

Abstract
Legionella pneumophila is a causative agent of a severe pneumonia, known as Legionnaires' disease. Legionella pathogenicity is mediated by specific virulence factors, called bacterial effectors, which are injected into the invaded host cell by the bacterial type IV secretion system. Bacterial effectors are involved in complex interactions with the components of the host cell immune and signaling pathways, which eventually lead to bacterial survival and replication inside the mammalian cell. Structural and functional studies of bacterial effectors are, therefore, crucial for elucidating the mechanisms of Legionella virulence. Here we describe the crystal structure of the LpiR1 (Lpg0634) effector protein and investigate the effects of its overexpression in mammalian cells. LpiR1 is an α-helical protein that consists of two similar domains aligned in an antiparallel fashion. The hydrophilic cleft between the domains might serve as a binding site for a potential host cell interaction partner. LpiR1 binds the phosphate group at a conserved site and is stabilized by Mn2+, Ca2+, or Mg2+ ions. When overexpressed in mammalian cells, a GFP-LpiR1 fusion protein is localized in the cytoplasm. Intracellular signaling antibody array analysis revealed small changes in the phosphorylation state of several components of the Akt signaling pathway in HEK293T cells overexpressing LpiR1.
Keywords: bacterial pathogenesis, cell signaling, crystal structure, protein evolution, protein-protein interaction, cellular localization, Legionella infection
Introduction
Legionella pneumophila is a facultative intracellular parasite of fresh water protozoans, which under certain conditions can gain access to the human respiratory system and cause severe pneumonia, known as Legionnaires' disease (1). L. pneumophila inhaled with contaminated aerosols are engulfed by pulmonary alveolar macrophages that serve as alternative host cells for the pathogen. Inside the eukaryotic cell L. pneumophila establishes a unique replicative niche called the Legionella-containing vacuole (2), which evades phagolysosomal fusion and thus supports infection progress. Legionella-containing vacuole formation and subversion of host cell immunity is mediated by injection of multiple virulence factors into the host cytoplasm via the type IV Dot/Icm secretion system (3, 4). Legionella has an unusually large and redundant repertoire of these virulence factors, called bacterial effectors. More than 300 bacterial effectors have been identified in Legionella (5); however, the cellular functions and biological activities of most Legionella effectors remain unknown, and structural information about these proteins is limited. Bacterial effectors are products of co-evolution of bacteria and their eukaryotic hosts, and many of them bear unique functional and structural aspects, which must be elucidated to understand the mechanisms of Legionella virulence.
LpiR1 (Legionella protein with an internal repeat, encoded by the lpg0634 gene) is a translocated effector (5) with as yet unknown function. Most recent analysis found LpiR1 in 21 of 41 analyzes genomes of Legionella isolates (6), Many of the isolates that do not contain this gene have not been associated with infection in humans. Moreover, our own BLAST searches identified LpiR1 orthologs in 37 Legionella species, supporting the notion that LpiR1 plays a supporting role in infection. LpiR1 is a 449-amino-acid-long protein predicted to consist of mostly α-helical regions (PSIPRED prediction server; Ref. 7). A search for paralogs of LpiR1 using DELTA-BLAST (8) identified only one protein, the 220-amino-acid-long Lpg1851, which shares ∼22% sequence identity with the N-terminal segment of LpiR1. Both LpiR1 and Lpg1851 are encoded by single-gene operons (9) located in different regions of the L. pneumophila Philadelphia 1 chromosome (10). Both LpiR1 and Lpg1851 were reported to be up-regulated in the post-exponential growth phase of the bacterium; however, individual knock-out of these genes had no effect on the growth of Legionella within U937 macrophages or protozoa (11). The knock-out strains were tested for the contact-dependent pore formation in membranes of sheep red blood cells (SRBCs) and only LpiR1 was found to partially contribute to the hemolysis of the SRBCs (11). The GC content of the LpiR1 and Lpg1851 genes is 38.7% and 36.2%, respectively, which is close to the average GC content of the L. pneumophila Philadelphia 1 chromosome (38%) (10), suggesting that they are ancient genes of the Legionella genus rather then acquired by a horizontal transfer from another organism. Recently, the crystal structure of Lpg1851 was solved and deposited in the Protein Data Bank (PDB code 4HFV)2; however, function of this protein is also unknown.
Here we present the crystal structure of LpiR1 and investigate its cellular localization in the host cell. The N- and C-terminal domains have the same fold, indicative of the occurrence of a gene duplication event during evolution. Despite its low sequence identity with Lpg1851, their structures show remarkable similarity. Extensive sequence comparison with homologs of these two proteins identified sequence conservation pattern, which when mapped onto the three-dimensional structure allowed us to identify functionally important regions of these effectors.
Experimental Procedures
Expression Vector Construction
The ligation-independent cloning method was used to generate the expression plasmids. The LpiR1 gene (Q5ZXU7_LEGPH) encodes a 449-amino-acid-long protein.
BLAST sequence alignment showed that the majority of closely related sequences start at Met-5 in lpg0634, and we chose this as the correct start of the gene. Nevertheless, we retained the numbering according to the sequence in the NCBI and UNIPROT databases. Prediction of disordered regions suggested that the C-terminal ∼40 residues show disordered characteristics (DISOPRED prediction server; Ref. 12). Therefore two versions were cloned, Thr-6–Val-449 and Thr-6–Glu-404. The constructs were PCR-amplified from the genomic DNA of L. pneumophila strain Philadelphia 1. All primers used for amplification are listed in Table 1. The PCR products were cloned into the ligation-independent cloning vector pMCSG7 (13) according to the standard protocol (13). The pMCSG7 vector encodes the N-terminal His6 tag, separated from the target gene by a cleavage site for tobacco etch virus protease. The obtained plasmids pMTB212 (amino acids 6–449) and pMTB213 (amino acids 6–404) were verified by DNA sequencing. Site-directed mutagenesis was performed with Q5 Site-Directed Mutagenesis kit (New England BioLabs, Ipswich, MA) according to the manufacturer's instructions.
TABLE 1.
