

Abstract
Zinc influences signaling pathways through controlled targeted zinc transport. Zinc transporter Zip14 KO mice display a phenotype that includes impaired intestinal barrier function with low grade chronic inflammation, hyperinsulinemia, and increased body fat, which are signatures of diet-induced diabetes (type 2 diabetes) and obesity in humans. Hyperglycemia in type 2 diabetes and obesity is caused by insulin resistance. Insulin resistance results in inhibition of glucose uptake by liver and other peripheral tissues, principally adipose and muscle and with concurrently higher hepatic glucose production. Therefore, modulation of hepatic glucose metabolism is an important target for antidiabetic treatment approaches. We demonstrate that during glucose uptake, cell surface abundance of zinc transporter ZIP14 and mediated zinc transport increases. Zinc is distributed to multiple sites in hepatocytes through sequential translocation of ZIP14 from plasma membrane to early and late endosomes. Endosomes from Zip14 KO mice were zinc-deficient because activities of the zinc-dependent insulin-degrading proteases insulin-degrading enzyme and cathepsin D were impaired; hence insulin receptor activity increased. Transient increases in cytosolic zinc levels are concurrent with glucose uptake and suppression of glycogen synthesis. In contrast, Zip14 KO mice exhibited greater hepatic glycogen synthesis and impaired gluconeogenesis and glycolysis related to low cytosolic zinc levels. We can conclude that ZIP14-mediated zinc transport contributes to regulation of endosomal insulin receptor activity and glucose homeostasis in hepatocytes. Therefore, modulation of ZIP14 transport activity presents a new target for management of diabetes and other glucose-related disorders.
Keywords: gluconeogenesis, glycogen, glycolysis, insulin, mitochondria, Insulin degrading enzyme
Introduction
The liver is the major metabolic organ responsible for both the utilization and production of glucose and energy balance (1). Impairment of hepatic glucose metabolism leads to postprandial hyperglycemia, which is associated with increased risk of death and major cardiovascular events (2). Therefore, hepatic glucose homeostasis is a critical focus for anti-diabetic therapies (3). Diabetic patients often display altered zinc homeostasis, and zinc supplementation has been investigated as an adjunct therapy in the management of diabetes. Meta-analysis of zinc supplementation trials with human subjects revealed that zinc produces a significant reduction in blood glucose concentration (4, 5). However, the mechanistic role of zinc in glucose metabolism remains unknown.
Zinc is an essential micronutrient that is involved in intracellular signaling (6–9). The potential for the involvement of zinc in carbohydrate metabolism has been shown from a limited number of studies. For example, dose-dependent zinc regulation of fructose 1,6-bisphosphatase, the key regulatory enzyme in gluconeogenesis, had been shown in vitro (10) and in vivo (11). In isolated rat hepatocytes, zinc-supplemented media enhanced glycolysis and glycogenolysis (12).
Zinc transporter proteins maintain zinc homeostasis. Zinc transporters are found within two gene families, ZIP (SLC39) and ZnT (SLC30), that function in a tissue-specific manner (13). ZIP transporters facilitate zinc influx into the cytosol, ZnT family transporters function in zinc efflux from the cytosol. Links between zinc transporter expression/function and insulin/glucose have been shown. Most notably ZnT8, which is almost exclusively expressed in pancreatic beta cells, has been shown to influence zinc incorporation into insulin (14, 15). A single nucleotide polymorphism (R325W) of ZnT8 is associated with a high risk of type 2 diabetes (16, 17). ZnT8 KO mice show abnormalities in insulin secretion, glucose tolerance, and body weight as well (18, 19). ZnT7 KO mice have decreased insulin secretion and increased insulin resistance in male mice (20).
In humans, postprandial plasma zinc and serum glucose levels are highly correlated (21), suggesting that zinc is taken up by the liver during postprandial glucose metabolism. However, the link between hepatic glucose metabolism and zinc transport activity has not been explored. ZIP14 is a SLC39 zinc transporter family member that traffics zinc into the cytoplasm. Phenotypically, Zip14 KO mice display enlarged pancreatic islets with hyperinsulinemia and greater body fat (22), conditions that resemble diet-induced diabetes (type 2 diabetes) and obesity. A low grade chronic inflammation (metabolic endotoxemia), which is a common feature of these models, is primarily due to impaired intestinal barrier function (23). In keeping with these characteristics, Zip14 KO mice also display impaired intestinal barrier function (24) with low grade chronic inflammation signatures. They precipitate the development of insulin resistance in adipose tissue from Zip14 KO mice (25). Interestingly, the liver of Zip14 KO mice is insulin-sensitive despite the low grade chronic inflammation (22).
Our novel finding that both hepatic zinc and glucose homeostasis are altered when zinc transporter Zip14 is deleted provides a suitable in vivo model for detailed mechanistic studies. Therefore, we conducted experiments using the Zip14 KO mouse model to evaluate a potential mechanism where zinc transport mediated by ZIP14 is involved in hepatic glucose metabolism.
Results
Ablation of Zip14 Prevents Hepatic Insulin Resistance
It has been shown in mice, pigs, and humans that a high fat diet (HFD)2 increases blood endotoxin levels leading to metabolic endotoxemia (23). Mice begin to display signs of metabolic endotoxemia (e.g. hyperinsulinemia, hyperglycemia, and weight gain) after as little as 6 weeks of HFD feeding (26). Zip14 KO mice fed a chow diet display impaired intestinal barrier function within weeks after weaning (24). Chow-fed Zip14 KO mice also have increased serum endotoxin (p < 0.001) and insulin (p < 0.05) levels that are enhanced in mice fed a HFD (Fig. 1A). This indicates that with the Zip14 KO mice, we successfully modeled metabolic endotoxemia that is induced by a high fat dietary intervention. Similarly, compared with chow-fed WT mice, expression of liver TNFα and IL1β transcripts were found to be greater and comparable in chow-fed Zip14 KO and HFD-fed WT and KO mice (data not shown). Weight gain was observed in both genotypes (p < 0.05) after HFD feeding (Fig. 1B).
FIGURE 1.

Ablation of Zip14 prevents metabolic endotoxemia-induced hepatic insulin insensitivity. The mice were fed either chow diet or HFD for 6 weeks. A, serum endotoxin and insulin were measured by LAL reagents and ELISA, respectively. B, body weight. C, following OGTT, the area under the curve (AUC) was calculated. D, following ITT, the AUC was calculated. E, abundance of ZIP14 and IR pathway proteins were compared in WT and KO mice that were fasted or fed chow or HFD. F, genotypic differences in complex formation between IR and GLUT2. The results are means ± S.E. (n = 5–10 mice). IP, immunoprecipitation; ns, not significant; KOHF, HFD-fed KO mice; WTHF, HFD-fed WT mice.
To investigate the physiological outcomes of Zip14 ablation, oral glucose and insulin tolerance tests were conducted. With both tests (Fig. 1, C and D), there was no difference detected in blood glucose levels between chow diet-fed WT and KO mice. Impaired glucose tolerance was detected in HFD-fed WT mice (p < 0.0.004), whereas no such effect was detected in the Zip14 KO mice. We have previously shown that adipose tissue of Zip14 KO mice were insulin-resistant (24). The phosphorylated insulin receptor (IR) (active form) was measured to test whether the Zip14 KO mice developed hepatic insulin resistance. Contrary to our expectation, enhanced IR phosphorylation was detected in the liver of both chow and HFD-fed Zip14 KO mice (Fig. 1E). Furthermore, glucose transporter 2 (GLUT2) protein levels were greater in fasted, chow-diet, and HFD-fed KO animals when compared with WT mice. A higher amount of IR and GLUT2 complex formation (27) was observed in Zip14 KO liver when proteins were immunoprecipitated with IR antibody (Fig. 1F), suggesting that hepatic glucose uptake will be greater in Zip14 KO mice. Together, these results led to our hypothesis that liver glucose metabolism is altered in Zip14 KO mice.
