

Abstract
Fetal overgrowth is common in obese women and is associated with perinatal complications and increased risk for the child to develop metabolic syndrome later in life. Placental nutrient transport capacity has been reported to be increased in obese women giving birth to large infants; however, the underlying mechanisms are not well established. Obesity in pregnancy is characterized by elevated maternal serum insulin and leptin, hormones that stimulate placental amino acid transporters in vitro. We hypothesized that maternal obesity activates placental insulin/IGF-I/mTOR and leptin signaling pathways. We tested this hypothesis in a mouse model of obesity in pregnancy that is associated with fetal overgrowth. C57BL/6J female mice were fed a control (C) or a high-fat/high-sugar (HF/HS) pelleted diet supplemented by ad libitum access to sucrose (20%) solution. Placentas were collected at embryonic day 18.5. Using Western blot analysis, placental mTOR activity was determined along with energy, inflammatory, leptin, and insulin signaling pathways (upstream modulators of mTOR). Phosphorylation of S6 ribosomal protein (S-235/236), 4E-BP1 (T-37/46), Insulin receptor substrate 1 (Y-608), Akt (T-308), and STAT-3 (Y-705) was increased in obese dams. In contrast, expression of placental caspase-1, IкBα, IL-1β, and phosphorylated-JNKp46/54-T183/Y185 was unaltered. Fetal amino acid availability is a key determinant of fetal growth. We propose that activation of placental insulin/IGF-I/mTOR and leptin signaling pathways in obese mice stimulates placental amino acid transport and contributes to increased fetal growth.
Keywords: fetal growth, intracellular signaling proteins, fetal programming, pregnancy, maternal-fetal exchange
maternal obesity during pregnancy is associated with a higher risk of pregnancy complications, as well as increased birth weight (55). Furthermore, children born to obese mothers are more likely to become obese themselves (58). Infants of obese mothers have increased visceral adiposity (10) and elevated risk for metabolic syndrome later in life (9, 22). Large-for-gestational age babies of overweight and obese mothers show increased lipolysis and a propensity for decreased insulin sensitivity already at birth (1) and are at particular risk to establish a metabolic trajectory leading to obesity, Type 2 diabetes, and cardiovascular disease in childhood and beyond (2, 45).
The mechanisms linking obesity in pregnancy to altered fetal growth and programming of adult disease are not well established, but there is an increasing awareness that changes in placental metabolism and nutrient transport capacity may contribute to altered fetal growth in maternal obesity (21). We recently generated a novel mouse model of maternal obesity by feeding a diet high in saturated fat, cholesterol, and simple sugars, resembling a diet common in Western societies (52). This resulted in fetal overgrowth associated with maternal metabolic alterations similar to that observed in pregnant women with high body mass index (BMI). Furthermore, the protein expression of specific glucose and amino acid transporter isoforms and amino acid transport activity were markedly elevated in placentas of obese dams (52), consistent with the possibility that fetal overgrowth in response to maternal obesity may be caused by increased placental nutrient transport. However, the placental signaling events linking maternal obesity to upregulation of placental nutrient transport remain to be established.
Mechanistic target of rapamycin (mTOR) is a serine/threonine kinase that integrates growth factor signaling with information on cellular levels of nutrients, oxygen, stress, and energy to regulate cell growth, mediated by effects on gene expression and protein translation. The mTOR protein is present in the cytoplasm in two complexes: mTOR complex 1 (mTORC1) and 2 (mTORC2) (37). mTORC1, the canonical target of rapamycin and it is analogs, is responsive to nutrient abundance, energy sufficiency, and growth factor signaling. mTORC1 signaling activity is commonly assessed by phosphorylation of downstream targets S6 kinase 1 (S6K1), ribosomal protein S6 (rpS6), and eukaryotic translation initiation factor 4E-binding protein 1 (4E-BP1). Phosphorylation of 4E-BP1 results in its dissociation from eukaryotic translation initiation factor 4E (eIF4E), allowing eIF4E to associate with the scaffold for the cap-binding complex, eukaryotic translation initiation factor 4G (or eIF4G). The fully assembled cap-binding complex, eIF4E, has the binding and helicase activities required to recruit mRNA to the ribosomes and initiate translation (14). mTORC2 has been reported to be involved in the regulation of actin cytoskeleton and cell survival through RAC-α serine/threonine-protein kinase (Akt) activation (60). Upstream regulators of mTOR activity include the insulin/phosphatidylinositide 3-kinases (PI3K)/Akt signal transduction pathway (24), the AMPK, leptin (46) and the ERK 1/2 pathway (33).
The placenta has been proposed to function as a nutrient sensor matching fetal growth to maternal nutrient availability by altering placental growth and nutrient transport capacity (28). Placental mTOR signaling in the placenta is believed to constitute a key component of the placental nutrient sensor (32, 51). Using the phosphorylation of well-established downstream targets as functional readouts, placental mTOR activity is positively correlated to fetal growth in animal models (3, 18, 32, 57) and in humans (27, 48). Moreover, mTOR is a positive regulator of System A and System L amino acid transporter in the placenta (53).
Obesity in nonpregnant individuals is associated with a low-grade chronic inflammation, which is believed to contribute to insulin resistance (41). Obese pregnant women also show signs of inflammation, including elevation of circulating cytokines such as TNF-α, monocyte chemoattractant protein-1 (MCP-1), IL-1, and IL-6 (5, 7, 11, 47), compared with pregnant women with normal BMI. In the placenta, inflammatory pathways may be activated in obese women in response to inflammatory stimuli or through infiltration of maternal immune cells with subsequent increased cytokine production (11). The key intracellular mediators of the inflammatory response include NF-κB, JNK, caspase-1, and p38-MAPK, which are activated by Toll-like receptor 4 signaling cascade and STAT3. The cellular effects of leptin are largely mediated by the JAK/STAT signaling pathway, which includes the phosphorylation of STAT3 at Tyr-705, resulting in STAT3 dimerization and migration to the nucleus, where transcription initiation is affected (17). However, Tyr-705-STAT3 phosphorylation can also be induced by other cytokines and hormones (36).