Primer used for construction of GFP and HA tagged LpiR1
F, forward; R, reverse.
| Primers | Sequence (5′-3′) | Restriction enzyme | Resulting plasmids |
|---|---|---|---|
| HA-F | GAACAAAGCTTATGGTCATGTACCCATACGAT | Hind III | pcDNA5/FRT/TO-HA |
| HA-R | TGGTAGGTACCAGCGTAATCTGGACCGTCATAT | Kpn I | |
| Lp0634 F | TAGCGGTACCACGTTTGTTTTAAAGGAATTTGATGCACT | Kpn I | pcDNA5/FRT/TO-HA-LpiR1 |
| Lp0634 R | TACCCTCGAGCTATACGGTCGGTTTGCTGGTCTCTTCT | Xho I | |
| Lp0634 GFP-F | TACCCTCGAGGCCACCATGACGTTTGTTTTAAAGGAATTTGATGCACT | XhoI | pEGFP-LpiR1 |
| Lp0634 GFP-R | GCTTGGTACCGTTACGGTCGGTTTGCTGGTCTCTTCT | KpnI | |
| Lpg1851 GFP-F | TACCCTCGAGATGTCATTTGAATTGGTCGCATATG | XhoI | pEGFP-Lpg1851 |
| Lpg1851 GFP-R | GCTTGGTACCGTAAGGCGATGATTTGCAACCTCAT | KpnI |
For protein localization studies of LpiR1 and Lpg1851 two sets of constructs were prepared having either a C-terminal GFP or an N-terminal HA3 tag. For the GFP-tagged constructs, the full-length LpiR1 and Lpg1851 were PCR-amplified from the genomic DNA of L. pneumophila strain Philadelphia 1. The amplified DNA segments were digested with XhoI and KpnI and ligated to the same restriction sites of pEGFP-N1 expression vector (Clontech Laboratories, Mountain View, CA).
For the HA-tagged constructs the vector pcDNA5/FRT/TO (14) was used. A triple hemagglutinin (HA3) tag was inserted into the pcDNA5/FRT/TO expression vector using HindIII and KpnI restriction sites. Restriction enzymes and T4 DNA ligase were purchased from New England BioLabs. The lpg0634 open reading frame was PCR-amplified to include KpnI and XhoI restriction sites for cloning purposes. The fragment was cloned into pcDNA5/FRT/TO-HA vector using the same restriction sites.
Expression and Purification of LpiR1
Competent BL21(DE3) cells were transformed with the expression vectors described in the previous section, and single colonies were used to inoculate LB media (50 ml, 100 μg/ml ampicillin, 0.4% glucose). Cells were grown at 37 °C overnight. The next day 1 liter of TB medium (100 μg/ml ampicillin) was inoculated with 50 ml of the overnight culture and grown at 37 °C with shaking at 220 rpm. Isopropyl β-d-1-thiogalactopyranoside (1 mm) was added when the A600 reached 2.0, and cells were cultivated for another 16 h at 18 °C and then harvested by centrifugation for 20 min at 5000 × g). The same expression strain, cultivated in conditions promoting Se-Met3 incorporation by metabolic inhibition of the methionine pathway (15), was used to produce the selenomethionine derivative of LpiR1(6–404).
Cell pellets were resuspended in buffer A (50 mm Tris, pH 7.7, 400 mm NaCl, 5% glycerol, 2 mm imidazole, 0.5 mm tris(2-carboxyethyl)phosphine (TCEP)) with 1 mm p-aminobenzamidine and 1 mm 4-(2-aminoethyl) benzenesulfonyl fluoride hydrochloride (AEBSF) as protease inhibitors and lysed with high pressure cell disrupter TS series Benchtop (Constant Systems Ltd, UK) at 35 p.s.i. After centrifugation for 1 h at 16,000 × g cleared cell lysate was loaded onto 5 ml of TALON Metal Affinity Resin (Clontech Laboratories). The protein was purified using batch protocol; after 1 h of incubation with cell lysate at 4 °C the resin was washed with 20 resin volumes of buffer A, and the protein was eluted with buffer A containing an additional 100 mm imidazole. The protein was dialyzed against 2 liters of buffer B (20 mm HEPES, pH 8.0, 150 mm NaCl, 0.5 mm tris(2-carboxyethyl)phosphine), cleaved with tobacco etch virus protease (1:50 (w:w) ratio, 16 h at 4 °C) to remove the His6 tag, and stored at −80 °C. Before crystallization the protein was subjected to gel filtration on a Superdex 200 10/300 GL column (GE Healthcare) in buffer C (15 mm HEPES, pH 7.7, 50 mm NaCl, 0.5 mm tris(2-carboxyethyl)phosphine), the peak fractions corresponding to purified protein were pooled and concentrated to 25 mg/ml.
Protein Melting Temperature
Experiments were performed with the fluorescent dye Sypro Orange (Molecular Probes) using Applied Biosystems StepOnePlus Real Time PCR Instrument (Life Technologies) according to the standard protocol developed by the manufacturer.
Isothermal Titration Calorimetry
Experiments were performed on the Nano ITC instrument (TA Instruments, New Castle, DE). 1 mm MnCl2 in 15 mm HEPES, pH 7.5, 0.1 m NaCl, 0.5 mm tris(2-carboxyethyl)phosphine was titrated into the calorimeter cell containing 0.1 mm LpiR1(6–449) in the same buffer. Experiments were performed at 20 °C, and data were analyzed with NanoAnalyze software (TA Instruments) using and independent model.
Crystallization
The initial crystallization conditions for LpiR1(6–449) and LpiR1(6–404) were identified with the pHClear screen (Qiagen, Toronto, ON) and were optimized using the hanging drop vapor diffusion method. Two different crystallization conditions produced diffraction quality crystals: (i) 10% polyethylene glycol (PEG) 6000, 0.1 m sodium citrate, pH 4.0, at 20 °C rendered crystals of both full-length and truncated LpiR1; (ii) 40% (±)-2-methyl-2,4-pentanediol, 0.1 m MES, pH 6.5, at 4 °C produced better diffracting crystals of truncated LpiR1 and its Se-Met derivative. For data collection, crystals were soaked in a cryo-protectant (reservoir solution supplemented with 25% ethylene glycol for the first condition or straight reservoir solution for the second condition) and flash-cooled in liquid nitrogen.
