

Abstract
Hypersensitivity pneumonitis (HP) is an immune-mediated interstitial lung disease that develops following repeated exposure to inhaled environmental antigens. The disease results in alveolitis and granuloma formation and may progress to a chronic form associated with fibrosis; a greater understanding of the immunopathogenic mechanisms leading to chronic HP is needed. We used the Saccharopolyspora rectivirgula (SR) mouse model of HP to determine the extent to which a switch to a Th2-type immune response is associated with chronic HP. Exposure of wild-type (WT) and tlr2/9−/− mice to SR for 14 wk resulted in neutrophilic and lymphocytic alveolitis that was not dependent on Toll-like receptors (TLRs) 2 and 9. Long-term exposure of WT mice to SR resulted in a significant increase in collagen deposition, protein leakage, and IL-1α accompanied by a decrease in quasistatic compliance and total lung capacity compared with unexposed mice. This was associated with an increase in IL-17 but not IL-4 production or recruitment of Th2 cells. tlr2/9−/− mice exhibited an increase in protein leakage but less IL-1α and collagen deposition in the lungs compared with WT mice, yet they still displayed a decrease in quasistatic compliance, although total lung capacity was not affected. These mice exhibited an increase in both IL-13 and IL-17, which suggests that IL-13 may ameliorate some of the lung damage caused by long-term SR exposure. Our results suggest that lung pathology following long-term SR exposure in WT mice is associated with the IL-17 response and that TLRs 2 and 9 may inhibit the development of the IL-13/Th2 response.
Keywords: hypersensitivity pneumonitis, fibrosis, Toll-like receptors 2 and 9
hypersensitivity pneumonitis (HP), also known as extrinsic allergic alveolitis, is an immune-mediated lung disease that develops following repeated exposure to a wide variety of inhaled environmental antigens (3, 18, 35–37). The environmental antigens that induce HP include organic dusts, vapors, fungi, bacteria, and molds as well as simple chemical compounds (2, 32, 45). The disease is characterized by a lymphocytic alveolitis, noncaseating granulomas, and, in some patients, it develops into a chronic form that is associated with fibrosis (12, 33, 35, 36, 45). The chronic form of the disease is associated with high morbidity and an ultimate 5-yr mortality rate of 27% (19, 42). The lack of information on disease pathogenesis has limited the development of successful therapies to treat the disease. The mainstay of treatment at present is avoidance of the inciting agent, which can improve survival in patients with chronic HP (14). Alternatively, corticosteroids such as mycophenolate acid and azathioprine, and other immunosuppressive agents, all with significant side effects, have also been used to treat patients with chronic HP when an inciting agent cannot be identified (23).
HP is a complex disease with components of both Type III (antibody-mediated) and Type IV (cell-mediated) hypersensitivity reactions. Farmer's lung disease (FLD) is one of the most common types of HP and is caused by repeated inhalation of the Gram-positive thermophile Saccharopolyspora rectivirgula (SR), which is commonly found in moldy hay (35). In mouse models and patients with HP there is induction of a proinflammatory cytokine response including TNF, IL-1β, IL-6, IL-8, IFNγ, IL-17, and a neutrophilic influx into the lung shortly after antigen exposure. Neutrophils isolated from the lungs of patients with a chronic form of FLD expressed high levels of gelatinase B and collagenase-2, which correlated with fibrosis (21, 31). Inhibition of neutrophil recruitment in a mouse model of HP resulted in a decrease in inflammation supporting the idea that neutrophils play a role in pathogenesis (29). As the time after antigen exposure increases, alveolitis becomes more lymphocytic with an increase in both CD8+ and CD4+ T cells. There have been conflicting reports of the CD4+/CD8+ T cell ratio in the bronchoalveolar lavage (BAL), which may be due to differences between the stage of disease, or the inciting antigen, or both (44). Although T cells are necessary for the disease, there are also conflicting reports on whether the disease is mediated by a Th1-, Th2-, or Th17-type response. In the FLD murine model there is an association between IL-17 and an increase in disease severity and collagen deposition (24, 39). Depletion of IL-17 either by antibody-mediated neutralization or genetic deficiency resulted in a reduced inflammatory response to SR (24, 39). Our previous studies demonstrated that mice with a deficiency in the Th1 transcription factor tbet exhibit an increase in Th17 cells and collagen production following SR exposure, confirming the role of IL-17 in pathogenesis and suggesting that Th17 cells are important in the disease process (1). However, these studies have been performed with relatively short periods of SR exposure and it is has been suggested that chronic HP, which is associated with significant mortality, is associated with a switch to a Th2 response. Barrera et al. (5) demonstrated a decrease in Th1 T cells and an increase in Th2 T cells in the BAL of patients with chronic HP compared with patients at the subacute stage of disease. These patients were diagnosed with pigeon breeders disease, another type of HP in which the chronic phase is characterized by fibrosis. Similarly, Mitaka et al. (28) used the murine model of pigeon breeders disease to demonstrate an association between fibrosis and the Th2 response. However, patients with chronic FLD develop both emphysematous and fibrotic changes in the lung, suggesting that the immunological mechanisms leading to lung damage in HP may differ depending on the environmental antigen triggering the disease (12). This may be important in the development of therapeutics to treat this stage of the disease.
Activation of the IL-17/neutrophil response is triggered through recognition of HP antigens by pattern recognition receptors (PRRs) expressed by innate immune cells. Our previous studies demonstrated that SR stimulates nuclear factor-κB activation through Toll-like receptor 2 (TLR2) but not TLRs 4, 5, or 7 in vitro (30). In vivo, stimulation through TLR2 is critical for neutrophil chemokine production and recruitment with a minor contribution from TLR9 (4, 30). Additionally, both TLRs 2 and 9 contributed to expression of IL-17A mRNA and Th17 cell generation following repeated SR exposure, although the response was not completely inhibited (4). Fong et al. (15) identified TLR6 as being important for expression of IL-17A in this model. TLR6 forms a heterodimer with TLR2 that enables binding to diacylated lipopeptides from bacterial membranes suggesting that it is the TLR2/6 heterodimer that is stimulated by SR. These studies suggest that TLRs 2 and 9 contribute to the neutrophil/IL-17 response during the acute and subacute phases of the disease; however, their contribution to IL-17 production and the chronic phase of disease are unknown.
The purpose of this study was to determine the extent to which the development of lung dysfunction and fibrosis in a model of FLD is associated with the neutrophil/IL-17 response or, alternatively, a switch to a Th2 response and the role of TLRs 2 and 9 in that response.
MATERIALS AND METHODS
Animals and S. rectivirgula exposure protocol.
C57BL/6 female mice were purchased from The Jackson Laboratory (Bar Harbor, ME) at 6 wk of age. tlr2/9−/− mice on a C57BL/6 background were bred at the University of Tennessee Health Science Center (UTHSC) and were in at least the 15th generation backcross. All animals were housed in sterile microisolator cages with sterile food and water ad libitum, and were maintained by the Division of Comparative Medicine at UTHSC according to the guidelines of the Animal Welfare Act. All animal care procedures were performed according to protocols approved by the UTHSC animal care and use committee.
S. rectivirgula (strain designation A1313-ATCC) was grown at 55°C in trypticase soy broth. The bacterial preparation was washed in endotoxin-free distilled water three times and lyophilized. The lyophilized preparation was reconstituted with endotoxin-free saline, and mice were exposed intranasally with 150 μg SR for 4 wk (three times per week for 3 wk and once in week 4) or similarly for 14–15 wk. Mice were analyzed 2–3 days after the last exposure.
BAL and lung cell isolation.