Maternal obesity is associated with elevated circulating levels of insulin and leptin (9, 11). These hormones have been shown to activate mTOR in nonplacental cells (17, 37). In cultured primary human trophoblast cells, insulin stimulates amino acid uptake mediated by a mTOR-dependent mechanism (50). Furthermore, maternal circulating levels of adiponectin are markedly decreased in obese pregnant women (26) and mice (52) compared with lean controls. Because maternal adiponectin inhibits insulin signaling in the placenta (4, 29, 54), the low maternal adiponectin associated with maternal obesity may contribute to activation of placental insulin signaling. In the current study, we used a novel mouse model of maternal obesity with fetal overgrowth and a maternal endocrine and metabolic profile similar to that of obese pregnant women (52) to test the hypothesis that maternal obesity activates placental insulin/IGF-I/mTOR and leptin signaling pathways.
MATERIALS AND METHODS
Animals and diets.
The Institutional Animal Care and Use Committee at the University of Texas Health Science Center San Antonio approved all of the study's protocols. Female ∼12-wk-old C57BL/6J mice (n = 24), which were proven breeders (one previous litter; The Jackson Laboratory, Bar Harbor, ME), were housed five per cage under controlled conditions (25°C, 12:12-h light-dark cycle). Starting at 13 wk of age, animals were fed ad libitum with a control (D12489B, 10.6 kcal% fat) or high-fat pellet diet (Western diet D12079B, 41 kcal% fat) supplemented with ad libitum access to sucrose (20%) solution (high fat/high sugar, HF/HS) (52). The sucrose solution was supplemented with vitamins (Vitamin Mix V10001, 10 g/4,000 kcal) and minerals (Mineral Mix S10001, 35 g/4,000 kcal). Diets and vitamin and mineral mixes were purchased from Research Diets (New Brunswick, NJ). All animals had free access to water. When females on the HF/HS diet had increased their body weight by 25%, which occurred after 4–6 wk on the diet, they, and age-matched females on the control diet, were mated by overnight housing with a male on a control diet. On the next morning, mating was confirmed by the presence of a vaginal plug [defined as embryonic day (E) 0.5], and dams were maintained on their respective diets throughout gestation. At E18.5, dams were euthanized for collection of tissue samples.
Collection of placental tissue.
Dams were fasted (4 h) and then euthanized at E18.5 by carbon dioxide inhalation. After laparotomy, fetuses and placentas were collected and quickly dried on blotting paper; any remaining fetal membranes were removed and weighed. All placentas in each litter were pooled and washed in PBS and transferred to 3 ml of buffer D [250 mM sucrose, 10 mM HEPES-Tris, and 1 mM EDTA (pH 7.4) at 4°C], protease and phosphatase inhibitor cocktail (Sigma-Aldrich, St. Louis, MO) was added at a dilution of 1:1,000, and the mixture was homogenized using a Polytron (Kinematica, Bohemia, NY), frozen in liquid nitrogen, and stored at −80°C until analysis.
Western blot analysis.
Western blot analysis was performed as previously described (32). In brief, 10 μg of total protein were loaded onto a SDS-PAGE, and electrophoresis was performed at a constant 100 V for 2 h. Proteins were transferred onto nitrocellulose membranes overnight at a constant 30 V. After the transfer, membranes were blocked in 5% blotting grade blocker nonfat dry milk (Bio-Rad, Hercules, CA) in TBS (wt/vol) plus 0.1% Tween 20 (vol/vol) for 1 h at room temperature. Membranes were incubated with primary antibodies overnight at 4°C. Subsequently, membranes were incubated with the appropriate secondary peroxidase-labeled antibodies for 1 h. After washing, bands were visualized using enhanced chemiluminescence detection reagents (GE Healthcare, Chalfont, St. Giles, Buckinghamshire, UK). Blots were stripped as described previously (51) and reprobed for β-actin as a loading control. Analysis of the blots was performed by densitometry using ImageJ software (National Institutes of Health, Bethesda, MD). For each protein target, the mean density of the control sample bands was assigned an arbitrary value of 1, and data are presented relative to control. The relative density of the target protein in each lane was divided by the density of the corresponding β-actin band as a loading control. Furthermore, β-actin did not differ between control and HF/HS group (data not shown).
Data presentation and statistics.
Data are presented as means ± SE. For fetal and placental data, means of each litter were calculated and used in the statistical analysis. Therefore, n represents the number of litters. Statistical significance of differences between control and HF/HS groups was assessed using Student's unpaired t-test. A P value <0.05 was considered significant.
RESULTS
Fetal and placental weights.
In the present study, placental samples were used from dams fed a control and HF/HS diet in which maternal metabolism and placental amino acid transport have been reported previously (52). Fetal weights were increased by 18% (P > 0.01; n = 12 in each group) at E18.5 in the HF/HS group compared with control (52). This was not due to a difference in litter size, which was essentially the same in the control (6.9 ± 0.14; n = 12) and HF/HS group (6.8 ± 0.32; n = 12). Placental weights were not different between groups.
Placental mTORC1 signaling is activated by maternal HF/HS diet.
To test the hypothesis that placental mTOR signaling is increased by feeding the dams a HF/HS diet, we determined the phosphorylation of rpS6 and 4E-BP1, two well-established functional readouts for mTORC1 signaling. The phosphorylation of ribosomal protein S6 (S-235/236), a component of the 40S ribosome and a physiologically relevant S6K1 substrate, was increased by more than 150% (P < 0.001, Fig. 1, A and B) in the HF/HS diet group compared with the control group. Similarly, feeding of HF/HS diet significantly increased the phosphorylation of 4E-BP1 at T-37/46 (+ 89%, P < 0.001, Fig. 2, A and B) compared with controls. There was no significant difference in the total S6 and 4E-BP1 expression level between control and HF/HS group placentas (Figs. 1 and 2, A and B).