X-ray Data Collection, Structure Solution, and Refinement
Crystals of native and Se-Met derivatized truncated LpiR1(6–404) produced in the second crystallization condition diffracted to 1.75 and 2.3 Å resolution, respectively, whereas the crystals of full-length LpiR1 diffracted to 2.4 Å resolution. The x-ray diffraction data were collected at 100 K at the 08ID-CMCF beamline at the Canadian Light Source (Saskatoon, Saskatchewan, Canada) (16). Diffraction data from the Se-Met crystals were collected at the selenium absorption edge wavelength 0.97949 Å The native and Se-Met datasets were processed and scaled in HKL-3000 (17). Initial phasing and model building were performed with the same software package. The model was then refined against a higher resolution native dataset using PHENIX software package (18). Iterative cycles of refinement were carried out using PHENIX, and visualization and manual rebuilding were performed with Coot (19). The final Rwork and Rfree values are 0.173 and 0.203, respectively. Pertinent details of data collection and refinement statistics are given in Table 2. We have also collected diffraction data for the crystal of full-length LpiR1(6–449) that diffracted only to 3.8 Å resolution. This structure was solved by molecular replacement method using LpiR1(6–404) as a search model. There was no interpretable electron density for the C-terminal segment beyond Glu-404, confirming that this segment is indeed disordered. The coordinates and structure factors of LpiR1(6–404) at 1.75 Å resolution were deposited in the PDB (entry 5FIA).
TABLE 2.
Summary of data collection and refinement statistics
The information for the highest resolution shell (1.82–1.75 Å) is given in parentheses. r.m.s.d., root mean squares deviation.
| LpiR1 form 1 (native) | LpiR1 form 1 (SeMet) | LpiR1 form2 | |
|---|---|---|---|
| Wavelength (Å) | 0.9795 | 0.9786 | 0.9795 |
| Resolution range (Å) | 50-1.75 (1.80-1.75) | 50-2.20 (2.28-2.20) | 50-2.4 (2.54-2.4) |
| Space group | P21 | P21 | P212121 |
| Cell parameters a, b, c (Å), β(○) | 81.4, 71.8, 94.2, 112.0 | 81.2, 72.0, 94.2, 112.1 | 83.8, 96.2, 151.4 |
| Total reflections | 46,4476 | 387,445 | 643,580 |
| Unique reflections | 100,375 | 51,325 | 48,632 |
| Multiplicity | 4.6 (4.7) | 7.5 (7.58) | 13.1 (13.0) |
| Completeness (%) | 98.7 (97.7) | 99.8 (99.7) | 99.7 (98.4) |
| Mean I/Sigma(I) | 17.39 (2.2) | 12.1 (2.53) | 13.7 (2.56) |
| Wilson B-factor (Å2) | 35.7 | 39.5 | 35.1 |
| Rmerge | 0.05 (0.86) | 0.125 (0.87) | 0.162 (1.0) |
| Rmeas | 0.057 (0.97) | 0.135 (0.94) | 0.175 (1.12) |
| CC1/2 | 0.999 (0.740) | 0.998 (0.835) | 0.998 (0.755) |
| Rwork | 0.170 | 0.171 | |
| Rfree | 0.201 | 0.213 | |
| B-factor (Å2) | |||
| Protein | 37.9 | 52.0 | |
| Solvent | 43.3 | 43.4 | |
| Ramachandran plot | |||
| Favored (%) | 98.9 | 98.8 | |
| Allowed (%) | 0.8 | 1.16 | |
| Outliers (%) | 0.3 | 0.0 | |
| Clash score | 3.24 | 4.29 | |
| r.m.s.d. | |||
| Bonds (Å) | 0.017 | 0.013 | |
| Angles (○) | 1.36 | 1.17 | |
| PDB code | 5FIA | 5JG4 |
The stereochemistry was validated with ADIT validation server at Research Collaboratory for Structural Bioinformatics, Rutgers University. Structure based sequence alignments were generated with BLAST (8). Structure superposition and calculation of root mean square deviation were performed with the program Swiss PDB Viewer v. 4.1 (20).
Phylogenetic Analysis
The N-terminal domain of LpiR1 was used as a template for the PSI-BLAST search. All the sequences with identity >20% and target coverage >80% were considered homologous. The sequences were aligned with PROMALS3D server (21) based on the structures of LpiR1 and Lpg1851 and the secondary structure predictions for all other proteins. The multiple sequence alignment was then used as an input for MEGA6 (22). The evolutionary history was inferred by using the Maximum Likelihood method based on the JTT matrix-based model (23). The topology of the phylogenetic tree was verified by bootstrap resampling analysis with 500 iterations (24).
Transient Transfection of HEK293T Cells
Human embryonic kidney cell line 293T (HEK293T) was cultured in Dulbecco's modified Eagle's medium (Sigma) supplemented with 10% fetal bovine serum (FBS) (Sigma) at 37 °C with 5% CO2. To express GFP-tagged LpiR1 effector in mammalian cells, the PCR-amplified gene was inserted into the pEGFP-N1 vector (Clontech Laboratories) rendering pEGFP-N1-LpiR1. DNA constructs were transfected into HEK293T cells using the X-treme GENE™ HP DNA Transfection Reagent (Roche Applied Science, catalog #06366236001) according to the manufacturer's instructions. Cells were visualized on a laser scanner confocal microscope (Zeiss LSM700).
Immunofluorescence
HA3-tagged LpiR1-expressing cells were grown on 12-mm diameter glass coverslips. Cells were fixed with 4% paraformaldehyde solution made up in PBS for 30 min at room temperature, washed in PBS, permeabilized with 0.5% Triton X-100 in PBS-Tween, and blocked in 5% normal horse serum for 20 min. Cells were incubated with primary anti-HA antibody (sc-7392, Santa Cruz Biotechnology Inc.) diluted 1:200 for 60 min at room temperature in the blocking solution. Cells were washed with PBS-Tween, and a secondary Alexa Fluor 488-conjugated goat anti-mouse IgG (Invitrogen) was overlaid on coverslips for 20 min at room temperature at a dilution of 1:2000 in blocking solution. Slides were mounted onto 1-mm glass slides and visualized on a Laser Scanner Confocal Microscope (Zeiss LSM700).
