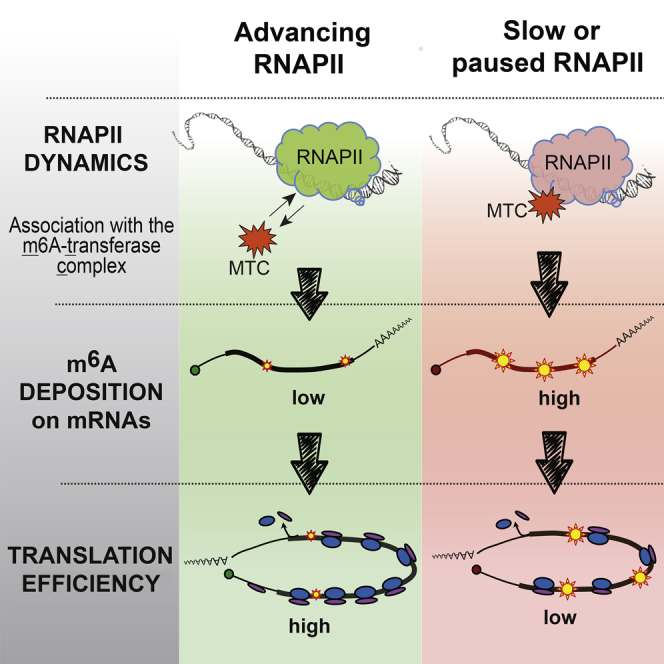
(H) Model of the transcription-dependent regulation of TE. Slow or paused RNAPII results in the enhanced interaction with MTC (step 1), which leads to enhanced deposition of m6A on mRNAs (step 2), negatively affecting translation efficiency (step 3).
To directly examine the role of m6A in translation, we knocked down two genes in the m6A pathway (METTL14 and YTHDH1, Figure S6F) and examined the effect on the expression of Rluc mRNA. Interestingly, knocking down either factor resulted in a significant boost in the expression of the non-induced Rluc, whereas the effect on the induced gene was moderate (Figure 6D). Importantly, while global polysome segregation analysis indicated only a slight effect of each knockdown on general translation (Figure S6G), possibly due to compensatory mechanisms, BPP assay verified a considerable increase in the TE of the non-induced Rluc mRNA in these conditions (Figure 6E). Given that methylation of coding regions (CDRs) has been recently suggested to attenuate translation (Choi et al., 2016), we re-examined the experiment presented in Figure 5A, in order to compare the relative changes in methylation of the different Rluc transcript regions. Remarkably, the highest gain of methylation upon transcriptional attenuation was observed throughout the CDR of Rluc (up to ∼11% near the end of the non-induced mRNA CDS), whereas both UTRs (i.e., 5′ and 3′) displayed the lowest levels (<1%, Figure 6F). This result therefore suggests a plausible explanation for the strong inhibitory effect of transcriptional repression on TE that we observed (Figure 3D). Lastly, we sought to directly examine the effect of m6A levels on protein production and produced Rluc mRNAs in vitro with different proportions of methylated adenosines. Strikingly, increased incorporation of m6A led to a progressive attenuation of the TE of Rluc (Figure 6H), suggesting an overall inhibitory effect of m6A modification on translation. Altogether, these results demonstrate that covalent m6A modification of mRNAs depends on the dynamics of the transcribing RNAPII, and tends to negatively affect TE. Therefore, we conclude that m6A links transcription and translation.
Discussion
In this study, we describe a direct and general flow of information from transcription to translation in human cells. We show that genes possessing strong transcriptional activity give rise to mRNAs with greater capacity to produce proteins. We propose that this link is mediated, at least in part, by the co-transcriptional N6-methylation of adenosines in mRNA—the m6A modification. Suboptimal transcription results in a relatively higher m6A content of transcripts and this, in turn, tends to reduce TE (Figure 6H).
Initially, we employed Pro-Lib, a library consisting of multiple human promoters that drive the transcription of a single reporter gene Rluc, and combined it with barcoded polysomal profiling procedure to examine TE of the reporter mRNAs. This approach allowed for several advantages. First, recombination-mediated single-copy genomic integration resulted in a balanced expression of the reporter constructs. Second, all vectors were integrated into the same genetic locus, minimizing the probability of an artificial effect of chromatin neighborhood on transcription. Third, establishing a pooled stable reporter population simplified the maintenance and treatments and increased experimental accuracy. Lastly, deep sequencing of the barcodes enabled a quantitative coverage of nearly all reporter transcripts in the polysomal fractions within a single experiment.
Using Pro-Lib, we observed that promoters possessing either confirmed or suspected TATA-box element (TATA) yield transcripts characterized by a higher capacity to accommodate ribosomes and produce proteins. This indicated that the presence of this element in a promoter not only boosts transcription, but also enhances mRNA translation, suggesting a positive effect of TATA-box promoter element on TE. Indeed, a recent genome-wide bioinformatics analysis has suggested that mammalian genes possessing TATA in their promoter tend to be translated more efficiently (Tamarkin-Ben-Harush et al., 2014), which was attributed to their characteristics (e.g., gene length, probability of upstream ORF, etc.). In this study, we support these observations and conclude, in addition, that a mere presence of intact TATA element in a promoter potentiates mRNA translation (Figure 2B).
The TATA element activates transcription (Xiao et al., 1995), resulting in elevated levels of mRNA. Indeed, we have identified a strong correlation between mRNA levels and translation in the Pro-Lib screen; additionally, we have also shown that this link is not unique to TATA, but also holds true for the global mammalian gene expression. We note that the genome-wide correlations between mRNA levels and TE are rather weak, probably reflecting multiple additional levels of regulation, but statistically significant. Interestingly, similar correlations were noticed in both animal cells and yeast (Schwanhäusser et al., 2011, Weinberg et al., 2016), likely indicating an evolutionarily conserved phenomenon. Importantly, while mRNA levels could be linked to TE via mechanisms that couple mRNA stability and translation (Radhakrishnan et al., 2016), we found that mRNA levels per se do not directly affect translation (Figures 3F–3H). Instead, we observed a global positive effect of transcription on TE. Impediment of transcription, either via CPT treatment or in the in vitro assays, demonstrated a role for transcriptional elongation in the determination of TE. Given that transcription initiation and elongation are related processes (Marbach-Bar et al., 2013), future studies are necessary in order to precisely appreciate the role of transcription initiation in the regulation of translation. Regarding mRNA translation, our observations suggest that it is responsive to transcriptional changes only when the initiation of translation involves 5′ cap. When this canonical initiation was bypassed by the introduction of an IRES, the translation machinery failed to respond to transcriptional fluctuations (Figures 4F–4H), indicating that this initial step, which is considered to be rate limiting and highly controlled, might exert additional aspects of translational regulation.