Fig. 1.
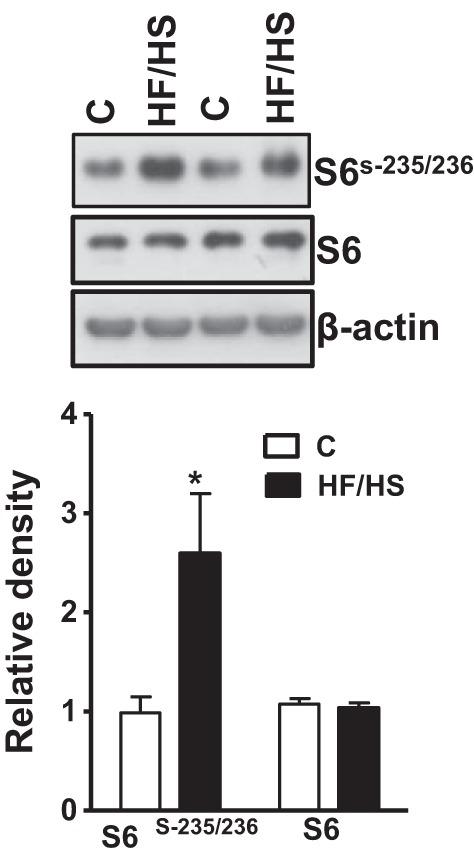
Placental S6 phosphorylation in mice fed a control (C) or high-fat/high-sugar (HF/HS) diet. A: representative Western blots of S6 (S-235/236) and total S6 in homogenates of mice placenta at E18.5. B: summary of the Western blot data. After normalization to β-actin, the mean density of control samples was assigned an arbitrary value of 1. Subsequently, individual control and HF/HS density values were expressed relative to this mean. Values are given as means ± SE; n = 12/each group, *P < 0.05 vs. control, using unpaired Student's t-test.
Fig. 2.
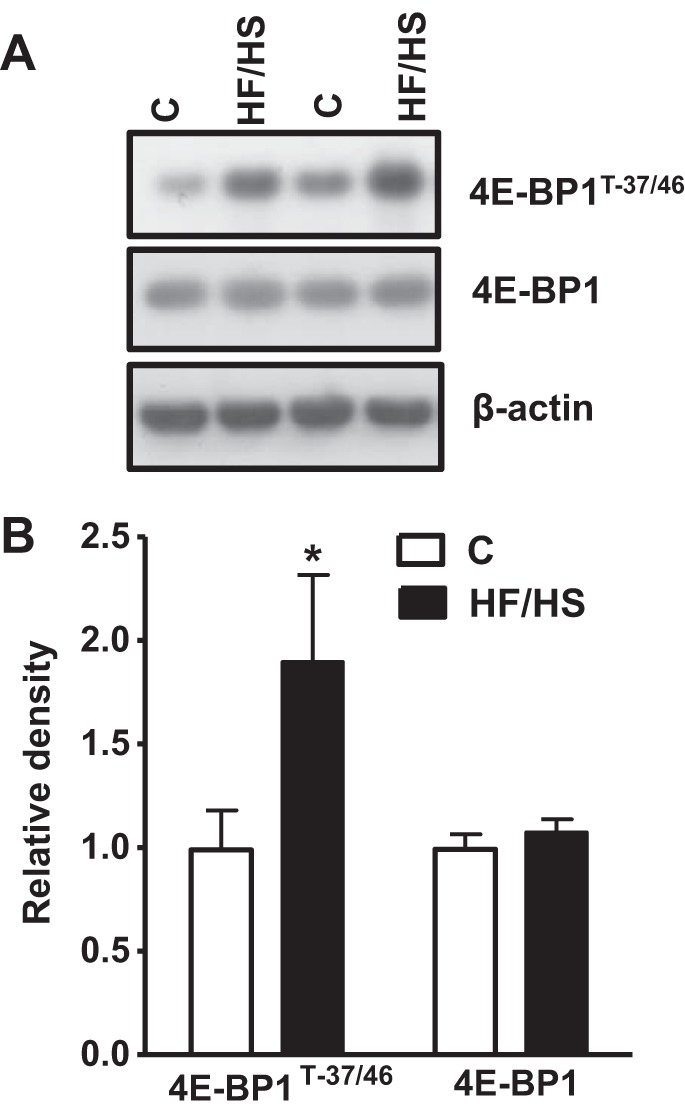
Placental 4E-BP1 phosphorylation in mice fed a control (C) or HF/HS diet. A: representative A: Western blots of 4E-BP1 (T-37/46) and total 4E-BP1 in homogenates of mice placenta at E18.5. B: summary of the Western blot data. After normalization to β-actin, the mean density of control samples was assigned an arbitrary value of 1. Subsequently, individual control and HF/HS density values were expressed relative to this mean. Values are given as means ± SE; n = 12/each group, *P < 0.05 vs. control, using unpaired Student's t-test.
Maternal HF/HS diet inhibits placental AMPK phosphorylation.
To investigate which upstream signaling pathways could influence mTORC1 activity in the placenta of dams fed an HF/HS diet, we measured AMPK activation. Phosphorylation of AMPKα at T-172, a functional readout of AMPK activity, was significantly lower in the placenta of dams fed a HF/HS diet (−75%, P < 0.0001). There was no significant difference in the total placental AMPK expression between control and HF/HS groups (Fig. 3, A and B).
Fig. 3.