Hepatic Insulin Sensitivity Is Enhanced in Zip14 KO Mice
Liver was collected following oral glucose administration. Enhanced phosphorylation of IR, PI3K, and AKT and a greater amount of IRS1 protein were detected in Zip14 KO mice at steady state and throughout the course of glucose uptake (Fig. 2A). Furthermore, greater GLUT2 protein were found in Zip14 KO mice (Fig. 2, A and B). Marked changes were observed at 90 min post-glucose gavage; therefore, most data are presented from 0 and 90 min post-gavage. Liver GLUT2 expression (p < 0.001) (Fig. 2B) and glucose content (p < 0.03) (Fig. 2C) were significantly greater in Zip14 KO mice when compared with WT. Although serum insulin concentration was greater in Zip14 KO mice, it was significantly lower following glucose administration (Fig. 2D). In WT mice, ZIP14 protein was up-regulated during hepatic glucose uptake (Fig. 2E). The liver zinc uptake was greater (p < 0.03) during glucose uptake as measured by 65Zn (Fig. 2F). Furthermore, Mt mRNA (a measure of available intracellular zinc) levels were up-regulated (p < 0.01). In contrast, the liver zinc uptake was less, and Mt mRNA did not change during glucose uptake in the KO mice.
FIGURE 2.

Hepatic ZIP14 and IR activity during glucose uptake. Following glucose gavage, blood and liver tissue were collected. A, an array of IR pathway proteins was examined by Western blot. B, Glut2 protein and mRNA were measured by Western blot and qPCR, respectively. C, liver glucose were measured by glucose oxidase assay. D, serum insulin concentration was measured by ELISA. E, ZIP14 Western blot. F, liver zinc and Mt mRNA. The results are means ± S.E. (n = 410 mice). *, p < 0.05; #, p < 0.01; ¥, p < 0.001.
ZIP14 Is Detected in the Endosomes of HepG2 Hepatocytes
To model the Zip14 KO phenotype of hyperinsulinemia, human HepG2 hepatocytes were treated with insulin and glucose following overnight serum and glucose starvation. ZIP14 was up-regulated along with P-IR and P-AKT in response to insulin and glucose treatment (Fig. 3A). When Zip14 was knocked down with siRNA, greater P-IR and P-AKT were detected as they were in Zip14 KO mice (Fig. 3A). Glucose uptake as measured with non-metabolizable fluorophore-labeled glucose (2NBDG) was greater (p < 0.02) in the Zip14 KD cells (Fig. 3A). In contrast, uptake was significantly lower in cells that were overexpressing Zip14 (data not shown). Zinc uptake was significantly (p < 0.03) impaired in the Zip14 KD cells (Fig. 3A). These results with HepG2 hepatocytes simulate the murine Zip14 KO phenotype.
FIGURE 3.

Subcellular ZIP14 localization and zinc redistribution during glucose uptake. After overnight serum starvation, HepG2 hepatocytes were treated with insulin (200 nm) and glucose (20 mm) and then harvested at the indicated points. Some HepG2 hepatocytes were transiently transfected with Zip14 siRNA, treated with insulin and glucose and harvested 3 h later. A, abundance of ZIP14 and IR pathway proteins in total cell lysate was examined by Western blot. Control and transfected cells were incubated with the non-metabolizable glucose analog 2NBDG along with insulin, and 2NBDG uptake was measured fluorometrically. Total zinc was measured by AAS. B, glucose uptake over time was determined by measuring both 2NBDG and total glucose. C, intracellular zinc concentration was assessed by AAS and labile zinc by fluorometry using FluoZin3-AM. D, ZIP14 abundance in PM and membrane rafts. E, ZIP14 abundance in cell surface. F, ZIP14 abundance in endosomes by Western blot and endosomal zinc concentrations were determined by fluorometry using FluoZin3-AM. The results are means ± S.E. (n = 3–5). *, p < 0.05; #, p < 0.01; ¥, p < 0.001. CSB, biotinylated cell surface proteins.
Glucose uptake on a temporal basis was measured both as a change in total glucose and with 2NBDG. Intracellular glucose increased for 3 h post-insulin/glucose treatment (Fig. 3B). By 5 h post-treatment, the intracellular glucose concentration declined, which suggests that glucose was being either stored or metabolized. In contrast, 2NBDG uptake continued throughout the treatment (Fig. 3B). Total cellular zinc concentrations measured by atomic absorption spectrophotometry (AAS) were significantly increased at 3 and 5 h post-insulin/glucose treatment. In contrast, zinc measured with FluoZin3-AM, which detects labile intracellular zinc, was significantly lower at both time points (Fig. 3C). Thus, we separated cytosolic fractions from total cell lysates following insulin and glucose treatment and found a gradual but significant increase in cytosolic zinc with time (Fig. 3C). To test whether changes in cellular zinc were specific to glucose treatment, we treated the hepatocytes with differing concentrations of glucose. We found dose-dependent increases and decreases in total and label zinc (data not shown). Furthermore, an increase in hepatic ZIP14 abundance was detected after glucose gavage, but there was no difference in ZIP14 abundance after insulin injection (data not shown).
To more closely establish a role for ZIP14 and zinc in metabolic events related to glucose uptake within specific subcellular compartments, the cytosol, plasma membrane (PM), membrane rafts, early endosomes (EEs), and late endosomes (LE) from HepG2 cell lysates were separated. ZIP14 was detected in all fractions except cytosol (data not shown). The markers were GAPDH (cytosol), NaK ATPase (PM), caveolin (membrane raft), EEA1 (EE), and rab7 (LE). The abundance of ZIP14 was highest in PM at one h after insulin/glucose treatment and decreased thereafter (Fig. 3D). In membrane rafts, ZIP14 abundance was the highest before treatment and decreased after insulin/treatment (Fig. 3D). Cell surface proteins were biotinylated, which revealed an increase in the amount of ZIP14 protein on the cell surface up to 1 h after insulin and glucose treatment. A reduction by 3 and 5 h thereafter (Fig. 3E) is in agreement with ZIP14 abundance at the PM (Fig. 3D). Time-dependent increases and decreases in ZIP14 protein were detected in EE and LE fractions, respectively (Fig. 3F). Surprisingly, in agreement with the changes in ZIP14 abundance, the labile zinc concentration was up-regulated and down-regulated in EE and LE, respectively (Fig. 3F). The correlated changes in ZIP14 and zinc in endosomal fractions suggest that ZIP14 is internalized by endocytosis.
ZIP14-mediated Zinc Transport Regulates Endosomal IR Activity
Upon activation the IR is internalized through either clathrin or caveolin-mediated endocytosis (28, 29). In Fig. 3 we have shown an increase in both abundance of ZIP14 and zinc concentration in EEs during glucose uptake. Therefore, we hypothesized that the endosomal zinc provided by internalized-ZIP14 might be involved in regulation of IR activation in EE. To test this possibility, we separated endosomes from the liver of WT and Zip14 KO mice at basal or 90 min post-glucose gavage following an overnight fast. In agreement with our in vitro data (Fig. 3F), after glucose gavage ZIP14 abundance was greater in EE and was lower LE (Fig. 4A). At 90 min after glucose gavage IR phosphorylation was greater in EE of Zip14 KO liver. Greater IR phosphorylation in EE of Zip14 KO could be related to a change in the internalization rate of the active IR or to endosomal processing of internalized IR. To test the first possibility, we separated subcellular fractions of liver at steady state (Fig. 4B). Lower amounts of P-IR were detected in PM in the liver of the Zip14 KO mice and were concurrent with a greater amount of P-IR in the EE. This suggests that in the absence of ZIP14, P-IR internalization was greater. Of note, GPR39, a PM membrane marker that does not undergo endocytic recycling, did not change (30). Furthermore, both hepatic EE and LE zinc concentrations were significantly lower (p < 0.04 and p < 0.03) when isolated from the Zip14 KO mice compared with the WT controls (Fig. 4C). These data were in agreement with earlier in vitro data (Fig. 3F) that showed a correlation between ZIP14 abundance and zinc concentration in endosomal fractions of HepG2 hepatocytes.
FIGURE 4.

Zip14 ablation alters internalized IR activity. Subcellular fractions from liver of WT and KO mice were separated by sucrose gradient ultracentrifugation. A, at 0 and 90 min post-glucose gavage, the amount of ZIP14 and P-IR in early and late endosomes were analyzed by Western blotting (endosomes from n = 4 mice combined per lane). B, at steady state, the amount of ZIP14 and P-IR along with appropriate site markers were analyzed by Western blotting (n = 6 mice combined per lane). C, endosomal zinc concentrations were determined by FluoZin3-AM fluorescence (n = 6). D, cathepsin D activity in endosomes (n = 10). E, IDE activity in endosomes (n = 10). F, proposed model that describes the proposed role of zinc in endosomal IR regulation. The results are means ± S.E. Cyto, cytosol.
