

Abstract
Rapid technological advances have led to the production of different types of biological data and enabled construction of complex networks with various types of interactions between diverse biological entities. Standard network data analysis methods were shown to be limited in dealing with such heterogeneous networked data and consequently, new methods for integrative data analyses have been proposed. The integrative methods can collectively mine multiple types of biological data and produce more holistic, systems-level biological insights. We survey recent methods for collective mining (integration) of various types of networked biological data. We compare different state-of-the-art methods for data integration and highlight their advantages and disadvantages in addressing important biological problems. We identify the important computational challenges of these methods and provide a general guideline for which methods are suited for specific biological problems, or specific data types. Moreover, we propose that recent non-negative matrix factorization-based approaches may become the integration methodology of choice, as they are well suited and accurate in dealing with heterogeneous data and have many opportunities for further development.
Keywords: data fusion, biological networks, non-negative matrix factorization, systems biology, omics data, heterogeneous data integration
1. Introduction
One of the most studied complex systems is the cell. However, its functioning is still largely unknown. It comprises diverse molecular structures, forming complex, dynamical molecular machinery, which can be naturally represented as a system of various types of interconnected molecular and functional networks (see figure 1 for an illustration). Recent technological advances in high-throughput biology have generated vast amounts of disparate biological data describing different aspects of cellular functioning also known as omics layers.
Figure 1.

A schematic illustration of the molecular information layers of a cell.
For example, yeast two-hybrid assays [1–7] and affinity purification with mass spectrometry [8,9] are the most widely used high-throughput methods for identifying physical interactions (bonds) among proteins. These interactions, along with the whole set of proteins, comprise the proteome layer. Other experimental technologies, such as next-generation sequencing [10–13], microarrays [14,15] and RNA-sequencing technologies [16–18], have enabled construction and analyses of other omics layers. Figure 1 illustrates these layers and their constituents: genes in the genome, mRNA in the transcriptome, proteins in the proteome, metabolites in the metabolome and phenotypes in the phenome. It illustrates that the mechanisms by which genes (in the genome layer) lead to complex phenotypes (in the phenome layer) depend on all intermediate layers and their mutual relationships (e.g. protein–DNA interactions).
It has largely been accepted that a comprehensive understanding of a biological system1 can come only from a joint analysis of all omics layers [19,20]. Such analysis is often referred as data (or network) integration. Data integration collectively analyses all datasets and builds a joint model that captures all datasets concurrently. A starting point of this analysis is to use a mathematical concept of networks to represents omics layers. A network (or a graph) consists of nodes (or vertices) and links (or edges). In biological networks, nodes usually represent discrete biological entities at a molecular (e.g. genes, proteins, metabolites, drugs, etc.) or phenotypic level (e.g. diseases), whereas edges represent physical, functional or chemical relationships between pairs of entities [21]. For the last couple of decades, networks have been one of the most widely used mathematical tools for modelling and analysing omics data [22]. In particular, these tools applied to studies of protein–protein interaction (PPI) networks [23,24], gene interaction (GI) networks [25–27], metabolic interaction (MI) networks [28–31] and gene co-expression (Co-Ex) networks [32,33] have extracted valuable biological information from these different views of molecular machinery inside a cell. However, a more complete understanding of a biological system is expected to be achieved by a joint, integrative analysis of all these networks. Constructing an integrated network representation that best captures all gene–gene associations by using all molecular networks has been one of the major challenges in network integration2 [34,35].
1.1. The need for data integration
Abundance of biological data has made data integration approaches increasingly popular over the past decade. Understanding cellular process and molecular interactions by integrating molecular networks has just been one of the challenges of data integration. In addition to molecular network data, accumulation of other data types has created a need for development of data integration methods that can address a wide range of biological problems and challenges. Some examples of these data include protein and genome sequence data [36], disease data from genome-wide association studies (GWAS) [12,13,37,38], mutation data from The Cancer Genome Atlas (TCGA) [39], copy number variation (CNV) data [40], functional annotation and ontology data, such as gene ontology (GO) [41] and disease ontology (DO) [42], protein structure data [43], drug chemical structure and drug–target interaction (DTI) data [44–46]. These data represent a valuable complement to the molecular networks and they are often incorporated into various data integration frameworks to increase reliability of newly discovered knowledge.
Because the literature on these topics is vast, we chose to mainly focus on the following currently foremost data integration problems:
— Network inference and functional linkage network (FLN) construction. Network inference is one of the major problems in systems biology aiming to understand GIs and their mutual influence [47]. It aims to construct network topology (or wiring between genes) based on the evidence from different data types. The special focus in the literature has been on the inference of gene regulatory networks (GRNs), whose nodes are genes and edges are regulatory interactions [48]. Standard methods for GRN inference have mostly been based on gene expression data. However, integration of expression data with other data types has been shown to improve the GRN inference process [49]. Unlike GRN, FLN captures all possible gene associations, constructed from multiple data types. FLN has demonstrated its potential in studying human diseases and predicting novel gene functions and interactions [35,50].
— Protein function and PPI prediction. Protein function (also known as protein annotation) prediction has been demonstrated to be a good alternative to the time-consuming experimental protein function characterization. It aims to computationally assign molecular functions to unannotated proteins. The accuracy of these methods has largely improved with the use of integration methods that can incorporate multiple different biological data conveying complementary information about protein functions [51–55]. Also, interactions between proteins are important for understanding intracellular signalling pathways and other cellular process. However, PPI network structure for many species is still largely unknown and therefore, many computational techniques for PPI interaction prediction have been proposed. Much attention has been paid to the data integration methods capable of inferring new PPIs by integrating various types of heterogeneous biological data [50,56,57].
— Disease gene prioritization and disease–disease association prediction. Prioritization of disease-causing genes is a problem of great importance in medicine. It deals with the identification of genes involved in a specific disease and providing a better understanding of gene aberrations and their roles in the formation of diseases [58]. However, a majority of diseases are characterized with a small number of known associated genes and experimental methods for discovering new disease-causing genes are expensive and time-consuming [59]. Therefore, computational methods for prioritization of disease genes have been proposed. Among the most popular methods are data integration methods owing to their ability to improve the reliability of prioritization by using multiple biological data sources [35,60]. Also, predicting associations between diseases is of great importance. Current disease associations are mainly derived from similarities between pathological profiles and clinical symptoms. However, the same disease could lead to different phenotype manifestations and thus, to inaccurate associations with other diseases. Integration of molecular data has been shown to lead to better and more accurate disease–disease associations [61].
— Drug repurposing. It aims to find new uses for existing drugs and thereby drastically reduce the cost and time for new drug discovery [62]. Accumulation of various biological data involving interactions between drugs, diseases and genes, and protein structural and functional similarities, provide us with new opportunities for data integration methods to generate new associations between diseases and existing drugs [63].
— Patient-specific data integration. It attempts to integrate patient-specific clinical (e.g. patient's history, laboratory analysis, etc.), genetic (e.g. somatic mutations) and genomic data (e.g. gene expression data from healthy and diseased tissues). Such approaches are contributing to the development of the nascent field of precision medicine, which aims to understand an individual patient's disease on the molecular level and hence, propose more precise therapies [64]. Data integration methods have started contributing to this growing field [65]. Here, we present data integration methods capable of jointly analysing clinical, patient- and disease-specific data to classify patients into groups with different clinical outcomes and prognoses, hence data integration methods contribute to improving therapies.
1.2. Computational challenges of data integration
The main goal of any data integration methodology is to extract additional biological knowledge from multiple datasets that cannot be gained from any single dataset alone. To reach this goal, data integration methodologies have to meet many computational challenges. These challenges arise owing to different sizes, formats and dimensionalities of the data being integrated, as well as owing to their complexity, noisiness, information content and mutual concordance (i.e. the level of agreement between datasets).
A number of current data integration methods meet some of these challenges to some extent, whereas the majority of them hardly meet any of them. A reason is that many data integration approaches are based on the methods designed for analysing one data type, and they are further adopted to deal with multiple data types. Thus, these methods often suffer from various limitations when applied to multiple data types. For example, in terms of network integration, standard methods for network analysis fail to simultaneously take into account connectivity structure (topology) of multiple different networks along with capturing the biological knowledge contained in them. They are based on different types of transformation methods to project, or merge multiple networks into a single, integrated network on which further analysis is performed [34,66–68]. Their limitations will be explained later in this article. However, more sophisticated network-based (NB) methods use either random walk or diffusion processes [69–73] to simultaneously explore connectivity structures (topologies) of multiple different networks and to infer integrated biological knowledge from all networks concurrently.
However, a majority of data integration studies are based on methods from machine learning (ML) owing to their ability to integrate diverse biological networks along with other biological data types. Namely, the basic strategy has been to use standard ML methods and extend them to incorporate disparate data types.
In this article, we provide a review of the methodologies for biological data integration and highlight their applications in various areas of biology and medicine. We compare these methods in terms of the following computational challenges:
— different size, format and dimensionality of datasets,
— presence of noise and data collection biases in datasets,
— effective selection of informative datasets,
— effective incorporation of concordant and discordant datasets, and
— scalability with the number and size of datasets.
We identify these computational challenges to be the most important, as every data integration methodology aims to address them at least to some extent.
1.3. Focus of this article
There are a number of review articles that cover related topics from different perspectives, or with a special focus on a particular biological problem. For example, Rider et al. [74] focus on methods for network inference with a special focus on probabilistic methods. The authors also argue about the need for standardized methods and datasets for proper evaluation of integrative methods. Kim et al. [75] focus on methods for construction and reconstruction of biological networks from multiple omics data, as well as on their statistical analysis and visualization tools. Bebek et al. [76] cover integrative approaches for identification of biomarkers and their implications in clinical science. They mostly focus on methods from network biology. Hamid et al. [77] proposе a conceptual framework for genomic and genetic data integration and review integration methods with a focus on statistical aspects. Kristensen et al. [78] review integrative methods applied in cancer research. They provide a comprehensive list of current tools and methods for genomic analyses in cancer.
In this paper, we focus on NB and to a large extent on ML data integration methods that have a wide range of applications in systems biology and a wide spectrum of data types that they can integrate. In particular, we focus on the state-of-the-art kernel-based (KB) methods [79], Bayesian networks (BNs) [80] and non-negative matrix factorization (NMF) [81] methods owing to their prominent applications in systems biology, as they can integrate large sets of diverse, heterogeneous biological data [82].
Note that there are many other data integration approaches that do not fall into the biological or methodological categories which we focus on in this review paper. Some of them include integration of multiple omics data for analysis of condition-specific pathways [83], integrative approaches for detecting modular structures in networks [84,85], integrated statistical analysis of multiple datasets [86], etc. Nevertheless, the methods and biological problems reviewed here cover a wide spectrum of foremost topics in systems biology. We also provide guidance on choosing suitable methods for integrating particular data. This is important as thus far there is no consensus (or guidelines) on what integration method should be used for a biological problem at study. Many of the existing review papers fail to provide answers to these questions. Here, we highlight the advantages and disadvantages of the most widely used data integration methods and provide an insight into which method should be used for which type of biological problem and applied on which type of homogeneous or heterogeneous data (see table 2 and §3 for more details). Some of these methods are also given as tools and can be useful to domain scientists. Please note that many of the existing reviews focus only on data integration methodologies in a specific biological domain, or on specific type of data. Thus, our review is comprehensive, as it covers a wide range of methodologies for data integration, as well as network and data types commonly used in data integration studies.
Table 2.
Summary of methods for data integration. See §§2 and 3 for abbreviations.
| method name | biological problem | data (network) types | approach | integration type | integration strategy | reference |
|---|---|---|---|---|---|---|
| NeXO | GO inference | PPI, GI, Co-Ex and YeastNet | NB | homogeneous | early | Dutkowski et al. [66] |
| GeneMANIA | gene function prediction | — | NB | homogeneous | early | Mostafavi & Morris [124] |
| MRLP | disease association prediction | DSN, GDA | NB | heterogeneous | early | Davis & Chawla [68] |
| PRINCE | disease–gene prioritization | PPI, DSN, GDA | NB | heterogeneous | intermediate | Vanunu et al. [72] |
| NBI | drug–target prediction | DCS, PSeqS, DTI | NB | heterogeneous | intermediate | Cheng et al. [70] |
| — | drug–disease association inference | DSN, DCS, Co-Ex | NB | heterogeneous | intermediate | Huang et al. [73] |
| — | gene-regulatory network inference | eQTL, GExD, TFBS and PPI | BN | heterogeneous | intermediate | Zhu et al. [125], Zhang et al. [126] |
| MAGIC | gene function prediction | PPI, GI, GExD and TFBS | BN | heterogeneous | intermediate | Troyanskaya et al. [54] |
| — | PPI prediction and FLN construction | PPI, Co-Ex, GO | BN | heterogeneous | late | Jansen et al. [127] |
| — | FLN construction | — | BN | heterogeneous | late | Linghu et al. [35], Lee et al. [50] |
| — | cancer prognosis prediction | clinical, GExD | BN | heterogeneous | intermediate | Gevaert et al. [128], van Vliet et al. [129] |
| PSDF | cancer prognosis prediction | GExD, CNV | BN | homogeneous | intermediate | Yuan et al. [130] |
| — | protein function prediction/classification | PSeqS, Co-Ex, PPI | KB | homogeneous | intermediate | Lanckriet et al. [51,52] |
| — | drug repurposing | DCS, DTI, PPI and GExD | KB | heterogeneous | intermediate | Napolitano et al. [63] |
| PreDR | drug repurposing | DCS, DTI, PPI and DSES | KB | heterogeneous | intermediate | Wang et al. [131] |
| — | network inference | GeXD, PPI, GO and PhylProf | KB | homogeneous | intermediate | Kato et al. [132] |
| KCCA | network inference | PPI, GexD, GO and PhylProf | KB | homogeneous | early | Yamanishi et al. [133] |
| — | cancer prognosis prediction | clinical, GExD | KB | heterogeneous | early | Daemen et al. [134] |
| DFMF | disease association prediction | PPI, GI, Co-Ex, CS, MI, DTI, GO, GA, DO, DSES and GDA | NMF | heterogeneous | intermediate | Žitnik et al. [61], Žitnik & Župan [135] |
| — | GO inference and gene function prediction | PPI, GI, Co-Ex, YeastNet, GO and GA | NMF | heterogeneous | intermediate | Gligorijević et al. [55] |
| — | PPI prediction | PStrS, GexF, PSeqS | NMF | homogeneous | intermediate | Wang et al. [57] |
| R-NMTF | GDA prediction | PPI, DSN, GDA | NMF | heterogeneous | intermediate | Hwang et al. [60] |
Furthermore, we identify deficiencies of particular methods when applied to multiple data types and point out possible mathematical and algorithmic improvements that can be undertaken to address the challenges listed in §1.2. Unlike other reviews, we also provide basic theoretical concepts of these methods that can familiarize the domain scientist with the basic computational concepts that the methods are based on and also serve as a starting point for possible methodological improvements. Moreover, to the best of our knowledge, this review is the first to present very recently proposed NMF methods for biological data integration and compare them with other ML data integration methods. We demonstrate many advantages of these methods over existing ML methods and propose their further improvement.
