

Summary
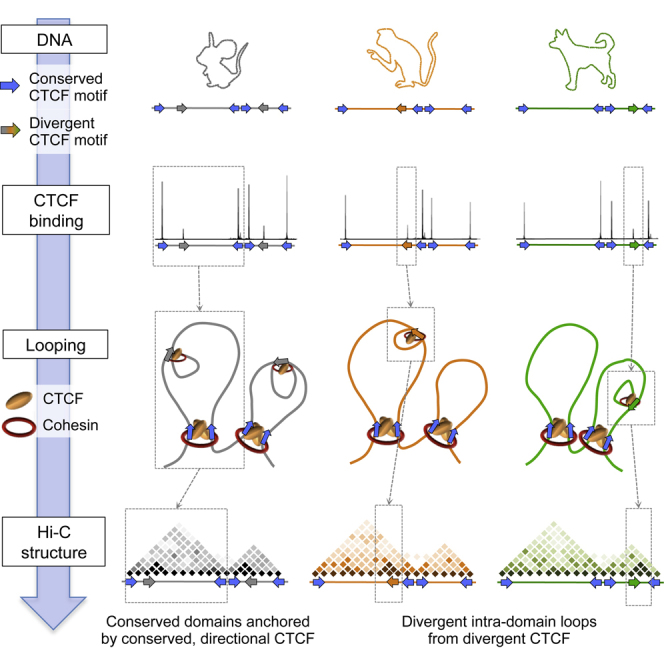
Topological domains are key architectural building blocks of chromosomes, but their functional importance and evolutionary dynamics are not well defined. We performed comparative high-throughput chromosome conformation capture (Hi-C) in four mammals and characterized the conservation and divergence of chromosomal contact insulation and the resulting domain architectures within distantly related genomes. We show that the modular organization of chromosomes is robustly conserved in syntenic regions and that this is compatible with conservation of the binding landscape of the insulator protein CTCF. Specifically, conserved CTCF sites are co-localized with cohesin, are enriched at strong topological domain borders, and bind to DNA motifs with orientations that define the directionality of CTCF’s long-range interactions. Conversely, divergent CTCF binding between species is correlated with divergence of internal domain structure, likely driven by local CTCF binding sequence changes, demonstrating how genome evolution can be linked to a continuous flux of local conformation changes. We also show that large-scale domains are reorganized during genome evolution as intact modules.
Graphical Abstract
Highlights
-
•
Multi-species Hi-C comparisons reveal robust conservation of chromosome organization
-
•
CTCF binding has evolved under two regimes with different effects on structure
-
•
Oriented CTCF motifs determine the directionality of CTCF-mediated interactions
To explore the mechanisms underlying the evolution of chromosomal domain structures, Vietri Rudan et al. compare four mammalian species and reveal a direct link between insulator site divergence and the evolution of chromatin domain structure. Their data point to a direct role for CTCF/cohesin in driving structural change in the genome.
Introduction
The discovery of a topological-domain-like three-dimensional organization in metazoan chromosomes (Sexton et al., 2012; Dixon et al., 2012; Nora et al., 2012; Hou et al., 2012) is re-shaping our understanding of genome structure and function. This new layer of large-scale genome organization provides insights into the way by which sparsely embedded regulatory elements could interact to drive long-range transcriptional regulation. However, the extent by which the multi-scale domain architecture facilitates long-range regulation or is implied by it, as well as the precise mechanisms organizing chromosomes into domains, is not truly understood.