IR deactivation occurs both at the PM and within endosomes through dephosphorylation either by phosphotyrosine protein phosphatase (PTP1B) activity (31) or insulin disassociation from IR (32). PTP1B activity is inhibited by zinc (8). Because we have shown that cytosolic zinc is lower in Zip14 KO mice, zinc inhibition of PTP1B phosphatase activity is unlikely. Therefore, we focused on endosome-associated enzymes such as insulin-degrading enzyme (IDE) and cathepsin D that are responsible for IR dephosphorylation through insulin disassociation and degradation (33, 34). We conducted IDE and cathepsin D enzyme assays using endosomal fractions from WT and Zip14 KO mouse liver. Both IDE and cathepsin D activities were significantly impaired (p < 0.02 and p < 0.05) in EE of Zip14 KO mice, whereas there was no difference in these activities detected in late endosomes between genotypes (Fig. 4, D and E). We interpret these data to indicate that because of a lower zinc concentration in EE of the Zip14 KO mice, the activities of these enzymes are impaired; thus, IR is maintained in its active state (Fig. 4F).
ZIP14-mediated Zinc Transport Could Provide a Negative Feedback for Glycogen Synthesis
Demonstrated here is a ZIP14-dependent increase in cytosolic zinc during insulin-induced glucose uptake in hepatocytes (Fig. 3C). To investigate the potential physiological consequences of this intracellular zinc redistribution, we first measured hepatocyte glycogen. A positive correlation between cytosolic zinc (Fig. 3C) and glycogen was found up to 3 h post-insulin/glucose treatment (Fig. 5A). At 5 h post-glucose/insulin, the glycogen concentration began to decline (p < 0.05), although cytosolic zinc continued to increase (Fig. 3C). This relationship suggests newly transported zinc produces a time-dependent negative feedback on glycogen synthesis (Fig. 5F).
FIGURE 5.

Zip14-mediated zinc transport influences hepatic glycogen. Glycogen was isolated from liver by ethanol precipitation, and following glycogen breakdown with amyloglucosidase, glucose was measured to calculate the glycogen content. A, glycogen concentration in HepG2 cells and an array of glycogen synthetic pathway proteins. B, glycogen concentration in HepG2 cells and glycogen synthetic pathway proteins after incubation with differing concentrations of zinc. C, liver glycogen and cytosolic zinc concentrations of WT and ZIP14 KO mice. D, glycogen synthetic pathway proteins in liver. E, liver glycogen concentration and an array of glycogen synthetic pathway proteins following a week of dietary zinc supplementation. F, proposed model for the influence of ZIP14-mediated transport of cytosolic zinc on glycogen synthesis. The results are means ± S.E. (n = 3–5 mice).
Glycogen synthase kinase 3 (GSK3) activity is negatively regulated by insulin through the IR-AKT pathway (35). Activation of AKT leads to the phosphorylation and inactivation of GSK3. A major substrate of GSK3 is glycogen synthase (GS), an enzyme that catalyzes the final step in glycogen synthesis. To determine the effect of zinc specifically on glycogen synthesis, we used both cytosolic fractions and total cell lysate of HepG2 hepatocytes to analyze proteins of this pathway. Following insulin and glucose treatment, time-dependent reductions in P-AKT (active) and P-GSK3 (inactive form) were shown by Western blot (Fig. 5A). The amount of P-GS (inactive) declined immediately after the insulin/glucose treatment; however, it increased thereafter, possibly because of enhanced GSK3 activity. Next, hepatocytes were treated with differing concentrations of zinc along with insulin and glucose for 3 h. A zinc-dependent reduction in total glycogen was produced (Fig. 5B). In agreement, reduced phosphorylation of AKT, GSK3 was shown to be zinc-dependent (Fig. 5B).
Zip14 KO mice have significantly lower (p < 0.001) cytosolic zinc concentrations in liver (Fig. 5C). To link low cytosolic zinc and glycogen synthesis in mice, we measured hepatic glycogen. Zip14 KO mice had significantly greater (p < 0.05) glycogen levels in the fed state (Fig. 5C). Higher amounts of phosphorylation of cytosolic AKT and GSK3, as well as increased amounts of GS, were found in livers of Zip14 KO mice (Fig. 5D). Furthermore, when mice were fed with zinc-supplemented diet (ZnS), liver glycogen was not elevated in Zip14 KO mice (Fig. 5E). Consistent with our in vitro data, lower GSK3 phosphorylation and GS abundance was found in ZnS-fed mice liver (Fig. 5E), showing that zinc provided by the diet can negatively regulate hepatic glycogen synthesis.
Impaired Gluconeogenesis in Zip14 KO Mice Is Due to Lower Hepatic Intracellular Zinc
In Fig. 5, we show that supplemental zinc provided by diet for mice or by the medium with hepatocytes caused inhibition of AKT signaling, whereas IR activity was not affected. Next we conducted insulin tolerance test (ITT), oral glucose tolerance test (OGTT), and pyruvate tolerance test (PTT) assays following zinc injections to evaluate hepatic gluconeogenic activity. Controls for effectiveness of zinc injections were the increase in serum and liver zinc concentrations and liver Mt mRNA expression (data not shown). There was no difference in blood glucose clearance between control and zinc-treated mice when the mice were injected with insulin, whereas higher blood glucose was detected in zinc-injected mice after both glucose and pyruvate administration (Fig. 6A). These results suggested that zinc may cause either impaired glucose uptake or enhanced gluconeogenesis. To test the former possibility, we measured Glut2 transcripts and protein and found no change in response to zinc, suggesting that zinc did not impair glucose uptake (Fig. 6B). Next, we measured the expression of rate-limiting enzymes of the gluconeogenic pathway, phosphoenolpyruvate carboxykinase (Pepck), and glucose-6-phosphatase (G6Pase). We found dose-dependent induction in zinc-injected mice in both transcripts and proteins of Pepck and G6Pase (Fig. 6B). Transcripts for Pepck and G6Pase in Zip14 KO were lower (p < 0.05 and p < 0.02) than WT mice when they fed both chow and zinc-adequate diet (ZnA). When mice were fed with ZnS diet, however, enhanced expression of hepatic Pepck and G6Pase transcripts (p < 0.01 and p < 0.01) and protein was detected (Fig. 6, C and D). Moreover, PTT results revealed that gluconeogenesis was slightly impaired in Zip14 KO mice and feeding with the ZnS diet enhanced (p < 0.001) gluconeogenesis in both genotypes (Fig. 6E). These data collectively indicate that zinc is a factor in regulation of hepatic gluconeogenesis and that impaired gluconeogenesis in Zip14 KO mice could be due to low intracellular zinc.
FIGURE 6.

Impaired hepatic gluconeogenesis in Zip14 KO mice. A, 2 h after a zinc injection (i.p.), ITT, OGTT, and PTT were conducted. B, 2 h after a zinc injection, liver was collected, and mRNA and protein abundance of Glut2, Pepck, and G6Pase was analyzed. C–E, mice were fed either chow, ZnA, or ZnS. C and D, hepatic mRNA and protein abundance of Pepck (C) and G6Pase (D) were analyzed. E, PTT in ZnA and ZnS-fed mice and AUC of PTT. The results are means ± S.E. (n = 5–10 mice).
ZIP14 and ZIP8 Influence ATP Synthesis via Regulating Mitochondrial and Cytosolic Zinc
Glucose metabolism is largely dependent on mitochondria to generate energy. Therefore, we sought to examine whether ZIP14 is also localized to the mitochondria. We purified mitochondria both from HepG2 cells and mouse liver by Nycodenz gradient ultracentrifugation and analyzed for ZIP14. Voltage-dependent anion-selective channel protein (VDAC) and cytochrome c were used as mitochondrial markers. Our data revealed that ZIP14 was localized to the mitochondria in HepG2 cells, and ZIP14 abundance was reduced during insulin and glucose treatment (Fig. 7A). We also examined the abundance of ZIP8 because it is the only ZIP transporter reported to have a mitochondrial localization (36). Abundance of mitochondrial ZIP8 increased with time in insulin/glucose-treated cells. It is of note that ZIP8 is the closest phylogenetic homolog to ZIP14 (37). The mitochondrial zinc content was reduced when the cells were treated with insulin and glucose (Fig. 7B). We interpreted these results to indicate that ZIP14 and ZIP8 might be working together, independently or as a heterodimer, to maintain an optimum zinc concentration in mitochondria. These data also implicate zinc in mitochondrial energy metabolism.