The paper is organized as follows. In §2, we introduce basic graph theoretic concepts for representing the data and publicly available data repositories. In §3, we survey the methods for data integration, with a detailed focus on NB, KB, BN and NMF methods. We first provide a brief introduction of these methods, followed by their extensions for data integration. We also highlight their advantages and disadvantages and provide directions for their further improvement. A discussion on future research directions is given in §4.
2. Biological data and network representation
Biological networks have revolutionized our view of biological systems and disease, as they enabled studies from a systems-level perspective. A network (or a graph), usually denoted as G = (V, E), consists of a set of nodes, V, and set of edges, E [87]. Depending on the type of data they represent, network edges can be directed or undirected, weighted or unweighted [88]. For example, an edge in a PPI network is undirected, as it represents a physical bond between two proteins, whereas an edge in a metabolic network is directed, as it represents a chemical reaction that converts one metabolite into another. Networks can also be used to model relations between different types of biological entities, such as genes and diseases. Such relations are usually represented by using bipartite networks. Namely, a bipartite (or in a more general case, a k-partite) network consists of two (or k) disjoint sets of nodes (partitions) and a set of edges connecting nodes between different partitions. For example, gene–disease associations (GDAs) are represented by a bipartite network. Combinations of general and bipartite network representations are usually used to link multiple types of networks into a single, complex, heterogeneous, multi-relational network. For example, in many network integration studies, a gene–gene association network and a disease–disease association network are linked by a GDA bipartite network, jointly forming a complex, heterogeneous, multi-relational network (see figure 2a for an illustration) [68].
Figure 2.

(a) An illustration of a heterogeneous network composed of a gene–gene interaction network (blue), a disease–disease association network (red) and a gene–disease association network (black edges). A simple integrated network is obtained via either gene, or disease projection method (see details in §3). The thickness of an edge in a projected network illustrates its weight. (b) An illustration of homogeneous gene–gene interaction networks. An integrated network is constructed by using a simple data merging method (see text in §3 for details).
A network connectivity pattern is often represented by an adjacency matrix [87]: for an undirected network G = (V, E), its adjacency matrix, A, is a square matrix of size 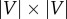 , where each row and column denotes a node and entries in the matrix are either Aij = 1, if nodes i and j are connected, or Aij = 0, otherwise. In the case of a weighted network, instead of binary values, entries in an adjacency matrix are real numbers representing the strengths of associations between the nodes. The Laplacian matrix of G, denoted as L, is another mathematical concept widely used in spectral graph theory [89] and semi-supervised ML problems [90]. It is defined as: L = D – A, where D is the diagonal degree matrix; that is, entries on the diagonal, Dii, are node degrees (the degree is the number of edges connected to a node), whereas off-diagonal entries, Dij, i ≠ j, are zeros.
, where each row and column denotes a node and entries in the matrix are either Aij = 1, if nodes i and j are connected, or Aij = 0, otherwise. In the case of a weighted network, instead of binary values, entries in an adjacency matrix are real numbers representing the strengths of associations between the nodes. The Laplacian matrix of G, denoted as L, is another mathematical concept widely used in spectral graph theory [89] and semi-supervised ML problems [90]. It is defined as: L = D – A, where D is the diagonal degree matrix; that is, entries on the diagonal, Dii, are node degrees (the degree is the number of edges connected to a node), whereas off-diagonal entries, Dij, i ≠ j, are zeros.
Network wiring (also called topology) has intensively been studied over the past couple of decades by using methods from graph theory and statistical physics [88]. The first studies of molecular networks have shown that many molecular networks are characterized by a complex, scale-free structure. Namely, they have a small number of highly connected nodes (hubs) whose removal disconnects the network and a large number of low-degree nodes [91]. Unlike the structure of random networks, such scale-free structures indicate that molecular networks emerge as a result of complex, dynamical processes taking place inside a cell. This property has been exploited in devising null models of these networks [92–94].
Over the past couple of decades, a variety of mathematical tools for extraction of biological knowledge from real-world molecular networks have been proposed. In this article, we do not provide a review of these methods because they are mainly used for single-type network analyses. For a recent review of these methods, we refer the reader to reference [95]. Here, we present a brief description of biological networks commonly used in network and data integration studies and the procedures for their construction. We refer the reader to reference [82] for more details.
Based on the criteria under which the links in the networks are constructed, we divide biological networks into the following three classes (see table 1 for a summary of data types and data repositories):
- Molecular interaction networks. They include the following network data types.
- — A PPI network consists of proteins (nodes) and physical bonds between them (undirected edges). A large number of studies have dealt with detection and analysis of these types of interactions in different species [23,105]. As proteins are coded by genes, a common way to denote nodes in a PPI network is by using gene notations. Such notations are more common, as they allow a universal representation of all molecular networks and enable their comparison and integration.
- — An MI network contains all possible biochemical reactions that convert one metabolite into another, as well as regulatory molecules, such as enzymes, that guide these metabolic reactions [106]. A common way to represent a metabolic network is by representing enzymes as nodes and two enzymes are linked if they catalyse (participate in) the same reaction [107]. Because enzymes are proteins, they can be denoted by a gene notation.
- — A DTI network is a bipartite network representing physical bonds between drug compounds in one partition and target proteins in the other [108]. Many databases containing curated DTIs from the scientific literature have been published.
- Functional association networks. They include the following network data types.
- — A GI network is a network of genes representing the effects of paired mutations onto the phenotype. Two genes are said to exhibit a positive (negative) GI if their concurrent mutations result in a better (worse) phenotype than expected by mutations of each one of the genes independently [21,27]. GIs may not represent physical ‘interactions' between the proteins, but their functional associations.
- — A GDA network is a bipartite network representing associations between diseases in one partition and disease-causing genes in the other partition.
- — Ontology networks (ONs) are valuable biological components in many network integration studies [61] and they are often integrated with other molecular network data. The two commonly used ontologies in network data integration literature are GO, which unifies the knowledge about functioning of genes and gene products [41], and DO, which unifies the knowledge about relationships between diseases [42]. The hierarchical structure of the ontologies is represented as a directed acyclic graph (DAG)—a graph with no cycles, where nodes represent GO terms (or DO terms) and edges represent parent–child semantic relations.
- Functional and structural similarity networks. They include the following network data types.
- — A gene Co-Ex network represents correlations between gene expression levels across different samples, or experimental conditions. Usually, Pearson correlation coefficient (PCC) between all pairs of genes is computed based on their vectors of expression levels across different samples, or experimental conditions. Then, using a statistical method, a significant value of PCC (threshold) is determined. Two genes are linked if their PCC is higher than the threshold value. The choice of the threshold greatly influences the resulting topology of the Co-Ex network. Therefore, it is essential to properly determine the appropriate threshold. For that purpose, a couple of methods have been proposed. Some methods use prior biological knowledge (e.g. known functional associations) to constrain the network construction [109]; others use statistical comparison with randomized expression data [110], or random matrix theory [111]. However, these studies do not account for the number of experimental conditions (the length of the vector of expression profiles), which has been shown to greatly influence the choice of the correlation threshold [112]. Hence, methods to overcome such limitations have been proposed. They rely on a combination of partial correlation [113] and information theory [114] approaches to determine a local correlation threshold for each pair of genes. Unlike the single correlation threshold applied across the entire network, this approach allows for identification of more meaningful gene-to-gene associations [112].
- — A drug chemical similarity (DCS) network represents similarities and differences between drugs' chemical structures. Each drug is composed of chemical substructures, which define its chemical fingerprint. These chemical fingerprints are usually represented by binary vectors whose coordinates encode the presence (‘1’) or absence (‘0’) of a particular substructure from a set of all known substructures. The chemical similarity between two drugs is computed based on the similarity between these vectors. Various measures for computing the similarity have been proposed [115,116]. A commonly used one is the Tanimoto coefficient [115]. The procedure for network construction is similar to that of Co-Ex networks. First, the statistically significant similarity threshold is determined, and then, based on the threshold, links between drugs are constructed. Other types of drug–drug similarity networks have also been used in the network data integration literature. A frequently considered one is the drug side-effect similarity (DSES) network. Namely, clinical side-effects of each drug have been collected and stored in numerous databases. The side-effects provide a drug with a profile and allow for construction of pairwise side-effect similarities between two drugs using the Jaccard index [117].
- — Protein similarity networks represent networks of proteins with similar sequences (PSeqS) [118] or structures (PStrS) [119]. Similarities between protein sequences are usually computed by using the BLAST algorithm [120], whereas the similarities between three-dimensional protein structures are usually computed by using the DaliLite algorithm [121].
Table 1.
Different types of biological data (the first two columns), types of biological entities and relations (interactions) between them (the second two columns) and databases containing the data (the last column).
| data type | network | entities/nodes | interactions/edges | data resource |
|---|---|---|---|---|
| molecular interactions | PPI | proteins | physical bonds | BioGRID [96] |
| MI | enzymes (proteins) | reaction catalysis | KEGG [97] | |
| DTI | drugs/targets | physical bonds | DrugBank [98], PubChem [46] | |
| functional associations | GI | genes (proteins) | genetic interactions | BioGRID [96] |
| GDA | genes/diseases | associations | OMIM [45], GWAS [99] PheWAS [100] |
|
| ON | GO (DO) term | hierarchical relations | GO [41], DO [42] | |
| functional/structural similarities | Co-Ex | genes | expression profile similarities | GEO [101], ArrayExpress [102], SMD [103] |
| DCS | drugs | structural similarities | DrugBank [98], PubChem [46] | |
| DSES | drugs | side-effect profile similarities | SIDER [104] | |
| PSeqS | proteins | protein sequence similarities | RefSeq [36] | |
| PStrS | proteins | structural similarities | PDB [43] |
Each of the above-described biological networks represents an important component of the system's cellular functioning and they often complement each other. For example, by comparing the number of common links between biological networks, many studies have reported a large overlap of the links between PPI and gene Co-Ex networks [122], whereas a small overlap of links has been observed between PPI and GI networks [123]. Hence, these studies have indicated that a GI network is a valuable complement to the other two biological networks and this has been confirmed in several network integration studies [55,61,66].
3. Computational methods for data integration
3.1. Types and strategies of data integration
Based on the type of data they integrate, integration methods can be divided into two types: homogeneous and heterogeneous integration methods (see table 2 for a detailed summary of the classification of methods into these types). Homogeneous integration deals with integration of networks with the same type of nodes (e.g. proteins), but different types of links between the nodes (e.g. GIs, PPIs, etc.). However, many biological data are heterogeneous, consisting of various types of biological entities and various types of relations. These data can be represented as collections of inter-related networks with various types of nodes and edges. For example, the GDA network along with the DCS network and the PPI network forms a heterogeneous network with multiple node and edge types. Heterogeneous data integration deals with a collective mining of these networks and the construction of a unified model.
The strategies for data integration can be divided into the following three categories (see table 2 for a detailed summary) [51,128,129,135–137]:
— Early (or full) data integration combines the datasets into a single dataset on which the data model is built. This often requires a transformation of the datasets into a common representation, which in some cases may result in information loss [51,135].
— Late (or decision) data integration builds models for each dataset separately, then it combines these models into a unified model. Building models from each dataset in isolation from others disregards their mutual relations, often resulting in reduced performance of the final model [128,135].
— Intermediate (or partial) data integration combines data through inference of a joint model. This strategy has often been preferred owing to its superior predictive accuracy reported in many studies (regardless of the chosen methods) [51,128,129,135,136], but there are some studies that report superiority of early and late integration strategy over the intermediate strategy [137]. This strategy does not require any data transformation and thus, it does not result in information loss.
3.2. Network-based methods
The majority of NB methods use very simple ways to integrate different types of network data and to create an integrated representation (or a model) of a set of networks. For example, in homogeneous network integration (see figure 2b for an illustration), where N different networks, 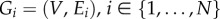 , with the same set of nodes, V, but different sets of links, Ei, are considered, a common way to construct an integrated network is by merging links of all networks over the same set of nodes (i.e.