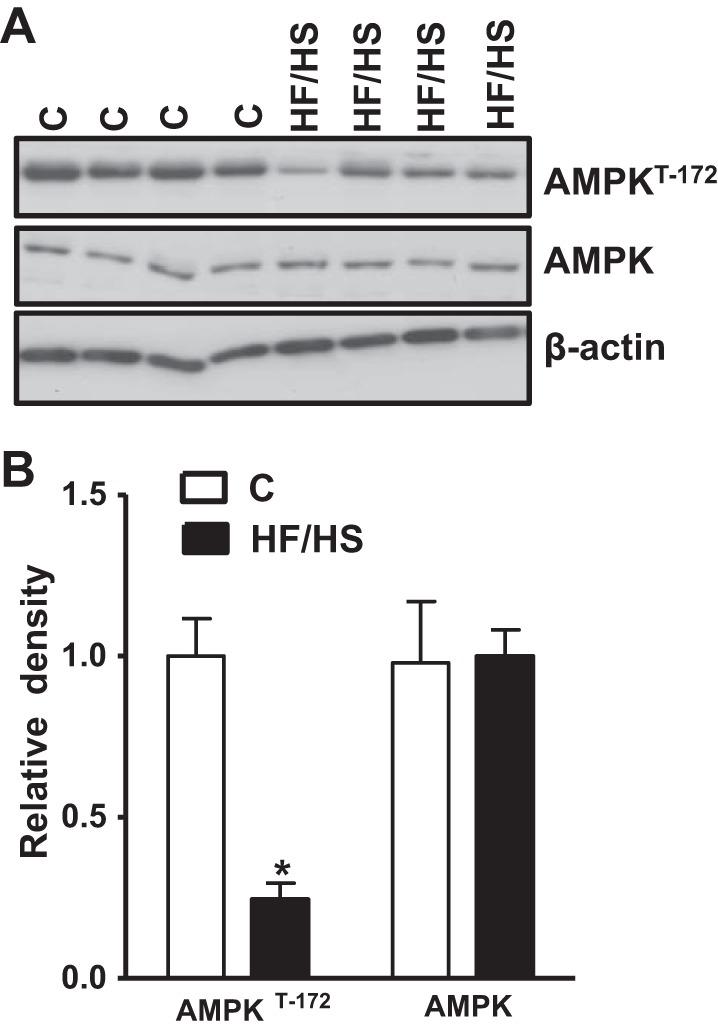
Placental AMPK phosphorylation in mice fed a control (C) or HF/HS diet. A: representative Western blots of AMPK (T-172) and total AMPK in homogenates of mice placenta at E18.5. B: summary of the Western blot data. After normalization to β-actin, the mean density of control samples was assigned an arbitrary value of 1. Subsequently, individual control and HF/HS density values were expressed relative to this mean. Values are given as means ± SE; n = 12/each group, *P < 0.05 vs. control, using unpaired Student's t-test.
Maternal HF/HS diet activates placental insulin/IGF-I signaling.
Placental insulin/IGF-I signaling activity was assessed by determining phosphorylation of insulin receptor substrate 1 (IRS-1) and Akt (Fig. 4, A and B). Phosphorylation of IRS-1 at Y-608 (+ 50%, P < 0.001) and Akt at T-308 (+ 90%, P < 0.001) was significantly higher in the placentas of dams fed a HF/HS diet compared with controls. There was no significant difference in the total IRS-1 or Akt expression between the two groups.
Fig. 4.

Placental IRS-1 and Akt phosphorylation in mice fed a control (C) or HF/HS diet. A: representative Western blots of IRS-1 (Y-608), Akt (T-308), and total IRS-1/Akt in homogenates of mice placenta at E18.5. B: summary of the Western blot data. After normalization to β-actin, the mean density of control samples was assigned an arbitrary value of 1. Subsequently, individual control and HF/HS density values were expressed relative to this mean. Values are given as means ± SE; n = 12/each group, *P < 0.05 vs. control, using unpaired Student's t-test.
Placental JNK, IκB, and caspase-1 inflammatory pathways are not regulated by maternal HF/HS diet.
We tested the hypothesis that feeding the dams a HF/HS diet leads to placental inflammation. We studied the expression of caspase-1 (analyzing 50- and 20-kDa bands together), IκBα, JNKp46/54-T183/Y185, and IL-1β in the placental homogenates of control and HF/HS groups. The phosphorylation or total expression of these targets did not differ between the two groups (Fig. 5, A and B).
Fig. 5.
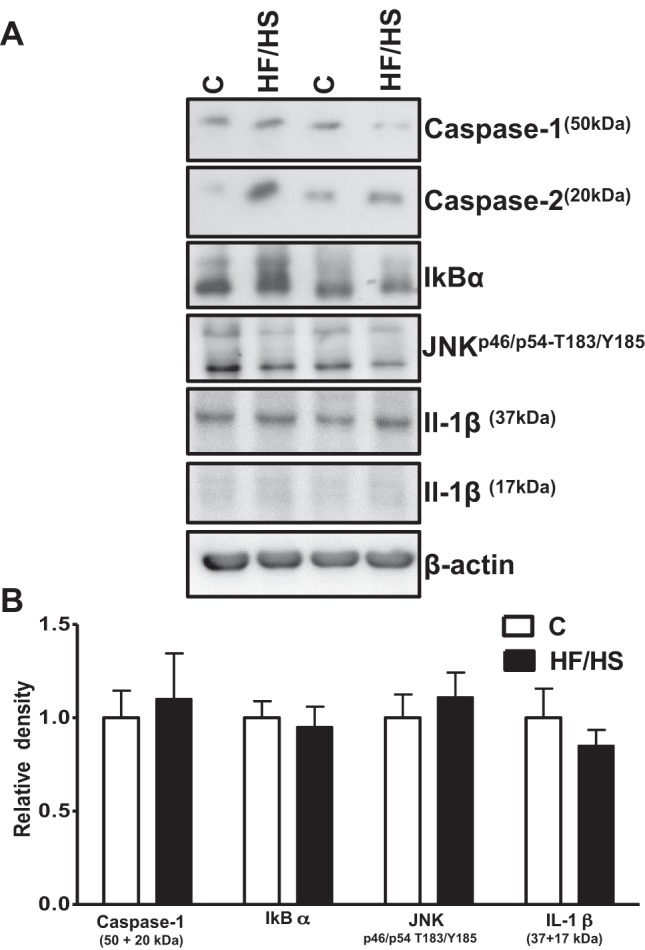
Placental of caspase-1, IκBα, JNKp46/54-T183/Y185 and IL-1β expression mice fed a control (C) or HF/HS diet. A: representative Western blots of caspase-1, IκBα, JNKp46/54-T183/Y185, and IL-1β in homogenates of mice placenta at E18.5. B: summary of the Western blot data. After normalization to β-actin, the mean density of control samples was assigned an arbitrary value of 1. Subsequently, individual control and HF/HS density values were expressed relative to this mean. Values are given as means ± SE; n = 12/each group.