Currently, the best-characterized mechanism for domain organization involves long-range interactions between insulator proteins (CCCTC-binding factor [CTCF] in mammals) and the cohesin complex (Phillips-Cremins et al., 2013; Sofueva et al., 2013; Zuin et al., 2014). CTCF is a DNA-binding protein that engages its 11 zinc fingers to bind to DNA at a large, information-rich consensus motif (Kim et al., 2007). CTCF is a critical transcriptional regulator, originally described as a repressor of the myc oncogene (Filippova et al., 1996) and subsequently shown to function as an enhancer blocker and an insulator element (Bell et al., 1999). The insulator activity of CTCF depends on cohesin (Parelho et al., 2008; Wendt et al., 2008), an essential protein complex required for sister chromatid cohesion during mitosis (Michaelis et al., 1997; Guacci et al., 1997), which also functions in gene regulation (Rollins et al., 1999; Pauli et al., 2008). Together, CTCF and cohesin exert their effects on gene regulation primarily through the formation or stabilization of long-range chromatin loops (Hadjur et al., 2009; Mishiro et al., 2009; Nativio et al., 2009; Seitan et al., 2011). Such CTCF/cohesin-anchored loops are distributed throughout the genome, creating a network of long-range contacts spanning multiple scales, including not only loops that define the borders of strongly demarcated topological domains but also loops within such domains (Phillips-Cremins et al., 2013; Seitan et al., 2013; Sofueva et al., 2013; Zuin et al., 2014). While CTCF binding specificity depends to a large extent on specific DNA sequence elements, the specificity and directionality of CTCF/cohesin long-range contacts (Sofueva et al., 2013) and the way by which specific sites are assembled to define topological domains are not fully understood.
The dependency of CTCF recruitment on DNA sequence elements and the role for this insulator in mediating long-range chromosomal organization suggest that CTCF may function as a key link between genome sequence and the evolution of chromosomal domain organization. Indeed, some conservation of chromosomal domain structures has been reported between human and mouse through both linear epigenomic analysis (Yaffe et al., 2010) and high-throughput chromosome conformation capture (Hi-C) comparisons (Dixon et al., 2012). Moreover, a comparative analysis of CTCF binding in several mammalian genomes suggests its evolutionary dynamics are context dependent, and conservation can be interrupted by mobile element activity (Schmidt et al., 2012). Despite these observations, a link between the evolutionary dynamics of CTCF binding and the evolution of chromosomal domain organization is yet to be explored.
Studies that have tracked the evolution of different transcription factor (TF) binding patterns have shown that sequence evolution alone is incapable of fully explaining the evolutionary dynamics of TF binding landscapes (Dermitzakis and Clark, 2001; Birney et al., 2007; Borneman et al., 2007; Schmidt et al., 2010). TF binding landscapes and large-scale chromosomal organization may function cooperatively to drive the evolution of genome regulation. These observations highlight the importance of multi-species comparative chromosomal structure analysis and its integration with insulator binding profiles across evolution. If the binding patterns of trans-factors such as CTCF are indeed strong drivers of domain organization, then their evolutionary dynamics should drive evolutionary conservation and divergence of chromosome domains.
With this in mind, we performed comparative Hi-C in non-cycling primary liver cells and analyzed the data together with CTCF binding profiles from the same species and tissue. Analysis of four mammalian Hi-C maps allowed us to explore how the evolution of CTCF binding profiles correlates, and in some cases likely drives, the evolution of chromosomal topologies. We find that the large-scale chromosomal domain structure is highly conserved between species, in a way that is correlated with the conservation of both the CTCF binding site and the orientation of its motif, resulting in directional long-range interactions that demarcate conserved domains. On the other hand, internal domain structure is observed to be more dynamic, and we discover remarkable correlation between evolutionary dynamics of CTCF sites and divergence of local insulation structure. Since the evolution of CTCF binding profiles is strongly driven at the nucleotide level within cis elements, our data suggest that internal domain structure can be modulated flexibly through local sequence evolution. Conversely, we show that interruption of large-scale domain structure is rare, and we suggest that instead of local sequence divergence, evolutionary manipulation of global chromosomal topologies is driven by processes involving duplications or rearrangements such as inversions, insertions/deletions, and translocations. We demonstrate this by charting cases of evolutionary domain shuffling in mouse and dog.