Intracellular Signaling Array
The cells transfected with the empty vector or vector containing HA3-LpiR1 were cultured with media containing 0.1% FBS for 24 h then treated with 100 ng/ml IGF-I (R&D systems, Minneapolis, MN) for 5 min. The cells were harvested and lysed on ice with 0.5 ml of lysis buffer (50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 1% Nonidet P-40, and 10% glycerol, 1 × complete protease inhibitor mixture) for 15 min. The lysates were centrifuged at 10,000 × g at 4 °C for 10 min. Intracellular signaling molecules were detected using a PathScan® intracellular signaling array kit (Cell Signaling Technology, #7744) according to the manufacturer's procedure. The fluorescent images of the slide were captured with Odyssey® Infrared Imaging System (LI-COR) and intensities of the spots were quantified using image studio analysis software.
Results and Discussion
Overall Structure of LpiR1
The LpiR1(6–449) yielded initial crystals diffracting only to 3.8 Å resolution. To enhance the quality of crystals we designed a truncated construct LpiR1(6–404) lacking the C-terminal region that was predicted to be partly disordered by DISOPRED server (supplemental Fig. S1) (12). This protein gave crystals diffracting to 1.75 Å resolution with (±)-2-methyl-2,4-pentanediol as a precipitant. At a later time we were able to optimize crystallization conditions for LpiR1(6–449) and have obtained crystals with PEG6000 that diffracted to 2.4 Å resolution. However, the last ∼50 residues were not visible in the electron density, confirming their predicted disorder. The visible part of LpiR1 was very similar to the higher resolution structure of truncated LpiR1. We speculate that the function of the C-terminal ∼50 residues might be in binding the chaperone essential for LpiR1 secretion.
LpiR1(6–404) crystals contain two molecules in the asymmetric unit. Superposition of the two chains revealed high overall similarity (root mean square deviation = 0.7 Å for 483 Cα atoms). The loop Pro-340–Thr-348 is well defined in molecule B but is disordered in molecule A. Fig. 1A shows the schematic representation of molecule B.
FIGURE 1.

The overall structure of LpiR1. A, schematic representation of LpiR1. The N-terminal domain is colored wheat, the C-terminal domain is magenta. B, the fold of the N-terminal domain is in rainbow colors, from blue at the N terminus to red at the C terminus. The up-and-down four helix bundle is shown on the left with α5-α6 packing on one side perpendicular to the helix bundle axis. C, superposition of the N- and C-terminal domains of LpiR1. Colors are as in panel A with the exception of the parts that differ in conformation in the N- and C-terminal domains, which are colored green (N-terminal domain) or blue (C-terminal domain). Structural representations were prepared with the program PyMOL.
LpiR1 is an α-helical protein and consists of two domains with a similar fold. Each domain contains six α-helices. The first four helices (α1-α4 and α7-α10) form an up-and-down antiparallel helical bundle, and the two C-terminal helices (α5-α6, α11-α12) pack nearly perpendicularly to the axis of the four-helix bundle and contact helices α2-α3 (Fig. 1B). The sixth helix (α6, α12) is the longest, with six (α6) and eight (α12) turns. The other equivalent helices in the two domains are of similar lengths. An ∼20-amino-acid-long linker connects the last N-terminal domain helix α6 to the first C-terminal domain helix α7. The two domains can be superimposed with root mean square deviation of 1.3 Å for 118 Cα atoms (of 182 common Cαs) (Fig. 1C), but their sequences show only 15.7% identity. The conformational differences between these two domains are restricted to the loop regions, in particular Leu-90–Ser-99 (Tyr-288–Thr-301 in C-domain), and a somewhat different bend of the C-terminal helices α6 and α12 (Fig. 1C).
The two domains associate in an antiparallel fashion with the main contacts formed between helices α6 and α12. Additional interdomain contacts are formed by the C-terminal parts of helices α5 and α11 and the loops following these helices. Helices α5, α6, α11, and α12 delineate a groove that extends across the entire width of the molecule and is ∼25 Å long and 8 Å wide, with α6 and α12 forming the floor and α5 and α11 forming the sides of the groove. One end of the groove is formed by the 340–349 loop that is flexible and observed in the electron density only in molecule B. The residues lining the groove have a largely hydrophilic character (Fig. 2A). Such a long and deep hydrophilic groove suggests a plausible binding site for an extended polypeptide presumably from the LpiR1 host target.
FIGURE 2.

Residue lining the interdomain groove and residue conservation in LpiR1 homologs. A, a close-up view of the residues lining the interdomain groove. The helices α5–6 and α11–12 are shown in schematic representation. Side chains pointing into the groove are shown in stick representation. The groove has a strongly hydrophilic character. B, surface representation of LpiR1 colored according to the level of conservation of residues, which was determined by the program CONSURF (31). The orientation is similar to that shown in A. The color assignment is indicated on the attached scale. Insets show conserved residues in Cluster 1 and Cluster 2 under a semitransparent surface. Dashed green lines represent hydrogen bonds, the red oval shows the location of Cluster 1, and the dashed white oval indicates the location of Cluster 2 on the underside of molecule as depicted in this view. C, the location of phosphate ion coordinated by residues from the conserved site 2. The difference electron density map is drawn at 7σ level and colored green. The map was calculated after refinement of the LpiR1 model and before the phosphate groups were added. The ion forms seven hydrogen bonds (dashed blue lines) to the protein, one additional through a bridging water molecule and to two other waters. D, docking of Thr(P) (left) or Ala-Tyr(P)-Pro tripeptide to the phosphate binding site. The phosphate groups were superimposed on the observed position of the phosphate. E, stereo view of the Mn2+ binding site. The initial difference electron density map is drawn at 3σ (green) and 7σ (magenta) level with the refined model near the cation binding site. Glu-141, Arg-143, and Asp-322 are from conserved site 1. Asp-320 forms a salt bridge with Arg-143.
Comparison with Known Structures
To identify proteins with similar fold in the Protein Data Bank we have used several servers including DALI (25), HHPRED (26), and PHYRE2 (27). Each search identified only one significant match over the entire domain, namely Lpg1851 (PDB code 4HFV) that mapped to one of the domains of LpiR1. No protein in the PDB contained both domains of LpiR1. Therefore, Lpg1851 and LpiR1 domains represent a new fold.