Here, we suggest that m6A, at least partially, mediates the communication between transcription and translation. Our results indicate that attenuated transcription leads to enhanced methylation of mRNAs. We present here biochemical evidence for a physical interaction between the METTL3 enzymatic subunit of the MTC and retarded RNAPII (Figure 5F). These results suggest that m6A modification is, at least in part, co-transcriptional and indicate that the affinity between MTC and RNAPII depends upon the dynamics of the latter. Although it remains unclear how exactly this interaction is induced, we propose that slow progression or frequent pausing of RNAPII increases the probability of MTC engagement. The global enrichment of modified nucleotides at the start of mRNAs and around STOP codons (Meyer et al., 2012, Dominissini et al., 2012) supports this hypothesis, as these are the locations of probable RNAPII pausing (Jonkers and Lis, 2015). Since a considerable proportion of genes experience slow or paused transcription (Core et al., 2008), it would be important to examine, in future studies, whether these genes are preferentially methylated. We note that, together with m6A, additional factors might bridge transcription and translation, such as precise sequence of 5’ UTR (Rojas-Duran and Gilbert, 2012, Tamarkin-Ben-Harush et al., 2017) and recruitment of mRNA coordinators (Harel-Sharvit et al., 2010).
Globally, we observed that enhanced mRNA methylation results in lower translation capacity. Interestingly, several recent studies have indeed linked m6A to translation, but report mainly a stimulatory role of this modification (Meyer et al., 2015, Wang et al., 2015, Zhou et al., 2015). We anticipate that the precise effect of m6A on translation depends on the location of the modified nucleotide within the transcript. Remarkably, most of the mentioned studies observed enhanced methylation within UTRs of transcripts. In contrast, methylation within CDRs reduced translation (Qi et al., 2016), probably due to the attenuated elongation (Hoernes et al., 2016, Choi et al., 2016). Given that most of the methylated residues are located within CDRs, we would anticipate an overall negative effect of m6A methylation on translation, which we indeed observed upon random incorporation of modified adenosines (Figure 6G).
Our study suggests that m6A modification of mRNAs is co-transcriptional and is used to epigenetically imprint mRNAs and control their future activity and fate. This way the dynamics of the transcription process could be recorded in the methylation profile of a given mRNA, which, in turn, could further impact multiple steps of mRNA biology.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-c-Myc (1:1000) | Cell Signaling | CAT#D84C12; RRID: AB_1903938 |
| Anti-GAPDH (1:2500) | Santa Cruz | CAT#sc-69778; RRID: AB_1124759 |
| Anti-METTL3 (1:1000) | Protein Tech | CAT#15073-1-AP; RRID: AB_2142033 |
| Anti-RNAPII (CTD4H8; 1:1000) | Millipore | CAT#05-623; RRID: AB_309852 |
| Polyclonal Goat Anti-Mouse Immunoglobulins/HRP | Dako | CAT#P0447; RRID: AB_2617137 |
| Polyclonal Goat Anti-Rabbit Immunoglobulins/HRP | Dako | CAT#P0448; RRID: AB_2617138 |
| Bacterial and Virus Strains | ||
| DH5α | Bioline | CAT#BIO-85026 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| RNA fragmentation reagent | Ambion | CAT#AM8740 |
| EpiMark N6-Methyladenosine Enrichment Kit | NEB | CAT#E1610S |
| Critical Commercial Assays | ||
| EpiQuik m6A RNA Methylation Quantification Kit | Epigenetek | CAT#P-9005-96 |
| 3′ RACE system for rapid amplification of cDNA ends | Invitrogen | CAT#18373019 |
| 5′ RACE System for Rapid Amplification of cDNA Ends, v2.0 | Invitrogen | CAT#18374058 |
| USB Poly(A) tail-length assay kit | Affymetrix | CAT#76455 1KT |
| Deposited Data | ||
| RNA-seq and Ribo-seq data | BJ cells | GEO: GSE45833 |
| PC9, H1933 cells | GEO: GSE96716 | |
| MCF7 (Nutlin) | GEO: GSE96714 | |
| MCF7 (CPT) | GEO: GSE96643 | |
| GRO-seq | BJ cells | GEO: GSE96717 |
| MCF7 cells | GEO: GSE96718 | |
| Experimental Models: Cell Lines | ||
| MCF7 | ATCC | ATCC HTB-22 |
| MCF7/FRT | This study | N/A |
| MCF7/FRT/TR | This study | N/A |
| HEK293 (N792D) | David Bentley | Fong et al., 2014 |
| HEK293 (C4/R749H) | David Bentley | Fong et al., 2014 |
| Oligonucleotides | ||
| DNA and RNA oligonucleotides | Table S4 | N/A |
| Recombinant DNA | ||
| List of cloned promoters and associated barcodes | Table S1 | N/A |
| List of used plasmids and detailed restriction sites | Table S3 | N/A |
| Software and Algorithms | ||
| Bowtie | Langmead et al., 2009 | http://bowtie-bio.sourceforge.net/index.shtml |
| HTseq | Anders et al., 2015 | http://www-huber.embl.de/HTSeq/doc/overview.html |
| EXPANDER package | Ulitsky et al., 2010 | http://acgt.cs.tau.ac.il/expander/ |
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Reuven Agami (r.agami@nki.nl).
Experimental Model and Subject Details
Human breast adenocarcinoma MCF7 cells (female) were purchased from ATCC and authenticated by detecting the deficiency of caspase-3 using western blotting. These cells were grown in DMEM media (GIBCO) supplemented with 10% bovine serum and penicillin/streptomycin (GIBCO) in 5%CO2-buffered incubators at 37°C. Cells were split twice per week and kept in culture for up to 8 weeks. To create cells compatible with FLP recombination system (ThermoFisher scientific), MCF7 cells were transfected with the pFRT/lacZeo and grown in the presence of Zeocin (400μg/ml) until stable colonies appeared. These cells were named MCF7/FRT and used for integration of Pro-Lib. To integrate Pro-Lib, these cells were transfected with Pro-Lib vectors along with pOG44 vector (ThermoFisher scientific) in quantities of 1.6μg+3μg respectively, per 35-mm well. After 48h, the cells were collected, re-plated onto 10-cm dishes, and selected for three weeks in the presence of Hygromycin (0.1 mg/ml f.c.). After appearance of the stable colonies, the cells were collected, frozen, and used for further experiments. To create cells compatible with T-REx inducible system (Invitrogen), MCF7/FRT cells were transfected with pcDNA6/TR vector encoding for the tetracycline-sensitive repressor and separate clones were selected for their resistance to Blasticidin S (5mg/ml). The induction efficiency was examined in separate clones by transient transfection with pcDNA4/TO-Rluc construct, incubation with Doxycycline (Sigma, 1 μg/ml final conc.) for 17 hr and measurement of Renilla protein activity. One suitable clone was chosen for further experiments and the cells were named MCF7/FRT/TR. These cells were stably transfected with pcDNA5/FRT/TO plasmid encoding for barcoded Renilla ORF (TRex-Rluc) in the presence of pOG44 vector (ThermoFisher scientific) and grown for three weeks in the presence of Hygromycin (0.1 mg/ml f.c.). Individual colonies were isolated and tested by treatment with Doxycycline (Sigma, 1 μg/ml final conc.). 293 cells bearing integrated alpha-amanitin resistant mutants of Rpb1(“wild-type” N792D, and “slow” C4/R749H), were a kind gift of D. Bentley. They were maintained in DMEM supplemented with 10% FBS, 1% penicillin/streptomycin, 200 μg/mL hygromycin, and 6.5 μg/mL blasticidin. To stimulate the expression of Rpb1 mutants, cells were induced with 2.0 μg/mL doxycycline for 16 hr and treated with 2.5 μg/mL α-amanitin for additional 42 hr.