BAL was performed by intratracheal injection of 1 ml of PBS with 2 mM EDTA into the lungs with immediate vacuum aspiration. The amount of fluid recovered was routinely around 70%. Cells were recovered from the BAL fluid (BALF) by centrifugation and counted using Trypan blue dye exclusion. The BALF was frozen at −80°C until used for protein determinations or ELISA assays for cytokine and chemokine measurement.
Lungs were perfused with PBS to remove blood, and lobes were removed. Lung tissue was digested with collagenase (20 U/ml) and deoxyribonuclease I (40 μg/ml) for 45 min at 37°C. Cells were freed by disruption in a Stomacher tissue processor and then isolated by centrifugation on a discontinuous Percoll gradient. Mononuclear cells were isolated at the 40/80% interface following density gradient centrifugation and used in the following assays.
ELISA.
Cytokines present in lung homogenates were measured by ELISA according to the manufacturer's instructions (Biolegend, San Diego, CA). Cytokine standards ranging from 15.6 to 4,000 pg/ml were prepared to determine the concentration of cytokine in the samples. For data analysis, a curve fit was applied to the standards and the sample concentrations were extrapolated from the standard curve using four-parameter logistic software (SoftMax Pro, Sunnyvale, CA).
Cell surface and intracellular flow cytometry.
Cell surface flow cytometry was performed on isolated BAL or lung cells using fluorochrome-conjugated antibodies to CD11b, CD11c, Gr-1, F4/80, CD45, CD4, CD8, βTcR, CD200, CD200R, and CD69 (BD Biosciences, San Jose, CA or ebiosciences, San Diego, CA). For intracellular cytokine staining, lung cells were incubated with PMA and ionomycin in the presence of brefeldin A (GolgiPlug; BD Biosciences). The cells were incubated with antibodies to CD45, CD4, and βTcR to identify CD4+ T cells and then fixed and permeabilized with 1% saponin followed by incubation with IL-17A, IL-4, or isotype control antibodies. A minimum of 10,000 events/sample were collected for BAL cells and 100,000 events/sample for lung cells on an LSRII cytometer (BD Biosciences). Expression of cell surface markers and intracellular cytokines was analyzed using DIVA software.
RNA isolation and qRT-PCR.
Total RNA was extracted from the upper right lobe of lung from individual mice using TRIzol (Invitrogen). Contaminating genomic DNA was removed by treatment with DNA-free (Ambion, Austin, TX) according to the manufacturer's directions. RNA (2 μg) was reverse transcribed into cDNA with a reverse transcription system (Promega, Madison, WI). Real-time PCR was performed with gene-specific primers to IL-17A, IL-4, IL-10, TNF, IFNγ, NLRP3, and caspase-1 using a LightCycler 480 real-time PCR thermal cycler (Roche Diagnostics, Indianapolis, IN). All primers and probes were chosen using the online software Universal Probe Library. Data were normalized to the HPRT gene and plotted as fold induction over unexposed control mice.
Lung function measurements.
Measurements of lung mechanics were made using the flexiVent system (SCIREQ, Montreal, QC, Canada). Mice were anesthetized using an intraperitoneal injection of ketamine/xylazine mixture (1:1, 0.15 ml/10 g body wt) and 1% inhaled isofluorane using a precision gas mixer (PEGAS 400; Columbus Instruments, Columbus, OH) and then tracheostomized with an 18-gauge needle and placed on the flexiVent. Quasistatic lung compliance was measured by obtaining pressure-volume (PV) curves. Each set of PV curves was preceded by two inflation maneuvers to total lung capacity to ensure equal volume history. Compliance was calculated by fitting data derived from the PV curves to the Salazar Knowles equation. Only measurements with a coefficient of determination (COD) greater than 0.9 were included in analysis. Measurements were repeated until at least two PV curves and three snapshot perturbations with acceptable CODs were obtained. The average measurement was calculated for each mouse.
Histology.
The left lobe of lung was removed and fixed with neutral-buffered formalin and embedded in paraffin. Sections (8 μm) were cut and stained with hematoxylin and eosin to analyze inflammation or Masson's trichrome to analyze lung fibrosis. Slides were digitized (Scanscope XT; Aperio, Vista, CA) and analyzed using Spectrum Imagescope software (v11.2, Aperio). Quantification of collagen staining in the trichrome-stained lung sections divided into 12 1-mm2 regions was performed using the positive pixel count algorithm (v9.1). The blue collagen was detected using a positive hue value of 0.65, a hue width of 0.2 (to include a range of color shades), and a color saturation threshold of 0.1, all other settings remained at default values. Data are reported as the average number of positive and strong positive pixels/lung section.
Statistical analysis.
Data from two groups were analyzed by a one-way or two-way Student's t-test using GraphPad Prism statistical software (GraphPad Software, San Diego, CA). Data from three groups were analyzed with ANOVA and a Tukey's post hoc test. Intracellular cytokine staining data were analyzed by two-way ANOVA with a Bonferonni post hoc test. Differences were considered significant at P < 0.05.
RESULTS
Mice with long-term exposure to SR developed a lymphocytic alveolitis that is independent of TLRs 2 and 9.
One of the defining features of patients with both acute and chronic HP is the development of lymphocytic alveolitis. To measure the extent to which T cells contribute to alveolitis during long-term SR exposure we exposed wild-type (WT) and tlr2/9−/− mice to SR for 4 or 14 wk and performed flow cytometry on cells isolated from the BALF 2 days following the last exposure (Table 1). The results demonstrate that WT mice exposed to SR for 14 wk exhibited an increased frequency of alveolar macrophages (AMs) (CD11c+/F4/80+) and T cells (βTcR+) compared with WT mice exposed for 4 wk. We did not observe a predominance of CD8+ T cells with long-term SR exposure; the ratio of CD4+ to CD8+ T cells was close to 1.0. Within the CD4+ and the CD8+ T cell population, the percentage of activated T cells (CD69+/βTcR+/CD4+ or CD8+) was not significantly different in WT mice with long-term exposure to SR compared with 4-wk exposure. Neutrophils were recruited into the lung within 6 h following SR exposure and their frequency dropped in the days following exposure (30). Two days following long-term SR exposure, the frequency of neutrophils in the alveolitis (∼15%) did not differ from that of the 4-wk SR-exposed mice. Despite the low frequency of neutrophils in the BAL at 2 days postexposure, long-term SR exposure is associated with a strong neutrophil influx at 1 day postexposure (% polymorphonuclear cells − WT unexposed = 0.6% ± 0.4; WT/SR exposed = 65% ± 10; tlr2/9−/−/SR = 59 ± 26).
Table 1.
Cellular composition of alveolitis following short- and long-term exposure of WT and tlr2/9−/− mice to SR
| PMN % | AM % | T cells % | CD4+ % | CD8+ % | CD4+/CD69+ % | CD8+/CD69+ % | |
|---|---|---|---|---|---|---|---|
| Unexposed | 0.02 ± 0.04 | 88.2 ± 3.7 | |||||
| WT SR 4 wk | 11.7* ± 6.1 | 9.0* ± 4.7 | 26.9 ± 2.4 | 53.2 ± 10.9 | 38 ± 11.6 | 79.4 ± 5.6 | 53.94 ± 15.7 |
| WT SR, 14 wk | 14.6* ± 2.6 | 29.6* ± 2.8 | 35.4 ± 4.2 | 43.9 ± 7.1 | 42.9 ± 5.5 | 73.5 ± 6.7 | 67.4 ± 3.5 |
| tlr2/9−/− SR, 4 wk | 6.2 ± 1.6 | 13.4* ± 3.9 | 34.4† ± 2.1 | 46 ± 6.2 | 42.4 ± 6.1 | 64.8† ± 4.4 | 56.43 ± 9.1 |
| tlr2/9−/− SR, 14 wk | 9.7* ± 2.7 | 25.0* ± 7.0 | 32.12 ± 2.9 | 48.6 ± 7.6 | 41.0 ± 7.9 | 65.1 ± 2.2 | 58.5 ± 8.8 |
Values are given as means ± SD of n = 4–5 animals/group. Significance was determined by 1-way ANOVA with Tukey's post hoc test. The percentage of neutrophils (PMNs; Gr-1+/F4/80−), AMs (F4/80+/CD11c+), CD4+ T cells (CD4+/βTcR+), and CD8+ T cells (CD8+/βTcR+) and activated CD4+ or CD8+ T cells (CD69+/βTcR+/CD4+ or CD8+) were measured by flow cytometry. PMNs, polymorphonuclear cells; AMs, alveolar macrophages; SR, Saccharopolyspora rectivirgula; WT, wild-type.