FIGURE 7.

Zip14 and Zip8 are involved with ATP synthesis. Mitochondria were purified from HepG2 hepatocytes and mouse liver by Nycodenz gradient ultracentrifugation. A, ZIP14 Western blot from HepG2 lysates. Abundance of ZIP14 and ZIP8 in HepG2 cells during glucose uptake. B, total and mitochondrial zinc and ATP concentrations in HepG2 cells. C, percentage of change in total and mitochondrial zinc and ATP. D, left panel, ZIP14 and ZIP8 abundance in liver. Middle and right panels, percentage of change in hepatic total and mitochondrial zinc and ATP. E, liver pyruvate concentrations. F, proposed model for zinc integration of transporters ZIP14 and ZIP8 and glycolysis and mitochondrial ATP synthesis. The results are means ± S.E. (n = 5). *, p < 0.05; #, p < 0.01; ¥, p < 0.001. P Mit, pure mitochondria; C Mit, crude mitochondria; Mito, mitochondria.
Next we evaluated the amount of ATP in total and mitochondrial fractions. Increased ATP production was shown in both total lysate and mitochondrial fractions following insulin/glucose treatment of the HepG2 cells (Fig. 7B). When Zip14 was knocked down by siRNA, whereas total zinc and ATP concentrations were lower in Zip14 KD cells, there was no difference in either zinc or ATP concentrations in mitochondria (Fig. 7C). In vivo we found slightly higher mitochondrial ZIP8 in liver of Zip14 KO (Fig. 7D). In agreement with our in vitro data, total liver zinc and ATP concentrations were lower (p < 0.03 and p < 0.03) in Zip14 KO mice, whereas there was no difference in mitochondrial zinc and ATP concentrations between genotypes (Fig. 7D).
The data above clearly showed that the hepatic mitochondrial zinc concentration and ATP synthesis were tightly regulated. Total zinc and ATP concentrations, however, were consistently low in Zip14 KD cells and Zip14 KO mice, raising the hypothesis that ZIP14 and ZIP14-mediated zinc transport might have a role in glycolysis. Therefore, we evaluated the amount of pyruvate in liver of WT and Zip14 KO mice. Pyruvate concentrations were lower in chow- (p < 0.02) and ZnA-fed (p < 0.04) Zip14 KO mice (Fig. 7E). Interestingly, feeding mice with ZnS diet caused a significant increase (p < 0.02) in hepatic pyruvate concentration in Zip14 KO, whereas there was no difference in WT mice (Fig. 7E). We interpret these data to indicate that zinc was required for hepatic glycolysis in hepatocytes, and because transport was impaired in Zip14 KO mice, an intracellular zinc deficiency was created (Fig. 7F). In support of our interpretation, Brand and Kleineke (12) showed that glycolysis was activated by zinc via stimulation of the glycolytic enzymes, pyruvate kinase, and phosphofructokinase.
Discussion
In this report, we demonstrate that in Zip14 KO mice, hepatic insulin sensitivity was enhanced despite a phenotype that includes low grade chronic inflammation (22). During insulin-induced hepatic glucose uptake, zinc is distributed into subcellular sites because of changes in ZIP14 abundance and localization. Specifically, ZIP14 is up-regulated at the PM then internalized to the endosomes, resulting in altered IR activity. Because of impaired hepatic zinc uptake, cytosolic zinc is lower in Zip14 KO mice and is concurrent with enhanced glycogen synthesis and impaired glycolysis and gluconeogenesis.
Upon internalization, insulin is released from IR and degraded by endosomal proteases. Insulin release from IR subsequently results in dephosphorylation (inactivation) of the receptor. The endosomal thiol zinc-metalloendopeptidase, IDE, is mainly responsible for disassociation and degradation of receptor-mediated internalized insulin (33). Catalytic activity of IDE is induced by zinc (38). The soluble aspartic acid protease, cathepsin D, has also been identified as the insulin-degrading enzyme of endosomal insulin within the liver (34, 39). Our findings show that IDE and cathepsin D activities were impaired in hepatic EE of Zip14 KO mice and was concurrent with a low zinc concentration (Fig. 4D). We propose that impaired IDE and cathepsin D activity potentiated insulin signaling in Zip14 KO liver by reducing the catabolism of internalized insulin. Inhibition of endosomal insulin catabolism is an attractive anti-diabetic approach (40). Therefore, our novel integration of ZIP14-transported zinc into the insulin signaling mechanism is a new potential target for the experimental manipulation of insulin signaling via IDE and cathepsin D activity.
GLUT2 is the primary hepatic liver transporter that facilitates bidirectional hepatic glucose under insulin regulation. Immunofluorescent confocal images revealed colocalization of GLUT2 and IR at the plasma membrane and cytosol of hepatocytes (27). Furthermore, a physical interaction between GLUT2 and IR has been shown by coimmunoprecipitation studies suggesting a mechanism that influence in glucose transport after insulin binding to IR. We have found a greater amount of GLUT2 in the liver of Zip14 KO mice (Fig. 1E). Furthermore, when we conducted immunoprecipitation using IR antibody, we detected a greater amount of GLUT2 in the liver lysates of Zip14 KO mice when compared WT (Fig. 1F). This finding suggests a greater hepatic glucose transport in Zip14 KO mice. Higher glucose concentrations in the liver of Zip14 KO mice further supported this finding (Fig. 2C).
We have reported here that ZIP14 abundance in endosomes correlated positively with the endosomal zinc concentration during glucose uptake. This was an unexpected finding because of the known nature of the ZIP family metal transport function. We propose that ZIP14-bound zinc is internalized by endocytosis, and because of the low pH environment (pH 6–6.5 in EE and pH 4–4.5 in LE), zinc is disassociated from ZIP14. This labile zinc could be involved in regulation of endosomal metalloproteases and thereby influence signal transduction of internalized receptors such as IR. Although the endosomal low-pH environment favors zinc disassociation from proteins, it may prevent zinc transport from endosomes to the cytosol because ZIP14-mediated zinc transport is pH-sensitive. In ZIP14-expressing Xenopus oocytes, the optimal pH for zinc transport was 7.5 (41). This proposal was supported by the in vivo data showing that endosomes of Zip14 KO mice were zinc-deficient (Fig. 4C). This is contrary to our previously published data suggesting that zinc was trapped in vesicles of intestine and adipose when Zip14 was deleted (24, 25). This difference could be explained in two possible ways: 1) ZIP14 is the only hepatic zinc transporter that controls endosomal zinc during glucose uptake, and 2) zinc transport efficiency is different in hepatocytes because endosomal pH varies between tissues (42, 43).
Our in vivo time course experiments revealed that the increase in ZIP14 abundance and subsequent changes in zinc concentration were found to be during the late postprandial state (Figs. 2 and 3). Furthermore, the amount of glycogen decreases when cytosolic zinc concentration was the highest at 5 h post-insulin/glucose treatment (Figs. 3 and 5A). Our interpretation of these data is that ZIP14-mediated zinc transport during glucose uptake is necessary to decrease glycogen synthesis in the late postprandial state. Zip14 KO liver glycogen was greater at the fed state (Fig. 5C) because the liver was zinc-deficient. The addition of zinc to media along with insulin/glucose treatment of cells in vitro (Fig. 5B) and after a week of ZnS feeding in vivo (Fig. 5E) reduced glycogen synthesis. In agreement with our findings, zinc inhibition of glycogen synthesis has been shown in liver of zinc-injected rats (44) and zinc-treated primary rat hepatocytes (45).