Maternal HF/HS diet activates placental STAT3 signaling.
We have previously reported increased circulating leptin levels in dams fed a HF/HS diet (52), and STAT3 is a key mediator of intracellular signaling downstream of the leptin receptor. Phosphorylation of STAT3 at Y-705 was significantly higher in the placenta of dams fed an HF diet (+ 60%, P < 0.001) compared with controls. Total STAT3 expression was not significantly different between the control and HF/HS groups (Fig. 6, A and B).
Fig. 6.

Placental phosphorylated STAT-3 expression in mice fed a control (C) or HF/HS diet. A: representative Western blots of STAT-3 (Y-705) and total STAT-3 in homogenates of mice placenta at E18.5. B: summary of the Western blot data. After normalization to β-actin, the mean density of control samples was assigned an arbitrary value of 1. Subsequently, individual control and HF/HS density values were expressed relative to this mean. Values are given as means ± SE; n = 12/each group. *P < 0.05 vs. control, using unpaired Student's t-test.
DISCUSSION
In this study, we demonstrate that placental insulin, mTOR, and STAT3 signaling pathways, all known to be positive regulators of placental amino acid transporters, are activated in a mouse model of obesity in pregnancy, which is associated with fetal overgrowth. We propose that these pathways stimulate placental amino acid transport and contribute to increased fetal growth.
We developed the mouse model of obesity in pregnancy used in the current study to resemble maternal obesity in women, and a number of previously published observations suggest that it is relevant for the human condition. First, maternal obesity was induced by feeding mice a high-fat diet supplemented with high sugar (52), which is comparable with the diet in overweight/obese women (reviewed in Refs. 19, 23, 43). Second, in this model, maternal obesity is associated with fetal overgrowth (52), which is a common clinical outcome in obese women (6, 16, 20, 39, 55). Third, maternal endocrine changes (increased fasting serum insulin and leptin, decreased adiponectin) and changes in placental nutrient transport (increased amino acid and glucose transport capacity) (52) replicate findings in obese women giving birth to large babies (27). Moreover, the activation of placental insulin and mTOR signaling and inhibition of AMPK signaling reported here are strikingly similar to changes in placentas of large infants of obese mothers (27). Therefore, this model addresses a critical need for an animal model of obesity in pregnancy that is clinically relevant.
Placental signaling pathways linking maternal nutrition and metabolism to changes in nutrient transport may include mTOR, which is regulated by a wide range of factors, including amino acids, glucose, oxygen, energy status, and insulin/IGF-I, leptin, and TNF-α signaling (59). In the current study, we show that phosphorylation of ribosomal protein S6 and 4E-BP1, representing well-established functional readouts of the mTORC1 signaling pathway, was increased in placentas of HF/HS-fed mice, consistent with an activation of placental mTOR signaling in obese women giving birth to larger babies (27). These findings are also in agreement with placental mTOR activation and increased fetal growth in a model of maternal overweight in the rat (18). We recently demonstrated that mTOR regulates system A and L amino acid transport activity by modulating cell surface abundance of sodium-dependent neutral amino acid transporter-1 and large neutral amino acid transporter 1 isoforms in cultured primary human trophoblast cells (53), providing one possible mechanism underlying the increase in expression of system A and L isoforms in the trophoblast plasma membrane in obese dams (52). To identify upstream signals activating mTOR signaling in placentas of obese dams, we studied AMPK, which is the primary cellular energy sensor and is phosphorylated at Thr-172 in response to increased AMP/ATP ratio. Activation of AMPK is a known negative regulator of mTOR signaling (37), and we have shown previously that placental AMPK is downregulated in a rat model of maternal overweight and increased fetal growth (18). In this study, we demonstrated that the activity of placental AMPK was markedly decreased in maternal obesity induced by HF/HS diet, in agreement with findings in obese women (27). Therefore, it is possible that the activation of placental mTORC1 signaling in placentas of obese mice is caused, in part, by AMPK inhibition.
The increased phosphorylation of placental IRS-1 and Akt, well-established functional readouts of insulin/IGF-I signaling, in response to maternal obesity is consistent with the elevated circulating levels of maternal insulin in obese dams that we reported previously (52). Maternal serum leptin levels are also increased in obese dams (52), which may contribute to the increased phosphorylation of STAT3 in placentas of obese dams observed in the present study. These signaling changes may contribute to the increased placental amino acid transport in maternal obesity (27, 52) because insulin stimulates placental system A and L amino acid transport in cultured primary human trophoblast cells (31, 49), and insulin and leptin have been reported to stimulate system A amino acid transport in placental villous fragments (25). Moreover, we have previously reported that activation of STAT3 by IL-6 stimulates system A amino acid transport in cultured trophoblast cells (30).
Activation of inflammatory pathways by TNF-α increases mTOR signaling activity in cell lines of nonplacental origin (38). Furthermore, we have reported previously that physiological concentrations of IL-6 and TNF-α stimulate amino acid transport in cultured primary human trophoblast cells (30). Some studies suggest that maternal obesity is associated with chronic low-grade inflammation (10). A variety of cytokines, including, IL-1β, IL-6, and IL-10; MCP1; interferon gamma; and TNF-α have been observed to be elevated in the maternal circulation in association with obesity in pregnancy in both humans and rodents (34, 35). We found only modest changes in circulating inflammatory markers in obese pregnant women (5). In addition, although maternal obesity in women was associated with increased placental STAT3 and p38-MAPK activity, classical inflammatory pathways, such as JNK and NF-κB, were not activated in the placenta (5). In our mouse model of obesity, we found little evidence of placental inflammation, suggesting that inflammatory signals are unlikely to cause activation of mTOR signaling or placental amino acid transport.