Sequence Conservation and Evolutionary Relationship between LpiR1 Orthologs
To gain insight into potential function of LpiR1, we have searched for its orthologs in bacteria using DELTA-BLAST (8). Proteins of similar length and with sequence identity exceeding ∼25% are from various Legionella strains. There is a second cluster of proteins detected by a BLAST search that are roughly half the size of LpiR1 and that show ∼22–28% sequence identity to either the N- or C-terminal domain of LpiR1. Lpg1851 belongs to this protein cluster. Multiple sequence alignment of LpiR1 orthologs with 25% or higher sequence identity showed 30 residues conserved in all these sequences (14 sequences, only one representative of proteins with 99% identity was included for analysis of the conservation pattern). Most of these residues are hydrophobic, located in the core of each domain. Several highly conserved side chains are solvent-accessible, and they cluster in two regions (Fig. 2B). The first cluster lies within one end of the groove described above and is formed by the side chains of Asp-141, Ser-142, Arg-143, Glu-322, Arg-381, and Ala-384. There are two salt bridges between these conserved side chains: Asp-141–Arg-143 and Glu-322–Arg-381 (Fig. 2B, Cluster 1). The arrangement of these conserved residues does not resemble known active sites. They contribute to the stabilization of the relative orientation of the N- and C-terminal domains preserving the shape of the groove. Indeed, the mutation of either of these two arginines (R143A or R381A) lowers the melting temperature, Tm, of LpiR1 (51.5 °C for a wild-type LpiR1) by ∼5 °C, whereas Tm of a double mutant drops to 40 °C. The second cluster is located on the surface of the C-terminal domain and distant from the groove (Fig. 2B, Cluster 2). Conserved residues are Glu-226, Lys-230, Arg-242, and Gln-245 (Fig. 2B, insert). They form two pairs connected by hydrogen bonds, namely Glu-226···Lys-230 and Arg-242···Gln-245, and their conservation, like that of several hydrophobic residues in the cores of both domains, has structural as well as functional significance.
Interestingly, in the structure of full-length LpiR1 we have observed nearby strong electron density (∼10σ difference map peak) that could be well fitted with a SO42− or PO42− ion. The oxygen atoms of this ion make seven hydrogen bonds with side chains of Glu-226, Lys-230, Arg-242, Thr-301, and NH of Val-302 and Ile-303, and one additional hydrogen bond through a bridging water to the carbonyl group of Ala-300 and to two additional waters (Fig. 2C). These multiple hydrogen bonds to conserved residues suggest a functional role for this site. We speculate that this is a phosphate binding site that recognizes either a phosphoserine/threonine/tyrosine (Fig. 2D) or a phospholipid head group. Indeed, docking Tyr(P)-containing tripeptide into this site can be done without collision with LpiR1. Binding Thr(P)-containing peptide might require small adjustments of LpiR1. The location of this site on the opposite face of LpiR1 to the surface canyon and the other conserved cluster of residues would suggest that binding through this site orients LpiR1 to expose the canyon for interaction with another target.
Stabilizing Effect of Divalent Cations
We have investigated the effect of metal ions and several other compounds on the stability of LpiR1 through monitoring the melting temperature. The strongest stabilizing effect was observed for Mn2+, which increased the LpiR1 Tm by ∼3.5 °C followed by Ca2+ and Mg2+, which increased Tm by ∼2 °C and 1 °C, respectively (supplemental Fig. S2). Co2+ and Ni2+ produced poorly interpretable melting curves, indicating that these metals might destabilize the structure of LpiR1. We also tested ATP and found no indication of binding. Isothermal titration calorimetry confirmed that Mn2+ binds to LpiR1 with Kd values of 9 μm (supplemental Fig. S3). We, therefore, soaked crystals of LpiR1 with either MnCl2 or MgCl2 and identified the metal binding site. Both metals bind at the same place on the protein surface in the C-terminal domain between the end of the loop leading from the C-to the N-terminal domain and the loop after helix α11. The metal ion has tetragonal bipyramidal coordination. The equatorial ligands are side-chain oxygens of Asp-316 and Asp-320 and two water molecules with distances of ∼2.0 Å whereas two axial ligands are water molecules with a distance of ∼2.4 Å (Fig. 2E). Two of these waters are hydrogen-bonded to Gln-208 and to carbonyl of Ile-205. Therefore, the increase of thermal stability relates to the strengthening of the connection between the two neighboring loops. Moreover, Asp-320 forms a salt bridge with Arg-143 from the conserved site 1 thus connecting this metal binding site with the conserved residue cluster.
LpiR1 and Lpg1851 Are Structural Paralogs
Lpg1851 was identified by sequence alignment as a potential paralog of LpiR1, albeit with a rather low sequence identity of ∼22%. Lpg1851 is another bacterial effector with as yet unknown function. It is only 220 amino acids long and aligns with one domain of LpiR1. Comparison of their structures shows that Lpg1851 has the same fold as each domain of LpiR1 (Fig. 3A). The helical bundles superimpose very well, but there are some differences in the orientation of helices α5-α6 relative to the bundle. Lpg1851 can be superimposed on the N- or C-terminal domains of LpiR1 with root mean square deviations of 1.35 Å for 67% and 1.53 Å for 61% of 191 Cα atoms, respectively. The structure-based alignment of 189 residues of Lpg1851 visible in the crystal structure shows 12 and 19% identity to the N- and C-domains of LpiR1, respectively (Fig. 3, A and B). Interestingly, Lpg1851 forms dimers through the α5-α6 interface in a manner very similar to the association of the two domains of LpiR1 (Fig. 3C). A groove similar to the one present at the interdomain interface in LpiR1 can also be seen at the interface of the Lpg1851 dimers. As a consequence of 2-fold symmetry and in variance with LpiR1, the groove is closed at both ends by a moderately conserved salt bridge Asp-12···Arg-144. Therefore, Lpg1851 displays a deep and long depression in the protein surface rather than a groove. The superposition of the Lpg1851 dimer and LpiR1 based only on helices α6 and α12 and their equivalences, which form the majority of the interdomain contacts, shows a moderate difference in the orientation of the two domains (Fig. 3C).