Results
Sequence-Driven Evolution of CTCF Binding Profiles
CTCF binding is strongly correlated with the topological architecture of mammalian chromosomes and participates in long-range chromatin loops, thereby underlying global contact insulation. We analyzed mouse (Mus musculus [Mmus]), dog (Canis familiaris [Cfam]), and macaque (Macaca mulatta [Mmul]) CTCF chromatin immunoprecipitation sequencing (ChIP-seq) profiles from primary liver cells (Schmidt et al., 2012), aiming to define how conservation and divergence of the insulator binding landscape co-evolve with chromosomal topology. Pairwise CTCF ChIP-seq analysis identified conserved or divergent CTCF binding sites within syntenic chromosomal regions (Figures 1A, 1B, and S1). Sites with the strongest CTCF binding intensities were highly conserved (77% of the top 0.1 percentile), while lower-intensity CTCF binding sites were enriched for divergent binding (57% of mouse-divergent sites) (Figure 1B). We computed the sequence affinity of the different classes of binding sites to the canonical CTCF consensus motif in mouse and found that the levels of motif affinity for conserved sites were overall higher than the level for the mouse-divergent sites (Figure S1).
Figure 1.

Evolution of CTCF Binding Correlates with Hi-C Domain Structure
(A) Representative CTCF ChIP-seq tracks on mouse (Mmus) chromosome 5; dog (Cfam) and macaque (Mmul) tracks are shown after liftOver to the mouse genome (mm10). Sites conserved across all three species (arrowhead) or specific to a single species (asterisks) are indicated as examples.
(B) A pairwise comparison of mouse CTCF ChIP and dog CTCF ChIP (liftOver track), identifying conserved or divergent binding sites (see Experimental Procedures).
(C) A comparison of the interspecies difference in CTCF ChIP signal against the difference in CTCF motif affinity in mouse versus dog. Scatterplots are highlighted for conserved (Mmus+/Cfam+, blue), mouse-divergent (Mmus+/Cfam−, black), and dog-divergent sites (Mmus−/Cfam+, green).
(D) A representative 2-Mb region from chromosome 5 of the mouse Hi-C contact maps. Also shown are the mouse (Mmus) CTCF ChIP track and conserved (Mmus+/Cfam+, blue) and divergent (Mmus+/Cfam−, gray) CTCF sites.
(E) Genome-wide relative position of conserved and divergent CTCF sites within mouse Hi-C domains.
(F) Contact insulation analysis for conserved (Mmus+/Cfam+) and mouse-divergent (Mmus+/Cfam−) CTCF sites in the mouse Hi-C data. See also Figure S1.
To understand the relationship between sequence affinity and CTCF binding at conserved or divergent sites, we correlated changes in CTCF binding with changes in CTCF sequence motif affinity among species. For this analysis, we used the canonical consensus motif from mouse that is the same in the other species (Schmidt et al., 2012). Remarkably, we found a direct association between sequence divergence and CTCF binding divergence. Conserved CTCF binding sites showed overall high motif affinities and a high degree of affinity conservation. Conversely, motifs underlying the divergent sites were evolutionarily dynamic and diverged in strong correlation to divergent binding intensity (Figures 1C and S1). The data show that when strong motifs in CTCF binding sites diverge, CTCF binding itself is concomitantly gained or lost. Interestingly, 65% of the sites that were conserved between mouse and dog were also conserved in macaque, while macaque-specific and dog-specific sites constituted another two populations of 775 and 891 sites, respectively, with weaker, more evolutionarily plastic motifs. Together, these data suggest that the CTCF insulator landscape is evolving under two regimes: the first involves a tight conservation of both sequence and binding landscape, and the second shows a dynamic interplay between divergence of specific cis elements and consequential evolution of the CTCF binding trait. The relatively direct influence of motif divergence on CTCF binding forms a potential link between sequence evolution and large-scale genome evolution.
CTCF Binding Site Evolution Is Correlated with the Mouse Hi-C Domain Structure
To investigate how the different classes of CTCF binding conservation correlate with chromosomal structure, we prepared Hi-C datasets on mouse liver cells (Figure S2). Filtering and normalization of the Hi-C ligation products was performed as before (Sofueva et al., 2013), revealing the characteristic chromosomal domain structure in these cells. Visualization of the CTCF occupancy groups with the Hi-C contact maps suggested that conserved CTCF binding sites were found at the borders of large-scale Hi-C domains while species-specific CTCF sites are located internal to domains (Figure 1D). This observation was supported with a genome-wide analysis whereby the relative position of conserved and divergent CTCF sites was determined with respect to all domains in the mouse genome (Figure 1E).