P < 0.05 compared with WT unexposed animals;
P < 0.05 compared with WT animals exposed to SR for 4 wk.
Our previous studies demonstrated that mice deficient in TLRs 2 and 9 had alterations in immune cell recruitment and activation following SR exposure for 3 wk. Specifically, neutrophil recruitment and production of the neutrophil chemokine CXCL2 was significantly reduced in tlr2/9−/− mice 1 day post-SR exposure (4). However, in this study examining long-term SR exposure, there was no significant difference in the frequency of neutrophils in the tlr2/9−/− mice compared with the WT mice at 2 days following 14 wk of SR exposure. Interestingly, levels of CXCL2 in the BALF were very low at this stage of the disease, whereas another neutrophil chemokine, CXCL1, was increased (Fig. 1). We observed a decrease in CD4+ T cells expressing the activation marker CD69 in tlr2/9−/− mice at 4 wk postexposure; however, by 14 wk of SR exposure there was no difference in the frequency of CD69+/CD4+ T cells. These results suggest that immune cell recruitment and activation is not dependent on TLRs 2 and 9 following long-term SR exposure, in contrast to short-term exposure.
Fig. 1.
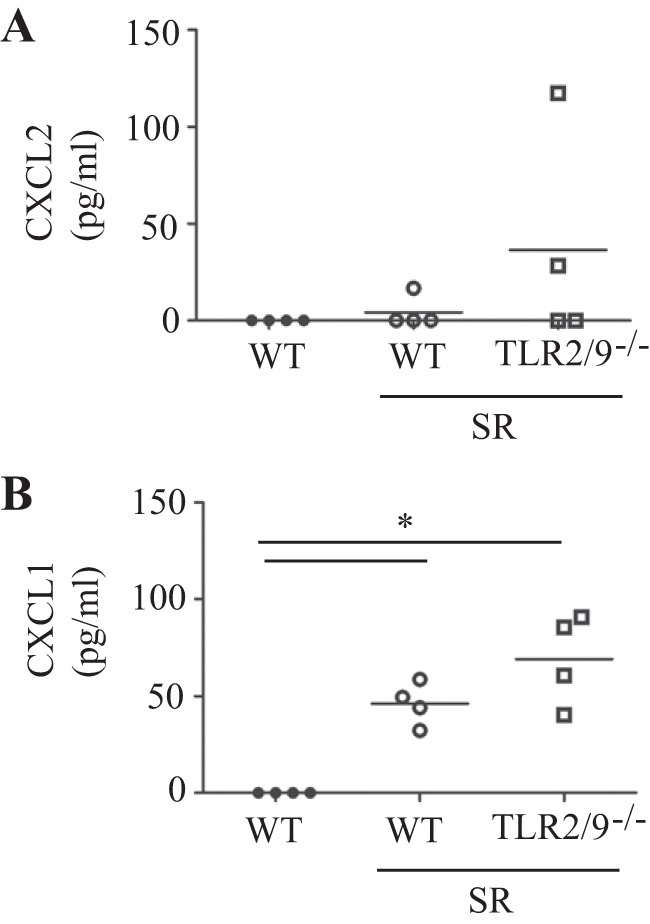
Neutrophil chemokine production in wild-type (WT) and tlr2/9−/− mice following long-term exposure to Saccharopolyspora rectivirgula (SR). WT and tlr2/9−/− mice were exposed to SR 3 times per week for 14 wk and analyzed 1 day after the last exposure. The cell-free bronchoalveolar lavage (BAL) fluid (BALF) was analyzed for CXCL1 and CXCL2 by a bead-based ELISA. Values are given as means ± SD (n = 4 per group). Significance was determined by 1-way ANOVA with a Tukey's post hoc test (*P < 0.05 compared with WT SR-exposed mice).
Long-term SR exposure caused an increase in CD200R expression by CD4+ T cell and AM populations.
The CD200R is upregulated on memory T cells and it has been suggested that its expression correlates with persistent antigen exposure (7, 34). To determine whether long-term exposure to SR led to an increase in CD200R expression or its ligand CD200, we performed flow cytometric analysis on cells isolated from the BALF of mice (Table 2). We measured an increase in CD200R expression by CD4+ T cells with 14 wk of SR exposure compared with 4 wk of SR exposure; the CD200R+ cells also expressed CD69, suggesting they had been activated. Expression of CD200 by CD4+ T cells was very low and did not change with increasing duration of exposure. There was no increase in CD200R or CD200 expression by CD8+ T cells. Expression of CD200R by CD4+ T cells was partially controlled by TLRs 2 and 9 because the tlr2/9−/− mice had a reduced expression of CD200R on CD4+ T cells compared with the WT mice.
Table 2.
CD200 and CD200R expression by AMs and T cells following short- and long-term SR exposure
| AM Gr1low |
CD4 T cells |
CD8 T cells |
||||||
|---|---|---|---|---|---|---|---|---|
| Gr1high, % | Gr1low, % | CD200R, % | CD200, % | CD200R, % | CD200, % | CD200R, % | CD200, % | |
| Unexposed | 0.0 ± 0.0 | 95.2 ± 0.58 | 0.48 ± 0.19 | 0.4 ± 0.07 | ND | ND | ND | ND |
| WT SR 4 wk | 32.7* ± 5.0 | 66.1* ± 5.3 | 10.6* ± 4.6 | 22.4* ± 7.9 | 6.4 ± 1.9 | 4.5 ± 2.1 | 0.68 ± 0.69 | 1.42 ± 1.12 |
| WT SR 14 wk | 23.7*† ± 2.6 | 75.8*† ± 2.8 | 39.5*† ± 5.9 | 46.8*† ± 7.7 | 16.8† ± 2.9 | 3.8 ± 1.6 | 1.44 ± 0.64 | 0.92 ± 0.32 |
| tlr2/9−/− SR 4 wk | 26.6* ± 2.89 | 72.0* ± 2.7 | 13.2* ± 2.1 | 10.7 ± 3.1 | 5.8 ± 1.9 | 9.9† ± 2.5 | 0.47 ± 0.12 | 3.3 ± 1.55 |
| tlr2/9−/− SR, 14 wk | 21.8* ± 3.8 | 77.3* ± 3.8 | 23.5*‡ ± 7.3 | 24.2*‡ ± 7.6 | 10.2‡ ± 4.5 | 6.2 ± 1.8 | 2.14 ± 0.49 | 1.92 ± 0.88 |
Values are given as means ± SD of n = 4–5 animals/group. Significance was determined by 1-way ANOVA with Tukey's post hoc test. WT and tlr2/9−/− mice were either not exposed or exposed to SR for 4 or 14 wk and bronchoalveolar lavage was analyzed 2 days after the last exposure. The percentage of alveolar macrophages (AMs; F4/80+/CD11c+) or CD4+ T cells expressing Gr1, CD200, or CD200R was measured by flow cytometry.