FIGURE 3.

Structural comparison of LpiR1 and Lpg1851. A, superposition of Lpg1851 (schematic representation, green) with N- and C-terminal domains of LpiR1 (Cα trace, wheat and magenta, respectively). B, sequence alignment of N and C domains of LpiR1 with Lpg1851 based on structural alignment. C, comparison of the LpiR1 monomer (wheat and magenta) with a dimer of Lpg1851 (light green and dark green), generated by the application of crystallographic symmetry, The superposition is based on the helices α6 and α12 and the equivalent helices in the Lpg1851 dimer, to emphasize small differences in the relative orientation of the two domains in these proteins. D, surface residue conservation of Lpg1851 in similar orientation to LpiR1 viewed from the side showing the interchain depression. Conservation was calculated by the program CONSURF. The inset shows the conserved residues in the largest surface exposed cluster, which is located on the underside of the dimer in the shown view and marked approximately by the white dashed oval.
The sequence conservation pattern of Lpg1851 and its homologs identified by DELTA-BLAST searches, which turned out also to be uniquely from Legionella strains, differs from that of LpiR1. The surface representation of the Lpg1851 dimer colored by the level of conservation based on alignment of 14 sequences with an identity >25% shows the most conserved surface-accessible residues are located at the edges of the dimer farthest away from the dimer interface and on the opposite side to the interdomain depression. The largest cluster of fully conserved surface residues is located in a shallow depression and includes Ser-45, Arg-48, Glu-51, Ser-89, Thr-90, and Tyr-91 (Fig. 3D). Arg-48 forms a salt bridge with Glu-51. This surface might be involved in binding to another molecule(s) rather than forming an active site, as it does not have the hallmarks of an active site for enzymatic reaction. In contrast to LpiR1, no residues lining the interchain depression are fully conserved, indicating that this area does not harbor an active site common to all orthologs.
When the sequences of homologs of LpiR1 and Lpg1851 are aligned together only three residues are fully conserved, Arg-42, Leu-49, and Leu-105 (LpiR1 numbering), and their roles appear to be related to maintaining structural integrity. Because neither LpiR1 nor Lpg1851 appear to be enzymes, we speculate that their function might be associated at least in part with the presence of a binding groove on their surface that is involved in binding to the host intracellular target and modifying its behavior. Because this groove is located at the domain interface, the dimeric form of the smaller Lpg1851 is likely its functional form, whereas the larger, two-domain LpiR1 acts as a monomer. Indeed, size exclusion chromatography confirmed the monomeric form of LpiR1 in solution (data not shown).
Evolutionary History
Analysis of protein structures available at the Protein Data Bank showed that the proteins with internal (pseudo)symmetrical repeats frequently have paralogs without (or with fewer copies of) the internal repeat but with a higher oligomerization state (28). Moreover, phylogenetic analysis of six families of proteins with internal repeats suggested that in the majority (but not all) of the cases they evolved from the ancestral protein without the repeat, as a result of gene duplication (28).
We have built a structure-based phylogenetic tree of all Lpg1851 and LpiR1 homologs in an attempt to trace the evolution of LpiR1. The obtained maximum-likelihood tree (Fig. 4) shows three major clusters with high support, corresponding to the homologs of Lpg1851 and each domain of LpiR1. There was also one weakly supported deep branch, consisting of Lpg1851 orthologs from Legionella oakridgensis and Legionella lansingensis but with a relatively low bootstrap value (51%), possibly resulting from a small range of sequences available for comparison. Extensive BLAST search yielded no LpiR1 orthologs present in these Legionella species. Consistently, the Legionella evolution tree based on 16S rRNA gene sequences showed that L. oakridgensis and L. lansingensis belong to a different clade than the Legionella spp. with both paralogs (29). The absence of another paralog in these species might indicate that the gene duplication that produced the two-domain ancestor of LpiR1 occurred after the divergence of these two Legionella clusters.
FIGURE 4.

Phylogenetic analysis of LpiR1 domains and their homologs by the Maximum Likelihood method. The percentage of replicate trees in which the associated proteins clustered together in the bootstrap test (500 replicates) are shown next to the branches. N-terminal and C-terminal domains of LpiR1 orthologs are indicated by N or C in front of the NCBI RefSeq protein accessions, respectively.
LpiR1 and Lpg1851 Are Localized in the Cytoplasm of Transfected Mammalian Cells
To investigate the subcellular localization of LpiR1 and Lpg1851, we tagged the C terminus of these proteins with GFP and overexpressed the fusion construct in the HEK293T cells. Cells transfected with a GFP-encoding vector served as a control. The subcellular localization of GFP, the LpiR1-GFP, and Lpg1851-GFP fusion proteins in HEK293T cells was examined using fluorescence microscopy. The GFP control was distributed throughout the cells, whereas LpiR1-GFP and Lpg1851-GFP were predominantly localized in the cell cytoplasm. The nucleus was stained with 4′,6-diamidino-2-phenylindole (DAPI). Merged images confirmed that LpiR1-GFP was evenly distributed in the cytoplasm (Fig. 5A). We subsequently tested localization of LpiR1 using a differently tagged construct, namely, the HA3-tagged LpiR1. The immunofluorescence confirmed localization of HA3-LpiR1 in the cytoplasm (Fig. 5B, lower panel), whereas HA3 tag alone was found in both nucleus and cytoplasm (Fig. 5B, upper panel). The localization of LpiR1 was also investigated in the HeLa cells (data not shown) with the same results.
FIGURE 5.