To further characterize chromosomal contacts around conserved and divergent CTCF sites, we analyzed the average contact distribution around these sites globally, measuring “contact insulation” by quantifying the decrease in contact probability between multiple elements separated by a CTCF site (Sofueva et al., 2013). Analysis of the composite contact insulation at multiple distance ranges, indicated strong insulation profiles for conserved CTCF sites, further supporting the idea that these conserved, high-intensity CTCF sites were co-occurring with the borders of large-scale domain (reminiscent of topological chromosomal domains) (Figure 1F, left panel). In comparison, the lower-intensity mouse-divergent sites showed a significantly weaker, more localized insulation profile (Figure 1F, right panel). Similar trends were also observed when classifying CTCF sites according to their conservation in macaque (Figure S1). In summary, we found strong correlation between the evolutionary dynamics of CTCF binding sites and mouse chromosome topology, indicating the possibility of a direct link between insulator site divergence and the evolution of topological domain structure.
Comparative Hi-C Reveals the Evolution of Chromosome Topologies
We used comparative Hi-C to examine conservation and divergence of chromosome topology and to test how evolution of CTCF binding sites might underpin this. We collected liver cells from macaque, rabbit (Oryctolagus cuniculus [Ocun]), and dog and processed them using the same approach applied to mouse, yielding chromosomal contact maps for each of these species (Figures S2 and S3). Evaluation of the overall topological structure within the three newly profiled species first indicated the integrity of their reference genome structures and generated a resource for future refinement of such assemblies. More importantly, the data showed that the chromosome topologies in macaque, dog, and rabbit are characterized by a chromosomal domain structure that is similar to the one inferred before for human and mouse (Dixon et al., 2012). For example, comparison of a 9-Mb syntenic region highlighted the extensive conservation of chromosomal structure across all species (Figure 2A). The maps also revealed evidence of intra-domain differences between species (Figure 2B). We quantified the extent of structural conservation genome-wide using a computational approach that allowed us to comprehensively describe domain structure at multiple scales. This pairwise approach revealed extensive genome-wide interspecies conservation of chromosome structure (Figures 2C and S3). A systematic analysis of paired domains in mouse and dog revealed that conserved domains are smaller in size compared to other domains and are classified as both active and passive clusters (Figure S4). Together, these data facilitated extensive analysis of the evolution of chromosomal topologies within regions that did not go through substantial genome rearrangement, allowing examination of the evolution of both large-scale domain borders and the insulation structure within domains.
Figure 2.

Chromosomal Domain Structure Is Robustly Conserved in Mammals
(A) Representative Hi-C contact maps of a 9-Mb syntenic region from mouse (Mmus), rabbit (Ocun), macaque (Mmul), and dog (Cfam). Maps are colored according to technically corrected contact enrichment. For scale purposes, the same size region is shown for all species, but in the macaque and dog genome, the syntenic region is reduced in size. The portion of the map that is outside of the synteny boundary is shaded out. Shown above each map is the quantification of contacts for distance bands of 160–480 kb (crossover 160 kb) and 80–240 kb (crossover 80 kb), as well as a track of orthologous genes, highlighting their conserved distribution in chromosomal domains.
(B) A zoom in of the domain highlighted with a white dashed box in (A) reveals differences in its internal organization across species.
(C) Global quantification of the correlation between the maps. Genome-wide contact enrichments between elements separated by 160–480 kb in rabbit, macaque, and dog were compared to the mouse genome after liftOver. Spearman correlation values are shown inside the plots. Axes units are contact enrichments [log2(observed/expected)].
See also Figures S2 and S3.