, with the same set of nodes, V, but different sets of links, Ei, are considered, a common way to construct an integrated network is by merging links of all networks over the same set of nodes (i.e. 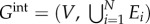 [66]. The adjacency matrix of the resulting integrated network is just a simple sum over the adjacency matrices representing individual networks:
[66]. The adjacency matrix of the resulting integrated network is just a simple sum over the adjacency matrices representing individual networks: 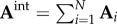 . In the case of weighted networks, the entries in the individual adjacency matrices, Ai, are scaled in the same range. However, this approach neglects the compatibility issues among individual networks in the construction of the integrated network. For example, by merging the links of modular3 and non-modular networks, the resulting network may not retain the modular structure. Other approaches that try to overcome this disadvantage create a weighted sum of adjacency matrices to construct the adjacency matrix of the merged network. Namely,
. In the case of weighted networks, the entries in the individual adjacency matrices, Ai, are scaled in the same range. However, this approach neglects the compatibility issues among individual networks in the construction of the integrated network. For example, by merging the links of modular3 and non-modular networks, the resulting network may not retain the modular structure. Other approaches that try to overcome this disadvantage create a weighted sum of adjacency matrices to construct the adjacency matrix of the merged network. Namely, 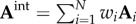 , where the weight wi ≥ 0 assigned to network i represents the contribution of network i to the quality of the inference4 (e.g. protein function prediction) on the merged network [34,124,138]. The weighting coefficients are obtained by solving a linear regression problem, which assigns lower weights to ‘less important’ networks. However, such weighting is problem-dependent, i.e. the structure of the resulting integrated network depends on the biological problem at study.
, where the weight wi ≥ 0 assigned to network i represents the contribution of network i to the quality of the inference4 (e.g. protein function prediction) on the merged network [34,124,138]. The weighting coefficients are obtained by solving a linear regression problem, which assigns lower weights to ‘less important’ networks. However, such weighting is problem-dependent, i.e. the structure of the resulting integrated network depends on the biological problem at study.
In heterogeneous network integration (see figure 2a for an illustration), the majority of studies have integrated networks containing different types of nodes and links by applying simple ‘projection’ methods [67,68,139]. Namely, they project network layers onto the one they are interested in. For example, in figure 2a, the GI network is projected onto the disease similarity network (DSN) by relating two diseases that have a gene in common. A weight of a link in the resulting disease–disease network represents the link multiplicity resulting from the projection. The disease–disease network is then further analysed by using standard NB methods. However, this projection method often results in information loss. Namely, by projecting networks onto a single node type network, the connectivity information of other node type networks is lost. That is, by projecting the gene–gene interaction network onto the disease–disease association network, the information about gene connections is lost along with the whole structure of the gene–gene interaction network. Therefore, by using these methods, we cannot analyse disease and gene connectivity patterns simultaneously.
More sophisticated methods capable of simultaneously analysing connectivity patterns of various networks are based on diffusion (information spreading across network links) over heterogeneous networks. They simultaneously explore the structure of each network and of their mutual relations; based on all this information, they create an integrated inference. Such approaches, also called network propagation methods, have been applied to biological problems, including gene–disease prioritization [69,72], drug–target prediction, drug repurposing [70,71] and drug–disease association prediction [73]. Although these approaches are mainly designed for a pair of inter-related networks, their further extensions to handle more networks are possible. For example, Huang et al. [73] extended the network propagation method to three inter-related networks. However, with the inclusion of multiple networks, the number of coupled iterative equations for information propagation (diffusion) grows and hence, the running time of the algorithm increases. Therefore, the scalability of these methods is limited.
3.3. Bayesian networks
BNs belong to the class of probabilistic graphical models that combine concepts from probability and graph theory to represent and model causal relations between random variables describing data [140]. A BN is a DAG, where nodes represent random variables (e.g. gene expression levels) and directed edges represent conditional probabilities between pairs of variables. For instance, a conditional probability distribution (CPD), between variables X and Y, denoted as p(X|Y), represents the probability of X given the value of Y. CPDs can model conditional dependencies between discrete or continuous variables, or a combination of both. For discrete variables, CPDs are given in the form of conditional probability tables containing values of probabilities that represent parameters of the model. For continuous variables, CPDs are usually modelled by using Gaussian distributions with the mean and standard deviation as model parameters (μ, σ). For example, CPD p(X|Y), with X being a continuous variable and Y being a discrete variable, can be represented as a set of parameters 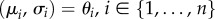 , each for a different value of
, each for a different value of 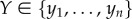 (i.e. (μi, σi) are the parameters of the Gaussian distribution p(x|yi)). BNs provide an elegant way to represent the structure of the data and their sparsity enables a compact representation and computation of the joint probability distribution (JPD) over the whole set of random variables. That is, the number of parameters to characterize the JPD is drastically reduced in the BN representation [80,140]; namely, a unique JPD of a BN containing n nodes (variables), x = (x1 , … , xn), can be formulated as:
(i.e. (μi, σi) are the parameters of the Gaussian distribution p(x|yi)). BNs provide an elegant way to represent the structure of the data and their sparsity enables a compact representation and computation of the joint probability distribution (JPD) over the whole set of random variables. That is, the number of parameters to characterize the JPD is drastically reduced in the BN representation [80,140]; namely, a unique JPD of a BN containing n nodes (variables), x = (x1 , … , xn), can be formulated as: 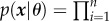
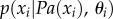 , where Pa(xi) denotes parents of variable xi and θ = (θ1 , … , θn) denotes the model parameters (i.e. θi = (μi,σi) denotes the set of parameters defining the CPD, p(xi|Pa(xi))).
, where Pa(xi) denotes parents of variable xi and θ = (θ1 , … , θn) denotes the model parameters (i.e. θi = (μi,σi) denotes the set of parameters defining the CPD, p(xi|Pa(xi))).
BNs have been applied to many tasks in systems biology, including modelling of protein signalling pathways [141], gene function prediction [54] and inference of cellular networks [142]. An illustration of a BN representing a GRN is shown in figure 3a. Each gene is represented by a variable denoting its expression. A state of each variable (gene expression) depends only on the states of its parents. This enables a factorization of a JPD into a product of CPDs describing each gene in terms of its parents.
Figure 3.
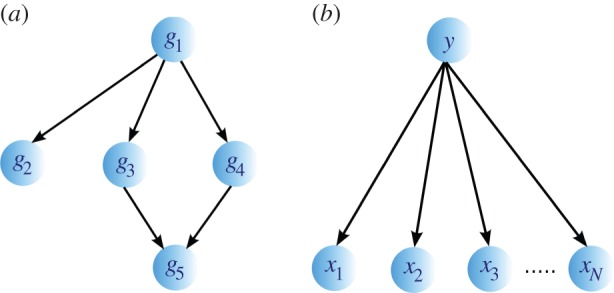
(a) A schematic illustration of a gene regulatory network modelled by BN. Genes are represented by nodes, whereas regulatory relations between genes are represented by directed edges. Gene g1 regulates the expression of genes g2, g3 and g4, and genes g3 and g4 regulate the expression of gene g5. Gene g1 is called a parent of g2, g3 and g4, whereas genes g2, g3 and g4 are called children of gene g1 (similar holds for other relations). A sparse representation implies that the expression level of a gene depends only on the expression levels of its regulators (parents in the network). The JPD of the system is 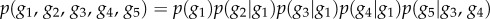 . (b) An example of a naive BN with a class node y being the parent to independent nodes x1, x2 , … , xN.
. (b) An example of a naive BN with a class node y being the parent to independent nodes x1, x2 , … , xN.
Constructing a BN describing the data consists of two steps: parameter learning and structure learning [80,140]. Because the number of possible structures of a BN grows super-exponentially with the number of nodes, the search for the BN that best describes the data is an NP-hard problem, and therefore heuristic (approximate) methods are used for solving it [143]. They usually start with some initial network structure and then gradually change it by adding, deleting or re-wiring some edges until the best scoring structure is obtained. For details about these and parameter estimation methods, we refer the reader to reference [80].
When the structure and parameters of a BN are learned (i.e. JPD is determined), an inference about dependencies between variables can be made. For example, assuming discrete values of variables describing genes as either expressed (on) or not (off) in figure 3a, we can ask what the likelihood of gene g5 being expressed is, given that gene g1 is expressed. This can be formulated as 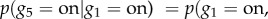
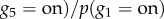 , where the numerator can be calculated by using the marginalization rule (i.e. by summing over all unknown (marginal) variables considering their possible values) [140]:
, where the numerator can be calculated by using the marginalization rule (i.e. by summing over all unknown (marginal) variables considering their possible values) [140]: 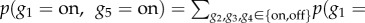
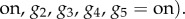 For large systems, with large numbers of variables, this summation becomes computationally intractable: the exact inference, or the summation of JPD over all possible values of unknown variables, is known to be an NP-hard problem [144]. Consequently, many approximation methods, such as variational methods and sampling methods, have been proposed [140].
For large systems, with large numbers of variables, this summation becomes computationally intractable: the exact inference, or the summation of JPD over all possible values of unknown variables, is known to be an NP-hard problem [144]. Consequently, many approximation methods, such as variational methods and sampling methods, have been proposed [140].
Recently, BNs have been used as a suitable framework for integration and modelling of various types of biological data. One of the biggest challenges in systems biology is a problem of network inference from disparate data sources—the construction of sparse networks where only important gene associations are present (strength of associations are represented by conditional probabilities) [74]. Disparate data sources can be incorporated in either one of two steps of BN construction—parameter learning or structure learning. Such networks play an important role in describing and predicting complex behaviour of a system supported by evidence from a variety of different biological data [145].
For example, Zhu et al. [125] combined gene expression data (GExD), expression of quantitative trait loci (eQTL),5 transcription factor binding site (TFBS) and PPI data to construct a causal, probabilistic network of yeast. In particular, they used the eQTL data to constrain the addition of edges in the probabilistic network, so that cis-eQTL acting genes are considered to be parents of trans-eQTL acting genes. They tested the performance of the constructed BN in predicting GO categories and they demonstrated that the predictive power of the integrated BN is significantly higher than that of the BN constructed solely from the gene expression data [125]. A similar procedure has also been applied by Zhang et al. [126], who constructed a gene-regulatory network by integrating data from brain tissues of late-onset Alzheimer's disease patients.
One of the first studies that integrated clinical and patient-specific omics data was presented by Gevaert et al. [128]. They integrated the gene expression data of tissues from breast cancer patients whose clinical outcome was known. They constructed the BN with genes and outcome variable (representing clinical data) as nodes and used it for a classification task: they classified patients into good and poor prognosis groups. They compared the performance of BN in reproducing the known outcomes in three different strategies, early, late and intermediate (see §3.1). They showed that the intermediate strategy was the most accurate one. A similar conclusion was drawn by van Vilet et al. [129].
Most studies have used the simplest BN, the so-called naive BN, for combining multiple heterogeneous biological data and constructing an integrated gene–gene association network (also called an FLN) [35,50,127,147]. The structure of a naive BN consists of a class node as a parent to all other independent nodes representing different data sources. Such a simple BN structure enables a much faster learning and inference. For example, in gene–gene association prediction, the class node may represent a set of interacting or non-interacting proteins, whereas the other variables in the naive BN represent input biological data, often in pairwise format (see §2). In addition to the input data, a gold standard data (e.g. gene pairs with known functional relations, usually from GO) is used for learning, i.e. for constructing the probability distributions. The basic assumption of a naive BN is that different data sources are conditionally independent, i.e. that the information in the datasets are independent given that a gene pair is either functionally associated or not.
Although simple, naive BNs have yielded good results in many data integration studies. They were initially proposed for data integration by Troyanskaya et al. [54], who developed a framework called multi-source association of genes by integration of clusters (MAGIC) for gene function prediction. They integrated systems-level PPI and GI data along with GExD and TFBS data of Saccharomyces cerevisiae. By using GO as the gold standard, they demonstrated an increased accuracy of their method applied on all datasets, compared with its performance on each input dataset separately. Furthermore, naive BNs have demonstrated usability in patient-specific data integration. Namely, a recent study used a naive BN to integrate gene expression and CNV6 data of prostate and breast cancer patients [130]. Unlike other methods, this method successfully detected a new subtype of prostate cancer patients with extremely poor survival outcome. Moreover, unlike many data integration studies that force incompatible data types to be fused, this is the first study that systematically considered compatibility of input data sources (see challenge 4 in §1.2). That is, the method was able to distinguish between concordant and discordant signals within each patient sample.
BNs are a good framework for biological network integration because of their ability to capture noisy conditional dependence between data variables through the construction of CPDs. However, they also have several disadvantages: (i) in the network inference problems, their sparse representation captures only important associations, whereas other associations are discarded; (ii) their acyclic representation cannot be used for modelling networks with loops, which are important in many biological networks, as they represent control mechanisms; (iii) the most important limitation of BNs is computational: as mentioned earlier, learning and inference processes of BNs are computationally intractable on large data, which is a major reason why many studies focus only on a small subset of nodes (genes) when constructing a BN.
3.4. Kernel-based methods
KB methods belong to the class of statistical ML methods used for data pattern analysis, e.g. for learning tasks, such as clustering, classification, regression, correlation, feature selection, etc. They work by mapping the original data to a higher dimensional space, called the feature space, in which the pattern analysis is performed. Such a mapping is represented by a kernel matrix [148]. A kernel matrix, K, is a symmetric, positive semi-definite matrix7 with entries Ki,j = k(xi, xj) representing similarities between all pairs of data points, xi, xj. The similarity between two data points k(xi,
xj) is computed as an inner product between their representations, ϕ(xi), ϕ(xj), in the feature space, 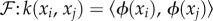 , where, ϕ maps data points from the input space,
, where, ϕ maps data points from the input space, 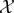 , to the feature space
, to the feature space 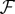 (a vector space where data points are represented as vectors), i.e.