P < 0.05 compared with WT unexposed group;
P < 0.05 compared with WT SR-exposed for 4 wk;
P < 0.05 compared with WT SR-exposed for 14 wk.
The inhibitory receptor:ligand pair CD200R:CD200 is also expressed on myeloid cells and is important in downregulating macrophage activation (22). Additionally, expression of CD200 by macrophages is believed to play a supporting role in fusion or multinucleation of macrophages (11). We measured expression of CD200 and CD200R on AMs by flow cytometry using CD11c+/F4/80+/autofluorescence+ as our criteria for identifying AMs in the BALF. Surprisingly, flow cytometric analysis revealed that this population could be divided into two subpopulations based on their expression of Gr1. Approximately 30% of AMs from WT mice exposed to SR for 4 or 14 wk were Gr1high, whereas the remaining AM population was Gr1low (Table 2). We considered the CD11c+/F4/80+/Gr1low subpopulation to be AMs, and analysis of CD200R/CD200 expression revealed an increase in AMs expressing either CD200R or CD200 at 14 wk compared with 4 wk of SR exposure. The increase in expression of both CD200R and CD200 was partially dependent on TLRs 2 and 9 because there was a significant reduction in their expression in the tlr2/9−/− mice compared with the WT mice with 14 wk of SR exposure. These results suggest that long-term exposure to SR correlates with an increase in the inhibitory receptor ligand pair CD200-CD200R, and that its regulation on AMs is partially controlled by TLRs 2 and 9.
Long-term SR exposure caused increased lung dysfunction.
To determine the extent to which long-term exposure to SR results in lung pathology we measured several parameters of disease severity. An increase in protein in BALF is a measure of decreased barrier integrity, and we detected an increase in protein in the BALF of WT mice with long-term exposure to SR compared with unexposed mice (Fig. 2A). Barrier dysfunction was also detected after long-term exposure in the mice with deficiency of TLRs 2 and 9. The alarmin IL-1α is present in epithelial cells lining the lung and is released by those cells when they are damaged or dying, initiating an inflammatory response (8, 11). We measured the level of IL-1α in the lungs of WT and tlr2/9−/− mice with long-term exposure to SR (Fig. 2B). The results demonstrate an increase in IL-1α in the lungs of WT mice compared with the tlr2/9−/− SR-exposed mice and unexposed mice. There was an increase in the level of IL-1α in tlr2/9−/− SR-exposed mice compared with unexposed mice but not to the level of the SR-exposed WT mice. To determine the level of inflammation following long-term SR exposure, we examined hematoxylin and eosin-stained lung sections from WT and mutant mice (Fig. 2C). The sections revealed an increase in neutrophils in the interstitial lung tissue surrounding the areas of granulomatous inflammation (Fig. 2C, arrow). In addition the appearance of multinucleate cells was observed in WT and tlr2/9−/− mice exposed to SR, a feature also common to patients with chronic HP. There were no differences in the inflammation observed in WT vs. tlr2/9−/− mice.
Fig. 2.

Long-term SR exposure results in decreased barrier integrity and increased inflammation in WT and tlr2/9−/− mice. WT and tlr2/9−/− mice were exposed to SR 3 times per week for 4 or 14 wk and analyzed 2 days after the last exposure. A: BAL was performed on mice and the concentration of total protein in the BALF was determined by the Bio-Rad protein assay. Significance was determined by 1-way ANOVA with a Tukey's post hoc test. *P < 0.05. B: lung homogenates were prepared and IL-1α expression was measured by ELISA. C: representative hematoxylin and eosin-stained lung sections from unexposed WT mice or 14-wk SR-exposed WT and tlr2/9−/− mice (original magnification ×10).
To measure the level of collagen deposition in the lungs of mice with long-term exposure to SR, lung sections were stained with Masson trichrome (Fig. 3). Compared with unexposed mice, lung sections from SR-exposed WT mice exhibited an increase in blue staining representing collagen deposition around the bronchi, within the granulomatous areas, and in the alveolar walls (Fig. 3, C and D). An increase in collagen deposition was also observed in lung sections from SR-exposed tlr2/9−/− mice (Fig. 3, E and F). To quantify the amount of collagen in the lung, the stained sections were analyzed using the positive pixel algorithm in the Spectrum Imagescope software (Fig. 3G). The results demonstrate a significant increase in the level of collagen deposition in lung sections from WT mice with long-term exposure to SR compared with unexposed WT mice. However, SR-exposed tlr2/9−/− mice did not have a significant increase in collagen deposition compared with WT-unexposed mice.
Fig. 3.
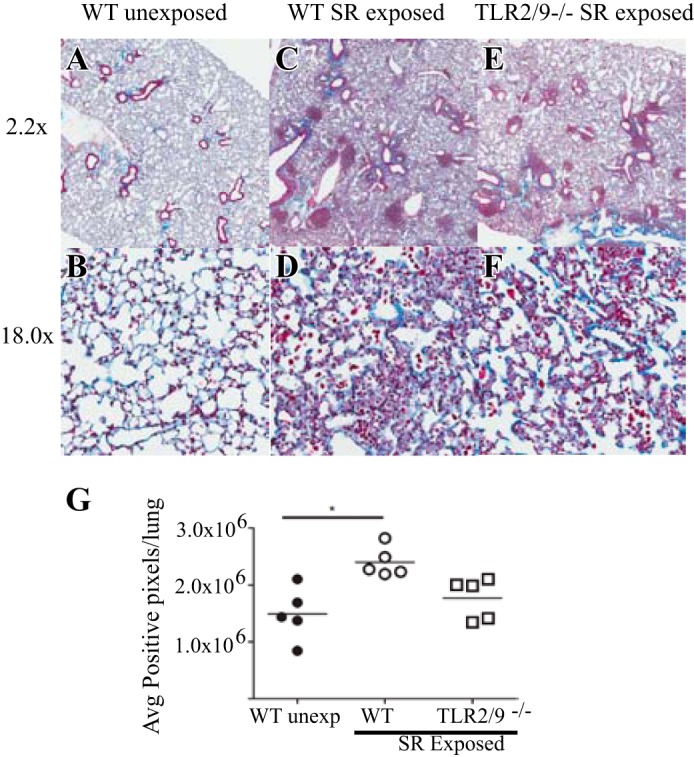
Long-term SR exposure results in an increase in collagen deposition in lungs of WT and tlr2/9−/− mice. Lung tissue from mice exposed to SR for 14 wk was harvested for histological examination. Lung slides were stained with Masson's trichrome, then digitized for image analysis. Blue-stained collagen pixels were quantified using the positive pixel algorithm. Representative images and the corresponding positive pixel markup image were captured at ×2.2 (A, C, and E) or ×18 (B, D, and F). Values are given as means ± SD of n = 5/group. Significance was determined by 1-way ANOVA with a Tukey's post hoc test. *P < 0.05.
To determine whether long-term SR exposure led to impaired lung function, we measured quasistatic compliance and total lung capacity in SR-exposed WT and tlr2/9−/− mice. Long-term exposure of both WT and tlr2/9−/− mice to SR resulted in a significant decrease in quasistatic compliance compared with unexposed mice (Fig. 4). Additionally, total lung capacity in SR-exposed WT mice were significantly decreased compared with that of unexposed mice (Fig. 4). Altogether, these results demonstrate that long-term exposure to SR caused barrier dysfunction and fibrosis resulting in a significant decrease in lung function.
Fig. 4.