Divergent CTCF Binding Drives Local Structural Change within Domains
Hi-C maps from liver cells of different species allowed us to ask how the evolutionary dynamics of CTCF correlate with conservation or divergence of domain structure. Analysis of specific loci showed that conserved CTCF sites were typically located at the borders of large-scale chromosomal domains that were themselves conserved between mouse and dog (Figure 3A). To test these observations globally, we computed the contact insulation profiles from either the mouse or dog Hi-C maps around conserved CTCF sites, showing that these sites indeed globally served as conserved insulation points (Figure 3B). Similar results were derived using a comparison of mouse and macaque (Figure S5). We also observed that conserved CTCF sites were strongly enriched for Rad21 in mouse (79% of conserved sites compared to 51% mouse-divergent sites co-localize with Rad21) and that CTCF/cohesin co-occupied sites exhibited strong contact insulation in all three species (data not shown).
Figure 3.

Conserved and Divergent CTCF Sites Show Differential Contact Insulation Behavior
(A) Representative Hi-C contact maps for a 2-Mb syntenic region in mouse (left panel) and dog (right panel). Also shown are the CTCF ChIP-seq tracks in each species as well as the conserved (Mmus+/Cfam+, blue) and divergent (Mmus+/Cfam−, gray or Mmus−/Cfam+, green) CTCF sites.
(B) Average contact insulation analysis at conserved (Mmus+/Cfam+) CTCF sites in mouse (leftmost panel) and dog (rightmost panel) Hi-C datasets. Arrows indicate liftOver of sites.
(C) Same as for (B), but for mouse-divergent (Mmus+/Cfam−) and dog-divergent (Mmus−/Cfam+) sites in both genomes. Divergent sites appear to mediate weaker, shorter-range insulation that disappears in the species where the site is not bound.
(D) Distribution of the difference in contact insulation between mouse and dog at conserved or divergent CTCF sites at 20–60 kb (left panel, 20-kb band) or 80–240 kb (right panel, 80-kb band) scales. Divergent sites exhibit a significant (Kolmogorov-Smirnov test, p < 1 × 10−12, marked by an asterisk) shift in their distributions in the 20-kb band, but not in the 80-kb band, compatible with them mediating insulation at a local scale, but not at higher ranges.
See also Figure S5.
In contrast to these highly stable sites, our data showed that divergent CTCF sites were located primarily within domains and exhibited local contact insulation. Comparative analysis of contact insulation at divergent CTCF sites revealed that indeed these sites correlated with divergent contact insulation profiles. For example, dog-divergent CTCF sites (Mmus−/Cfam+) exhibited local contact insulation specifically in the dog genome, whereas these same sites exhibited background levels of contact insulation when examined in the mouse Hi-C data (Figures 3C and S5). Importantly, the change in insulation following CTCF binding site evolution was stronger at the local (20-kb) scale, but was not significant at the higher (80-kb) scale (Figure 3D), suggesting that large-scale domain changes either are not affected by CTCF evolution or are under strong negative selection and are therefore not observed. These observations were further strengthened when we examined CTCF binding sites that were “partially” conserved. CTCF sites that were bound in mouse and dog, but lost in macaque, were associated with reduced contact insulation in the macaque genome. Thus, the data demonstrate a relationship between CTCF binding divergence and divergence of local insulation structure and therefore point to a role for CTCF in driving structural change in the genome.
The continuous evolutionary dynamics of intra-domain looping can play a key role in tuning promoter-enhancer contacts within domains. Consistent with this, we observe long-range contacts between divergent CTCF sites and enhancers or transcription start sites (TSSs) (Figure S6). Furthermore, analysis of transcription data from mouse, dog, and macaque liver reveals that divergent CTCF sites are contacting differentially expressed genes with a greater frequency than non-differentially expressed genes (Kolmogorov-Smirnov test, p < 0.05) (Figure S6). Together, these data support the hypothesis that emergence of divergent CTCF binding sites can contribute to changes in gene expression.