(a vector space where data points are represented as vectors), i.e. 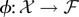 [79,148]. Function k(xi, xj) is called a kernel function and its explicit definition is the only requirement of the kernel method, whereas the mapping function ϕ and the properties of the feature space
[79,148]. Function k(xi, xj) is called a kernel function and its explicit definition is the only requirement of the kernel method, whereas the mapping function ϕ and the properties of the feature space 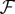 do not need to be explicitly specified. For example, given a set of proteins and their amino acid sequences, entries in the kernel matrix are usually generated by using the BLAST pairwise sequence alignment algorithm [120]. Hence, computing the protein embedding function, ϕ, and constructing the feature space,
do not need to be explicitly specified. For example, given a set of proteins and their amino acid sequences, entries in the kernel matrix are usually generated by using the BLAST pairwise sequence alignment algorithm [120]. Hence, computing the protein embedding function, ϕ, and constructing the feature space, 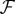 , is not necessary. Other kernel functions for measuring similarities between two proteins include the spectrum kernel [150,151], the motif kernel [152] and the Pfam kernel [153]. In addition to string data, molecular network data are frequently used in KB integration studies. Molecular networks are usually represented by diffusion kernels, which encode similarities between the nodes of the network [154], defined as:
, is not necessary. Other kernel functions for measuring similarities between two proteins include the spectrum kernel [150,151], the motif kernel [152] and the Pfam kernel [153]. In addition to string data, molecular network data are frequently used in KB integration studies. Molecular networks are usually represented by diffusion kernels, which encode similarities between the nodes of the network [154], defined as: 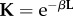 , where β > 0 is the parameter that quantifies the degree of the diffusion and L is the network Laplacian (see §2 for further details). The elements of the diffusion kernel matrix, Kij, quantify the closeness between nodes i and j in the network.
, where β > 0 is the parameter that quantifies the degree of the diffusion and L is the network Laplacian (see §2 for further details). The elements of the diffusion kernel matrix, Kij, quantify the closeness between nodes i and j in the network.
The methods using kernel matrices of data include: support vector machines (SVMs) [155], principal component analysis (PCA) [156], canonical correlation analysis (CCA) [157]. SVM classifiers have frequently been used for prediction tasks in computational biology owing to their high accuracy [158]. They were originally used for binary classification problems and can be defined as follows: given two classes of data items, a positive and a negative class labelled as 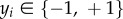 , and a dataset consisting of n classified data points (xi, yi), where data point xi belongs to a class yi, a classification task consists of correctly predicting a class membership of a new, unlabelled data point, xnew [159]. This can be formulated as an optimization problem: given the kernel matrix constructed between all pairs of data points, K(xi, xj), and labels yi, construct a hyperplane in the high-dimensional feature space that separates –1 class from +1 class by maximizing the margin, that is, the distance from the hyperplane to any data point from any class. Having obtained the hyperplane, the membership of a new data point is then easily predicted by localizing the data point in the feature space with respect to the separating hyperplane. For a more detailed description and application of this method, we refer the reader to review papers [160,161].
, and a dataset consisting of n classified data points (xi, yi), where data point xi belongs to a class yi, a classification task consists of correctly predicting a class membership of a new, unlabelled data point, xnew [159]. This can be formulated as an optimization problem: given the kernel matrix constructed between all pairs of data points, K(xi, xj), and labels yi, construct a hyperplane in the high-dimensional feature space that separates –1 class from +1 class by maximizing the margin, that is, the distance from the hyperplane to any data point from any class. Having obtained the hyperplane, the membership of a new data point is then easily predicted by localizing the data point in the feature space with respect to the separating hyperplane. For a more detailed description and application of this method, we refer the reader to review papers [160,161].
Kernel matrices represent a principled framework for representing different types of data including strings, sets, vectors, time series and graphs [79]. This property provides the KB methods with an advantage over the BN-based methods, as the BN-based methods are mostly suited to pairwise datasets owing to easier construction of conditional probabilities in that case. However, choosing the ‘right’ kernel function is not straightforward. Therefore, instead of constructing a single kernel, usually multiple candidate kernels for a single dataset are constructed using different measures of similarity [162]. These kernels are then linearly combined into one kernel that is then used for further analysis. Such an approach is called a multiple kernel learning (MKL) and has been shown to lead to a better performance than single KB methods, especially in genomic data analysis [163]. The mathematical basis for this approach is in the closure property of kernel matrices [79]. Namely, all elementary mathematical operations (e.g. addition, subtraction, multiplication) between two kernel matrices do not change the property of the final matrix (i.e. the positive semi-definite property stays preserved). This property provides the foundation for using KB methods for data integration by MKL. Namely, given a set of kernel matrices, {K1 , … , Kn}, representing n different datasets, the single kernel matrix representing integrated data is obtained by a linear combination, 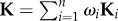 , where non-negative coefficients, ωi, determine the weights of each dataset and they are obtained through an optimization procedure of the KB method [52,164]. Namely, the non-negativity constraint imposed on weights reduces the optimization problem to a quadratically constrained quadratic problem [51]. The solution of this optimization problem gives the weighting coefficients. Thus, the obtained weights, ωi, allow us to distinguish between more and less informative datasets. Note that all kernel matrices, Ki, representing different datasets, need to be constructed over the same feature space to be correctly combined. In the case of heterogeneous data integration, this often requires data to be transformed, or projected onto the same feature space, which often results in information loss. This is the biggest drawback of KB data integration methods.
, where non-negative coefficients, ωi, determine the weights of each dataset and they are obtained through an optimization procedure of the KB method [52,164]. Namely, the non-negativity constraint imposed on weights reduces the optimization problem to a quadratically constrained quadratic problem [51]. The solution of this optimization problem gives the weighting coefficients. Thus, the obtained weights, ωi, allow us to distinguish between more and less informative datasets. Note that all kernel matrices, Ki, representing different datasets, need to be constructed over the same feature space to be correctly combined. In the case of heterogeneous data integration, this often requires data to be transformed, or projected onto the same feature space, which often results in information loss. This is the biggest drawback of KB data integration methods.
KB methods have first been proposed as a technique for data integration by Lanckriet et al. [51]. In that paper, they trained a 1-norm soft margin8 SVM to classify proteins into membrane or ribosomal groups. They constructed seven different kernels representing three different types of data: amino acid sequences, gene expression and PPI data. They showed that the performance of their classifier is the highest when all datasets are taken into account. A similar approach was applied for function prediction of baker's yeast proteins [52] and demonstrated that an SVM classifier trained on all data performs better than a classifier trained on any single type of data.
KB methods have also demonstrated their power in integrating molecular, structural and phenotypic data for drug repurposing. A recent study integrates three different layers of information represented in a drug-centred feature space [63]. Their kernel matrices represent (i) drug chemical similarities based on their structures (see §2 for details about measures of chemical similarity); (ii) drug similarities based on the positions of their targets in the PPI network; and (iii) drug similarities based on the correlations between gene profiles under the drug influence. Based on the combination of these three kernel matrices and the existing drug classification, the authors trained the classifier and proposed the top misclassified drugs as new candidates for repurposing [63]. In a similar study, a method called PreDR (predict drug repurposing) for predicting novel drug–disease associations was proposed. An SVM classier was trained on integrated kernel matrices of drugs based on their chemical structure, target proteins and side-effect profiles [131].
Although SVM methods are the most popular KB methods for regression and classification tasks in computational biology, other learning methods can also be applied on kernel matrices for performing different tasks. For example, a special application of KB methods (as well as of BN methods) is in the network inference problem. Unlike BN methods, which reconstruct the whole network without any prior knowledge about its structure, KB methods assume that a part of the network is known. For instance, Kato et al. [132] use a small part of the PPI network and the three different types of protein data to complete the whole PPI network. Namely, they construct a small, incomplete kernel matrix representing a known part of the PPI network and define kernel matrix completion problem that uses a weighted combination of three kernel matrices representing similarities between gene expression profiles, phylogenetic profiles (PhylProf) and amino acid sequences to complete the small kernel matrix. The authors report the high accuracy of their method in the reconstruction of the PPI network, and metabolic networks, and outline the ability of the method to selectively integrate different biological data. Another study uses the kernel CCA method to infer the PPI network and to identify features indicative of PPIs by detecting correlations between heterogeneous datasets (gene expression, protein localization and phylogenetic profiles) and the PPI network [133]. The inference process is done in a supervised manner, as it uses part of the true protein network as the gold standard.
Along with BN methods, KB methods can also be used for integration of clinical data with genomic data. An example study that integrates gene expression and clinical data of breast cancer patients is demonstrated by Daement et al. [134]. They applied a least-square SVM [165] on the combination of two patient-centric kernel matrices: the first was constructed based on patients' gene expression similarities, whereas the second was constructed based on similarities between patients' clinical variables. The authors reported 70% accuracy of their approach to correctly classify patients according to the appearance of distant subclinical metastases based on the primary tumour. They also demonstrated that both datasets, the expression and the clinical data, contribute to the performance of the classification.
From these examples, we can see that data integration by using KB methods has several advantages. The main advantage is that they can integrate a wide range of data types. Moreover, a liner combination of kernels provides a selective way of accounting for datasets, by assigning lower weights to less informative and noisier datasets. Hence, this integration method meets challenges 2 and 3 listed in §1.2. However, a big disadvantage is that heterogeneous datasets need to be transformed into a common feature space to be properly integrated, which can lead to information loss (see example shown in figure 4a,b). In the case of heterogeneous networks, data transformation prevents modelling of multi-relational data, i.e. a simultaneous modelling of different types of relations in the data.
Figure 4.

(a) Heterogeneous networks of genes (PPI, GI and MI) and drugs (chemical similarities) and links between drugs and genes (DTI). Intertype relations are represented by drug–target interaction (DTI) network, whereas intratype connections are represented by four networks: protein–protein interaction (PPI), genetic interaction (GI) and metabolic interaction (MI) molecular networks of genes, and the chemical similarity network of drugs (see §2 for further details about these networks and their construction). (b) An illustration of a KB data integration method for drug clustering. All kernel matrices are expressed in the drug similarity feature space based on the closeness between their targets (proteins) in each molecular network (K1, K2 and K3) and based on the similarity between their chemical structures (K4). All kernel matrices are linearly combined into a resulting kernel matrix K, on which the drug clustering is performed by using KB clustering methods. (c) An illustration of an NMTF-based data integration method for drug clustering: factorization of the DTI relation matrix under the guidance of molecular and chemical connectivity constraints represented by the constraint matrices. Drugs are assigned to clusters based on the entries in obtained G2 cluster indicator matrix.
3.5. Non-negative matrix factorization
NMF is an ML method commonly used for dimensionality reduction and clustering problems. It aims to find two low-dimensional, non-negative matrices, 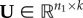 and
and 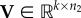 , whose product provides a good approximation of the input non-negative data matrix,
, whose product provides a good approximation of the input non-negative data matrix, 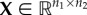 , i.e. X ≈ UV. The method was originally introduced by Lee & Seung [166] for parts-based decomposition of images. The non-negativity constraints provide matrix factors, U and V, with a more meaningful interpretation than previous approaches, such as PCA [156] and vector quantization [167], whereas the choice of parameter k ≪ min{n1, n2} (also called the rank parameter) provides a dimensionality reduction [168].
, i.e. X ≈ UV. The method was originally introduced by Lee & Seung [166] for parts-based decomposition of images. The non-negativity constraints provide matrix factors, U and V, with a more meaningful interpretation than previous approaches, such as PCA [156] and vector quantization [167], whereas the choice of parameter k ≪ min{n1, n2} (also called the rank parameter) provides a dimensionality reduction [168].
In recent years, there has been a significant increase in the number of studies using NMF owing to it being a relaxed form of K-means clustering, one the most widely used unsupervised learning algorithms [169]. Namely, NMF can be applied to clustering as follows: a set of n data points represented by d-dimensional vectors can be placed into columns of a d × n data matrix X. This matrix is then approximately factorized into two non-negative matrices, U and V, where matrix  is the cluster indicator matrix, that is, based on its entries, n data points are assigned to k clusters, whereas U is the basis matrix. In particular, each data point, j, is assigned to cluster, i, if Vij is the maximum value in column j of matrix V. This procedure is called a hard clustering, as each data point belongs to exactly one cluster [170]. For recent advances on using NMF methods for other clustering problems, we refer the reader to a recent book chapter [171].
is the cluster indicator matrix, that is, based on its entries, n data points are assigned to k clusters, whereas U is the basis matrix. In particular, each data point, j, is assigned to cluster, i, if Vij is the maximum value in column j of matrix V. This procedure is called a hard clustering, as each data point belongs to exactly one cluster [170]. For recent advances on using NMF methods for other clustering problems, we refer the reader to a recent book chapter [171].
The NMF method has found applications in many areas, including computer vision [166,172], document clustering [173,174], signal processing [175,176], bioinformatics [57,177,178], recommendation systems [179,180] and social sciences [181,182]. This is due to the fact that NMF can cover nearly all categories of ML problems. Nevertheless, the biggest application comes with the extension of NMF to heterogeneous data. Namely, the above-described NMF can only be used for homogeneous data clustering. Therefore, the formalism was further extended by Ding et al. [183] to co-cluster heterogeneous data by defining non-negative matrix tri-factorization (NMTF). Given a data matrix, R12, encoding relations between two sets of objects of different types (e.g. adjacency matrix of DTI bipartite network representing interactions between n1 genes and n2 drugs, see example in figure 4a,c), NMTF, decompose matrix  into three non-negative matrix factors as follows:
into three non-negative matrix factors as follows: 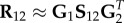 , where
, where 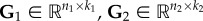 are the cluster indicator matrices of the first and the second dataset, respectively, and
are the cluster indicator matrices of the first and the second dataset, respectively, and  is a low-dimensional representation of the initial matrix. In analogy with NMF method, rank parameters k1 and k2 correspond to numbers of clusters in the first and the second dataset. In addition to co-clustering, NMTF can also be used for matrix completion [184]. Namely, after obtaining low-dimensional matrix factors, the reconstructed data matrix
is a low-dimensional representation of the initial matrix. In analogy with NMF method, rank parameters k1 and k2 correspond to numbers of clusters in the first and the second dataset. In addition to co-clustering, NMTF can also be used for matrix completion [184]. Namely, after obtaining low-dimensional matrix factors, the reconstructed data matrix
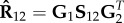 is more complete than the initial data matrix, R12, featuring new entries, unobserved in the data, that emerged from the latent structure captured by the low-dimensional matrix factors. Therefore, NMTF provides a unique approach for modelling multi-relational heterogeneous network data and predicting new, previously unobserved links.
is more complete than the initial data matrix, R12, featuring new entries, unobserved in the data, that emerged from the latent structure captured by the low-dimensional matrix factors. Therefore, NMTF provides a unique approach for modelling multi-relational heterogeneous network data and predicting new, previously unobserved links.