We also report a potential role for ZIP14 and ZIP14-mediated zinc transport to regulate gluconeogenesis. Our data revealed that both injection of zinc and dietary supplementation with zinc induced expression of two rate-limiting gluconeogenic enzymes: Pepck and G6Pase (Fig. 6, C and D). Furthermore, gluconeogenesis was enhanced in zinc-supplemented mice when pyruvate was given as a substrate (Fig. 6E). Gluconeogenic enzymes are regulated at the transcriptional level (46). Among the many transcription factors that regulate the genes of these gluconeogenic enzymes (47), sterol regulatory element-binding protein 1c (Srebp1c), a repressor, and cAMP response element binding protein (CREB), an activator, were of the interest because we previously found increased hepatic expression of Srebp1c in Zip14 KO (22) and dose-dependent zinc regulation of CREB phosphorylation (48).
In summary, our data collectively reveal that ZIP14-mediated hepatic zinc transport influences regulation of IR activity, glucose uptake, glycogen synthesis, and gluconeogenesis (Fig. 8). Therefore, ZIP14 and zinc are essential for hepatic glucose homeostasis. These new insights into the fundamental role of a zinc transporter in hepatic glucose homeostasis suggest a new drug target for pharmacological management of diabetes and other glucose-related disorders.
FIGURE 8.
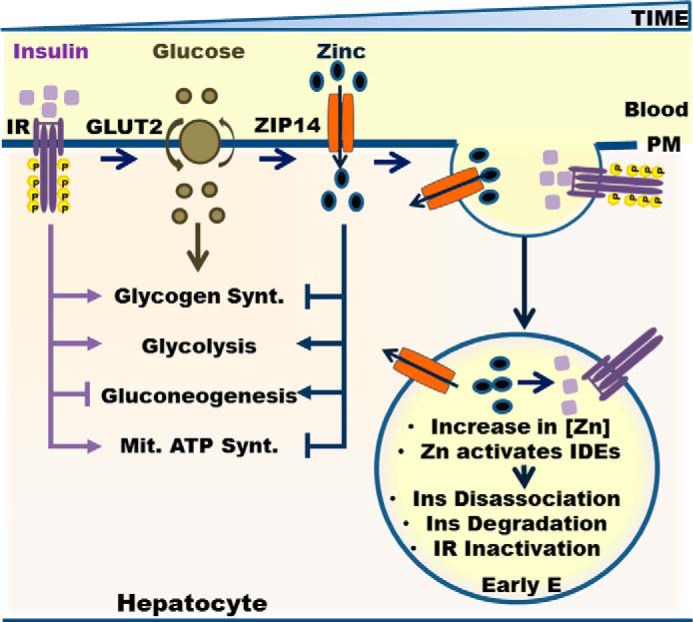
Role of ZIP14 and zinc in hepatic glucose metabolism. Ins, insulin; Mit., mitochondria; Synt., synthesis.
Experimental Procedures
Murine Strain, Diets, and Treatments
Heterozygous (ZIP14+/−) mice of a mixed C57BL6; 129S5 strain were obtained to establish a breeding colony to generate both homozygous WT and homozygous KO mice. WT and KO mice were used between 8 and 16 weeks of age (22). The mice were maintained using standard rodent husbandry and received a commercial, irradiated chow diet (Harlan Teklad 7912; Indianapolis, IN, with 60 mg Zn/kg as ZnO) and tap water. Specific dietary treatments with purified diets were either AIN76a-based diet (Research Diets, New Brunswick, NJ) containing 30 (ZnA) and 180 (ZnS) mg Zn/kg, respectively, or chow diet and a HFD (D12492, 60% kal/kg; Research Diets, New Brunswick, NJ). For the controlled dietary zinc studies, the mice were fed a ZnA for 4 days and the ZnA or ZnS diets for 1 week. For the HFD feeding studies, the mice were fed with the HFD or chow for 6 weeks. The mice were singly caged in shoebox cages for the dietary feeding studies.
Blood glucose levels were measured by tail bleeding using the OneTouch® Ultra Blood glucose monitoring system immediately prior to and at 15, 30, 90, and 180 min after 0.75 unit of insulin/kg of administration (i.p.) for the ITT, 3 mg of glucose/kg administration (gavage) for the OGTT, and 2 mg of pyruvate/kg administration (i.p.) for the PTT. The mice were fasted 4–5 h for ITT and overnight for OGTT and PTT. In some experiments, at selected time points, the mice were sacrificed for the purposes of collecting blood and tissue following oral glucose administration. To evaluate the temporal relationship of zinc uptake to glucose administration 65Zn was provided by gavage (2 μCi/mouse in 250 μl of saline) to fasted mice. One hour later, glucose was administered by gavage. Liver tissue was collected at 0, 15, 90, and 180 min to assess 65Zn uptake. The mice were anesthetized by isofluorane (Baxter, Deerfield, IL) inhalation for injections, gavage, and euthanasia by cardiac puncture. Protocols were approved by the University of Florida's Institutional Animal Care and Use Committee.
RNA Isolation and qPCR
For RNA isolation, tissues were placed in TRI reagent (Ambion-Thermo Fisher Scientific, Waltham, MA) and homogenized with a bullet blender (Next Advance, Averill Park, NY). Total RNA was treated with Turbo DNA-free reagents (Ambion-Thermo Fisher Scientific, Waltham, MA). Primer/probe sequences for the PCRs were either designed in-house using Primer Express® software (Applied Biosystems, Carlsbad, CA) (8) or TaqMan gene expression assays (Glut2, Pepck, and GPase), purchased from Applied Biosystems. One-step reverse transcriptase reactions (Applied Biosystems) were used for qPCR. TBP (TATA-binding protein) mRNA was the normalizer for relative quantitation.
Western blotting and Immunoprecipitation
Tissue samples were immediately flash frozen in liquid nitrogen at collection. Tissues were homogenized with a bullet blender (Next Advance, Averill Park, NY) in lysis buffer (20 mm Tris-HCl, 137 mm NaCl, 10% glycerol, 1% Triton X-100, 2 mm EDTA) containing protease and phosphatase inhibitors and PMSF. For immunoprecipitation, lysates that contained 1 mg/ml protein were incubated overnight with respective antibody and then for 4 h with protein A/G-agarose beads (Pierce-Thermo Fisher Scientific). The proteins were separated by SDS-PAGE and transferred to nitrocellulose membranes. ZIP14 and ZIP8 antibodies were made in house (8). P-IR, IR, P-PI3K, PI3K, P-AKT, AKT, E-CADHERIN, EEA1, RAB7, caveolin, clathrin heavy chain, GABDH, P-GSK3, GSK3, P-GS, GS, VDAC, and cytochrome c antibodies were purchased from Cell Signaling Technology (Danvers, MA). IRS1, PEPCK, and GPase antibodies were purchased from Santa Cruz (Dallas, TX). GPR39 and tubulin antibodies were purchased from Abcam (Cambridge, MA). Immunoblots were visualized with enhanced chemiluminescence (SuperSignalTM, West Pico; Life Technologies) to measure abundance by digital imaging (Protein Simple, San Jose, CA).
Cell Culture and Treatments
HepG2 hepatocytes were maintained at 37 °C in 5% CO2 in DMEM (Invitrogen) containing 10% FBS, penicillin, and streptomycin (Corning, Corning, NY). For experiments the hepatocytes were seeded at the density of 4 × 105 cells/ml in DMEM. After 24 h, the cells were incubated with serum- and glucose-free DMEM (Sigma) containing NaHCO3, C3H3NaO3, l-glutamine, and penicillin/streptomycin for 20 h. To test effects of glucose on gene/protein expression and cellular zinc concentration, the cells were treated with 20 mm d-glucose (Sigma) and 200 nm insulin (Sigma) from 5 min to a 5-h time period. In some experiments, the cells were cotreated with 5–100 μm ZnSO4. For the glucose uptake experiments following serum/glucose starvation, the cells were incubated with 1 mm 2NBDG (Cayman, Ann Arbor, MI) and 200 nm insulin. At the end of the incubation period, fluorescence was measured at 485/535 excitation/emission as a measure of glucose uptake. Labile zinc measurements were conducted by using a fluorescent probe FluoZin3-AM (Invitrogen). Following treatments, the cells were washed with Locke's buffer (154 mm NaCl, 5.6 mm KCl, 3.6 mm NaHCO3, 1.2 mm MgSO4, 5.6 mm glucose, 2.5 mm CaCl2, 10 mm HEPES), Next the cells were incubated at 37 °C with serum-free DMEM containing 5 μm FluoZin3-AM for 30 min and an additional 30 min with Locke's buffer. Fluorescence was measured at 494/516 excitation/emission.