Conserved CTCF Sites Are Directional and Interact with Other Conserved Sites
While it is known that CTCF binding specificity greatly depends on its specific DNA consensus sequence, the specificity and directionality of CTCF/cohesin long-range contacts (Sofueva et al., 2013) and the way by which specific sites are assembled to define topological domains are not fully understood. As our data indicated that conserved CTCF binding sites have conserved motif affinities (Figure 1C), and because it is known that the CTCF consensus motif is non-symmetric, we asked whether conserved sites could also be conserved for the orientation of the CTCF motif. Indeed, 94% (3,265/3,483) of CTCF binding sites that are conserved between mouse and dog are also conserved in their orientation. To explore this further, we profiled contact insulation around conserved CTCF binding sites grouped according to the strand that the consensus motif was found on. We observed an asymmetric insulation behavior that was mirrored when the orientation of the motif was reversed (Figure 4A). This analysis uncoupled “insulation” (blue) from “preferential contacts” (red) and revealed that preferential contacts are made on one side of the oriented CTCF binding site, indicating that the orientation of the motif likely contributes to directionality of CTCFs long-range interactions. Consistent with this, we profiled the genome-wide relative position within chromosomal domains of Mmus+/Cfam+ conserved CTCF sites grouped according to the orientation of their binding motif as above. We observed that the conserved CTCF binding sites that are enriched at the edges of conserved domains (Figure 1E) have a specific orientation of their motif relative to chromosomal domains (Figure 4B). These observations were replicated when we compared mouse and macaque.
Figure 4.

Conserved CTCF Sites Engage in Strong, Directional Interactions with Other Conserved Sites
(A) Average contact insulation analysis in both Mmus and Cfam genomes for conserved (Mmus+/Cfam+) CTCF binding sites grouped according to the orientation of their binding motif. The consensus motif shown was generated from mouse binding sites.
(B) Genome-wide relative position within chromosomal domains of Mmus+/Cfam+ conserved CTCF sites grouped according to the orientation of their binding motif as above.
(C and D) (C) 4C-seq analysis and Hi-C maps of a 1.2-Mb syntenic region in the mouse and (D) dog genomes. Shown are 4C-seq viewpoints designed to four conserved (Mmus+/Cfam+) CTCF binding sites proximal to Hi-C domain borders and one 4C-seq viewpoint located at a mouse-divergent (Mmus+/Cfam−) CTCF site. The symbol above each 4C-seq bait indicates the strand (and orientation) of each viewpoint. Each 4C-seq experiment is represented by the median normalized 4C-seq coverage in a sliding window of 5 kb (top) and a multi-scale domainogram indicating normalized mean coverage in windows ranging between 2 kb and 50 kb.
(E) Relative intra-domain contact enrichment between CTCF sites (solid lines) as a function of distance when the two sites are <5 kb away from a CTCF binding site (solid lines) or where only one site is <5 kb away from a CTCF site (dotted lines). Shown are the relative contact enrichments within repressive domains for conserved (Mmus+/Cfam+, blue), mouse-divergent (Mmus+/Cfam−, gray) and dog-divergent (Mmus−/Cfam+, green) CTCF sites in mouse Hi-C (left) and dog Hi-C (right).
To characterize the contact relationship between evolutionarily stable or flexible CTCF sites and to further understand how they contribute to the evolution of chromosome domain structure, we performed a high-resolution high-throughput circular chromosome conformation capture (4C-seq) study. We designed four 4C-seq viewpoints to a series of neighboring conserved CTCF binding sites bordering conserved domains in the mouse and dog as well as to a mouse-specific site. The results showed that each conserved CTCF site engages in very strong and directional interactions with neighboring conserved CTCF sites (Figure 4C). Remarkably, the specific interactions mediated by conserved sites in the mouse genome were themselves precisely conserved in the dog genome (Figure 4D) and define the underlying domain structure. In each case, the long-range interaction was anchored by a pair of conserved CTCF sites whereby one CTCF site had an orientation on the “+” strand and the other on the “−” strand and could provide the basis for the observed directionality of CTCF-mediated interactions. Moreover, a viewpoint designed to a mouse-divergent site exhibited weak interactions within the mouse domain, analogous to the local insulation behavior observed in Figure 3B (Figure 4C). Importantly, the mouse-divergent viewpoint had no prominent interactions in the dog genome, confirming the specificity of its interaction network.