The problem of finding optimal low-rank non-negative matrices whose product is equal to the initial data matrix is known to be NP-hard [185]. Thus, heuristic algorithms for finding approximate solutions have been proposed [186]. They involve solving an optimization problem that minimizes the distance between the input data matrix and the product of low-dimensional matrix factors. The most common measure of the distance used in construction of the objective (cost) function is the Frobenius norm (also called the Euclidean norm) [149]. Hence, the objective function to be minimized can be defined as follows 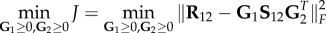 . Note that it is not necessary to impose the non-negativity constraint to the S12 matrix, as only the non-negativity of G1 and G2 is required for co-clustering problems. This is also known as a semi-NMTF problem [187]. Low-dimensional matrix factors, G1, G2 and S12, are computed by using iterative update rules derived by applying standard procedures from constrained optimization theory [188]. These update rules ensure decreasing behaviour of the objective function, J, over iterations. The most popular rules are multiplicative update rules, which preserve the non-negative property of the matrix factors through update iterations. They start with randomly initialized matrix factors and iteratively update them until the convergence criterion is met [183,189]. For more details about the convergence criterion, other update rules and initialization strategies, we refer the reader to references [186,190].
. Note that it is not necessary to impose the non-negativity constraint to the S12 matrix, as only the non-negativity of G1 and G2 is required for co-clustering problems. This is also known as a semi-NMTF problem [187]. Low-dimensional matrix factors, G1, G2 and S12, are computed by using iterative update rules derived by applying standard procedures from constrained optimization theory [188]. These update rules ensure decreasing behaviour of the objective function, J, over iterations. The most popular rules are multiplicative update rules, which preserve the non-negative property of the matrix factors through update iterations. They start with randomly initialized matrix factors and iteratively update them until the convergence criterion is met [183,189]. For more details about the convergence criterion, other update rules and initialization strategies, we refer the reader to references [186,190].
Note that the NMF optimization problems belong to the group of non-convex optimization problems (i.e. the objective function, J, is a non-convex function of its variables) [186]. Unlike convex optimization problems, which are characterized by the global minimum solution and whose algorithms scale well with the problem size [191], non-convex optimization problems face a range of difficulties, including finding the global minimum (and thus the unique solution) and a very slow convergence to a local minimum. Nevertheless, even a local minimum solution of NMF has been shown to have meaningful properties in many data mining applications [186]. Using this method for data integration is based on penalized non-negative matrix tri-factorization (PNMTF), which was originally designed for co-clustering heterogeneous relational data [192,193]. Applicability of PNMTF to data integration problems comes from the fact that it can easily be extended to any number, N, of datasets mutually related by relation matrices
Rij (e.g. sets of genes, drugs, diseases, etc.) [135], where indices, i ≠ j, 1 ≤ i, j ≤ N, denote different datasets. The relation matrices are simultaneously decomposed into low-dimensional factors, Gi, Gj and Sij, within the same optimization function. The key ingredients of this approach are low-dimensional factors, Gi, 1 ≤ i ≤ N, that are shared across the decomposition of all relation matrices, ensuring the influence of all datasets on the resulting model. For example, matrix G3 is shared in the decomposition of all relation matrices Ri3 and R3j,  , and therefore, the clustering assignment obtained from matrix G3 is influenced by all datasets represented by these relation matrices. Similarly, for instance, the reconstruction of matrix
, and therefore, the clustering assignment obtained from matrix G3 is influenced by all datasets represented by these relation matrices. Similarly, for instance, the reconstruction of matrix 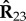 is influenced by all datasets represented by matrices Rij, i ≠ 2 and j ≠ 3, whose factorizations include either matrix G2 or G3.
is influenced by all datasets represented by matrices Rij, i ≠ 2 and j ≠ 3, whose factorizations include either matrix G2 or G3.
Moreover, the method can further be extended as a semi-supervised method that incorporates additional, prior information into the objective function to guide the co-clustering. Namely, in many studies, the datasets itself can have their internal structures represented by networks. For example, in figure 4a,c, in addition to intertype drug–gene relations represented by relation matrix R12, both datasets, drugs and genes are characterized by intratype connections represented by different networks, molecular networks connecting genes and a chemical similarity network connecting drugs. These connections are encoded in the form of Laplacian matrices, Li, and they are incorporated into the objective function as constraints (hence the name constraint matrices) to guide the co-clustering, by enforcing two connected drugs or genes to belong to the same cluster. For instance, the last two terms in the formula displayed in figure 4c represent penalty terms through which these constraint matrices are incorporated into the objective function. These terms are also known as graph regularization terms [194,195]. For more details about the construction of the objective function and derivation of the multiplicative update rules, we refer the reader to references [135,192,193].
Hence, NMTF provides a principled framework for integration of any number, type and size of heterogeneous molecular network data. Given that this method has only recently begun to be used for data integration, there are very few papers that use it. A pioneering application is for predicting new disease–disease associations [61]. A large heterogeneous network system is modelled, consisting of four different inter-related types of objects: genes, diseases, GO terms and drugs. Intertype relations representing gene–DO-term, gene–drug and gene–GO-term associations are represented by relation matrices, whereas intratype relations representing five different molecular networks connecting genes (PPI, GI, MI, Co-Ex and cellular signalling (CS)), a network of side-effect similarity connecting drugs, and GO and DO semantic relations connecting GO and DO terms, respectively, are represented by constraint matrices. After computing low-dimensional matrix factors, the cluster indicator matrix of DO terms (diseases) is used to group diseases into different classes and to predict new disease–disease associations that are not present in the current DO. The authors also estimate the influence of each data source onto the model prediction accuracy and find that the GI network contributes the most to the quality of the integrated model. A similar study demonstrates the potential of the method to reconstruct GO and to predict new GO term associations and gene annotations (GAs) [55] by using evidence from four different types of molecular networks of baker's yeast. Another study uses an NMTF matrix completion approach to predict new GDAs by factorizing known GDAs under prior knowledge from the DSN and the PPI network [60]. The method has also been used for predicting PPIs from the existing PPI network and other biological data sources, including protein sequence, structure and gene expression data [57].
Using NMTF for network data integration has numerous advantages over the other two methods outlined in this section. First, it does not require any data transformation, or any special matrix construction, but instead, it integrates networks naturally represented by adjacency matrices. This drastically reduces chances for information loss. Second, the great accuracy of the method, whose superiority over KB has been demonstrated, stems from the intermediate integration strategy [135]. Finally, the biggest advantage of the method is in its ability to simultaneously model all types of relations in the data, i.e. to simultaneously cluster and create predictive models of all types of data without any data transformation. In contrast, KB methods can only model only one type of data at a time by transforming all data sources into a common feature space. In figure 4, we illustrate the differences between these two methods. Unlike KB, NMTF can be used to co-cluster genes and drugs simultaneously, as well to create a model of gene–drug relations, by using evidence from all available networks. In contrast, KB methods can only be used to classify one entity at the time (either drugs or genes or their relations).
Even though the performance of NMTF is superior to the other two methods, it also has disadvantages. In particular, mathematical limitations owing to non-convex optimization result in time intensive convergence for large-scale datasets. Moreover, unlike KB methods, NMTF methods integrate data in a non-adaptive way, i.e. there are no weighting approaches to combine datasets that can weight more and less informative datasets. Also, the method cannot model the intratype relations, as it is designed for factorizing intertype relations. Hence, there is plenty of room for methodological improvements.
4. Discussion and further challenges
Experimental technologies have enabled us to measure and analyse an ever-increasing volume and diversity of biological and medical data. With an increasing number and type of these data available, there is an increasing need for developing adequate computational methods for their analysis, modelling and integration. Data integration methods have provided a way of comprehensively and simultaneously analysing data towards a more complete understanding of biological systems.
Here, we have reviewed the current, state-of-the-art methods most commonly used in many data integration studies. We have highlighted their advantages and disadvantages and also provided some ideas for their further improvement.
As presented above, the current integration methods hardly meet the challenges listed in §1.2. Given their shortcomings, we provide general guidelines about which methods are more suited for specific biological problems or data types. In that manner, BNs are more suited for small-size datasets (e.g. for reconstruction of disease-specific networks or pathways with small numbers of nodes and edges), due to their inability to handle large-scale datasets. On the other hand, KB methods can handle large-scale datasets, but not the heterogeneous datasets effectively. NMF methods have been shown to be more superior in handling heterogeneous data. In terms of the integration strategy, integration methods relying on intermediate data integration strategies have been shown to result in the best performance accuracy.
Given the growing size and complexity of the data, coupled with computational intractability of many problems underlying analyses of biological data, developing computational methods for data integration that meet all the challenges is very difficult and a subject of active research. Most of the methods are not able to distinguish between concordant and discordant signals in the data. Moreover, all but KB methods lack computational means for automatically selecting informative datasets. Hence, data weighting approaches in KB methods could be similarly defined for NMF methods to automatically select informative matrices to be factorized. Such weighting approaches could be incorporated into the NMF objective function (see §3.5).
Another problem in data integration studies is that there are no standardized measures and a common body of data for validation and assessment of the quality of the integration methods and thus, there are no proper means by which two methods can be compared. For example, many studies dealing with the same problems integrate different types and amounts of data. Hence, a standardized assessment and validation approaches for data integration methods have yet to be proposed.
Although imperfect, the methods applied to different data integration studies have already yielded good results and further developments are promising to open up new avenues and yield crucial advancements in the field. Other research areas, such as economics, climatology, neuroscience and social science, which are also faced with a flood of data, will also benefit from these methods.
Endnotes
Henceforth, the term biological system will refer to a cell.
Because most of the data that we focus on in this paper can be represented as networks (see §2); henceforth, we will be using terms network integration and data integration interchangeably.
A modular network is a network whose nodes can be partitioned into groups (communities) and whose edges are very dense between nodes within a community and very sparse between nodes of different communities [88].
Inference is a process of prediction of unseen events based on observed evidence.
eQTLs are genomic loci (specific locations on a gene) that regulate expression levels of mRNA. eQTLs that regulate expression of their gene-of-origin are referred as cis-eQTL, whereas eQTLs that regulate expression of distant genes are referred as trans-eQTL [146].
CNVs are regions in the genome having significantly more or less copies than the reference human genome sequence.
Positive semi-definite matrix is a matrix with non-negative eigenvalues [149].
Soft margin refers to the SVM method that allows for data points to be mislabelled. It is used when the hyperplane cannot cleanly separate data points into –1 and +1 classes. In that case, the soft margin method will choose a hyperplane that separates data points as cleanly as possible by introducing non-negative slack variables that measure the degree of misclassification. 1-norm refers to the norm of slack variables introduced into the SVM objective function as penalization terms [158].
Competing interests
We declare we have no competing interests.
Funding
This work was supported by the European Research Council (ERC) Starting Independent Researcher grant no. 278212, the National Science Foundation (NSF) Cyber-Enabled Discovery and Innovation (CDI) OIA-1028394, the Serbian Ministry of Education and Science Project III44006, and ARRS project J1-5454.