WT and tlr2/9−/− mice exposed long term to SR develop reduced compliance and total lung capacity. Mice were exposed to SR for 15 wk, and lung function measured by flexiVent 3 days postexposure. Quasistatic compliance and total lung capacity were determined by a pressure-volume curve. Each point represents the average value of 2 measurements for each mouse. Values are given as means ± SD of n = 4–6/group. Significance was determined by 1-way ANOVA with Tukey's post hoc test. *P < 0.05.
Long-term exposure to SR by WT mice was associated with an IL-17 response but not a shift to an IL-4 response.
There is evidence in chronic pigeon breeders disease that the development of fibrosis correlates with a switch from a Th1-type response to a Th2-type immune response. However, studies with earlier time points in the SR model of FLD have demonstrated that IL-17 is associated with increased disease severity. To determine whether mice exposed to SR for a long time switch from a Th17-type response to the more profibrotic Th2-type response, we performed quantitative RT-PCR (qRT-PCR) and measured the expression of cytokine mRNA in the lungs of exposed and control mice. SR exposure resulted in a strong upregulation of IL-17A expression, which was further increased in tlr2/9−/− mice compared with unexposed mice (Fig. 5A). We measured the level of IL-17 in the BALF 1 day after SR exposure and tlr2/9−/− mice had a significant increase in IL-17 compared with both WT and unexposed controls (Fig. 5C). There was no increase in IL-17 in the BALF of exposed WT mice. Expression of IL-4 was not detected by qRT-PCR and no bands were observed when the PCR product was run on a gel (Fig. 5C). However, IL-13 mRNA was detected in WT SR-exposed mice but tlr2/9−/− mice demonstrated a significant increase in IL-13 mRNA compared with both WT exposed and unexposed mice. Expression of the neutrophil chemokine CXCL2, IL-10, and IFNγ mRNA were also increased in mice following long-term SR exposure compared with unexposed mice, but there were no differences between WT and tlr2/9−/− mice exposed to SR.
Fig. 5.

Long-term SR exposure correlates with an IL-17 response and not IL-4. WT and tlr2/9−/− mice were exposed to SR for 14 wk and analyzed 3 days after exposure. A: expression of mRNA for cytokines was determined by quantitative RT-PCR on RNA isolated from individual lung lobes (n = 5–7 mice per group) and expressed as fold induction over WT unexposed mice. Values are given as means ± SD of duplicate samples; significance was determined by 1-way ANOVA with a Tukey's post hoc test (*P < 0.05 compared with WT SR-exposed mice). B: PCR products for IL-4 were electrophoresed on an agarose gel and stained with ethidium bromide to visualize the bands. C: cell-free BALF was analyzed for IL-17A by a bead-based ELISA. Values are given as means ± SD (n = 5 per group); significance was determined by 1-way ANOVA with Tukey's post hoc test (*P < 0.05 compared with WT SR-exposed mice). D and E: lungs were harvested from mice exposed to SR for 14 wk and digested to obtain a single cell suspension. The cells were stimulated with media or PMA/ionomycin to induce cytokine production. D: representative gating strategy for intracellular cytokine staining: a lymphocyte gate was set based on forward and side scatter characteristics, followed by expression of CD45 to gate out nonhematopoietic lung cells. The CD45+ T cells were gated on the basis of expression of the β chain of the TcR and lack of expression of F4/80. Isotype controls were used to set the gates to determine the contribution of CD4+ and CD4− T cells to IL-4 or IL-17 production. E: the percentage of CD45+/CD4+/βTcR+ T cells expressing IL-17 (Th17) or IL-4 (Th2) was determined by intracellular flow cytometry. Values are given as means ± SD of n = 5/group. Significance was determined by 2-way ANOVA with a Bonferroni post hoc test. *P < 0.05.
We performed intracellular cytokine staining on cells isolated from the lungs of SR-exposed (15 wk) mice to measure levels of Th17 and Th2 cells. The cells were stimulated ex vivo with PMA and ionomycin and the results demonstrate an increase in IL-17+/CD4+ Th cells in WT mice and a significant increase in tlr2/9−/− mice compared with unexposed mice (Fig. 5, D and E). There were no detectable IL-4+/CD4+ Th cells at this time point in either WT or tlr2/9−/− mice exposed to SR, suggesting that there was no switch to a Th2 response. These results suggest that in WT mice there was no shift to a Th2 response with long-term SR exposure and the disease remains one that is predominantly IL-17-mediated. However, the increase in IL-13 mRNA in tlr2/9−/− mice suggests that a Th2 environment may be developing in those mice.
Long-term exposure to SR caused an increase in IL-1β.
IL-1β plays key roles in IL-17-mediated responses; IL-1β and IL-23 induce IL-17 production by γδ T cells (41). IL-1β can further amplify the response by increasing IL-23 production, which in turn, stimulates IL-17 production (27). IL-1β also contributes to Th17 cell differentiation, in part through the production of IL-6 and is closely associated with IL-17-mediated pathology (9, 10). We measured the level of IL-1β in lung homogenates from mice with long-term exposure to SR and found that was increased in both WT and tlr2/9−/− mice (Fig. 6A). The canonical pathway for IL-1β production is activation of the inflammasome resulting in activation of caspase-1 and cleavage of pro-IL-1β to its mature form. We used qRT-PCR to measure the level of inflammasome components in the lungs of long-term SR-exposed mice. The results demonstrate an increase in mRNA for NLRP3, one of the NLR family members capable of initiating inflammasome formation, and caspase-1 in WT and tlr2/9−/− mice (Fig. 6B). Stimulation of WT bone marrow-derived macrophages (BMDMs) with SR plus ATP resulted in release of IL-1β into the culture supernatant suggesting that SR can induce pro-IL-1β production and in the presence of an NLRP3 activator (ATP) result in IL-1β release (Fig. 6C). However, pro-IL-1β production is dependent on TLRs 2 and 9 because BMDMs derived from tlr2/9−/− mice did not produce IL-1β following SR-plus-ATP stimulation. These results suggest an association between amplification of the IL-17 response and IL-1β. In addition, activation of PRRs that control inflammasome formation may play a role in lung pathology associated with long-term exposure to SR.
Fig. 6.

Expression of inflammasome components in the lung following long-term exposure to SR. WT and tlr2/9−/− mice were exposed to SR 3 times per week for 14 wk and analyzed 2 days after the last exposure. A: lung homogenates were prepared and IL-1β expression was measured by ELISA. B: expression of mRNA for NLRP3 and caspase-1 was determined by qRT-PCR on RNA isolated from individual lung lobes (n = 5 mice per group) and is expressed as fold induction over WT unexposed mice. C: bone marrow-derived macrophages were prepared from WT and tlr2/9−/− mice and stimulated with LPS or SR for 3 h followed by ATP for 45 min. Culture supernatants were collected and IL-1β was measured by ELISA. Significance was determined by 2-way ANOVA with a Tukey's post hoc test. *P < 0.05.
DISCUSSION
The development of chronic HP is associated with significant morbidity and mortality, and the lack of treatment options makes research of disease critical. Our studies are novel because they focus on long-term exposure to SR, which resulted in fibrosis and barrier dysfunction significant enough to cause a measurable decrease in lung compliance and lung volume. The immunological changes that occur with chronic inflammation are likely critical for disease pathogenesis and are unlikely to be observed with shorter exposure periods. The results of these studies identify potential new targets for therapeutic interventions at this phase of the disease.