Global analysis of Hi-C contacts between pairs of CTCF binding sites stratified according to genomic distances in cis (Sofueva et al., 2013) confirmed the 4C-seq observation systematically (Figure 4E). Consistent with the high-resolution 4C-seq profiles, Hi-C trends showed that conserved CTCF sites strongly contacted one another within the same domain. Divergent CTCF sites engaged in significantly weaker contacts with other divergent sites, even when stratifying thoroughly for genomic distances. Importantly, little to no contact was observed in the mouse genome between dog-divergent sites. These results show that evolutionarily stable CTCF sites are engaged in strong contacts with one another and suggest that in so doing, they create an interaction network that may support the conservation of domain structure. On the other hand, divergent CTCF sites are involved in weaker interactions, perhaps reflecting the evolutionary flexibility of the binding sites themselves.
Domains Maintain Their Integrity during Chromosomal Rearrangements
Our data suggested that large-scale domain re-organization does not typically occur following insulator divergence. How then can it still be observed? Our interspecies comparative Hi-C data allowed us to ask what happens to the integrity of conserved chromosomal domains when genomes are challenged by structural rearrangements. If chromosomal domains act as modular units (e.g., to regulate gene expression), then large-scale rearrangements would be expected to occur at domain borders, so as to maintain the integrity of these structures. We scanned the mouse and dog genomes for differences in the distance between contiguous orthologous genes in the two species. Our analysis uncovered a number of complex rearrangements between the mouse and dog genomes involving insertions, inversions, and duplications. In each case, we discovered that the rearrangement occurred at the border between two chromosomal domains. This is exemplified in the Hi-C map from chromosome 15 in dog (Figure 5). Here, we found two domains, one containing the Slc5a9 gene and the other containing the Trabd2b gene (highlighted by red dots). Comparison of this region to the mouse genome revealed that a 2-Mb insertion occurred in the mouse genome that contains the Skint gene cluster, which is rapidly evolving and unique to the mouse lineage (Boyden et al., 2008). Remarkably, the insertion occurred directly between two neighboring dog domains in such a way as to perfectly maintain their integrity. A similar rearrangement event occurred at the Mrgpr gene cluster in the mouse genome (Dong et al., 2001), again preserving the structure of the neighboring domains (Figure S7). In another example, we observed a large-scale 5.5-Mb insertion in the dog genome containing multiple domains, and again, the domains on either side of the insertion have been maintained intact (Figure S8). These examples suggest that domains function as modular units and are selected against breakage during genome rearrangements.
Figure 5.

Chromosomal Domains Evolve as Modular Units
(A) A schematic of the rearrangement between mouse (gray) and dog (green).
(B) Hi-C maps from a syntenic region in mouse (left) and dog (right). Shown also are CTCF ChIP tracks, conserved and divergent sites, and genes in the region. A cluster of non-orthologous Skint genes (black dots) has been inserted in mouse between the orthologous genes Slc5a9 and Trabd2b (highlighted as red dots). The inserted region (bordered in yellow) forms its own nested chromosomal domain structure, probably as a result of gene-duplication events. Highlighted in blue are other orthologous genes in the region.
See also Figures S7 and S8.
Discussion
In this study, we examined Hi-C contact maps and CTCF binding profiles from four mammalian species to understand the relationship between the evolution of CTCF binding sites and chromosome structures. Our data reveal that CTCF binding sites have evolved under two regimes, whereby some CTCF elements are constrained both at the level of DNA sequence as well as in their binding while other CTCF elements exhibit significantly more flexibility. While both groups can mediate contact insulation, conserved CTCF elements are enriched at large-scale domain borders that tend to be themselves conserved. Meanwhile, evolutionarily flexible CTCF sites tend to be located internal of large-scale domains and mediate local structural change uniquely in that lineage. Our data thereby point to a strong correlation between the evolution of CTCF binding and chromosomal structure and extend on our current understanding of context-dependent CTCF binding sites and their specific roles in chromosomal domain architecture (Dixon et al., 2012; Dowen et al., 2014). Importantly, since CTCF binding information is encoded in high specificity cis elements, the intra-domain insulator dynamics we observe directly link local sequence evolution with chromosomal architectures. This direct linkage has strong implications for the study of CTCF and genome function and for our understanding of the evolutionary dynamics in complex genomes.