Sex differences in the rate of fetal growth have long been recognized (40) and the sex of the fetus may affect the ability of the placenta to respond to adverse stimuli (12, 42, 44, 56). Because placentas were pooled for isolation of trophoblast plasma membranes for subsequent analysis of amino acid transport activity (52), we were not able to separate our findings, according to fetal sex or study placental morphology. One potential limitation of this study is, therefore, that sex-specific effects of maternal obesity on placental function and morphology were not examined. Furthermore, the current study does not address the question of whether the marked effects on placental signaling are caused by the high-fat diet, the obesogenic metabolic environment, or a combination of these factors. In addition, it remains to be demonstrated that fetuses of HF/HS dams have increased adiposity. As with all animal studies, extrapolation of findings in this mouse model to pregnant women with obesity has to be done with caution. The mouse has a large litter and differences in maturity at birth (13), and placental structure (8) between humans and mice introduce some limitations in using the mouse as a model for human pregnancy. However, the extensive functional similarities between the mouse and human placentas (15) suggest that this animal model is relevant for the human.
Perspectives and Significance
Our findings indicate that HF/HS diet-induced maternal obesity in mice activates placental insulin/mTOR and STAT3 signaling. Given the well-established role of mTOR as a positive regulator of placental amino acid transport (53), we propose that the activation of placental mTOR in our HF/HS model increases placental amino acid transport capacity (52). We have previously reported that this mouse model of maternal obesity induced by an HF/HS diet shares many characteristics with human pregnancies complicated by obesity, including high dietary fat and sugar intake, maternal hyperleptinemia, hyperinsulinemia, and increased placental amino acid nutrient transfer and fetal overgrowth (52). In this study, we demonstrate that our mouse model of maternal obesity and fetal overgrowth is associated with changes in placental signaling that closely resembles the signaling changes in placentas of obese women giving birth to large babies (27). Obesity and metabolic syndrome may, in part, originate in fetal life. In particular, babies of mothers with obesity are often large at birth and have increased adiposity, which predisposes them for the development of metabolic disease later in life (9, 22). This model will help us better understand the impact of maternal overweight/obesity on placental function and pregnancy outcomes.
GRANTS
This study was supported by National Institutes of Health Grant OD016724.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: F.J.R., T.L.P., and T.J. conception and design of research; F.J.R. performed experiments; F.J.R. analyzed data; F.J.R., T.L.P., and T.J. interpreted results of experiments; F.J.R. prepared figures; F.J.R., T.L.P., and T.J. drafted manuscript; F.J.R., T.L.P., and T.J. edited and revised manuscript; F.J.R., T.L.P., and T.J. approved final version of manuscript.
REFERENCES
- 1.Ahlsson FS, Diderholm B, Ewald U, Gustafsson J. Lipolysis and insulin sensitivity at birth in infants who are large for gestational age. Pediatrics 120: 958–965, 2007. [DOI] [PubMed] [Google Scholar]
- 2.Alfaradhi MZ, Ozanne SE. Developmental programming in response to maternal overnutrition. Front Genet 2: 27, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arroyo JA, Brown LD, Galan HL. Placental mammalian target of rapamycin and related signaling pathways in an ovine model of intrauterine growth restriction. Am J Obstet Gynecol 201: 616e 611–617, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aye IL, Gao X, Weintraub ST, Jansson T, Powell TL. Adiponectin inhibits insulin function in primary trophoblasts by PPARα-mediated ceramide synthesis. Mol Endocrinol 28: 512–524, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aye IL, Lager S, Ramirez VI, Gaccioli F, Dudley DJ, Jansson T, Powell TL. Increasing maternal body mass index is associated with systemic inflammation in the mother and the activation of distinct placental inflammatory pathways. Biol Reprod 90: 129, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baeten JM, Bukusi EA, Lambe M. Pregnancy complications and outcomes among overweight and obese nulliparous women. Am J Publ Health 91: 436–440, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Basu S, Haghiac M, Surace P, Challier JC, Guerre-Millo M, Singh K, Waters T, Minium J, Presley L, Catalano PM, Hauguel-de Mouzon S. Pregravid obesity associates with increased maternal endotoxemia and metabolic inflammation. Obesity (Silver Spring) 19: 476–482, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carter AM. Animal models of human placentation—a review. Placenta 28 Suppl A: S41–S47, 2007. [DOI] [PubMed] [Google Scholar]
- 9.Catalano PM, Farrell K, Thomas A, Huston-Presley L, Mencin P, de Mouzon SH, Amini SB. Perinatal risk factors for childhood obesity and metabolic dysregulation. Am J Clin Nutr 90: 1303–1313, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Catalano PM, Presley L, Minium J, Hauguel-de Mouzon S. Fetuses of obese mothers develop insulin resistance in utero. Diabetes Care 32: 1076–1080, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Challier JC, Basu S, Bintein T, Minium J, Hotmire K, Catalano PM, Haguel-de Mouzon S. Obesity in pregnancy stimulates macrophage accumulation and inflammation in the placenta. Placenta 29: 274–281, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clifton VL, Hodyl NA, Murphy VE, Giles WB, Baxter RC, Smith R. Effect of maternal asthma, inhaled glucocorticoids and cigarette use during pregnancy on the newborn insulin-like growth factor axis. Growth Horm IGF Res 20: 39–48, 2010. [DOI] [PubMed] [Google Scholar]