Long-term exposure to SR resulted in neutrophilic and lymphocytic infiltration into the airways similar to that observed with shorter exposure times. The main difference between short-and long-term SR exposure was an increase in the percent of AMs recovered in the BALF of both WT and tlr2/9−/− mice exposed to SR for 14 wk. There have been conflicting reports on whether chronic HP is associated with a decrease in the CD4/CD8 T cell ratio in the BALF; however, our studies did not identify a change in this ratio between short- and long-term exposure. Both CD4+ and CD8+ T cells recovered from the BALF of mice with long-term exposure were activated as demonstrated by an increase in CD69 expression, and the deficiency in TLRs 2 and 9 did not affect CD69 expression. Interestingly, there was also an increase in CD4+ T cells expressing the inhibitory receptor CD200R. It is not known whether CD200R on T cells acts in an inhibitory manner as it does in myeloid cells or whether it plays a role in effector T cell function. Some studies have demonstrated an association between CD200R expression by CD4+ T cells and chronic antigen exposure. There was an increase in CD200R expression on polarized effector CD4+ T cells in a mouse model of chronic helminth infection (7). In our study, CD200R was coexpressed with CD69 on CD4+ T cells, suggesting that it is associated with the activated/effector T cell population in this model also and may have value as a biomarker of antigen-driven inflammation in patients. There is evidence that the CD200:CD200R pathway may shift the cytokine balance in favor of IL-4 and IL-10 production and may inhibit Th17 cell differentiation, however, it is unclear whether these are direct effects of CD200R expression by CD4+ T cells or myeloid cells (17, 26). We did not measure expression of CD200 or CD200R (or CD69) on T cells isolated from the lung and therefore we cannot speculate on the role of T cell expression of CD200:CD200R in contributing to the changes in interstitial tissue. However, these studies suggest that chronic exposure to SR may lead to changes in T cell phenotype that are critical to the development of fibrosis and further studies at this stage of the disease are critical.
It has been clearly demonstrated that CD200R acts as an inhibitory receptor on myeloid cells. Mice that are deficient in CD200 exhibit an increase in activated macrophages and greater susceptibility to autoimmune diseases such as experimental autoimmune encephalomyelitis and collagen-induced arthritis (22). We observed an upregulation of both CD200 and CD200R on AMs with long-term SR exposure, although they were not coexpressed on the same cells. The increase in CD200 expression correlates with an increase in multinucleate cells in the BALF and lung tissue, and thus CD200 may play a role in triggering fusion, as has been observed with other models (11). Expression of CD200R by AMs suggests that AM activation during chronic HP may be regulated by soluble or cell surface-bound CD200. The results from our studies suggest that the CD200R/CD200 system is partially regulated by TLRs 2 and 9 because there was less of an increase in their expression with long-term SR exposure. It is not clear whether this was due to direct regulation of CD200R or CD200 by these TLRs or indirectly due to the decreased inflammation that is observed in these mice early in the disease. The possibility of exploiting the CD200/CD200R pathway for diagnostic or therapeutic uses warrants further investigation of its role in disease.
There is considerable heterogeneity in the clinical presentation of HP in part because of difficulty in classifying the stages: acute, subacute, and chronic. Patients with fibrotic disease often present with restrictive lung disease and a decrease in carbon dioxide diffusion capacity (37). This is the first study to measure decreased lung function in a mouse model of HP. Our results demonstrate that WT and tlr2/9−/− mice exposed to SR for 14 wk exhibited a significant decrease in quasistatic lung compliance compared with nonexposed mice. The reduced lung compliance can be affected by fluid leakage (indicated by protein in the BALF), inflammatory cell recruitment into the lung, and fibrosis. Our results demonstrate that both WT and tlr2/9−/− mice exhibited a significant increase in total protein in the BALF compared with unexposed mice. In WT mice exposed to SR for 14 wk, the decrease in quasistatic lung compliance was also associated with an increase in fibrosis. Gross examination of the Masson trichrome-stained lung sections from tlr2/9−/− mice revealed increased collagen (blue staining) compared with lungs from nonexposed mice. However, quantitative analysis of the lung sections did not reveal a significant increase in collagen deposition in tlr2/9−/− mice compared with WT nonexposed mice. In addition, compared with unexposed mice, total lung capacity in tlr2/9−/− mice was not significantly reduced. These results suggest that fibrosis is not solely responsible for the decrease in quasistatic compliance observed in the mice; protein leakage and inflammatory cell recruitment must also contribute to reduced compliance.
It has been proposed that the development of fibrosis in chronic HP is due to a switch from a Th1-type response to the more profibrotic Th2-type response. However, despite the increase in fibrosis in WT mice, we were unable to detect Th2 cells or IL-4 mRNA in the lungs of WT mice with long-term exposure to SR. It is possible that IL-4 is present but at levels too low for us to measure in whole lung extracts, and we did detect a low level of IL-13 mRNA in the lungs of long-term SR-exposed WT mice. However, the lack of a large eosinophilic influx along with the neutrophilic/IL-17 response at a time point that corresponds to lung dysfunction and fibrosis suggest that a strong Th2 response is not necessary for development of fibrosis in this model. We were able to detect both Th17 cells in the lung by intracellular cytokine staining and IL-17A mRNA by qRT-PCR. That we did not observe a large percentage of Th17 cells in the lungs of exposed mice despite high levels of IL-17 mRNA suggests that other cells also contribute to its production. Hasan et al. (20) showed that neutrophils and macrophages were the main cell types that expressed IL-17A in mice exposed to SR for 3 wk. It is possible that neutrophils and macrophages contribute to IL-17 production during the chronic stage of FLD. Although our results suggest that Th17 cells predominate in the chronic phase of FLD we did not undertake a complete analysis of all the Th subsets including Tregs, and additional studies are necessary to fully characterize the T cell response at this stage of the disease.
Functionally, IL-17 may play several roles in contributing to lung pathology during the chronic phase of HP. It is a strong inducer of neutrophil chemokines and neutrophil recruitment, and there is a positive correlation between lung neutrophils and lung fibrosis in patients with HP and an increase in gelatinase B and collagenase-2 activity that may contribute to the fibrotic response (21, 31). Additionally, the bleomycin and IL-1β-mediated lung fibrosis models have found that IL-17 production by Th17 and γδ T cells is associated with increased lung fibrosis (43). Our results are supported by previous studies that examined the earlier stages of HP. IL-17ra−/− mice exhibited a decrease in lung inflammation and collagen deposition in the lung following 4 wk of SR exposure (39). However, our results conflict with studies in patients with pigeon breeders disease in whom an increase in Th2 cells over Th1 cells is observed during the chronic phase, although the authors of that study did not measure the level of Th17 cells (5). The causative agent or agents of pigeon breeders disease are proteins found in bird droppings and feathers, moieties distinct from the pathogen-associated molecular patterns contained in SR, which is a Gram-positive member of the actinomycete family. It is likely that differences in PRRs (and combinations of PRRs) stimulated by these different inciting agents dictates the extent to which a Th1, Th2, or Th17 response ensues. However, it is likely that an exaggerated T cell response, despite whether it is Th2 or Th17, will cause some type of lung pathology.