Method Details
Manipulations in Cells
To induce autophagy, MCF7 cells were incubated in EBSS (GIBCO) medium for 3 hr. To inhibit mTORC1, Torin1 was added to the medium for 2 hr (250 nM; Tocris Bioscience, Bristol, UK). Cells were irradiated at 10Gy dose and then incubated for additional 18 hr at normal conditions. To arrest cells in mitosis, Taxol (Sigma-Aldrich, 1 μM f.c. for 17 hr) was added to the growth medium. FuGENE6 (Roche) was used for transfection of DNA plasmids, DharmaFECT1 (Dharmacone) for transfection of siRNAs and in-vitro transcribed mRNAs, according to the manufacturers’ instructions. To impede the dynamics of transcribing RNA polII, cells were treated with Camptothecin (CPT, Sigma) to the final conc. of 6 μM for the indicated times.
DNA Cloning
Psi-Check2 vector with cloned FRT sequence and hygromycin-resistance gene served as the basic vector for the construction of Pro-Lib. To clone human promoters upstream of the Renilla ORF in Pro-Lib, putative promoter regions were cloned between BglII and NheI sites of the basic Pro-Lib vector (Figure S1A) after addition of random barcodes and removal of the SV40 promoter. All barcodes were sequenced as well as the proximal parts of the cloned promoter regions to confirm their identity. All other cloning procedures were performed using standard procedures; DNA was extracted from gel using Agarose Gel DNA Extraction Kit (Roche); all constructs were sequenced. The list of the plasmids used in the study together with their details is available in Table S3.
Luciferase Assay
Luciferase assay was measured using Dual-Luciferase Reporter assay kit (Promega). Cultured cells were lysed with passive lysis buffer for 15 min at room temperature. Renilla (Rluc) and Firefly (Fluc) enzymatic activities were assayed with the substrates supplied with the kit using Centro XS3 LB960 machine (Berthold technologies). Typically, expression of Fluc was used to normalize the Rluc expression. Myc activity was measured using Myc-reporter constructs (pGL3-M4 and pGL3-M4-mut, (Nagel et al., 2008). MCF7 cells (1x105 cells) were plated in 12-well plates and transfected with 200ng of the reporter constructs using Fugene-6 (Promega). Luciferase activity was measured 36 hr after transfection.
Barcoded Polysomal Profiling
Sucrose gradients for separation of polysomes were usually prepared by gentle sequential addition of 2.2ml of the different sucrose solutions (e.i., 47, 37, 27, 17 and 7% in Tris-HCl pH = 7.5 (f.c. 20mM), MgCl2 (f.c. 10mM) and KCl (f.c. 100mM), supplemented with 2mM DTT, Ribosafe RNase inhibitor (Bioline, 1 μl/ml) and CHX (100 μg/ml) into a 12 mL tube (Beckman, 9/16 × 3 1/2 in.) and left overnight at 4°C to achieve continuous gradient prior to the centrifugation. Cells growing in subconfluent conditions (typically ≈80%–85% confluency) were washed once with ice-cold PBS, collected into 15 mL tubes and incubated with PBS/cycloheximide (CHX, 100 μg/ml) for 5 min on ice. After sedimentation (400xg for 3min), the cells were lysed in lysis buffer (20 mM Tris-HCl (pH 7.5), 10 mM MgCl2, 100 mM KCl, 1% NP40) supplemented with EDTA-free cOmplete Protease Inhibitors (Roche), 2mM DTT, Ribosafe RNase inhibitor (Bioline, 1 μl/ml) and CHX (100 μg/ml) for 10min on ice. To resolve cell aggregations, the lysates were passed three times through a 25G needle. The lysates were centrifuged 1300xg for 10 min at 4°C and the supernatants were transferred into new tubes. From the cleared lysates, 500 μL were loaded on top of each gradient, mounted on SW41TI rotor and centrifuged at 36000rpm for 2 hr at 4°C. Following the centrifugation, each gradient was split into 15 equal fractions of 760 μl, which were subjected to RNA isolation using TRIsure (Bioline) according to the manufacturer’s instructions. RNA was precipitated from 750 μL of the collected material, using 2 μL of GlycoBlue (Ambion) and 750 μL of isopropanol, washed once with 70% of ethanol and reconstituted in 25 μL of water. Typically, 7 μL of total RNA were taken as a template for reverse transcription reactions using Tetro cDNA synthesis kit (Bioline) and oligo dT primers. Barcodes of the Pro-Lib transcripts detected by qRT-PCR using specific primers. To construct libraries ready for deep sequencing, barcodes were PCR amplificed using indexed primers bearing P5, P7 and Illuseq sequences, while each collected fraction was associated with a unique index. The PCR products were cleaned using QIAquick PCR purification kit (QIAGEN), size selected on E-Gel SizeSelect 2% gels (Invitrogen) and sequenced using Illumina Hi-seq 2000 or 2500.
RNA Manipulations
RNA was isolated from cultured cells using TRI-sure reagent (Bioline) according to the manual, using Glycoblue (Ambion) as a carrier. RNA was typically reconstituted in 30 μL of sterile nuclease-free water (GIBCO) and stored at −20°C. To analyze RNA on agarose gels, a portion of isolated RNA was mixed with equal volume of 2xRNA-loading dye (Thermo Fisher Scientific), supplemented with 20ng/ml Ethidium bromide, heated at 70°C for 5min and loaded on 1.5% agarose gel. After separation at 100mV for 30min, gels were visualized and documented. Reverse transcription was done using Tetro cDNA synthesis kit (Bioline) according to the manual. Typically, 7 μL of total RNA was taken for a single RT reaction. Real-time PCR experiments were performed using SensiFAST CYBR (Bioline) reagent and LightCycler 480II (Roche) using standard conditions.