- 13.Demetrius L. Aging in mouse and human systems: a comparative study. Ann NY Acad Sci 1067: 66–82, 2006. [DOI] [PubMed] [Google Scholar]
- 14.Dever TE. Gene-specific regulation by general translation factors. Cell 108: 545–556, 2002. [DOI] [PubMed] [Google Scholar]
- 15.Dilworth MR, Sibley CP. Review: Transport across the placenta of mice and women. Placenta 34 Suppl: S34–S39, 2013. [DOI] [PubMed] [Google Scholar]
- 16.Ehrenberg HM, Mercer BM, Catalano PM. The influence of obesity and diabetes on the prevalence of macrosomia. Am J Obstet Gynecol 191: 964–968, 2004. [DOI] [PubMed] [Google Scholar]
- 17.Frühbeck G. Intracellular signalling pathways activated by leptin. Biochem J 393: 7–20, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gaccioli F, White V, Capobianco E, Powell TL, Jawerbaum A, Jansson T. Maternal overweight induced by a diet with high content of saturated fat activates placental mTOR and eIF2α signaling and increases fetal growth in rats. Biol Reprod 89: 96, 2013. [DOI] [PubMed] [Google Scholar]
- 19.Gilbert PA, Khokhar S. Changing dietary habits of ethnic groups in Europe and implications for health. Nutr Rev 66: 203–215, 2008. [DOI] [PubMed] [Google Scholar]
- 20.Group HSCR. Hyperglycaemia and adverse pregnancy outcome (HAPO) study: associations with maternal body mass index. BJOG 117: 575–584, 2010. [DOI] [PubMed] [Google Scholar]
- 21.Higgins L, Greenwood SL, Wareing M, Sibley CP, Mills TA. Obesity and the placenta: A consideration of nutrient exchange mechanisms in relation to aberrant fetal growth. Placenta 32: 1–7, 2011. [DOI] [PubMed] [Google Scholar]
- 22.Hirschler V, Roque MI, Calcagno ML, Gonzalez C, Aranda C. Maternal waist circumference and the prediction of children's metabolic syndrome. Arch Pediatr Adolesc Med 161: 1205–1210, 2007. [DOI] [PubMed] [Google Scholar]
- 23.Hui AL, Back L, Ludwig S, Gardiner P, Sevenhuysen G, Dean HJ, Sellers E, McGavock J, Morris M, Jiang D, Shen GX. Effects of lifestyle intervention on dietary intake, physical activity level, and gestational weight gain in pregnant women with different pre-pregnancy body mass index in a randomized control trial. BMC Pregnancy Childbirth 14: 331, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Inoki K, Kim J, Guan KL. AMPK and mTOR in cellular energy homeostasis and drug targets. Annu Rev Pharmacol Toxicol 52: 381–400, 2012. [DOI] [PubMed] [Google Scholar]
- 25.Jansson N, Greenwood SL, Johansson BR, Powell TL, Jansson T. Leptin stimulates the activity of the system A amino acid transporter in human placental villous fragments. J Clin Endocrinol Metab 88: 1205–1211, 2003. [DOI] [PubMed] [Google Scholar]
- 26.Jansson N, Nilsfelt A, Gellerstedt M, Wennergren M, Hulthen-Rossander L, Powell TL, Jansson T. Maternal hormones linking maternal bodymass index and dietary intake to birth weight. Am J Clin Nutr 87: 1743–1749, 2008. [DOI] [PubMed] [Google Scholar]
- 27.Jansson N, Rosario FJ, Gaccioli F, Lager S, Jones HN, Roos S, Jansson T, Powell TL. Activation of placental mTOR signaling and amino acid transporters in obese women giving birth to large babies. J Clin Endocrinol Metab 98: 105–113, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jansson T, Powell TL. IFPA 2005 Award in Placentology Lecture. Human placental transport in altered fetal growth: does the placenta function as a nutrient sensor? A review. Placenta 27 Suppl A S91–S97, 2006. [DOI] [PubMed] [Google Scholar]
- 29.Jones HN, Jansson T, Powell TL. Full-length adiponectin attenuates insulin signaling and inhibits insulin-stimulated amino acid transport in human primary trophoblast cells. Diabetes 59: 1161–1170, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jones HN, Jansson T, Powell TL. IL-6 stimulates system A amino acid transporter activity in trophoblast cells through STAT3 and increased expression of SNAT2. Am J Physiol Cell Physiol 297: C1228–C1235, 2009. [DOI] [PubMed] [Google Scholar]
- 31.Karl PI. Insulin-like growth factor-1 stimulates amino acid uptake by the cultured human placental trophoblast. J Cell Physiol 165: 83–88, 1995. [DOI] [PubMed] [Google Scholar]
- 32.Kavitha JV, Rosario FJ, Nijland MJ, McDonald TJ, Wu G, Kanai Y, Powell TL, Nathanielsz PW, Jansson T. Down-regulation of placental mTOR, insulin/IGF-I signaling, and nutrient transporters in response to maternal nutrient restriction in the baboon. FASEB J 28: 1294–1305, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kawabata K, Murakami A, Ohigashi H. Citrus auraptene targets translation of MMP-7 (matrilysin) via ERK1/2-dependent and mTOR-independent mechanism. FEBS Lett 580: 5288–5294, 2006. [DOI] [PubMed] [Google Scholar]
- 34.Kepczynska MA, Wargent ET, Cawthorne MA, Arch JR, O'Dowd JF, Stocker CJ. Circulating levels of the cytokines IL10, IFNγ and resistin in an obese mouse model of developmental programming. J Dev Orig Health Dis 4: 491–498, 2013. [DOI] [PubMed] [Google Scholar]
- 35.Kim DW, Young SL, Grattan DR, Jasoni CL. Obesity during pregnancy disrupts placental morphology, cell proliferation, and inflammation in a sex-specific manner across gestation in the mouse. Biol Reprod 90: 130, 2014. [DOI] [PubMed] [Google Scholar]
- 36.Kisseleva T, Bhattacharya S, Braunstein J, Schindler CW. Signaling through the JAK/STAT pathway, recent advances and future challenges. Gene 285: 1–24, 2002. [DOI] [PubMed] [Google Scholar]