In contrast to the WT mice, there was a significant increase in both IL-17 and IL-13 mRNA in tlr2/9−/− mice with long-term exposure to SR, although we did not detect IL-4 mRNA expression or an increase in Th2 cells in these mice. The increase in IL-17 is in contrast to the earlier stages of the disease in which IL-17 was decreased in SR-exposed tlr2/9−/− mice (4). It is likely that additional PRRs are being activated during the chronic stage of FLD and may contribute to IL-17 production during this phase. The increase in IL-17 (mRNA and protein) in the lungs of tlr2/9−/− mice despite lower levels of fibrosis runs counter to our hypothesis that IL-17 mediates pathology during chronic FLD. Surprisingly, these lungs also demonstrated an increase in IL-13 mRNA; IL-13 has wound-healing properties, however, when produced in excess it has been associated with fibrosis. These results suggest that a Th2-type response develops in tlr2/9−/− mice and that this may be protective in FLD. Although surprising, earlier studies using the SR model of HP suggested that the Th2 response was protective during initiation of disease. Strains of mice such as C57BL/6 that are biased toward Th1 responses are more susceptible to disease, whereas strains of mice such as DBA/2 that are biased toward Th2 responses, are resistant to disease (32, 33). The difference in the Th1 vs. Th2 bias between the strains in response to SR was shown to be due to increased stability of IL-4 and IL-13 mRNA in the DBA/2 strain compared with C57Bl/6 (6). The protective role of IL-4 was demonstrated by Ghadirian et al. (16) when mice treated with recombinant IL-4 exhibited reduced lung inflammation following SR exposure. Our results suggest that IL-13 may also be protective during chronic FLD because compared with WT-exposed mice, tlr2/9−/− mice exhibited a significant reduction in levels of the alarmin IL-1α and fibrosis in the lungs. It is possible that levels of IL-13 present contribute to normal wound healing in tlr2/9−/− mice. An alternative possibility is that the inflammatory response in tlr2/9−/− mice is simply delayed compared with that in WT mice and that with more exposure we would observe an increase in fibrosis and other markers of lung damage similar to that observed in WT mice.
The ability of IL-17 to mediate pathology typically occurs in synergy with other cytokines such as TNF or IL-1β (38). Both cytokines are induced during HP, but IL-1β has numerous functions that may contribute to amplifying the neutrophil/IL-17 response during HP. The importance of IL-1β in IL-17-mediated diseases is illustrated in IL-1Ra−/− mice, which have unopposed IL-1 activity and develop IL-17-dependent rheumatoid arthritis (27). However, in addition to inducing the IL-17 response, IL-1β also synergizes with IL-17 in mediating its pathogenic effects (38). We found an increase in IL-1β in the lungs of SR-exposed mice, which may contribute both to driving IL-17 production and neutrophil recruitment. BALF obtained from patients with HP also reveals an increase in IL-1β. The canonical pathway of IL-1β production is inflammasome activation resulting in activation of caspase-1 and cleavage of pro-IL-1β into the mature form. Each type of inflammasome is formed in response to stimulation of a PRR, typically one of the NLR family members. To this end we identified an increase in NLRP3 and caspase-1 mRNA in the lungs of mice with long-term exposure to SR, suggesting that this signaling pathway may contribute to production of IL-1β. SR may directly activate one of the inflammasomes or, alternatively, the generation of danger-associated molecular patterns (DAMPs) due to lung injury may result in activation of the NLRP3 inflammasome amplifying the inflammatory response.
It is likely that the differences in the chronic stage of the various types of HP are a result of differences in PRR recognition of the inciting antigens. As the disease becomes chronic, the combination of PRRs activated directly via the inciting agent and PRRs activated by DAMPs generated as a result of chronic inflammation may lead to distinct outcomes (i.e., emphysema vs. fibrosis) for various types of HP. Overall, we have demonstrated that long-term exposure to SR results in the development of a predominantly IL-17-mediated response that is largely independent of TLRs 2 and 9.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grants HL-084172 to E.A. Fitzpatrick and HL-094366 and HL-123540 to C.M. Waters.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
K.A., C.M.W., and E.A.F. conception and design of research; K.A., M.C.G., A.S., G.R., C.L.L., and E.A.F. performed experiments; K.A., C.M.W., and E.A.F. analyzed data; C.M.W. and E.A.F. interpreted results of experiments; E.A.F. prepared figures; K.A. drafted manuscript; K.A., C.M.W., and E.A.F. edited and revised manuscript; K.A., M.C.G., A.S., G.R., C.L.L., C.M.W., and E.A.F. approved final version of manuscript.
REFERENCES
- 1.Abdelsamed HA, Desai M, Nance SC, Fitzpatrick EA. T-bet controls severity of hypersensitivity pneumonitis. J Inflamm (Lond) 8: 15, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Agostini C, Trentin L, Facco M, Semenzato G. New aspects of hypersensitivity pneumonitis. Curr Opin Pulm Med 10: 378–382, 2004. [DOI] [PubMed] [Google Scholar]
- 3.Ando M, Suga M. Hypersensitivity pneumonitis. Curr Opin Pulm Med 3: 391–395, 1997. [DOI] [PubMed] [Google Scholar]
- 4.Andrews K, Abdelsamed H, Yi AK, Miller MA, Fitzpatrick EA. TLR2 regulates neutrophil recruitment and cytokine production with minor contributions from TLR9 during hypersensitivity pneumonitis. PLoS One 8: e73143, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barrera L, Mendoza F, Zuniga J, Estrada A, Zamora AC, Melendro EI, Ramirez R, Pardo A, Selman M. Functional diversity of T cell subpopulations in subacute and chronic hypersensitivity pneumonitis. Am J Respir Crit Care Med 177: 44–55, 2008. [DOI] [PubMed] [Google Scholar]
- 6.Butler NS, Monick MM, Yarovinsky TO, Powers LS, Hunninghake GW. Altered IL-4 mRNA stability correlates with Th1 and Th2 bias and susceptibility to hypersensitivity pneumonitis in two inbred strains of mice. J Immunol 169: 3700–3709, 2002. [DOI] [PubMed] [Google Scholar]
- 7.Caserta S, Nausch N, Sawtell A, Drummond R, Barr T, Macdonald AS, Mutapi F, Zamoyska R. Chronic infection drives expression of the inhibitory receptor CD200R, and its ligand CD200, by mouse and human CD4 T cells. PLoS One 7: e35466, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen CJ, Kono H, Golenbock D, Reed G, Akira S, Rock KL. Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nat Med 13: 851–856, 2007. [DOI] [PubMed] [Google Scholar]
- 9.Chizzolini C, Chicheportiche R, Alvarez M, de Rham C, Roux-Lombard P, Ferrari-Lacraz S, Dayer JM. Prostaglandin E2 synergistically with interleukin-23 favors human Th17 expansion. Blood 112: 3696–3703, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chung Y, Chang SH, Martinez GJ, Yang XO, Nurieva R, Kang HS, Ma L, Watowich SS, Jetten AM, Tian Q, Dong C. Critical regulation of early Th17 cell differentiation by interleukin-1 signaling. Immunity 30: 576–587, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cui W, Cuartas E, Ke J, Zhang Q, Einarsson HB, Sedgwick JD, Li J, Vignery A. CD200 and its receptor, CD200R, modulate bone mass via the differentiation of osteoclasts. Proc Natl Acad Sci USA 104: 14436–14441, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Erkinjuntti-Pekkanen R, Rytkonen H, Kokkarinen JI, Tukiainen HO, Partanen K, Terho EO. Long-term risk of emphysema in patients with farmer's lung and matched control farmers. Am J Respir Crit Care Med 158: 662–665, 1998. [DOI] [PubMed] [Google Scholar]