In Vitro Synthesis of mRNA
To synthesize Renilla mRNA using nuclear extract, we used HeLaScribe Nuclear Extract in vitro Transcription System (Promega) according to the manufacturer’s protocol. Briefly, DNA templates including promoter, ORF, UTRs and polyA site were PCR-amplified from the appropriate pGEM-T vectors, isolated on agarose gels and cleaned. For a single reaction, 200ng of a clean template were transcribed with 7 μL of nuclear extract, in a buffer containing 1.5mM MgCl2 in a siliconized RNase-free 1.5 mL eppendorf tube (Ambion) at 30°C for 1.5 hr. The reaction was stopped by addition of TRIsure compound (Bioline) and RNA was isolated, analyzed on agarose gels, quantified using NanoDrop, compared using ImageJ and stored at −20°C. Transfection was done in 96-wells plates using Dharmafect1 agent (Thermo Scientific), according to the manufacturer’s instructions. Approximately 10ng of recovered mRNA were transfected as x1-amount. Rluc activity was measured ≈24 hr following transfection. For m6A measurements, reconstituted synthetic RNA was furthermore cleaned using RNeasy kit (QIAGEN). To synthesize Renilla mRNA using T7 RNA polymerase, we used HiScribe T7 High Yield RNA Synthesis kit (NEB) according to the manufacturer’s instruction. Briefly, PCR-produced DNA fragments encoding for Renilla ORF were transcribed in presence of Ribo m7G Cap Analog (Promega, 8mM f.c.) at 37°C for 2 hr followed by DNase treatment for 15 min and isolation of RNA. To produce transcripts with methylated content, N6-Methyladenosine-5′-Triphosphate (Trilink, 100mM stock conc.) was added together with ATP (supplied in the kit) at the indicated proportions. After recovery, the RNAs were polyadenylated using E.Coli Poly(A) polymerase (NEB) and purified. After recovery and analysis on gels, RNAs were transfected into living cells as described above or subjected to in-vitro translation.
In vitro translation of mRNAs
To translate mRNAs encoding for Rluc protein, rabbit reticulocytes system, nuclease treated (Promega) was employed, according to the manufacturer’s protocol. All reactions were done in volume of 25 μL at 30°C for 10 min. To quantify the produced Rluc protein, aliquots of 5 μL were taken for luciferase measurements using Dual-Luciferase Reporter assay kit (Promega).
Northern Blotting
Total RNA was extracted from growing MCF7 cells as described and enriched for mRNA using Oligotex kit (QIAGEN). Equal volumes of reconstituted RNA were separated on 1.2% agarose gel supplemented with 0.66% (vol/vol) formaldehyde and transferred onto Amersham Hybond-N+ membrane (GE Healthcare). RNA probes targeting Renilla or Firefly mRNAs were produced by in vitro transcription (T7 high yield kit, NEB) of PCR fragments encoding for sequences complementary to these transcripts (see Table S4 for used oligos) using 32P(α)-UTP. Following cleaning of the probes using MicroSpin G-50 columns (GE Healthcare), hybridization was performed in ULTRAhyb Hybridization buffer (Ambion) rotating at 68°C for 4 hr. The membrane was washed twice with 2xSSC buffer supplemented with 0.1%SDS and twice with 0.2xSSC buffer supplemented with 0.1%SDS. The signal was detected by FujiFilm FLA-3000 PhosphorImager and analyzed using ImageJ.
Measurement of m6A Content
To estimate the relative m6A content of RNA, EpiQuik m6A RNA Methylation Quantification Kit (Epigentek) was used. Briefly, equal volumes of RNA solution (4-8 μl) were processed according to the manufacturer’s instructions, aside with the negative, positive, and standards controls.
5′ RACE Analysis
Generally, 5′RACE experiments followed the procedure established for 5′ RACE System for Rapid Amplification of cDNA Ends, v2.0 (Invitrogen). Briefly, total RNA was extracted with TRIsure (Bioline) and subsequently poly-A selected with Oligotex Direct mRNA mini kit (QIAGEN). For each 5′ RACE assay, 500ng of poly-A selected mRNA were used for cDNA synthesis with the following primer: CCCTTCTCCTTGAATG. Synthesized cDNA were purified with QIAquick PCR purification kit (QIAGEN). Then the cDNA were dC-tailed with TdT for 10min at 37°C and a first round PCR was performed with a gene specific primer (GSP2), AGTTTCCGCATGATCTTGCTTG, and Abridged Anchor Primer (AAP). A nested PCR was performed with a nested gene specific primer (GSP1), GTTGATGAAGGAGTCCAGCACGTT, and Abridged Universal Amplification primer (AUAP). For determination of the sequences of 5′ RACE PCR products, the nested PCR products were ligated into pGEM-T vector (Promega) and transfected into competent bacteria. Unless otherwise indicated, at least 20 different bacterial colonies from each ligation were subsequently analyzed using Sanger sequencing.
3′ RACE Analysis
The protocol was adapted from 3′RACE system for rapid amplification of cDNA ends (Invitrogen). Briefly, total RNA was poly-A selected and reverse transcribed with oligo(dT)-containing adaptor primer (AP) GGCCACGCGTCGACTAGTACTTTTTTTTTTTTTTTTT. Synthesized cDNA was treated with Rnase H to remove residual RNA templates and PCR-amplified with Rluc specific primer CTGAGAGTGTCGTGGACGTGA and Abridged Anchor Primer (AAP). A nested PCR was performed with a nested Rluc primer GCCTAAGATGTTCATCGAGTCC and AAP.
Sequencing Library Preparation of 5′ and 3′ RACE
PCR products from 5′ and 3′ RACE preparations were PCR-amplified with Illumina sequencing compatible primers (p5 primer, 5′RACE-p7 primer, and 3′RACE-p7 primer). PCR products were then purified with CleanPCR magnetic beads (CleanNA), and pooled with equal molar, ran on Illumina Miseq platform.
Poly(A) Tail Analysis
Determination of poly(A) tail-length was performed with USB® Poly(A) tail-length assay kit (Affymetrix) following the manufactor’s protocol. In short, in-vitro transcribed RNA was G/I tailed, and reverse-transcribed with specific adaptor primer (sequence not provided). The poly G/I tailed cDNA was then amplified with a Rluc specific primer (GCGTGCTGAAGAACGAGCAGTAA) and a provided universal PCR reverse primer. Finally, PCR products were resolved on a 2.5% agarose gel with ethidium bromide.
Lentiviral Infection
To generate viruses with the Renilla-expressing plasmid, HEK293T cells were plated 24h prior transfection. For virus production, 3.5 μg of pVSV-G, 5 μg of pMDL-RRE and 2.5 μg of pRSV-REV was transfected with 10 μg of the expression plasmid pLenti-puro-Rluc. Lentivirus-containing supernatant was harvested 48h after the transfection, filtered through a 0.45 μm membrane (Milipore Steriflip HV/PVDF) and kept at −80°C. Subsequently, to infect MCF7 cells, 1x106 cells were plated in 10-cm dish one day prior infection. For low MOI infection, cells were infected with 20 μL of the virus-containing supernatant; for high MOI infection, cells were infected with 900 μL (45-fold) of the supernatant. Cells were refreshed with DMEM 18h after infection and subsequently recovered for 24h. Afterward, low MOI-infected MCF7 cells were selected with 1 mg/mL puromycin and high MOI-infected MCF7 cells were selected with higher concentrations of puromycin (up to 25ug/mL) for 1 week.