References
- 1.Ito T, et al. 2000. Toward a protein–protein interaction map of the budding yeast: a comprehensive system to examine two-hybrid interactions in all possible combinations between the yeast proteins. Proc. Natl Acad. Sci. USA 97, 1143–1147. ( 10.1073/pnas.97.3.1143) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Uetz P, et al. 2000. A comprehensive analysis of protein–protein interactions in Saccharomyces cerevisiae. Nature 403, 623–627. ( 10.1038/35001009) [DOI] [PubMed] [Google Scholar]
- 3.Giot L, et al. 2003. A protein interaction map of Drosophila melanogaster. Science 302, 1727–1736. ( 10.1126/science.1090289) [DOI] [PubMed] [Google Scholar]
- 4.Li S, et al. 2004. A map of the interactome network of the metazoan C. elegans. Science 303, 540–543. ( 10.1126/science.1091403) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stelzl U, et al. 2005. A human protein–protein interaction network: a resource for annotating the proteome. Cell 122, 957–968. ( 10.1016/j.cell.2005.08.029) [DOI] [PubMed] [Google Scholar]
- 6.Simonis N, et al. 2009. Empirically controlled mapping of the Caenorhabditis elegans protein–protein interactome network. Nat. Methods 6, 47–54. ( 10.1038/nmeth.1279) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Consortium AIM. 2011. Evidence for network evolution in an Arabidopsis interactome map. Science 333, 601–607. ( 10.1126/science.1203877) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gavin A, et al. 2006. Proteome survey reveals modularity of the yeast cell machinery. Nature 440, 631–636. ( 10.1038/nature04532) [DOI] [PubMed] [Google Scholar]
- 9.Krogan N, et al. 2006. Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature 440, 637–643. ( 10.1038/nature04670) [DOI] [PubMed] [Google Scholar]
- 10.Hawkins RD, Hon GC, Ren B. 2010. Next-generation genomics: an integrative approach. Nat. Rev. Genet. 11, 476–486. ( 10.1038/nrg2795) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nielsen R, Paul JS, Albrechtsen A, Song YS. 2011. Genotype and SNP calling from next-generation sequencing data. Nat. Rev. Genet. 12, 443–451. ( 10.1038/nrg2986) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hirschhorn JN, Daly MJ. 2005. Genome-wide association studies for common diseases and complex traits. Nat. Rev. Genet. 6, 95–108. ( 10.1038/nrg1521) [DOI] [PubMed] [Google Scholar]
- 13.Duerr RH, et al. 2006. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science 314, 1461–1463. ( 10.1126/science.1135245) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Quackenbush J. 2001. Computational analysis of microarray data. Nat. Rev. Genet. 2, 418–427. ( 10.1038/35076576) [DOI] [PubMed] [Google Scholar]
- 15.Dahlquist KD, Salomonis N, Vranizan K, Lawlor SC, Conklin BR. 2002. GenMAPP, a new tool for viewing and analyzing microarray data on biological pathways. Nat. Genet. 31, 19–20. ( 10.1038/ng0502-19) [DOI] [PubMed] [Google Scholar]
- 16.Marioni JC, Mason CE, Mane SM, Stephens M, Gilad Y. 2008. RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 18, 1509–1517. ( 10.1101/gr.079558.108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. 2008. Mapping and quantifying mammalian transcriptomes by RNA-seq. Nat. Methods 5, 621–628. ( 10.1038/nmeth.1226) [DOI] [PubMed] [Google Scholar]
- 18.Wang Z, Gerstein M, Snyder M. 2009. RNA-seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 10, 57–63. ( 10.1038/nrg2484) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Joyce AR, Palsson BØ. 2006. The model organism as a system: integrating omics data sets. Nat. Rev. Mol. Cell Biol. 7, 198–210. ( 10.1038/nrm1857) [DOI] [PubMed] [Google Scholar]
- 20.Gomez-Cabrero D, et al. 2014. Data integration in the era of omics: current and future challenges. BMC Syst. Biol. 8, I1 ( 10.1186/1752-0509-8-S2-I1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vidal M, Cusick ME, Barabási A-L. 2011. Interactome networks and human disease. Cell 144, 986–998. ( 10.1016/j.cell.2011.02.016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aittokallio T, Schwikowski B. 2006. Graph-based methods for analysing networks in cell biology. Brief Bioinformatics 7, 243–255. ( 10.1093/bib/bbl022) [DOI] [PubMed] [Google Scholar]
- 23.Pržulj N. 2011. Protein–protein interactions: making sense of networks via graph-theoretic modeling. BioEssays 33, 115–123. ( 10.1002/bies.201000044) [DOI] [PubMed] [Google Scholar]
- 24.Hakes L, Pinney JW, Robertson DL, Lovell SC. 2008. Protein–protein interaction networks and biology—what's the connection? Nat. Biotechnol. 26, 69–72. ( 10.1038/nbt0108-69) [DOI] [PubMed] [Google Scholar]
- 25.Tong AHY, et al. 2004. Global mapping of the yeast genetic interaction network. Science 303, 808–813. ( 10.1126/science.1091317) [DOI] [PubMed] [Google Scholar]
- 26.Dixon SJ, Costanzo M, Baryshnikova A, Andrews B, Boone C. 2009. Systematic mapping of genetic interaction networks. Annu. Rev. Genet. 43, 601–625. ( 10.1146/annurev.genet.39.073003.114751) [DOI] [PubMed] [Google Scholar]
- 27.Costanzo M, et al. 2010. The genetic landscape of a cell. Science 327, 425–431. ( 10.1126/science.1180823) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tanaka R. 2005. Scale-rich metabolic networks. Phys. Rev. Lett. 94, 168101 ( 10.1103/PhysRevLett.94.168101) [DOI] [PubMed] [Google Scholar]
- 29.Ravasz E, Somera AL, Mongru DA, Oltvai ZN, Barabási A-L. 2002. Hierarchical organization of modularity in metabolic networks. Science 297, 1551–1555. ( 10.1126/science.1073374) [DOI] [PubMed] [Google Scholar]
- 30.Ma H, Zeng A-P. 2003. Reconstruction of metabolic networks from genome data and analysis of their global structure for various organisms. Bioinformatics 19, 270–277. ( 10.1093/bioinformatics/19.2.270) [DOI] [PubMed] [Google Scholar]
- 31.Wagner A, Fell DA. 2001. The small world inside large metabolic networks. Proc. R. Soc. Lond. B 268, 1803–1810. ( 10.1098/rspb.2001.1711) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Prieto C, Risueo A, Fontanillo C, De Las Rivas J. 2008. Human gene coexpression landscape: confident network derived from tissue transcriptomic profiles. PLoS ONE 3, e3911 ( 10.1371/journal.pone.0003911) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stuart JM, Segal E, Koller D, Kim SK. 2003. A gene-coexpression network for global discovery of conserved genetic modules. Science 302, 249–255. ( 10.1126/science.1087447) [DOI] [PubMed] [Google Scholar]
- 34.Mostafavi S, Ray D, Warde-Farley D, Grouios C, Morris Q. 2008. GeneMANIA: a real-time multiple association network integration algorithm for predicting gene function. Genome Biol. 9, S4 ( 10.1186/gb-2008-9-s1-s4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Linghu B, Snitkin E, Hu Z, Xia Y, DeLisi C. 2009. Genome-wide prioritization of disease genes and identification of disease–disease associations from an integrated human functional linkage network. Genome Biol. 10, R91 ( 10.1186/gb-2009-10-9-r91) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pruitt KD, Tatusova T, Brown GR, Maglott DR. 2012. NCBI reference sequences (RefSeq): current status, new features and genome annotation policy. Nucleic Acids Res. 40, D130–D135. ( 10.1093/nar/gkr1079) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sladek R, et al. 2007. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature 445, 881–885 ( 10.1038/nature05616) [DOI] [PubMed] [Google Scholar]
- 38.The Wellcome Trust Case Control Consortium. 2007. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 447, 661–678. ( 10.1038/nature05911) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weinstein JN, et al. 2013. The cancer genome atlas pan-cancer analysis project. Nat. Genet. 45, 1113–1120. ( 10.1038/ng.2764) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zarrei M, Merico D, Scherer SW. 2015. A copy number variation map of the human genome. Nat. Rev. Genet. 16, 172–183. ( 10.1038/nrg3871) [DOI] [PubMed] [Google Scholar]
- 41.Ashburner M, et al. 2000. Gene ontology: tool for the unification of biology. Nat. Genet. 25, 25–29. ( 10.1038/75556) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schriml LM, Arze C, Nadendla S, Chang Y-WW, Mazaitis M, Felix V, Feng G, Kibbe WA. 2012. Disease ontology: a backbone for disease semantic integration. Nucleic Acids Res. 40, D940–D946. ( 10.1093/nar/gkr972) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat T, Weissig H, Shindyalov IN, Bourne PE. 2000. The protein data bank. Nucleic Acids Res. 28, 235–242. ( 10.1093/nar/28.1.235) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wishart DS, Knox C, Guo AC, Cheng D, Shrivastava S, Tzur D, Gautam B, Hassanali M. 2008. Drugbank: a knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res. 36(Suppl 1), D901–D906. ( 10.1093/nar/gkm958) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Davis AP, et al. 2013. The comparative toxicogenomics database: update 2013. Nucleic Acids Res. 41, D1104–D1114. ( 10.1093/nar/gks994) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bolton EE, Wang Y, Thiessen PA, Bryant SH. 2008. PubChem: integrated platform of small molecules and biological activities. Annu. Rep. Comput. Chem. 4, 217–241. ( 10.1016/S1574-1400(08)00012-1) [DOI] [Google Scholar]
- 47.Albert R. 2007. Network inference, analysis, and modeling in systems biology. Plant Cell 19, 3327–3338. ( 10.1105/tpc.107.054700) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee TI, et al. 2002. Transcriptional regulatory networks in Saccharomyces cerevisiae. Science 298, 799–804. ( 10.1126/science.1075090) [DOI] [PubMed] [Google Scholar]
- 49.Hecker M, Lambeck S, Toepfer S, Van Someren E, Guthke R. 2009. Gene regulatory network inference: data integration in dynamic models—a review. Biosystems 96, 86–103. ( 10.1016/j.biosystems.2008.12.004) [DOI] [PubMed] [Google Scholar]
- 50.Lee I, Date SV, Adai AT, Marcotte EM. 2004. A probabilistic functional network of yeast genes. Science 306, 1555–1558. ( 10.1126/science.1099511) [DOI] [PubMed] [Google Scholar]
- 51.Lanckriet GRG, De Bie T, Cristianini N, Jordan MI, Noble WS. 2004. A statistical framework for genomic data fusion. Bioinformatics 20, 2626–2635. ( 10.1093/bioinformatics/bth294) [DOI] [PubMed] [Google Scholar]
- 52.Lanckriet G, Deng M, Cristianini N, Jordan M, Noble W. 2004. Kernel-based data fusion and its application to protein function prediction in yeast. In Biocomputing 2004, Proc. the Pacific Symp., Hawaii, USA, 6–10 January, pp. 300–311. [DOI] [PubMed]
- 53.Ma X, Chen T, Sun F. 2013. Integrative approaches for predicting protein function and prioritizing genes for complex phenotypes using protein interaction networks. Brief. Bioinformatics 15, 685–698. ( 10.1093/bib/bbt041) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Troyanskaya OG, Dolinski K, Owen AB, Altman RB, Botstein D. 2003. A Bayesian framework for combining heterogeneous data sources for gene function prediction (in Saccharomyces cerevisiae). Proc. Natl Acad. Sci. USA 100, 8348–8353. ( 10.1073/pnas.0832373100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gligorijević V, Janjić V, Pržulj N. 2014. Integration of molecular network data reconstruct gene ontology. Bioinformatics 30, i594–i600. ( 10.1093/bioinformatics/btu470) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nariai N, Kolaczyk ED, Kasif S. 2007. Probabilistic protein function prediction from heterogeneous genome-wide data. PLoS ONE 2, e337 ( 10.1371/journal.pone.0000337) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang H, Huang H, Ding C, Nie F. 2013. Predicting protein–protein interactions from multimodal biological data sources via nonnegative matrix tri-factorization. J. Comput. Biol. 20, 344–358. ( 10.1089/cmb.2012.0273) [DOI] [PubMed] [Google Scholar]
- 58.Köhler S, Bauer S, Horn D, Robinson PN. 2008. Walking the interactome for prioritization of candidate disease genes. Am. J. Hum. Genet. 82, 949–958. ( 10.1016/j.ajhg.2008.02.013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bromberg Y. 2013. Chapter 15: disease gene prioritization. PLoS Comput. Biol. 9, e1002902 ( 10.1371/journal.pcbi.1002902) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hwang T, Atluri G, Xie M, Dey S, Hong C, Kumar V, Kuang R. 2012. Co-clustering phenomegenome for phenotype classification and disease gene discovery. Nucleic Acids Res. 40, e146 ( 10.1093/nar/gks615) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Žitnik M, Janjić V, Chris L et al. 2013. Discovering disease–disease associations by fusing systems-level molecular data. Sci. Rep. 3, 3202 ( 10.1038/srep03202) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ashburn TT, Thor KB. 2004. Drug repositioning: identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 3, 673–683. ( 10.1038/nrd1468) [DOI] [PubMed] [Google Scholar]
- 63.Napolitano F, Zhao Y, Moreira VM, Tagliaferri R, Kere J, D'Amato M, Greco D. 2013. Drug repositioning: a machine learning approach through data integration. J. Cheminform. 5, 30 ( 10.1186/1758-2946-5-30) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hamburg MA, Collins FS. 2010. The path to personalized medicine. N. Engl. J. Med. 363, 301–304. ( 10.1056/NEJMp1006304) [DOI] [PubMed] [Google Scholar]
- 65.Ritchie MD, de Andrade M, Kuivaniemi H. 2015. The foundation of precision medicine: integration of electronic health records with genomics through basic, clinical, and translational research. Front. Genet. 6, 1–4. ( 10.3389/fgene.2015.00104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dutkowski J, Kramer M, Surma MA, Balakrishnan R, Cherry JM, Krogan NJ, Ideker T. 2013. A gene ontology inferred from molecular networks. Nat. Biotechnol. 31, 38–45. ( 10.1038/nbt.2463) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sun K, Buchan N, Larminie C, Pržulj N. 2014. The integrated disease network. Integr. Biol. 6, 1069–1079. ( 10.1039/c4ib00122b) [DOI] [PubMed] [Google Scholar]
- 68.Davis DA, Chawla NV. 2011. Exploring and exploiting disease interactions from multirelational gene and phenotype networks. PLoS ONE 6, e22670 ( 10.1371/journal.pone.0022670) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Guo X, Gao L, Wei C, Yang X, Zhao Y, Dong A. 2011. A computational method based on the integration of heterogeneous networks for predicting disease–gene associations. PLoS ONE 6, e24171 ( 10.1371/journal.pone.0024171) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cheng F, et al. 2012. Prediction of drug–target interactions and drug repositioning via network-based inference. PLoS Comput. Biol. 8, e1002503 ( 10.1371/journal.pcbi.1002503) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Emig D, Ivliev A, Pustovalova O, Lancashire L, Bureeva S, Nikolsky Y, Bessarabova M. 2013. Drug target prediction and repositioning using an integrated network-based approach. PLoS ONE 8, e60618 ( 10.1371/journal.pone.0060618) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vanunu O, Magger O, Ruppin E, Shlomi T, Sharan R. 2010. Associating genes and protein complexes with disease via network propagation. PLoS Comput. Biol. 6, e1000641 ( 10.1371/journal.pcbi.1000641) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Huang Y-F, Yeh H-Y, Soo V-W. 2013. Inferring drug–disease associations from integration of chemical, genomic and phenotype data using network propagation. BMC Med. Genomics 6, 1–14. ( 10.1186/1755-8794-6-S3-S4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rider AK, Chawla NV, Emrich SJ. 2013. A survey of current integrative network algorithms for systems biology, pp. 479–495. Amsterdam, The Netherlands: Springer. [Google Scholar]