- 14.Fernández Pérez ER, Swigris JJ, Forssén AV, Tourin O, Solomon JJ, Huie TJ, Olson AL, Brown KK. Identifying an inciting antigen is associated with improved survival in patients with chronic hypersensitivity pneumonitis. Chest 144: 1644–1651, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fong DJ, Hogaboam CM, Matsuno Y, Akira S, Uematsu S, Joshi AD. Toll-like receptor 6 drives interleukin-17A expression during experimental hypersensitivity pneumonitis. Immunology 130: 125–136, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ghadirian E, Denis M. Murine hypersensitivity pneumonitis: interleukin-4 administration partially abrogates the disease process. Microb Pathog 12: 377–382, 1992. [DOI] [PubMed] [Google Scholar]
- 17.Gorczynski RM, Cattral MS, Chen Z, Hu J, Lei J, Min WP, Yu G, Ni J. An immunoadhesin incorporating the molecule OX-2 is a potent immunosuppressant that prolongs allo- and xenograft survival. J Immunol 163: 1654–1660, 1999. [PubMed] [Google Scholar]
- 18.Gurney JW. Hypersensitivity pneumonitis. Radiol Clin North Am 30: 1219–1230, 1992. [PubMed] [Google Scholar]
- 19.Hanak V, Golbin JM, Hartman TE, Ryu JH. High-resolution CT findings of parenchymal fibrosis correlate with prognosis in hypersensitivity pneumonitis. Chest 134: 133–138, 2008. [DOI] [PubMed] [Google Scholar]
- 20.Hasan SA, Eksteen B, Reid D, Paine HV, Alansary A, Johannson K, Gwozd C, Goring KA, Vo T, Proud D, Kelly MM. Role of IL-17A and neutrophils in fibrosis in experimental hypersensitivity pneumonitis. J Allergy Clin Immunol 131: 1663–1673, 2013. [DOI] [PubMed] [Google Scholar]
- 21.Haslam PL. Bronchoalveolar lavage in extrinsic allergic alveolitis. Eur J Respir Dis Suppl 154: 120–135, 1987. [PubMed] [Google Scholar]
- 22.Hoek RM, Ruuls SR, Murphy CA, Wright GJ, Goddard R, Zurawski SM, Blom B, Homola ME, Streit WJ, Brown MH, Barclay AN, Sedgwick JD. Down-regulation of the macrophage lineage through interaction with OX2 (CD200). Science 290: 1768–1771, 2000. [DOI] [PubMed] [Google Scholar]
- 23.Johannson K, Ryerson CJ. Making an accurate diagnosis of chronic hypersensitivity pneumonitis. Can Respir J 21: 371–370, 2014. [DOI] [PubMed] [Google Scholar]
- 24.Joshi AD, Fong DJ, Oak SR, Trujillo G, Flaherty KR, Martinez FJ, Hogaboam CM. Interleukin-17-mediated immunopathogenesis in experimental hypersensitivity pneumonitis. Am J Respir Crit Care Med 179: 705–716, 2009. [DOI] [PubMed] [Google Scholar]
- 26.Li Y, Zhao LD, Tong LS, Qian SN, Ren Y, Zhang L, Ding X, Chen Y, Wang YX, Zhang W, Zeng XF, Zhang FC, Tang FL, Zhang X, Ba DN, He W, Cao XT, Lipsky PE. Aberrant CD200/CD200R1 expression and function in systemic lupus erythematosus contributes to abnormal T cell responsiveness and dendritic cell activity. Arthritis Res Ther 14: R123, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu FL, Chen CH, Chu SJ, Chen JH, Lai JH, Sytwu HK, Chang DM. Interleukin (IL)-23 p19 expression induced by IL-1beta in human fibroblast-like synoviocytes with rheumatoid arthritis via active nuclear factor-kappaB and AP-1 dependent pathway. Rheumatology (Oxford) 46: 1266–1273, 2007. [DOI] [PubMed] [Google Scholar]
- 28.Mitaka K, Miyazaki Y, Yasui M, Furuie M, Miyake S, Inase N, Yoshizawa Y. Th2-biased immune responses are important in a murine model of chronic hypersensitivity pneumonitis. Int Arch Allergy Immunol 154: 264–274, 2011. [DOI] [PubMed] [Google Scholar]
- 29.Nance S, Cross R, Yi AK, Fitzpatrick EA. IFN-gamma production by innate immune cells is sufficient for development of hypersensitivity pneumonitis. Eur J Immunol 35: 1928–1938, 2005. [DOI] [PubMed] [Google Scholar]
- 30.Nance SC, Yi AK, Re FC, Fitzpatrick EA. MyD88 is necessary for neutrophil recruitment in hypersensitivity pneumonitis. J Leukoc Biol 83: 1207–1217, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pardo A, Barrios R, Gaxiola M, Segura-Valdez L, Carrillo G, Estrada A, Mejia M, Selman M. Increase of lung neutrophils in hypersensitivity pneumonitis is associated with lung fibrosis. Am J Respir Crit Care Med 161: 1698–1704, 2000. [DOI] [PubMed] [Google Scholar]
- 32.Patel AM, Ryu JH, Reed CE. Hypersensitivity pneumonitis: current concepts and future questions. J Allergy Clin Immunol 108: 661–670, 2001. [DOI] [PubMed] [Google Scholar]
- 33.Remy-Jardin M, Remy J, Wallaert B, Muller NL. Subacute and chronic bird breeder hypersensitivity pneumonitis: sequential evaluation with CT and correlation with lung function tests and bronchoalveolar lavage. Radiology 189: 111–118, 1993. [DOI] [PubMed] [Google Scholar]
- 34.Rijkers ES, de Ruiter T, Baridi A, Veninga H, Hoek RM, Meyaard L. The inhibitory CD200R is differentially expressed on human and mouse T and B lymphocytes. Mol Immunol 45: 1126–1135, 2008. [DOI] [PubMed] [Google Scholar]
- 35.Salvaggio JE, deShazo RD. Pathogenesis of hypersensitivity pneumonitis. Chest 89: 190S–193S, 1986. [DOI] [PubMed] [Google Scholar]
- 36.Sharma OP, Fujimura N. Hypersensitivity pneumonitis: a noninfectious granulomatosis. Semin Respir Infect 10: 96–106, 1995. [PubMed] [Google Scholar]
- 37.Sharma SK, Pande JN, Verma K, Guleria JS. Bronchoalveolar lavage fluid (BALF) analysis in interstitial lung diseases—a 7-year experience. Indian J Chest Dis Allied Sci 31: 187–196, 1989. [PubMed] [Google Scholar]
- 38.Shen F, Gaffen SL. Structure-function relationships in the IL-17 receptor: implications for signal transduction and therapy. Cytokine 41: 92–104, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Simonian PL, Roark CL, Wehrmann F, Lanham AK, Diaz del Valle F, Born WK, O'Brien RL, Fontenot AP. Th17-polarized immune response in a murine model of hypersensitivity pneumonitis and lung fibrosis. J Immunol 182: 657–665, 2009. [PMC free article] [PubMed] [Google Scholar]
- 41.Sutton CE, Lalor SJ, Sweeney CM, Brereton CF, Lavelle EC, Mills KH. Interleukin-1 and IL-23 induce innate IL-17 production from gammadelta T cells, amplifying Th17 responses and autoimmunity. Immunity 31: 331–341, 2009. [DOI] [PubMed] [Google Scholar]
- 42.Vourlekis JS, Schwarz MI, Cherniack RM, Curran-Everett D, Cool CD, Tuder RM, King TE Jr, Brown KK. The effect of pulmonary fibrosis on survival in patients with hypersensitivity pneumonitis. Am J Med 116: 662–668, 2004. [DOI] [PubMed] [Google Scholar]
- 43.Wilson MS, Madala SK, Ramalingam TR, Gochuico BR, Rosas IO, Cheever AW, Wynn TA. Bleomycin and IL-1beta-mediated pulmonary fibrosis is IL-17A dependent. J Exp Med 207: 535–552, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yoshizawa Y, Ohtani Y, Hayakawa H, Sato A, Suga M, Ando M. Chronic hypersensitivity pneumonitis in Japan: a nationwide epidemiologic survey. J Allergy Clin Immunol 103: 315–320, 1999. [DOI] [PubMed] [Google Scholar]
- 45.Zacharisen MC, Fink JN. Hypersensitivity pneumonitis and related conditions in the work environment. Immunol Allergy Clin North Am 31: 769–786, vii, 2011. [DOI] [PubMed] [Google Scholar]