CRISPR/Cas9-Mediated Mutagenesis
For the mutagenesis of c-Myc TATA box sequence, lentivirus containing sgRNAs targeting the TATA box sequence were generated and subsequently used for infecting MCF7 cells as described above. After puromycin (2 μg/ml) selection, cells were plated in a 96-well plate following serial dilutions in order to achieve single cell clones of the Cas9-generated mutants. Several single cell clones were collected, expanded, and sequenced. To sequence the indel mutations within the c-Myc locus, genomic DNA was extracted from the grown colonies using DNeasy Blood and Tissue Kit (QIAGEN), PCR amplified using c-Myc promoter-specific primers, sub-cloned into pGEM-T vector (Promega) and sequenced.
Western Blotting
MCF7 cells were washed with ice-cold PBS and scraped off the plate. Cells were centrifuged at 1000xg for 10min at 4°C. Subsequently, cell pellets were lysed, by adding an appropriate amount of ice-cold lysis buffer (20mM Tris-HCl at pH 7.5, 150mM NaCl, 1.8mM MgCl2 and 0.5% NP40) supplemented with cOmplete protease inhibitor cocktail (Roche) and 1mM DTT. Cell lysates were incubated on ice for 20min with occasional vortexing followed by centrifugation at 15000xg for 15min at 4°C. Protein concentrations were determined using Pierce BCA protein assay kit (Thermo Scientific). Proteins were separated on SDS-PAGE gels and transferred onto Nitrocellulose membrane (Bio-rad, 0.2 μM), which was blocked in 5% Blotting-grade blocker and probed with the indicated antibodies. All antibodies were diluted in PBS-0.25% Tween solution. For detection, blots were reacted with the SuperSigna West Dura Extended Duration Substrate (ThermoFisher). Images were captured with Chemidoc XRS+ (Biorad) and analyzed with Image Lab software; quantifications were done using ImageJ.
m6A RNA Immunoprecipitation (MeRIP)
Immunoprecipitation of m6A was adapted from the protocol of EpiMark® N6-Methyladenosine Enrichment Kit (New England Biolabs). Total RNA was isolated as mentioned above and enriched for mRNA using Oligotex mRNA kit (QIAGEN) following manufacturer’s instruction. The isolated mRNA was fragmented with RNA fragmentation reagent (Ambion) for 5min, 94°C and purified through ethanol precipitation. To bind antibody to the beads, protein G beads (Invitrogen) were pre-incubated with 1 μL of anti-m6Aantibody (Neb, Cat. E1610S) in IPP buffer (150 mM NaCl, 10 mM Tris-HCl, pH 7.5, 0.1% NP-40) at 4°C. Subsequently, the beads were washed twice in IPP buffer and incubated with RNA for 3h with head-to-tail rotation at 4°C (10% of the material were kept as input control). Afterward, beads were washed twice in IPP buffer, twice in low salt buffer (50 mM NaCl, 10 mM Tris-HCl, pH 7.5, 0.1% NP-40), and twice in high salt buffer (500 mM NaCl, 10 mM Tris-HCl, pH 7.5, 0.1% NP-40). RNA was eluted with 30 μL buffer RLT (QIAGEN, Cat. 79216) for 5min and subsquently purified through ethanol precipitation. For quantification of precipitation, both input samples and IP eluates were examined by qRT-PCR as described above.
RNAPII Immunoprecipitation
MCF7 cells growing on 15-cm dishes were treated with CPT (Sigma, 6 μM f.c.) or DMSO (as a control) for 5 hr. After collection on ice, the cells were incubated in 8ml of PBS supplemented with 1% formaldehyde for 10 min at room temperature in constant rotation. After quenching with glycine (0.125M f.c.) for 5 min, the cells were spun, washed once with PBS, flash-frozen in liquid nitrogen and kept at −80°C. After thawing in 1ml of lysis buffer (50 mM HEPES-NaOH-pH7.5, 150 mM NaCl, 5 mM MgCl2, 0.1% NP40) supplemented with EDTA-free cOmplete Protease Inhibitors and PhosSTOP (Roche), the lysate was divided into three equal 2 mL eppendorf tubes and sonicated at 4°C using Bioraptor (Diagenode) for 10 cycles (each cycle including 30sec of high-power sonication followed by 30sec pause). The lysates were centrifuged for 10 min. at 1800xg at 4°C to remove unbroken cells. Equal volumes of supernatants were added to 20 μl of Dynabeads-Protein G suspension (Life Technologies) pre-coupled with 5 μg of anti-RNA polymerase II antibodies (Millipore, clone CTD4H8) or normal mouse IgG (Santa Cruz) in PBS and 1% BSA for 2 hr at room temperature. The IP reaction was carried out for 2 hr at 4°C with constant rotating. After that, the beads were washed once with the regular lysis buffer and three times with stringent lysis buffer (as detailed above, but with 300 mM NaCl). The last washing was done with PBS, and the beads were transferred into new 1.5 mL tubes and the bound proteins were eluted and reverse-crosslinked by incubation with 50 μL of 1x Laemmli sample buffer at 100°C for 10min. After collection, the eluates were resolved on 8% SDS-PAGE and probed with the indicated antibodies.