Zip14 Knockdown by siRNA and Overexpression
HepG2 hepatocytes were transfected either with 10 nm Zip14 siRNA (Ambion-Thermo Fisher Scientific) by using HiPerfect transfection reagent (Qiagen) or 1 μg pCMV-SupportZip14 vector by using Effectene transfection reagent (Qiagen). Following transfection, the cells were incubated in normal growth conditions for 72 h and then serum- and glucose-starved for 20 h. Following the starvation period, secondary treatments were applied.
Cell Surface Biotinylation
HepG2 hepatocytes were incubated with an ice-cold solution of Sulfo-NHS-SS-biotin) in PBS (250 μg/ml) for 1 h with gentle rocking as previously described (49). The reaction was quenched by the addition of Tris-Cl; and after collection, the cells were pelleted by centrifugation. The cell pellet was resuspended in ND lysis buffer, and biotinylated proteins were separated by using Sepharose-conjugated streptavidin bead for immunoprecipitation.
Analytical Procedures
Blood was collected by cardiac puncture, and serum was obtained by centrifugation at 3000 × g for 15 min. Liver tissue was collected (∼50 mg) and digested in 2 ml of HNO3 for 3 h at 90 °C. HepG2 hepatocytes were washed and placed into the ice-cold PBS. The cell pellets (3000 rpm for 5 min) were digested in 500 μl of HNO3 for 2 h at 80 °C. Zinc concentrations were measured by flame AAS. Tissue and cellular zinc values were normalized to the wet tissue weight and total cell number or protein concentrations, respectively. To measure liver glucose, proteins in homogenates were removed by TCA precipitation. Supernatants were either used to measure total liver glucose with glucose oxidase assay (Sigma) spectrophotometrically or for glycogen precipitation by ethanol. Following precipitation, glycogen was hydrolyzed to glucose with an amyloglucosidase (Sigma), which was measured with the glucose oxidase assay. Liver ATP was isolated by Phenol-TE and measured with ATP detection reagents (Life Technologies). Liver pyruvate was measured with pyruvate detection reagents (Cayman, Ann Arbor, MI). Serum insulin was measured by ELISA (Mercodia, Uppsala, Sweden). Serum endotoxin was determined by using the limunus amoebocyte lysate (LAL) assay (Thermo Fisher Scientific). Diluted serum was heated at 70 °C for 10 min. Heat-inactivated serum samples were combined with LAL reagent and incubated at 37 °C. Absorbance was read at 405 nm. IDE (Anaspec, Fremont, CA) and cathepsin D (RayBio, Norcross, GA) enzymatic activities in liver endosomes were measured fluorometrically at excitation/emission 490/520 and 328/460, respectively.
Subcellular Fractionation
Buffers were supplemented with protease and phosphatase inhibitors and all steps were carried on in the cold. For plasma membrane, isolation from HepG2 hepatocytes was done by using specific reagents (BioVision, Milpitas, CA). A rat PM separation method (50) was modified for the mouse. Briefly liver tissue was homogenized in IB-1 buffer (225 mm mannitol, 75 mm sucrose, 30 mm Tris-Cl, pH 7.4, 0.5% BSA, 0.1 mm EDTA) with Potter-Elvehjem homogenizer. Supernatants from a centrifugation at 10,000 × g for 10 min were centrifuged at 25,000 × g for 20 min to obtain a crude PM pellet. Supernatant was centrifuged at 95,000 × g for 2.5 h to obtain a cytosol fraction, and the pellet was resuspended in SB (225 mm mannitol, 75 mm sucrose, 30 mm Tris-Cl, pH 7.4) and centrifuged at 25,000 × g for 20 min. The resulting pellets were resuspended in PMRB (5 mm BisTris, 0.2 mm EDTA, pH 6) for centrifugation using a discontinues sucrose gradient of 38, 43, and 53% containing PMRB. Following centrifugation at 95,000 × g for 2.5 h, PM fraction was collected from the interface between 53 and 43% sucrose and was diluted with SB, and PM proteins were pelleted at 95,000 × g for 1 h.
For membrane raft isolation, isolation from HepG2 hepatocytes was done by using specific reagents (G-biosciences, St. Louis, MO). For endosome separation, the separation protocol was modified from Refs. 51 and 52. HepG2 hepatocytes were resuspended in HBA (3 mm imidazole, pH 7.4, 1 mm EDTA). Livers were homogenized in fresh HBA as above. HepG2 hepatocytes or liver cell suspensions in HBA were kept on ice for 20 min. After an addition of equal amounts of HBB (500 mm sucrose, 3 mm imidazole, pH 7.4, 1 mm EDTA, 0.06 mm cycloheximide), 50 strokes with glass/glass pestle (A) was applied. Thereafter, HB1 (250 mm sucrose, 3 mm imidazole, pH 7.4) was added and centrifuged at 2,500 × g for 10 min to obtain post-nuclear supernatant. For the isolation of endosomal fractions, the sucrose concentration of the post-nuclear supernatant was adjusted to 40.6% by adding 62% sucrose, loaded on the bottom of an ultracentrifuge tube (Beckman), and overlaid sequentially with 35% sucrose, 25% sucrose and finally filling up the rest of the tube with HB2. The samples were centrifuged at 210,000 × g for 1.5 h. Late and early endosomal fractions were collected from the interphase between 25% sucrose and HB2 and interphase between 35 and 25% sucrose, respectively. Endosomal proteins were pelleted by centrifugation at 100,000 × g for 1 h after the addition of HBA.
For mitochondria separation, the mitochondrial separation protocol was modified from Refs. 53 and 54. To obtain crude mitochondria, HepG2 hepatocytes were scraped into PBS collected by centrifugation at 200 × g for 5 min. The pellets were gently resuspended in RSB hypo buffer (10 mm NaCl, 1.5 mm MgCl2, 10 mm Tris-HCl, pH 7.5). Livers were washed with RSB hypo buffer at the time of collection and then homogenized in fresh RSB hypo buffer with a Teflon pestle (glass/Teflon Potter-Elvehjem) by using three strokes. Following a 10-min incubation on ice, 10 strokes with a Teflon pestle were applied. The addition of MS homogenization buffer (525 mm mannitol, 175 mm sucrose, 12.5 mm Tris-HCl, pH 7.5, 2.5 mm EDTA, pH 7.5) was followed by a 5-min centrifugation at 13,000 × g. The supernatant was centrifuged at 17,000 × g for 15 min. The pellet (crude mitochondria) resuspended in 4 ml 25% Nycodenz and was placed on the following discontinuous Nycodenz gradients: 1.7 ml of 34% and 2.7 ml of 30% of Nycodenz, and this was topped off with 2.7 ml of 23% and finally 1 ml of 20% Nycodenz. The tubes were centrifuged for 90 min at 52,000 × g. The mitochondria were collected from the 25/30% Nycodenz interface and diluted with the same volume of resuspending buffer (200 mm mannitol, 50 mm sucrose, 1 mm EDTA, 0.5 mm EGTA, 10 mm Tris-HCl, pH 7.4) and then centrifuged at 15,000 × g for 20 min. The pellet was considered to contain pure mitochondria.
Statistical Analyses
All data are shown as the means ± S.E. The means were compared by using, where appropriate, Student's paired t test or one-way analysis of variance with Tukey-Kramer test. A p value <0.05 was considered statistically significant. Analyses were performed by using both GraphPad InStat and SAS software.
Author Contributions
T. B. A. and R. J. C. designed the experiments and analyzed the data. R. J. C., T. B. A., and C. T. wrote the manuscript. T. B. A. and C. T. collected the samples. T. B. A. performed the experiments. M.-H. K. provided technical assistance. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Oriana Teran and Jinhee Kim for helpful discussions and Matthew Beke for help with the manuscript.