- 37.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell 149: 274–293, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee DF, Kuo HP, Chen CT, Hsu JM, Chou CK, Wei Y, Sun HL, Li LY, Ping B, Huang WC, He X, Hung JY, Lai CC, Ding Q, Su JL, Yang JY, Sahin AA, Hortobagyi GN, Tsai FJ, Tsai CH, Hung MC. IKK beta suppression of TSC1 links inflammation and tumor angiogenesis via the mTOR pathway. Cell 130: 440–455, 2007. [DOI] [PubMed] [Google Scholar]
- 39.Li N, Liu E, Guo J, Pan L, Li B, Wang P, Liu J, Wang Y, Liu G, Baccarelli AA, Hou L, Hu G. Maternal prepregnancy body mass index and gestational weight gain on pregnancy outcomes. PloS One 8: e82310, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lubchenco LO, Hansman C, Dressler M, Boyd E. Intrauterine growth as estimated from liveborn birth-weight data at 24 to 42 weeks of gestation. Pediatrics 32: 793–800, 1963. [PubMed] [Google Scholar]
- 41.Lumeng CN, Saltiel AR. Inflammatory links between obesity and metabolic disease. J Clin Invest 121: 2111–2117, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mao J, Zhang X, Sieli PT, Falduto MT, Torres KE, Rosenfeld CS. Contrasting effects of different maternal diets on sexually dimorphic gene expression in the murine placenta. Proc Natl Acad Sci USA 107: 5557–5562, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moran LJ, Sui Z, Cramp CS, Dodd JM. A decrease in diet quality occurs during pregnancy in overweight and obese women which is maintained post-partum. Int J Obes (Lond) 37: 704–711, 2013. [DOI] [PubMed] [Google Scholar]
- 44.Muralimanoharan S, Guo C, Myatt L, Maloyan A. Sexual dimorphism in miR-210 expression and mitochondrial dysfunction in the placenta with maternal obesity. Int J Obes (Lond) 39: 1274–1281, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Poston L. Developmental programming and diabetes. The human experience and insight from animal models. Best Pract Res Clin Endocrinol Metab 24: 541–552, 2010. [DOI] [PubMed] [Google Scholar]
- 46.Procaccini C, De Rosa V, Galgani M, Carbone F, Cassano S, Greco D, Qian K, Auvinen P, Cali G, Stallone G, Formisano L, La Cava A, Matarese G. Leptin-induced mTOR activation defines a specific molecular and transcriptional signature controlling CD4+ effector T-cell responses. J Immunol 189: 2941–2953, 2012. [DOI] [PubMed] [Google Scholar]
- 47.Roberts KA, Riley SC, Reynolds RM, Barr S, Evans M, Statham A, Hor K, Jabbour HN, Norman JE, Denison FC. Placental structure and inflammation in pregnancies associated with obesity. Placenta 32: 247–254, 2011. [DOI] [PubMed] [Google Scholar]
- 48.Roos S, Jansson N, Palmberg I, Saljo K, Powell TL, Jansson T. Mammalian target of rapamycin in the human placenta regulates leucine transport and is down-regulated in restricted fetal growth. J Physiol 582: 449–459, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Roos S, Lagerlöf O, Wennergren M, Powell TL, Jansson T. Regulation of amino acid transporters by glucose and growth factors in cultured primary human trophoblast cells is mediated by mTOR signaling. Am J Physiol Cell Physiol 297: C723–C731, 2009. [DOI] [PubMed] [Google Scholar]
- 50.Roos S, Powell TL, Jansson T. Placental mTOR links maternal nutrient availability to fetal growth. Biochem Soc Trans 37: 295–298, 2009. [DOI] [PubMed] [Google Scholar]
- 51.Rosario FJ, Jansson N, Kanai Y, Prasad PD, Powell TL, Jansson T. Maternal protein restriction in the rat inhibits placental insulin, mTOR, and STAT3 signaling and down-regulates placental amino acid transporters. Endocrinology 152: 1119–1129, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rosario FJ, Kanai Y, Powell TL, Jansson T. Increased placental nutrient transport in a novel mouse model of maternal obesity with fetal overgrowth. Obesity (Silver Spring) 23: 1663–1670, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rosario FJ, Kanai Y, Powell TL, Jansson T. Mammalian target of rapamycin signalling modulates amino acid uptake by regulating transporter cell surface abundance in primary human trophoblast cells. J Physiol 591: 609–625, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rosario FJ, Schumacher MA, Jiang J, Kanai Y, Powell TL, Jansson T. Chronic maternal infusion of full-length adiponectin in pregnant mice down-regulates placental amino acid transporter activity and expression and decreases fetal growth. J Physiol 590: 1495–1509, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sebire NJ, Jolly M, Harris JP, Wadsworth J, Joffe M, Beard RW, Regan L, Robinson S. Maternal obesity and pregnancy outcome: a study of 287,213 pregnancies in London. Int J Obes Relat Metab Disord 25: 1175–1182, 2001. [DOI] [PubMed] [Google Scholar]
- 56.Stark MJ, Wright IM, Clifton VL. Sex-specific alterations in placental 11β-hydroxysteroid dehydrogenase 2 activity and early postnatal clinical course following antenatal betamethasone. Am J Physiol Regul Integr Comp Physiol 297: R510–R514, 2009. [DOI] [PubMed] [Google Scholar]
- 57.Vaughan OR, Fisher HM, Dionelis KN, Jefferies EC, Higgins JS, Musial B, Sferruzzi-Perri AN, Fowden AL. Corticosterone alters materno-fetal glucose partitioning and insulin signalling in pregnant mice. J Physiol 593: 1307–1321, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Whitaker RC. Predicting preschooler obesity at birth: the role of maternal obesity in early pregnancy. Pediatrics 114: e29–e36, 2004. [DOI] [PubMed] [Google Scholar]
- 59.Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell 124: 471–484, 2006. [DOI] [PubMed] [Google Scholar]
- 60.Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol 12: 21–35, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]