Ribosomal Profiling
Ribosome profiling (RP) was performed as previously described (Loayza-Puch et al., 2016). Briefly, CHX (100 μg/ml) was added to the medium of growing cells and incubated for 5min at 37°C. Approximately 30x106 cells were collected in ice-cold PBS and lysed in lysis buffer (20 mM Tris-HCl pH 7.8, 10 mM MgCl2, 100 mM KCl, 1% Triton X-100, 2 mM DTT, 100 μg/ml CHX, cOmplete protease inhibitors (Roche). Lysates were passed through a 26G needle (BD bioscience) for further homogenization, and centrifuged at 4°C 1300xg for 10 min. Supernatants were treated with 2.5U/μl of RNase I (Ambion) for 45 min at room temperature in gentle constant rotation. Lysates were loaded onto a linear sucrose gradient (7%–47%, as detailed above), and fractionated by ultracentrifugation, using a SW-41Ti rotor at 36000 rpm (221632.5g) during 2 hr. The sucrose gradient was divided into 14 fractions of 830 μl. The monosome-enriched sucrose fractions (7 to 10) were collected and treated with proteinase K (PCR grade, Roche), in presence of 1% SDS. The so-released ribosome protected fragments (RPFs) were purified using TRIsure reagent (Bioline), following the manufacturer’s instructions. RPFs between 30 and 33 nucleotides in length were size-selected in 10% acrylamide gel and isolated. The 3′ ends of the RPFs were dephosphorylated by treatment with T4 polynucleotide kinase (New England Biolabs) for 6 hr at 37°C in MES-NaOH buffer (100mM MES-NaOH, pH 5.5, 10mM MgCl2, 10mM β-mercaptoethanol, 300mM NaCl). 3′ adaptor (RA3) was ligated using T4 RNA ligase 1 (New England Biolabs) for 3.5 hr at 37°C, in absence of ATP. Ligation products were size selected in 10% acrylamide gel, and the 5′ ends were phosphorylated by treatment with T4 polynucleotide kinase for 30 min at 37°C, in presence of 1mM ATP. 5′ adaptor (RA5) was ligated using T4 RNA ligase 1 (New England Biolabs) during 2.5h at 37°C, and the ligation products were selected in 10% acrylamide gel. Ribosomal RNA depletion was performed by biotin-streptavidin affinity purification using biotinylated ribosomal RNA probes and streptavidin dynabeads. Retro-transcription of the ligation products into cDNA was performed using Super Script III reverse-transcriptase (Invitrogen) following the manufacturer’s instructions, and the primer RTP. PCR amplification was performed using the forward primer RP1, and the reverse primer RPI that contained a hexanucleotide index used to multiplex different samples during next generation sequencing (NGS). PCR products were size selected by E-Gel electrophoresis (Invitrogen), and submitted to NGS using HiSeq 2500 System (Illumina). The sequence of 3′ and 5′ adapters, RTP, RP1 and different RPI primers is available in Table S5.
RNA-Seq
Total RNA Isolation
Total RNA was extracted using TRIzol reagent (15596-018, Ambion life technologies) according to the manufactures protocol. Briefly, 0.2x volumes of chloroform (Chloroform stab./Amylene, Biosolve) was added to the Trizol homogenate and the tube(s) (Falcon, 15mL) were shaken vigorously. The tube(s) were incubated for 2-3 min at room temperature and centrifuged (Hettich, rotanta 46 RS) for 1 hr at 4°C. Approximately 70% of the upper aqueous phase was transferred to a clean 15 mL tube and 0.5x volume of isopropanol (33539, Sigma-Aldrich,) was added. The tube(s) were incubated overnight at −20°C and centrifuged for 30 min at 4°C. The supernatant was removed and the pellet was washed twice with 80% ethanol (32221-2.5L, Sigma-Aldrich). The total RNA pellet was air-dried for 8 min and dissolved in an appropriate volume of nuclease free water (Ambion) and quantified using Nanodrop UV-VIS Spectrophotometer. The total RNA was further purified using the MinElute Cleanup Kit (74204, QIAGEN) according to the manufactures instructions. Quality and quantity of the total RNA was assessed by the 2100 Bioanalyzer using a Nano chip (Agilent, Santa Clara, CA). Total RNA samples having RIN > 8 were subjected to library generation.
TruSeq Stranded mRNA Sample Preparation
Strand-specific libraries were generated using the TruSeq Stranded mRNA sample preparation kit (Illumina, RS-122-2101/2) according to the manufacturer’s instructions (Illumina, Part # 15031047 Rev. E). Briefly, polyadenylated RNA from 1000ng intact total RNA was purified using oligo-dT beads. Following purification the RNA was fragmented, random primed and reverse transcribed using SuperScript II Reverse Transcriptase (Invitrogen) with the addition of Actinomycin D. Second strand synthesis was performed using Polymerase I and RNaseH with replacement of dTTP for dUTP. The generated cDNA fragments were 3′ end adenylated and ligated to Illumina Paired-end sequencing adapters and subsequently amplified by 12 cycles of PCR. The libraries were analyzed on a 2100 Bioanalyzer using a 7500 chip (Agilent, Santa Clara, CA), diluted and pooled equimolar into a 10nM multiplex sequencing pool.
Sequencing
The libraries were sequenced with 65 base single reads on a HiSeq2500 using V4 chemistry (Illumina).
Quantification and Statistical Analysis
While mentioned, n represents the number of biological repeats of the same experiment. Data are represented as mean ± SEM. p values presented in Figures 3A,E; 4A,D; S5B,C; S6A,B were calculated using Wilcoxon’s test. p values mentioned in other figures were calculated using t test (one-tail distribution, homoscedastic variance). In all figures, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.005, ∗∗∗∗p < 0.001.
Polysomal Data Analysis and Preparation of Heatmaps
Polysomal fractions and promoter vectors were identified by their unique indexes and barcodes, respectively. Expression count matrix was derived by counting the frequency of each barcode in each fraction. To avoid fluctuations due to low coverage, only vectors covered by at least 10,000 counts (summation over all fractions) were included in the analyses. For heatmap display, vector counts (rows) were standardized to mean = 0 and SD = 1. Heatmaps were generated using the EXPANDER package (Ulitsky et al., 2010). Order of promoter vectors in the heatmap was determined by hierarchical clustering of the rows.
RNA-Seq and Ribo-Seq Analysis
Sequenced reads were aligned to a reference set of human curated protein-coding transcripts (plus the five human rRNA transcripts) using Bowtie (Langmead et al., 2009). This reference set of transcripts was based on Ensembl’s gene annotations (release 69). For genes with multiple isoforms, the one with longest coding DNA sequence (CDS) region and, in case not unique, the one with longest UTRs among the ones with the longest CDS, was selected to represent the gene. Only uniquely mapped reads were used in subsequent analyses. RNA expression levels and ribosome occupancy were estimated by calculating reads per kilobase of mRNA per million reads (RPKM) per transcript, taking into account either all reads that map to the transcript (for estimation of RNA levels using RNA-seq data) or only those mapping to its CDS (for estimation of ribosome occupancy). In estimation of ribosome occupancy in CDS, 5′ ends of reads were offset 12 nucleotides to the 3′ direction to match the P-site location of ribosome (Ingolia et al., 2009). Translation efficiency (TE) was estimated by the (log2) ratio between ribosome occupancy and mRNA level. Only genes with at least 20 reads in RNA-seq and Ribo-seq samples were included in analyses.
GRO-Seq Analysis
Sequenced reads were aligned to the human genome (hg19) using bowtie2, and number of reads mapping to annotated genes (Ensembl v69) were counted by HTseq (Anders et al., 2015). Quantile normalization was then applied to allow comparisons between different samples. Only genes covered by at least 20 reads were included in the analysis.
Data and Software Availability
The deep sequencing datasets generated in this study have been deposited in the GEO database under accession number GEO: GSE96643.