This work was supported by National Institutes of Health Grant R01-DK-94244 and the Boston Family Endowment of the University of Florida Foundation (to R. J. C.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- HFD
- high fat diet
- IR
- insulin receptor
- PM
- plasma membrane
- EE
- early endosome
- LE
- late endosome
- IDE
- insulin-degrading enzyme
- GSK3
- glycogen synthase kinase 3
- GS
- glycogen synthase
- ZnA
- zinc-adequate diet
- ZnS
- zinc-supplemented diet
- ITT
- insulin tolerance test
- OGTT
- oral glucose tolerance test
- PTT
- pyruvate tolerance test
- qPCR
- quantitative PCR
- AAS
- atomic absorption spectrophotometry
- LAL
- limunus amoebocyte lysate
- AUC
- area under the curve
- 2NBDG
- 2-deoxy-2-[(7-nitro-2,1,3-benzoxadiazol-4-yl)amino]-d-glucose.
References
- 1. Moore M. C., Coate K. C., Winnick J. J., An Z., and Cherrington A. D. (2012) Regulation of hepatic glucose uptake and storage in vivo. Adv. Nutr. 3, 286–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mannucci E., Monami M., Lamanna C., and Adalsteinsson J. E. (2012) Post-prandial glucose and diabetic complications: systematic review of observational studies. Acta Diabetol. 49, 307–314 [DOI] [PubMed] [Google Scholar]
- 3. Nathan D. M., Buse J. B., Davidson M. B., Ferrannini E., Holman R. R., Sherwin R., and Zinman B. (2009) Medical management of hyperglycemia in type 2 diabetes: a consensus algorithm for the initiation and adjustment of therapy: a consensus statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care 32, 193–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Capdor J., Foster M., Petocz P., and Samman S. (2013) Zinc and glycemic control: a meta-analysis of randomised placebo controlled supplementation trials in humans. J. Trace Elem. Med. Biol. 27, 137–142 [DOI] [PubMed] [Google Scholar]
- 5. Jayawardena R., Ranasinghe P., Galappatthy P., Malkanthi R., Constantine G., and Katulanda P. (2012) Effects of zinc supplementation on diabetes mellitus: a systematic review and meta-analysis. Diabetol. Metab. Syndr. 4, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cousins R. J., Liuzzi J. P., and Lichten L. A. (2006) Mammalian zinc transport, trafficking, and signals. J. Biol. Chem. 281, 24085–24089 [DOI] [PubMed] [Google Scholar]
- 7. Jeong J., and Eide D. J. (2013) The SLC39 family of zinc transporters. Mol. Aspects Med. 34, 612–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Aydemir T. B., Sitren H. S., and Cousins R. J. (2012) The zinc transporter Zip14 influences c-met phosphorylation and hepatocyte proliferation during liver regeneration in mice. Gastroenterology 142, 1536–1546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Beyersmann D., and Haase H. (2001) Functions of zinc in signaling, proliferation and differentiation of mammalian cells. Biometals 14, 331–341 [DOI] [PubMed] [Google Scholar]
- 10. Pedrosa F. O., Pontremoli S., and Horecker B. L. (1977) Binding of Zn2+ to rat liver fructose-1,6-bisphosphatase and its effect on the catalytic properties. Proc. Natl. Acad. Sci. U.S.A. 74, 2742–2745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cowen L. A., Bell D. E., Hoadley J. E., and Cousins R. J. (1986) Influence of dietary zinc deficiency and parenteral zinc on rat liver fructose 1,6-bisphosphatase activity. Biochem. Biophys. Res. Commun. 134, 944–950 [DOI] [PubMed] [Google Scholar]
- 12. Brand I. A., and Kleineke J. (1996) Intracellular zinc movement and its effect on the carbohydrate metabolism of isolated rat hepatocytes. J. Biol. Chem. 271, 1941–1949 [DOI] [PubMed] [Google Scholar]
- 13. Kambe T., Tsuji T., Hashimoto A., and Itsumura N. (2015) The physiological, biochemical, and molecular roles of zinc transporters in zinc homeostasis and metabolism. Physiol. Rev. 95, 749–784 [DOI] [PubMed] [Google Scholar]
- 14. Chimienti F., Devergnas S., Favier A., and Seve M. (2004) Identification and cloning of a beta-cell-specific zinc transporter, ZnT-8, localized into insulin secretory granules. Diabetes 53, 2330–2337 [DOI] [PubMed] [Google Scholar]
- 15. Mitchell R. K., Hu M., Chabosseau P. L., Cane M. C., Meur G., Bellomo E. A., Carzaniga R., Collinson L. M., Li W. H., Hodson D. J., and Rutter G. A. (2016) Molecular genetic regulation of Slc30a8/ZnT8 reveals a positive association with glucose tolerance. Mol. Endocrinol. 30, 77–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sladek R., Rocheleau G., Rung J., Dina C., Shen L., Serre D., Boutin P., Vincent D., Belisle A., Hadjadj S., Balkau B., Heude B., Charpentier G., Hudson T. J., Montpetit A., et al. (2007) A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature 445, 881–885 [DOI] [PubMed] [Google Scholar]
- 17. Boesgaard T. W., Zilinskaite J., Vänttinen M., Laakso M., Jansson P. A., Hammarstedt A., Smith U., Stefan N., Fritsche A., Häring H., Hribal M., Sesti G., Zobel D. P., Pedersen O., Hansen T., et al. (2008) The common SLC30A8 Arg325Trp variant is associated with reduced first-phase insulin release in 846 non-diabetic offspring of type 2 diabetes patients: the EUGENE2 study. Diabetologia 51, 816–820 [DOI] [PubMed] [Google Scholar]
- 18. Nicolson T. J., Bellomo E. A., Wijesekara N., Loder M. K., Baldwin J. M., Gyulkhandanyan A. V., Koshkin V., Tarasov A. I., Carzaniga R., Kronenberger K., Taneja T. K., da Silva Xavier G., Libert S., Froguel P., Scharfmann R., et al. (2009) Insulin storage and glucose homeostasis in mice null for the granule zinc transporter ZnT8 and studies of the type 2 diabetes-associated variants. Diabetes 58, 2070–2083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hardy A. B., Wijesekara N., Genkin I., Prentice K. J., Bhattacharjee A., Kong D., Chimienti F., and Wheeler M. B. (2012) Effects of high-fat diet feeding on Znt8-null mice: differences between beta-cell and global knockout of Znt8. Am. J. Physiol. Endocrinol. Metab. 302, E1084–E1096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Huang L., Kirschke C. P., Lay Y. A., Levy L. B., Lamirande D. E., and Zhang P. H. (2012) Znt7-null mice are more susceptible to diet-induced glucose intolerance and insulin resistance. J. Biol. Chem. 287, 33883–33896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. King J. C., Hambidge K. M., Westcott J. L., Kern D. L., and Marshall G. (1994) Daily variation in plasma zinc concentrations in women fed meals at six-hour intervals. J. Nutr. 124, 508–516 [DOI] [PubMed] [Google Scholar]
- 22. Aydemir T. B., Chang S. M., Guthrie G. J., Maki A. B., Ryu M. S., Karabiyik A., and Cousins R. J. (2012) Zinc transporter ZIP14 functions in hepatic zinc, iron and glucose homeostasis during the innate immune response (endotoxemia). PLoS One 7, e48679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cani P. D., Amar J., Iglesias M. A., Poggi M., Knauf C., Bastelica D., Neyrinck A. M., Fava F., Tuohy K. M., Chabo C., Waget A., Delmée E., Cousin B., Sulpice T., Chamontin B., et al. (2007) Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 56, 1761–1772 [DOI] [PubMed] [Google Scholar]
- 24. Guthrie G. J., Aydemir T. B., Troche C., Martin A. B., Chang S. M., and Cousins R. J. (2015) Influence of ZIP14 (slc39A14) on intestinal zinc processing and barrier function. Am. J. Physiol. Gastrointest. Liver Physiol. 308, G171–G178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Troche C., Aydemir T. B., and Cousins R. J. (2016) Zinc transporter Slc39a14 regulates inflammatory signaling associated with hypertrophic adiposity. Am. J. Physiol. Endocrinol. Metab. 310, E258–E268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ozcan U., Cao Q., Yilmaz E., Lee A. H., Iwakoshi N. N., Ozdelen E., Tuncman G., Görgün C., Glimcher L. H., and Hotamisligil G. S. (2004) Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 306, 457–461 [DOI] [PubMed] [Google Scholar]