- 75.Kim TY, Kim HU, Lee SY. 2010. Data integration and analysis of biological networks. Curr. Opin. Biotechnol. 21, 78–84. ( 10.1016/j.copbio.2010.01.003) [DOI] [PubMed] [Google Scholar]
- 76.Bebek G, Koyutuerk M, Price ND, Chance MR. 2012. Network biology methods integrating biological data for translational science. Brief. Bioinformatics 13, 446–459. ( 10.1093/bib/bbr075) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hamid JS, Hu P, Roslin NM, Ling V, Greenwood CMT, Beyene J. 2009. Data integration in genetics and genomics: methods and challenges. Hum. Genomics Proteomics 2009, 869093 ( 10.4061/2009/869093) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kristensen VN, Lingjærde OC, Russnes HG, Vollan HKM, Frigessi A, Børresen-Dale A-L. 2014. Principles and methods of integrative genomic analyses in cancer. Nat. Rev. Cancer 14, 299–313. ( 10.1038/nrc3721) [DOI] [PubMed] [Google Scholar]
- 79.Borgwardt K. 2011. Kernel methods in bioinformatics. In Handbook of statistical bioinformatics (eds Lu HH-S, Schlkopf B, Zhao H), Springer Handbooks of Computational Statistics, pp. 317–334. Berlin, Germany: Springer. [Google Scholar]
- 80.Needham CJ, Bradford JR, Bulpitt AJ, Westhead DR. 2007. A primer on learning in Bayesian networks for computational biology. PLoS Comput. Biol. 3, e129 ( 10.1371/journal.pcbi.0030129) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Devarajan K. 2008. Nonnegative matrix factorization: an analytical and interpretive tool in computational biology. PLoS Comput. Biol. 4, e1000029 ( 10.1371/journal.pcbi.1000029) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gligorijević V, Pržulj N. 2015. Computational methods for integration of biological data. In Personalised medicine: a new medical and social challenge (eds N Bodiroga-Vukobrat, K Pavelic, D Rukavina, GG Sander) Berlin, Germany: Springer. [Google Scholar]
- 83.Alcaraz N, Pauling J, Batra R, Barbosa E, Junge A, Christensen A, Azevedo V, Ditzel HJ, Baumbach J. 2014. Keypathwayminer 4.0: condition-specific pathway analysis by combining multiple omics studies and networks with cytoscape. BMC Syst. Biol. 8, 99 ( 10.1186/s12918-014-0099-x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mitra K, Carvunis A-R, Ramesh SK, Ideker T. 2013. Integrative approaches for finding modular structure in biological networks. Nat. Rev. Genet. 14, 719–732. ( 10.1038/nrg3552) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Jha A, et al. 2015. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 42, 419–430. ( 10.1016/j.immuni.2015.02.005) [DOI] [PubMed] [Google Scholar]
- 86.Hwang D, et al. 2005. A data integration methodology for systems biology. Proc. Natl Acad. Sci. USA 102, 17 296–17 301. ( 10.1073/pnas.0508647102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.West DB. 2000. Introduction to graph theory, 2nd edn New York, NY: Prentice Hall. [Google Scholar]
- 88.Newman M. 2010. Networks: an introduction. New York, NY: Oxford University Press, Inc. [Google Scholar]
- 89.Das K. 2004. The Laplacian spectrum of a graph. Comput. Math. Appl. 48, 715–724. ( 10.1016/j.camwa.2004.05.005) [DOI] [Google Scholar]
- 90.Belkin M, Niyogi P, Sindhwani V. 2006. Manifold regularization: a geometric framework for learning from labeled and unlabeled examples. J. Mach. Learn. Res. 7, 2399–2434. [Google Scholar]
- 91.Albert R. 2005. Scale-free networks in cell biology. J. Cell Sci. 118, 4947–4957. ( 10.1242/jcs.02714) [DOI] [PubMed] [Google Scholar]
- 92.Barabasi A-L, Oltvai ZN. 2004. Network biology: understanding the cell's functional organization. Nat. Rev. Genet. 5, 101–113. ( 10.1038/nrg1272) [DOI] [PubMed] [Google Scholar]
- 93.Pržulj N, Corneil DG, Jurisica I. 2004. Modeling interactome: scale-free or geometric? Bioinformatics 20, 3508–3515. ( 10.1093/bioinformatics/bth436) [DOI] [PubMed] [Google Scholar]
- 94.Higham DJ, Rašajski M, Pržulj N. 2008. Fitting a geometric graph to a protein–protein interaction network. Bioinformatics 24, 1093–1099. ( 10.1093/bioinformatics/btn079) [DOI] [PubMed] [Google Scholar]
- 95.Winterbach W, Mieghem PV, Reinders M, Wang H, Ridder DD. 2013. Topology of molecular interaction networks. BMC Syst. Biol. 7, 90 ( 10.1186/1752-0509-7-90) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chatr-Aryamontri A, et al. 2013. The BioGRID interaction database. Nucleic Acids Res. 41, D816–D823. ( 10.1093/nar/gks1158) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M. 2012. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 40, D109–D114. ( 10.1093/nar/gkr988) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Knox C, et al. 2011. DrugBank 3.0: a comprehensive resource for ‘omics’ research on drugs. Nucleic Acids Res. 39(Suppl 1), D1035–D1041. ( 10.1093/nar/gkq1126) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Li MJ, et al. 2012. GWASdb: a database for human genetic variants identified by genome-wide association studies. Nucleic Acids Res. 40, D1047–D1054. ( 10.1093/nar/gkr1182) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Denny JC, et al. 2013. Systematic comparison of phenome-wide association study of electronic medical record data and genome-wide association study data. Nat. Biotechnol. 31, 1102–1111. ( 10.1038/nbt.2749) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Barrett T, et al. 2007. NCBI GEO: mining tens of millions of expression profiles database and tools update. Nucleic Acids Res. 35(Suppl 1), D760–D765. ( 10.1093/nar/gkl887) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Parkinson H, et al. 2005. ArrayExpress—a public repository for microarray gene expression data at the EBI. Nucleic Acids Res. 33(Suppl 1), D553–D555. ( 10.1093/nar/gki056) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hubble J, et al. 2009. Implementation of GenePattern within the Stanford microarray database. Nucleic Acids Res. 37, D898–D901. ( 10.1093/nar/gkn786) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kuhn M, Campillos M, Letunic I, Jensen LJ, Bork P. 2010. A side effect resource to capture phenotypic effects of drugs. Mol. Syst. Biol. 6, 343 ( 10.1038/msb.2009.98) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Phizicky EM, Fields S. 1995. Protein–protein interactions: methods for detection and analysis. Microbiol. Rev. 59, 94–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Jeong H, Tombor B, Albert R, Oltvai ZN, Barabási A-L. 2000. The large-scale organization of metabolic networks. Nature 407, 651–654. ( 10.1038/35036627) [DOI] [PubMed] [Google Scholar]
- 107.Zhou T. 2013. Computational reconstruction of metabolic networks from Kegg. In Computational toxicology, vol. 930 (eds Reisfeld B, Mayeno AN), pp. 235–249. New York, NY: Humana Press. [DOI] [PubMed] [Google Scholar]
- 108.Yildirim MA, Goh KI, Cusick ME, Barabási AL, Vidal M. 2007. Drug–target network. Nat. Biotechnol. 25, 1119–1126. ( 10.1038/nbt1338) [DOI] [PubMed] [Google Scholar]
- 109.Ziv ZB, et al. 2003. Computational discovery of gene modules and regulatory networks. Nat. Biotechnol. 21, 1337–1342. ( 10.1038/nbt890) [DOI] [PubMed] [Google Scholar]
- 110.De Smet F, Mathys J, Marchal K, Thijs G, De Moor B, Moreau Y. 2002. Adaptive quality-based clustering of gene expression profiles. Bioinformatics 18, 735–746. ( 10.1093/bioinformatics/18.5.735) [DOI] [PubMed] [Google Scholar]
- 111.Luo F, Yang Y, Zhong J, Gao H, Khan L, Thompson D, Zhou J. 2007. Constructing gene co-expression networks and predicting functions of unknown genes by random matrix theory. BMC Bioinformatics 8, 299 ( 10.1186/1471-2105-8-299) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Reverter A, Chan EK. 2008. Combining partial correlation and an information theory approach to the reversed engineering of gene co-expression networks. Bioinformatics 24, 2491–2497. ( 10.1093/bioinformatics/btn482) [DOI] [PubMed] [Google Scholar]
- 113.Baba K, Shibata R, Sibuya M. 2004. Partial correlation and conditional correlation as measures of conditional independence. Aust. N.Z. J. Stat. 46, 657–664. ( 10.1111/j.1467-842X.2004.00360.x) [DOI] [Google Scholar]
- 114.Cover TM, Thomas JA. 2012. Elements of information theory. New York, NY: John Wiley & Sons. [Google Scholar]
- 115.Willett P, Barnard JM, Downs GM. 1998. Chemical similarity searching. J. Chem. Inf. Comput. Sci. 38, 983–996. ( 10.1021/ci9800211) [DOI] [Google Scholar]
- 116.Nikolova N, Jaworska J. 2003. Approaches to measure chemical similarity—a review. QSAR Combin. Sci. 22, 1006–1026. ( 10.1002/qsar.200330831) [DOI] [Google Scholar]
- 117.Zhang P, Agarwal P, Obradovic Z. 2013. Computational drug repositioning by ranking and integrating multiple data sources, vol. 8190 of Lecture Notes in Computer Science, pp. 579–594: Berlin, Germany: Springer. [Google Scholar]
- 118.Atkinson HJ, Morris JH, Ferrin TE, Babbitt PC. 2009. Using sequence similarity networks for visualization of relationships across diverse protein superfamilies. PLoS ONE 4, e4345 ( 10.1371/journal.pone.0004345) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Valavanis I, Spyrou G, Nikita K. 2010. A similarity network approach for the analysis and comparison of protein sequence/structure sets. J. Biomed. Inform. 43, 257–267. ( 10.1016/j.jbi.2010.01.005) [DOI] [PubMed] [Google Scholar]
- 120.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J. Mol. Biol. 215, 403–410. ( 10.1006/jmbi.1990.9999) [DOI] [PubMed] [Google Scholar]
- 121.Holm L, Park J. 2000. DaliLite workbench for protein structure comparison. Bioinformatics 16, 566–567. ( 10.1093/bioinformatics/16.6.566) [DOI] [PubMed] [Google Scholar]
- 122.Ge H, Walhout AJ, Vidal M. 2003. Integrating omic information: a bridge between genomics and systems biology. Trends Genet. 19, 551–560. ( 10.1016/j.tig.2003.08.009) [DOI] [PubMed] [Google Scholar]
- 123.Mani R, St.Onge RP, Hartman JL, Giaever G, Roth FP. 2008. Defining genetic interaction. Proc. Natl Acad. Sci. USA 105, 3461–3466. ( 10.1073/pnas.0712255105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Mostafavi S, Morris Q. 2012. Combining many interaction networks to predict gene function and analyze gene lists. Proteomics 12, 1687–1696. ( 10.1002/pmic.201100607) [DOI] [PubMed] [Google Scholar]
- 125.Zhu J, et al. 2008. Integrating large-scale functional genomic data to dissect the complexity of yeast regulatory networks. Nat. Genet. 40, 854–861. ( 10.1038/ng.167) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Zhang B, et al. 2013. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimers disease. Cell 153, 707–720. ( 10.1016/j.cell.2013.03.030) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Jansen R, et al. 2003. A Bayesian networks approach for predicting protein–protein interactions from genomic data. Science 302, 449–453. ( 10.1126/science.1087361) [DOI] [PubMed] [Google Scholar]
- 128.Gevaert O, Smet FD, Timmerman D, Moreau Y, Moor BD. 2006. Predicting the prognosis of breast cancer by integrating clinical and microarray data with Bayesian networks. Bioinformatics 22, e184–e190. ( 10.1093/bioinformatics/btl230) [DOI] [PubMed] [Google Scholar]
- 129.van Vliet MH, Horlings HM, van de Vijver MJ, Reinders MJ, Wessels LF. 2012. Integration of clinical and gene expression data has a synergetic effect on predicting breast cancer outcome. PLoS ONE 7, e40358 ( 10.1371/journal.pone.0040358) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Yuan Y, Savage RS, Markowetz F. 2011. Patient-specific data fusion defines prognostic cancer subtypes. PLoS Comput. Biol. 7, e1002227 ( 10.1371/journal.pcbi.1002227) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Wang Y, Chen S, Deng N, Wang Y. 2013. Drug repositioning by kernel-based integration of molecular structure, molecular activity, and phenotype data. PLoS ONE 8, e78518 ( 10.1371/journal.pone.0078518) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kato T, Tsuda K, Asai K. 2005. Selective integration of multiple biological data for supervised network inference. Bioinformatics 21, 2488–2495. ( 10.1093/bioinformatics/bti339) [DOI] [PubMed] [Google Scholar]
- 133.Yamanishi Y, Vert J-P, Kanehisa M. 2004. Protein network inference from multiple genomic data: a supervised approach. Bioinformatics 20(Suppl. 1), i363–i370. ( 10.1093/bioinformatics/bth910) [DOI] [PubMed] [Google Scholar]
- 134.Daemen A, Gevaert O, De Moor B. 2007. Integration of clinical and microarray data with kernel methods. In Engineering in medicine and biology society, 2007. EMBS 2007. 29th Annual Int. Conf. of the IEEE, pp. 5411–5415. Piscataway, NJ: IEEE. [DOI] [PubMed] [Google Scholar]
- 135.Žitnik M, Župan B. 2015. Data fusion by matrix factorization. IEEE Trans. Pattern Anal. Mach. Intell. 37, 41–53. ( 10.1109/TPAMI.2014.2343973) [DOI] [PubMed] [Google Scholar]
- 136.Pavlidis P, Cai J, Weston J, Noble WS. 2002. Learning gene functional classifications from multiple data types. J. Comput. Biol. 9, 401–411. ( 10.1089/10665270252935539) [DOI] [PubMed] [Google Scholar]
- 137.Ozen A, Gonen M, Alpaydin E, Haliloglu T. 2009. Machine learning integration for predicting the effect of single amino acid substitutions on protein stability. BMC Struct. Biol. 9, 66 ( 10.1186/1472-6807-9-66) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Chen Y, Hao J, Jiang W, He T, Zhang X, Jiang T, Jiang R. 2013. Identifying potential cancer driver genes by genomic data integration. Sci. Rep. 3, 66 ( 10.1038/srep03538) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Goh K-I, Cusick ME, Valle D, Childs B, Vidal M, Barabsi A-L. 2007. The human disease network. Proc. Natl Acad. Sci. USA 104, 8685–8690. ( 10.1073/pnas.0701361104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Ben-Gal I. 2008. Bayesian networks. New York, NY: John Wiley & Sons, Ltd. [Google Scholar]
- 141.Sachs K, Perez O, Pe'er D, Lauffenburger DA, Nolan GP. 2005. Causal proteinsignaling networks derived from multiparameter single-cell data. Science 308, 523–529. ( 10.1126/science.1105809) [DOI] [PubMed] [Google Scholar]
- 142.Friedman N. 2004. Inferring cellular networks using probabilistic graphical models. Science 303, 799–805. ( 10.1126/science.1094068) [DOI] [PubMed] [Google Scholar]
- 143.Chickering DM. 1996. Learning Bayesian networks is NP-complete. In Learning from data, pp. 121–130. Berlin, Germany: Springer. [Google Scholar]
- 144.Cooper GF. 1990. The computational complexity of probabilistic inference using Bayesian belief networks. Artif. Intell. 42, 393–405. ( 10.1016/0004-3702(90)90060-D) [DOI] [Google Scholar]
- 145.Schadt E, Friend S, Shaywitz D. 2009. A network view of disease and compound screening. Nat. Rev. Drug Discov. 8, 286–295. ( 10.1038/nrd2826) [DOI] [PubMed] [Google Scholar]
- 146.Rockman MV, Kruglyak L. 2006. Genetics of global gene expression. Nat. Rev. Genet. 7, 862–872. ( 10.1038/nrg1964) [DOI] [PubMed] [Google Scholar]
- 147.Franceschini A, et al. 2013. STRING v9.1: protein–protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 41, D808–D815. ( 10.1093/nar/gks1094) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Schölkopf B, Tsuda K, Vert J-P. 2004. Kernel methods in computational biology. Cambridge, MA: MIT Press. [Google Scholar]
- 149.Golub GH, Van Loan CF. 2012. Matrix computations, vol. 3 Baltimore, MD: JHU Press. [Google Scholar]
- 150.Leslie CS, Eskin E, Noble WS.2002. The spectrum kernel: a string kernel for SVM protein classification. In Pacific Symp. on Biocomputing, 3–7 January, pp. 566–575.