Author Contributions
B.S. and R.A. planned and designed the experiments, analyzed the results, and wrote the manuscript; B.S., R.H., and J.A.F.O.V. cloned Pro-Lib; B.S., R.H., F.L.-P., and V.C. performed the experiments; R.E. performed all bioinformatical tests. All authors read and approved the manuscript.
Acknowledgments
We thank Pieter van Breugel, Alejandro Piñeiro Ugalde, Behzad Moumbeini, and Nicolas Léveillé for the technical assistance and fruitful discussions and greatly appreciate the help of Ron Kerkhoven and the Netherlands Cancer Institute (NKI-AVL) genomics core facility in sequencing. We thank David Bentley for kindly providing us the cell lines expressing α-amanitin-resistant versions of RNAPII. R.E. is a Faculty Fellow of the Edmond J. Safra Center for Bioinformatics at Tel Aviv University. This work was supported by funds from the enhReg ERC-AdV (ERC-2012-AdG; grant agreement no. 322493) and NWO (NGI 93512001/2012) programs to R.A.
Published: April 6, 2017
Footnotes
Supplemental Information includes four tables and can be found with this article online at http://dx.doi.org/10.1016/j.cell.2017.03.031.
Contributor Information
Boris Slobodin, Email: boris.slobodin@gmail.com.
Ran Elkon, Email: ranel@tauex.tau.ac.il.
Reuven Agami, Email: r.agami@nki.nl.
Supplemental Information
References
- Anders S., Pyl P.T., Huber W. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–1699. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne B.J., Davis M.S., Yamaguchi J., Bergsma D.J., Subramanian K.N. Definition of the simian virus 40 early promoter region and demonstration of a host range bias in the enhancement effect of the simian virus 40 72-base-pair repeat. Proc. Natl. Acad. Sci. USA. 1983;80:721–725. doi: 10.1073/pnas.80.3.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J., Ieong K.W., Demirci H., Chen J., Petrov A., Prabhakar A., O’Leary S.E., Dominissini D., Rechavi G., Soltis S.M. N(6)-methyladenosine in mRNA disrupts tRNA selection and translation-elongation dynamics. Nat. Struct. Mol. Biol. 2016;23:110–115. doi: 10.1038/nsmb.3148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Core L.J., Waterfall J.J., Lis J.T. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science. 2008;322:1845–1848. doi: 10.1126/science.1162228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominissini D., Moshitch-Moshkovitz S., Schwartz S., Salmon-Divon M., Ungar L., Osenberg S., Cesarkas K., Jacob-Hirsch J., Amariglio N., Kupiec M. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485:201–206. doi: 10.1038/nature11112. [DOI] [PubMed] [Google Scholar]
- Dori-Bachash M., Shalem O., Manor Y.S., Pilpel Y., Tirosh I. Widespread promoter-mediated coordination of transcription and mRNA degradation. Genome Biol. 2012;13:R114. doi: 10.1186/gb-2012-13-12-r114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dujardin G., Lafaille C., de la Mata M., Marasco L.E., Muñoz M.J., Le Jossic-Corcos C., Corcos L., Kornblihtt A.R. How slow RNA polymerase II elongation favors alternative exon skipping. Mol. Cell. 2014;54:683–690. doi: 10.1016/j.molcel.2014.03.044. [DOI] [PubMed] [Google Scholar]
- Elfakess R., Dikstein R. A translation initiation element specific to mRNAs with very short 5’UTR that also regulates transcription. PLoS ONE. 2008;3:e3094. doi: 10.1371/journal.pone.0003094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong N., Kim H., Zhou Y., Ji X., Qiu J., Saldi T., Diener K., Jones K., Fu X.D., Bentley D.L. Pre-mRNA splicing is facilitated by an optimal RNA polymerase II elongation rate. Genes Dev. 2014;28:2663–2676. doi: 10.1101/gad.252106.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haimovich G., Choder M., Singer R.H., Trcek T. The fate of the messenger is pre-determined: a new model for regulation of gene expression. Biochim. Biophys. Acta. 2013;1829:643–653. doi: 10.1016/j.bbagrm.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harel-Sharvit L., Eldad N., Haimovich G., Barkai O., Duek L., Choder M. RNA polymerase II subunits link transcription and mRNA decay to translation. Cell. 2010;143:552–563. doi: 10.1016/j.cell.2010.10.033. [DOI] [PubMed] [Google Scholar]
- Hoernes T.P., Clementi N., Faserl K., Glasner H., Breuker K., Lindner H., Hüttenhofer A., Erlacher M.D. Nucleotide modifications within bacterial messenger RNAs regulate their translation and are able to rewire the genetic code. Nucleic Acids Res. 2016;44:852–862. doi: 10.1093/nar/gkv1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia N.T., Ghaemmaghami S., Newman J.R., Weissman J.S. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009;324:218–223. doi: 10.1126/science.1168978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonkers I., Lis J.T. Getting up to speed with transcription elongation by RNA polymerase II. Nat. Rev. Mol. Cell Biol. 2015;16:167–177. doi: 10.1038/nrm3953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korkmaz G., Lopes R., Ugalde A.P., Nevedomskaya E., Han R., Myacheva K., Zwart W., Elkon R., Agami R. Functional genetic screens for enhancer elements in the human genome using CRISPR-Cas9. Nat. Biotechnol. 2016;34:192–198. doi: 10.1038/nbt.3450. [DOI] [PubMed] [Google Scholar]
- Langmead B., Trapnell C., Pop M., Salzberg S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Léveillé N., Melo C.A., Rooijers K., Díaz-Lagares A., Melo S.A., Korkmaz G., Lopes R., Akbari Moqadam F., Maia A.R., Wijchers P.J. Genome-wide profiling of p53-regulated enhancer RNAs uncovers a subset of enhancers controlled by a lncRNA. Nat. Commun. 2015;6:6520. doi: 10.1038/ncomms7520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loayza-Puch F., Drost J., Rooijers K., Lopes R., Elkon R., Agami R. p53 induces transcriptional and translational programs to suppress cell proliferation and growth. Genome Biol. 2013;14:R32. doi: 10.1186/gb-2013-14-4-r32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loayza-Puch F., Rooijers K., Buil L.C., Zijlstra J., Oude Vrielink J.F., Lopes R., Ugalde A.P., van Breugel P., Hofland I., Wesseling J. Tumour-specific proline vulnerability uncovered by differential ribosome codon reading. Nature. 2016;530:490–494. doi: 10.1038/nature16982. [DOI] [PubMed] [Google Scholar]
- Marbach-Bar N., Ben-Noon A., Ashkenazi S., Tamarkin-Ben Harush A., Avnit-Sagi T., Walker M.D., Dikstein R. Disparity between microRNA levels and promoter strength is associated with initiation rate and Pol II pausing. Nat. Commun. 2013;4:2118. doi: 10.1038/ncomms3118. [DOI] [PubMed] [Google Scholar]