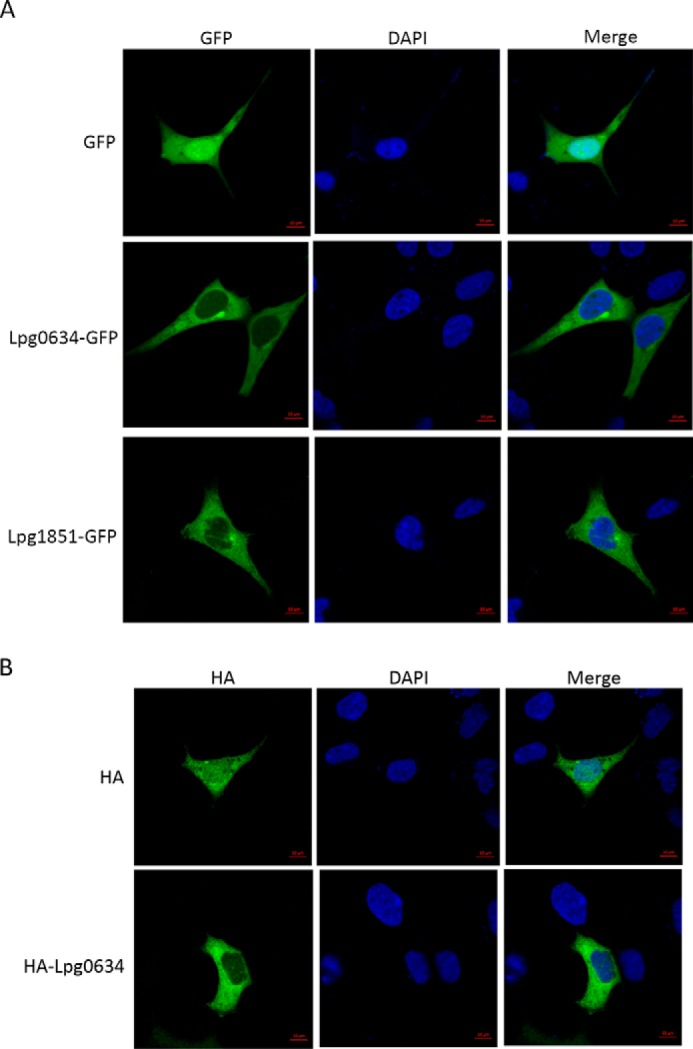
Subcellular localization of LpiR1-GFP and Lpg1851-GFP. A, confocal microscopy images of HEK293T cells 24 h after transfection with pEGFP-N1 (upper panel) or LpiR1-GFP (middle panel) or Lpg1851-GFP (lower panel). Green fluorescence indicates the position of GFP or LpiR1-GFP or Lpg1851-GFP, whereas the blue fluorescence (DAPI) indicates the position of the nucleus. B, immunofluorescence staining for HA3 (green, upper panel) or HA3-LpiR1 (green, lower panel) in HEK293T cells. The blue fluorescence (DAPI) indicates the position of the nucleus.
Effect of LpiR1 Overexpression on Intracellular Signaling Pathways
To identify the host signaling pathways that might be affected by overexpression of LpiR1, we have used the intracellular signaling antibody array kit PathScan® (Cell Signaling Technology, #7744). No significant changes in ERK1/2 and p38 pathways were detected. A small decrease in phosphorylation of some components of Akt pathway (mTOR-Ser-2448, Akt-Thr-308, STAT1-Tyr-701) was detected in IGF-stimulated cells transiently transfected with LpiR1; however, the observed effect was not stable (reproduced in four replicates of five; data not shown).
According to our localization studies, LpiR1 is found mainly in the cytoplasm. The protein is not toxic to mammalian cells and on its own triggers small or transient changes in the cellular signaling cascades. Based on the structural insight indicating the lack of enzymatic function, these effects are indirect and likely associated with modulating host protein(s) through direct binding. Binding of the phosphate ion to the conserved site 2 of LpiR1 and collision-free docking of Thr(P) or Tyr(P) suggest a functional role of this site as a recognition/binding site of phosphoprotein(s) or phospholipids that would orient the grove located on the opposite surface of LpiR1 and containing conserved site 1 for further functional interactions. Moreover, the micromolar binding constant for Mn2+ and Mg2+ suggests that additional stabilization of the protein occurs in the host cell. Of the many effector proteins identified in pathogenic bacteria, only a small number are absolutely essential for the infection process (30). The contribution of other effectors is cumulative and reflects to some extent their partially redundant nature, which increases the fitness of the pathogen. The relatively small effects we have observed are in line with these observations. The novel fold observed in LpiR1 and lpg1851 extends the known repertoire of protein folds and adds to the protein-protein interaction scaffolds.
Author Contributions
K. A. B., D. H. A., and M. C. designed the study and wrote the paper. M. T. B. designed and constructed vectors for expression of LpiR1 constructs. K. A. B. purified and crystallized the LpiR1 protein and determined its x-ray structure with the help of K. v S. L. L. performed all cell-based experiments and PathScan array assays. All authors analyzed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We acknowledge the Protein Characterization and Crystallization Facility, College of Medicine, University of Saskatchewan for access to the crystallization robot and the 08ID-1 beamline at the Canadian Light Source (CLS) for diffraction data collection. Canadian Light Source is supported by the Canada Foundation for Innovation, Natural Sciences and Engineering Research Council of Canada, the University of Saskatchewan, the Government of Saskatchewan, Western Economic Diversification Canada, the National Research Council Canada, and the Canadian Institutes of Health Research. We thank Dr. Wei Xiao and Dr. Zhaojia Wu (Department of Microbiology and Immunology, University of Saskatchewan) for providing pcDNA4/TO-HA and pEGF-N1 plasmids and mammalian cells used in this work. We also thank Dr. Alla Gagarinova for helpful discussions and Dr. Alexander Grigoryan (College of Agriculture and Bioresources, University of Saskatchewan) for help with the phylogenetic analysis.
This work was supported by Canadian Institutes of Health Research Grant MOP 48370 (to M. C.). The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Figs. 1–3.
The atomic coordinates and structure factors (codes 5FIA and 5JG4) have been deposited in the Protein Data Bank (http://wwpdb.org/).
K. Michalska et al., unpublished information.
- SeMet
- selenomethionine.