- 151.Leslie CS, Eskin E, Cohen A, Weston J, Noble WS. 2004. Mismatch string kernels for discriminative protein classification. Bioinformatics 20, 467–476. ( 10.1093/bioinformatics/btg431) [DOI] [PubMed] [Google Scholar]
- 152.Ben-Hur A, Brutlag D. 2003. Remote homology detection: a motif based approach. Bioinformatics 19(Suppl. 1), 26–i33. ( 10.1093/bioinformatics/btg1002) [DOI] [PubMed] [Google Scholar]
- 153.Gomez SM, Noble WS, Rzhetsky A. 2003. Learning to predict protein–protein interactions from protein sequences. Bioinformatics 19, 1875–1881. ( 10.1093/bioinformatics/btg352) [DOI] [PubMed] [Google Scholar]
- 154.Kondor RI, Lafferty J.2002. Diffusion kernels on graphs and other discrete structures. In Proc. the ICML, 8–12 July, pp. 315–322.
- 155.Hearst MA, Dumais ST, Osman E, Platt J, Scholkopf B. 1998. Support vector machines. IEEE Intell. Syst. Appl. 13, 18–28. ( 10.1109/5254.708428) [DOI] [Google Scholar]
- 156.Jolliffe I. 2005. Principal component analysis. New York, NY: Wiley Online Library. [Google Scholar]
- 157.Hardoon D, Szedmak S, Shawe-Taylor J. 2004. Canonical correlation analysis: an overview with application to learning methods. Neural Comput 16, 2639–2664. ( 10.1162/0899766042321814) [DOI] [PubMed] [Google Scholar]
- 158.Vapnik VN, Vapnik V. 1998. Statistical learning theory, vol. 1 New York, NY: Wiley. [Google Scholar]
- 159.Boser BE, Guyon IM, Vapnik VN. 1992. A training algorithm for optimal margin classifiers. In Proc. the Fifth Annual Workshop on Computational Learning Theory, 27–29 July, pp. 144–152. New York, NY: ACM. [Google Scholar]
- 160.Ben-Hur A, Ong CS, Sonnenburg S, Schölkopf B, Rätsch G. 2008. Support vector machines and kernels for computational biology. PLoS Comput. Biol. 4, e1000173 ( 10.1371/journal.pcbi.1000173) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Noble WS, et al. 2004. Support vector machine applications in computational biology. In Kernel methods in computational biology (eds B Schoelkopf, K Tsuda, J-P Vert), pp. 71–92. Cambridge, MA: MIT Press. [Google Scholar]
- 162.Gönen M, Alpaydın E. 2011. Multiple kernel learning algorithms. J. Mach. Learn. Res. 12, 2211–2268. [Google Scholar]
- 163.Wang X, Xing EP, Schaid DJ. 2014. Kernel methods for large-scale genomic data analysis. Brief. Bioinformatics 16, 183–192. ( 10.1093/bib/bbu024) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Yu S, Tranchevent L.-C, Moor BD, Moreau Y. 2011. Kernel-based data fusion for machine learning— methods and applications in bioinformatics and text mining, vol. 345 of Studies in Computational Intelligence. Berlin, Germany: Springer. [Google Scholar]
- 165.Suykens JA, Vandewalle J. 1999. Least squares support vector machine classifiers. Neural Process. Lett. 9, 293–300. ( 10.1023/A:1018628609742) [DOI] [Google Scholar]
- 166.Lee DD, Seung HS. 1999. Learning the parts of objects by non-negative matrix factorization. Nature 401, 788–791. ( 10.1038/44565) [DOI] [PubMed] [Google Scholar]
- 167.Gersho A, Gray RM. 1992. Vector quantization and signal compression. Berlin, Germany: Springer Science & Business Media. [Google Scholar]
- 168.Cichocki A, Zdunek R, Phan AH, Amari S-I. 2009. Nonnegative matrix and tensor factorizations: applications to exploratory multi-way data analysis and blind source separation. New York, NY: John Wiley & Sons. [Google Scholar]
- 169.Ding C, He X, Simon HD.2005. On the equivalence of nonnegative matrix factorization and spectral clustering. In Proc. the 2005 SIAM Int. Conf. on Data Mining, 21–23 April, pp. 606–610.
- 170.Zass R, Shashua A. 2005. A unifying approach to hard and probabilistic clustering. In Tenth IEEE Int. Conf. on Computer vision, 2005. ICCV 2005, vol. 1, pp. 294–301. Piscataway, NJ: IEEE. [Google Scholar]
- 171.Li T, Ding CHQ. 2013. Nonnegative matrix factorizations for clustering: a survey. In Data clustering: algorithms and applications, pp. 149–176. New York, NY: Chapman & Hall/CRC. [Google Scholar]
- 172.Liu W, Zheng N. 2004. Non-negative matrix factorization based methods for object recognition. Pattern Recogn. Lett. 25, 893–897. ( 10.1016/j.patrec.2004.02.002) [DOI] [Google Scholar]
- 173.Xu W, Liu X, Gong Y. 2003. Document clustering based on non-negative matrix factorization. In Proc. the 26th Annual Int. ACM Sigir Conf. on Research and Development in Information Retrieval, 28 July–1 August, pp. 267–273. New York, NY: ACM. [Google Scholar]
- 174.Shahnaz F, Berry MW, Pauca VP, Plemmons RJ. 2006. Document clustering using nonnegative matrix factorization. Inf. Process. Manage. 42, 373–386. ( 10.1016/j.ipm.2004.11.005) [DOI] [Google Scholar]
- 175.Smaragdis P, Brown JC. 2003. Non-negative matrix factorization for polyphonic music transcription. In Applications of signal processing to audio and acoustics, pp. 177–180. Piscataway, NJ: IEEE. [Google Scholar]
- 176.Virtanen T. 2007. Monaural sound source separation by nonnegative matrix factorization with temporal continuity and sparseness criteria. IEEE Trans. Audio, Speech Lang. Process. 15, 1066–1074. ( 10.1109/TASL.2006.885253) [DOI] [Google Scholar]
- 177.Brunet J-P, Tamayo P, Golub TR et al. 2004. Metagenes and molecular pattern discovery using matrix factorization. Proc. Natl Acad. Sci. USA 101, 4164–4169. ( 10.1073/pnas.0308531101) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Hutchins LN, Murphy SM, Singh P, Graber JH. 2008. Position-dependent motif characterization using non-negative matrix factorization. Bioinformatics 24, 2684–2690. ( 10.1093/bioinformatics/btn526) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Koren Y, Bell R, Volinsky C. 2009. Matrix factorization techniques for recommender systems. Computer 42, 30–37. ( 10.1109/MC.2009.263) [DOI] [Google Scholar]
- 180.Zhang S, Wang W, Ford J, Makedon F. 2006. Learning from incomplete ratings using non-negative matrix factorization. In SDM, pp. 549–553. Bethesda, MD: SIAM. [Google Scholar]
- 181.Cheng C, Yang H, King I, Lyu MR. 2012. Fused matrix factorization with geographical and social influence in location-based social networks. In Aaai'12, 22–26 July, pp. 1–1. [Google Scholar]
- 182.Li T, Zhang Y, Sindhwani V.2009. A non-negative matrix tri-factorization approach to sentiment classification with lexical prior knowledge. In Proc. the Joint Conf. of the 47th Annual Meeting of the ACL and the 4th Int. Joint Conf. on Natural Language Processing of the AFNLP, 2–7 August, pp. 244–252. Association for Computational Linguistics.
- 183.Ding C, et al. 2006. Orthogonal nonnegative matrix Tri-factorizations for clustering. In Proc. the 12th ACM SIGKDD Int. Conf. On Knowledge Discovery and Data Mining, KDD ’06, 20–23 August, pp. 126–135. New York, NY: ACM.
- 184.Johnson CR. 1990. Matrix completion problems: a survey. In Matrix theory and applications, vol. 40 of Proceedings of Symposia in Applied Mathematics, pp. 171–198. Providence, RI: AMS. [Google Scholar]
- 185.Vavasis SA. 2009. On the complexity of nonnegative matrix factorization. SIAM J. Optim. 20, 1364–1377. ( 10.1137/070709967) [DOI] [Google Scholar]
- 186.Berry MW, Browne M, Langville AN, Pauca VP, Plemmons RJ. 2007. Algorithms and applications for approximate nonnegative matrix factorization. Comput. Stat Data Anal. 52, 155–173. ( 10.1016/j.csda.2006.11.006) [DOI] [Google Scholar]
- 187.Ding C, Li T, Jordan MI. 2010. Convex and semi-nonnegative matrix factorizations. IEEE Trans. Pattern Anal. Mach. Intell. 32, 45–55. ( 10.1109/TPAMI.2008.277) [DOI] [PubMed] [Google Scholar]
- 188.Sun W, Yuan Y.-X. 2006. Optimization theory and methods: nonlinear programming, vol. 1 Berlin, Germany: Springer Science & Business Media. [Google Scholar]
- 189.Lee DD, Seung HS. 2001. Algorithms for non-negative matrix factorization. In Advances in neural information processing systems, pp. 556–562. Cambdrige, MA: MIT Press. [Google Scholar]
- 190.Albright R, Cox J, Duling D, Langville A, Meyer C. 2006. Algorithms, initializations, and convergence for the nonnegative matrix factorization. Tech. Rep, 81706. North Carolina State University, Raleigh, N.C.
- 191.Boyd S, Vandenberghe L. 2004. Convex optimization. New York, NY: Cambridge University Press. [Google Scholar]
- 192.Wang H, Huang H, Ding C. 2011. Simultaneous clustering of multi-type relational data via symmetric nonnegative matrix tri-factorization. In Proc. the 20th ACM Int. Conf. on Information and Knowledge Management, CIKM ’11, pp. 279–284. New York, NY: ACM. [Google Scholar]
- 193.Wang F, Li T, Zhang C. 2008. Semi-supervised clustering via matrix factorization. In SDM, pp. 1–12. Atlanta, GA: SIAM. [Google Scholar]
- 194.Cai D, He X, Han J, Huang TS. 2011. Graph regularized nonnegative matrix factorization for data representation. IEEE Trans. Pattern Anal. Mach. Intell. 33, 1548–1560. ( 10.1109/TPAMI.2010.231) [DOI] [PubMed] [Google Scholar]
- 195.Shang F, Jiao L, Wang F. 2012. Graph dual regularization non-negative matrix factorization for co-clustering. Pattern Recognit. 45, 2237–2250. ( 10.1016/j.patcog.2011.12.015) [DOI] [Google Scholar]