- Meyer K.D., Saletore Y., Zumbo P., Elemento O., Mason C.E., Jaffrey S.R. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell. 2012;149:1635–1646. doi: 10.1016/j.cell.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer K.D., Patil D.P., Zhou J., Zinoviev A., Skabkin M.A., Elemento O., Pestova T.V., Qian S.B., Jaffrey S.R. 5′ UTR m(6)A Promotes Cap-Independent Translation. Cell. 2015;163:999–1010. doi: 10.1016/j.cell.2015.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagel R., le Sage C., Diosdado B., van der Waal M., Oude Vrielink J.A., Bolijn A., Meijer G.A., Agami R. Regulation of the adenomatous polyposis coli gene by the miR-135 family in colorectal cancer. Cancer Res. 2008;68:5795–5802. doi: 10.1158/0008-5472.CAN-08-0951. [DOI] [PubMed] [Google Scholar]
- Nott A., Le Hir H., Moore M.J. Splicing enhances translation in mammalian cells: an additional function of the exon junction complex. Genes Dev. 2004;18:210–222. doi: 10.1101/gad.1163204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oktaba K., Zhang W., Lotz T.S., Jun D.J., Lemke S.B., Ng S.P., Esposito E., Levine M., Hilgers V. ELAV links paused Pol II to alternative polyadenylation in the Drosophila nervous system. Mol. Cell. 2015;57:341–348. doi: 10.1016/j.molcel.2014.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi S.T., Ma J.Y., Wang Z.B., Guo L., Hou Y., Sun Q.Y. N6-Methyladenosine Sequencing Highlights the Involvement of mRNA Methylation in Oocyte Meiotic Maturation and Embryo Development by Regulating Translation in Xenopus laevis. J. Biol. Chem. 2016;291:23020–23026. doi: 10.1074/jbc.M116.748889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radhakrishnan A., Chen Y.H., Martin S., Alhusaini N., Green R., Coller J. The DEAD-box protein Dhh1p couples mRNA decay and translation by monitoring codon optimality. Cell. 2016;167:122–132. doi: 10.1016/j.cell.2016.08.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter J.D., Coller J. Pausing on Polyribosomes: Make Way for Elongation in Translational Control. Cell. 2015;163:292–300. doi: 10.1016/j.cell.2015.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas-Duran M.F., Gilbert W.V. Alternative transcription start site selection leads to large differences in translation activity in yeast. RNA. 2012;18:2299–2305. doi: 10.1261/rna.035865.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwanhäusser B., Busse D., Li N., Dittmar G., Schuchhardt J., Wolf J., Chen W., Selbach M. Global quantification of mammalian gene expression control. Nature. 2011;473:337–342. doi: 10.1038/nature10098. [DOI] [PubMed] [Google Scholar]
- Shimba S., Bokar J.A., Rottman F., Reddy R. Accurate and efficient N-6-adenosine methylation in spliceosomal U6 small nuclear RNA by HeLa cell extract in vitro. Nucleic Acids Res. 1995;23:2421–2426. doi: 10.1093/nar/23.13.2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonenberg N., Hinnebusch A.G. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell. 2009;136:731–745. doi: 10.1016/j.cell.2009.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamarkin-Ben-Harush A., Schechtman E., Dikstein R. Co-occurrence of transcription and translation gene regulatory features underlies coordinated mRNA and protein synthesis. BMC Genomics. 2014;15:688. doi: 10.1186/1471-2164-15-688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamarkin-Ben-Harush A., Vasseur J.J., Debart F., Ulitsky I., Dikstein R. Cap-proximal nucleotides via differential eIF4E binding and alternative promoter usage mediate translational response to energy stress. eLife. 2017;6 doi: 10.7554/eLife.21907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoreen C.C., Chantranupong L., Keys H.R., Wang T., Gray N.S., Sabatini D.M. A unifying model for mTORC1-mediated regulation of mRNA translation. Nature. 2012;485:109–113. doi: 10.1038/nature11083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulitsky I., Maron-Katz A., Shavit S., Sagir D., Linhart C., Elkon R., Tanay A., Sharan R., Shiloh Y., Shamir R. Expander: from expression microarrays to networks and functions. Nat. Protoc. 2010;5:303–322. doi: 10.1038/nprot.2009.230. [DOI] [PubMed] [Google Scholar]
- Wang X., Lu Z., Gomez A., Hon G.C., Yue Y., Han D., Fu Y., Parisien M., Dai Q., Jia G. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014;505:117–120. doi: 10.1038/nature12730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Zhao B.S., Roundtree I.A., Lu Z., Han D., Ma H., Weng X., Chen K., Shi H., He C. N(6)-methyladenosine modulates messenger RNA translation efficiency. Cell. 2015;161:1388–1399. doi: 10.1016/j.cell.2015.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Feng J., Xue Y., Guan Z., Zhang D., Liu Z., Gong Z., Wang Q., Huang J., Tang C. Structural basis of N(6)-adenosine methylation by the METTL3-METTL14 complex. Nature. 2016;534:575–578. doi: 10.1038/nature18298. [DOI] [PubMed] [Google Scholar]
- Weinberg D.E., Shah P., Eichhorn S.W., Hussmann J.A., Plotkin J.B., Bartel D.P. Improved ribosome-footprint and mrna measurements provide insights into dynamics and regulation of yeast translation. Cell Rep. 2016;14:1787–1799. doi: 10.1016/j.celrep.2016.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wildeman A.G., Sassone-Corsi P., Grundström T., Zenke M., Chambon P. Stimulation of in vitro transcription from the SV40 early promoter by the enhancer involves a specific trans-acting factor. EMBO J. 1984;3:3129–3133. doi: 10.1002/j.1460-2075.1984.tb02269.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao H., Friesen J.D., Lis J.T. Recruiting TATA-binding protein to a promoter: transcriptional activation without an upstream activator. Mol. Cell. Biol. 1995;15:5757–5761. doi: 10.1128/mcb.15.10.5757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao W., Adhikari S., Dahal U., Chen Y.S., Hao Y.J., Sun B.F., Sun H.Y., Li A., Ping X.L., Lai W.Y. Nuclear m(6)A reader YTHDC1 regulates mRNA splicing. Mol. Cell. 2016;61:507–519. doi: 10.1016/j.molcel.2016.01.012. [DOI] [PubMed] [Google Scholar]
- Zhou J., Wan J., Gao X., Zhang X., Jaffrey S.R., Qian S.B. Dynamic m(6)A mRNA methylation directs translational control of heat shock response. Nature. 2015;526:591–594. doi: 10.1038/nature15377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zid B.M., O’Shea E.K. Promoter sequences direct cytoplasmic localization and translation of mRNAs during starvation in yeast. Nature. 2014;514:117–121. doi: 10.1038/nature13578. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.