References
- 1. Fields B. S., Benson R. F., and Besser R. E. (2002) Legionella and Legionnaires' disease: 25 years of investigation. Clin. Microbiol. Rev. 15, 506–526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Isberg R. R., O'Connor T. J., and Heidtman M. (2009) The Legionella pneumophila replication vacuole: making a cosy niche inside host cells. Nat. Rev. Microbiol. 7, 13–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Vogel J. P., Andrews H. L., Wong S. K., and Isberg R. R. (1998) Conjugative transfer by the virulence system of Legionella pneumophila. Science 279, 873–876 [DOI] [PubMed] [Google Scholar]
- 4. Vincent C. D., Friedman J. R., Jeong K. C., Buford E. C., Miller J. L., and Vogel J. P. (2006) Identification of the core transmembrane complex of the Legionella Dot/Icm type IV secretion system. Mol. Microbiol. 62, 1278–1291 [DOI] [PubMed] [Google Scholar]
- 5. Zhu W., Banga S., Tan Y., Zheng C., Stephenson R., Gately J., and Luo Z.-Q. (2011) Comprehensive identification of protein substrates of the Dot/Icm Type IV transporter of Legionella pneumophila. PLoS ONE 6, e17638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Burstein D., Amaro F., Zusman T., Lifshitz Z., Cohen O., Gilbert J. A., Pupko T., Shuman H. A., and Segal G. (2016) Genomic analysis of 38 Legionella species identifies large and diverse effector repertoires. Nat. Genet. 48, 167–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Buchan D. W., Minneci F., Nugent T. C., Bryson K., and Jones D. T. (2013) Scalable web services for the PSIPRED Protein Analysis Workbench. Nucleic Acids Res. 41, W349–W357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Boratyn G. M., Schäffer A. A., Agarwala R., Altschul S. F., Lipman D. J., and Madden T. L. (2012) Domain enhanced lookup time accelerated BLAST. Biol. Direct 7, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mao F., Dam P., Chou J., Olman V., and Xu Y. (2009) DOOR: a database for prokaryotic operons. Nucleic Acids Res. 37, D459–D463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chien M., Morozova I., Shi S., Sheng H., Chen J., Gomez S. M., Asamani G., Hill K., Nuara J., Feder M., Rineer J., Greenberg J. J., Steshenko V., Park S. H., Zhao B., et al. (2004) The genomic sequence of the accidental pathogen Legionella pneumophila. Science 305, 1966–1968 [DOI] [PubMed] [Google Scholar]
- 11. Hayashi T., Nakamichi M., Naitou H., Ohashi N., Imai Y., and Miyake M. (2010) Proteomic analysis of growth phase-dependent expression of Legionella pneumophila proteins which involves regulation of bacterial virulence traits. PLoS ONE 5, e11718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ward J. J., McGuffin L. J., Bryson K., Buxton B. F., and Jones D. T. (2004) The DISOPRED server for the prediction of protein disorder. Bioinformatics 20, 2138–2139 [DOI] [PubMed] [Google Scholar]
- 13. Stols L., Gu M., Dieckman L., Raffen R., Collart F. R., and Donnelly M. I. (2002) A new vector for high-throughput, ligation-independent cloning encoding a tobacco etch virus protease cleavage site. Protein Expr. Purif. 25, 8–15 [DOI] [PubMed] [Google Scholar]
- 14. Qin Z., Lu M., Xu X., Hanna M., Shiomi N., and Xiao W. (2013) DNA-damage tolerance mediated by PCNA*Ub fusions in human cells is dependent on Rev1 but not Poleta. Nucleic Acids Res. 41, 7356–7369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Doublié S. (1997) Preparation of selenomethionyl proteins for phase determination. Methods Enzymol. 276, 523–530 [PubMed] [Google Scholar]
- 16. Grochulski P., Fodje M. N., Gorin J., Labiuk S. L., and Berg R. (2011) Beamline 08ID-1, the prime beamline of the canadian macromolecular crystallography facility. J. Synchrotron Radiat. 18, 681–684 [DOI] [PubMed] [Google Scholar]
- 17. Minor W., Cymborowski M., Otwinowski Z., and Chruszcz M. (2006) HKL-3000: the integration of data reduction and structure solution: from diffraction images to an initial model in minutes. Acta Crystallogr. D 62, 859–866 [DOI] [PubMed] [Google Scholar]
- 18. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Echols N., Headd J. J., Hung L. W., Jain S., Kapral G. J., Grosse Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R. D., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., and Zwart P. H. (2011) The Phenix software for automated determination of macromolecular structures. Methods 55, 94–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 20. Guex N., and Peitsch M. C. (1997) SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 18, 2714–2723 [DOI] [PubMed] [Google Scholar]
- 21. Pei J., Kim B. H., and Grishin N. V. (2008) PROMALS3D: a tool for multiple protein sequence and structure alignments. Nucleic Acids Res. 36, 2295–2300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tamura K., Stecher G., Peterson D., Filipski A., and Kumar S. (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jones D. T., Taylor W. R., and Thornton J. M. (1992) The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 8, 275–282 [DOI] [PubMed] [Google Scholar]
- 24. Felsenstein J. (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39, 783–791 [DOI] [PubMed] [Google Scholar]
- 25. Holm L., and Rosenström P. (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Söding J., Biegert A., and Lupas A. N. (2005) The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 33, W244–W248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kelley L. A., Mezulis S., Yates C. M., Wass M. N., and Sternberg M. J. (2015) The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Abraham A. L., Pothier J., and Rocha E. P. (2009) Alternative to homo-oligomerisation: the creation of local symmetry in proteins by internal amplification. J. Mol. Biol. 394, 522–534 [DOI] [PubMed] [Google Scholar]
- 29. Ko K. S., Lee H. K., Park M. Y., Lee K. H., Yun Y. J., Woo S. Y., Miyamoto H., and Kook Y. H. (2002) Application of RNA polymerase beta-subunit gene (rpoB) sequences for the molecular differentiation of Legionella species. J. Clin. Microbiol. 40, 2653–2658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Brüggemann H., Cazalet C., and Buchrieser C. (2006) Adaptation of Legionella pneumophila to the host environment: role of protein secretion, effectors and eukaryotic-like proteins. Curr. Opin. Microbiol. 9, 86–94 [DOI] [PubMed] [Google Scholar]
- 31. Landau M., Mayrose I., Rosenberg Y., Glaser F., Martz E., Pupko T., and Ben-Tal N. (2005) ConSurf 2005: the projection of evolutionary conservation scores of residues on protein structures. Nucleic Acids Res. 33, W299–W302 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.