- 27. Eisenberg M. L., Maker A. V., Slezak L. A., Nathan J. D., Sritharan K. C., Jena B. P., Geibel J. P., and Andersen D. K. (2005) Insulin receptor (IR) and glucose transporter 2 (GLUT2) proteins form a complex on the rat hepatocyte membrane. Cell. Physiol. Biochem. 15, 51–58 [DOI] [PubMed] [Google Scholar]
- 28. Ceresa B. P., Kao A. W., Santeler S. R., and Pessin J. E. (1998) Inhibition of clathrin-mediated endocytosis selectively attenuates specific insulin receptor signal transduction pathways. Mol. Cell. Biol. 18, 3862–3870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fagerholm S., Ortegren U., Karlsson M., Ruishalme I., and Strålfors P. (2009) Rapid insulin-dependent endocytosis of the insulin receptor by caveolae in primary adipocytes. PLoS One 4, e5985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Holliday N. D., Holst B., Rodionova E. A., Schwartz T. W., and Cox H. M. (2007) Importance of constitutive activity and arrestin-independent mechanisms for intracellular trafficking of the ghrelin receptor. Mol. Endocrinol. 21, 3100–3112 [DOI] [PubMed] [Google Scholar]
- 31. Elchebly M., Payette P., Michaliszyn E., Cromlish W., Collins S., Loy A. L., Normandin D., Cheng A., Himms-Hagen J., Chan C. C., Ramachandran C., Gresser M. J., Tremblay M. L., and Kennedy B. P. (1999) Increased insulin sensitivity and obesity resistance in mice lacking the protein tyrosine phosphatase-1B gene. Science 283, 1544–1548 [DOI] [PubMed] [Google Scholar]
- 32. Tamaki M., Fujitani Y., Hara A., Uchida T., Tamura Y., Takeno K., Kawaguchi M., Watanabe T., Ogihara T., Fukunaka A., Shimizu T., Mita T., Kanazawa A., Imaizumi M. O., Abe T., et al. (2013) The diabetes-susceptible gene SLC30A8/ZnT8 regulates hepatic insulin clearance. J. Clin. Invest. 123, 4513–4524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shen Y., Joachimiak A., Rosner M. R., and Tang W. J. (2006) Structures of human insulin-degrading enzyme reveal a new substrate recognition mechanism. Nature 443, 870–874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Authier F., Metioui M., Fabrega S., Kouach M., and Briand G. (2002) Endosomal proteolysis of internalized insulin at the C-terminal region of the B chain by cathepsin D. J. Biol. Chem. 277, 9437–9446 [DOI] [PubMed] [Google Scholar]
- 35. Welsh G. I., and Proud C. G. (1993) Glycogen synthase kinase-3 is rapidly inactivated in response to insulin and phosphorylates eukaryotic initiation factor eIF-2B. Biochem. J. 294, 625–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Besecker B., Bao S., Bohacova B., Papp A., Sadee W., and Knoell D. L. (2008) The human zinc transporter SLC39A8 (Zip8) is critical in zinc-mediated cytoprotection in lung epithelia. Am. J. Physiol. Lung Cell. Mol. Physiol. 294, L1127–L1136 [DOI] [PubMed] [Google Scholar]
- 37. Schmitt-Ulms G., Ehsani S., Watts J. C., Westaway D., and Wille H. (2009) Evolutionary descent of prion genes from the ZIP family of metal ion transporters. PLoS One 4, e7208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Grasso G., Pietropaolo A., Spoto G., Pappalardo G., Tundo G. R., Ciaccio C., Coletta M., and Rizzarelli E. (2011) Copper(I) and copper(II) inhibit Aβ peptides proteolysis by insulin-degrading enzyme differently: implications for metallostasis alteration in Alzheimer's disease. Chemistry 17, 2752–2762 [DOI] [PubMed] [Google Scholar]
- 39. Araki N., Yokota S., Takashima Y., and Ogawa K. (1995) The distribution of cathepsin D in two types of lysosomal or endosomal profiles of rat hepatocytes as revealed by combined immunocytochemistry and acid phosphatase enzyme cytochemistry. Exp. Cell Res. 217, 469–476 [DOI] [PubMed] [Google Scholar]
- 40. Leissring M. A., Malito E., Hedouin S., Reinstatler L., Sahara T., Abdul-Hay S. O., Choudhry S., Maharvi G. M., Fauq A. H., Huzarska M., May P. S., Choi S., Logan T. P., Turk B. E., Cantley L. C., et al. (2010) Designed inhibitors of insulin-degrading enzyme regulate the catabolism and activity of insulin. PLoS One 5, e10504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pinilla-Tenas J. J., Sparkman B. K., Shawki A., Illing A. C., Mitchell C. J., Zhao N., Liuzzi J. P., Cousins R. J., Knutson M. D., and Mackenzie B. (2011) Zip14 is a complex broad-scope metal-ion transporter whose functional properties support roles in the cellular uptake of zinc and nontransferrin-bound iron. Am. J. Physiol. Cell Physiol. 301, C862–C871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Murphy R. F. (1988) Processing of endocytosed material. In Advances in Molecular and Cell Biology (Kenneth R. M., ed) pp. 159–180, Elsevier [Google Scholar]
- 43. Sipe D. M., Jesurum A., and Murphy R. F. (1991) Absence of Na+,K+-ATPase regulation of endosomal acidification in K562 erythroleukemia cells: analysis via inhibition of transferrin recycling by low temperatures. J. Biol. Chem. 266, 3469–3474 [PubMed] [Google Scholar]
- 44. Etzel K. R., and Cousins R. J. (1983) Hyperglycemic action of zinc in rats. J. Nutr. 113, 1657–1663 [DOI] [PubMed] [Google Scholar]
- 45. Rognstad R. (1984) Inhibition of glycogen synthesis in rat hepatocytes by medium Zn2+. Biochem. Biophys. Res. Commun. 122, 726–733 [DOI] [PubMed] [Google Scholar]
- 46. Yabaluri N., and Bashyam M. D. (2010) Hormonal regulation of gluconeogenic gene transcription in the liver. J. Biosci. 35, 473–484 [DOI] [PubMed] [Google Scholar]
- 47. Jitrapakdee S. (2012) Transcription factors and coactivators controlling nutrient and hormonal regulation of hepatic gluconeogenesis. Int. J. Biochem. Cell Biol. 44, 33–45 [DOI] [PubMed] [Google Scholar]
- 48. Aydemir T. B., Liuzzi J. P., McClellan S., and Cousins R. J. (2009) Zinc transporter ZIP8 (SLC39A8) and zinc influence IFN-γ expression in activated human T cells. J. Leukocyte Biol. 86, 337–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Elia G. (2012) Cell surface protein biotinylation for SDS-PAGE analysis. Methods Mol. Biol. 869, 361–372 [DOI] [PubMed] [Google Scholar]
- 50. Suski J. M., Lebiedzinska M., Wojtala A., Duszynski J., Giorgi C., Pinton P., and Wieckowski M. R. (2014) Isolation of plasma membrane-associated membranes from rat liver. Nat. Protoc. 9, 312–322 [DOI] [PubMed] [Google Scholar]
- 51. de Araùjo M. E., Huber L. A., and Stasyk T. (2008) Isolation of endocitic organelles by density gradient centrifugation. Methods Mol. Biol. 424, 317–331 [DOI] [PubMed] [Google Scholar]
- 52. Stasyk T., Schiefermeier N., Skvortsov S., Zwierzina H., Peränen J., Bonn G. K., and Huber L. A. (2007) Identification of endosomal epidermal growth factor receptor signaling targets by functional organelle proteomics. Mol. Cell. Proteomics 6, 908–922 [DOI] [PubMed] [Google Scholar]
- 53. Clayton D. A., and Shadel G. S. (2014) Isolation of mitochondria from cells and tissues. Cold Spring Harbor Protoc. pdb.top074542 [DOI] [PubMed] [Google Scholar]
- 54. Song Y., Hao Y., Sun A., Li T., Li W., Guo L., Yan Y., Geng C., Chen N., Zhong F., Wei H., Jiang Y., and He F. (2006) Sample preparation project for the subcellular proteome of mouse liver. Proteomics 6, 5269–5277 [DOI] [PubMed] [Google Scholar]