A central causal role for CTCF/cohesin in establishing domain structure is widely hypothesized, but direct experimental evidence has proven difficult to attain. Previous studies have observed a correlation between insulator binding and domain borders (Sexton et al., 2012; Dixon et al., 2012; Nora et al., 2012; Hou et al., 2012), and knockout experiments have suggested a quantitative link between loss of chromosomal looping structure and loss of the CTCF/cohesin binding landscapes (Sofueva et al., 2013; Zuin et al., 2014; Seitan et al., 2013). Given the pervasive impact of CTCF/cohesin on nuclear organization and gene regulation, it is difficult to identify the mechanisms of their action through classic genetic perturbation. Instead, the evolutionary comparison used here offers us thousands of naturally occurring genomic perturbations that can be identified and characterized at both the sequence and chromosomal topology levels. This strategy has yielded strong evidence of a direct link between the gain/loss of CTCF binding sites and a corresponding gain/loss of local domain insulation. Our comparative Hi-C analysis therefore strongly supports the idea that CTCF is causally connected to chromosomal looping structures.
The comparative chromosomal domain analysis described here has revealed a spectrum of evolutionary consequences, ranging from the conservation of essential large-scale chromosomal domains to the flexibility of continuous genomic adaptation. CTCF and cohesin complexes are deeply evolutionarily conserved, and the data here show that their role in mediating chromosome topologies and, even more remarkably, the large-scale building blocks of such topologies are also highly conserved. Our data suggest that the orientation of the CTCF motif may underlie the observed directionality of CTCF/cohesin-mediated long-range contacts and provide a rationale by which specific sites are assembled to define topological domains. Given that CTCF binding is strongly influenced by its consensus sequence, our data suggest that the assembly of domain structure is “hardwired” in the genome. This also has implications for further understanding the nature of the relationship between CTCF and cohesin, since biochemical studies have revealed that cohesin subunits interact with CTCF primarily through its C-terminal tail (Xiao et al., 2011), placing cohesin on a particular side of the chromosomal domain.
Interestingly, while we were able to observe cases whereby local sequence evolution perturbed CTCF binding and disrupted chromosomal looping, the structures that were affected due to this insulator divergence were primarily local loops. Cases of large-scale topological domains that were split or fused due to insulator divergence were not observed. We hypothesize that this stability is achieved by a combination of both local purifying selection on key CTCF binding sites and by buffering of major topological loops by additional factors. Strikingly, the cases of large-scale domain divergence that we were able to characterize were all linked with evolutionary genome rearrangements and revealed a mechanism that can reshuffle whole domains such that the rearranged chromosomal modules are aligned with existing domain borders. It is still, however, formally possible that rearrangements take place between CTCF sites that are mediating strong interactions.
In addition to the importance of topological domain and insulator conservation described here, the evolutionary dynamics that couple intra-domain CTCF divergence with changes in the local domain structure emerge as potentially fundamental for genome regulation. Loops contained within domains link enhancers (and their bound trans-factors) to target gene promoters. While it is still unclear how such targeting is regulated and how evolution can manipulate it, based on our data, we hypothesize that flexible CTCF binding sites within domains can influence looping from the promoter or enhancer as well by demarcating the implicated functional elements. As CTCF sites are sufficiently sequence-specific to be directly tunable by local nucleotide substitutions, it is intriguing to speculate that the intra-domain looping structure is a key and evolvable feature affecting gene regulation. Such a trait, if indeed quantitatively important, should be further studied between and within populations and species.
Experimental Procedures
Liver Homogenization and Fixation
Fresh or frozen liver from mouse, rabbit, macaque, and dog were processed for Hi-C or 4C-seq libraries. With the exception of mouse, the samples used for the Hi-C libraries were the same as the material used for CTCF ChIP-seq (Schmidt et al., 2012). Livers were fixed in 10% formalin for 20 min, and ∼1 g was cut and processed with a Dounce homogenizer (ten strokes with a loose pestle followed by ten strokes with the tight pestle). After filtration through a 70-μm nylon cell strainer, the sample was washed twice with PBS, spinning down at 852 rcf for 5 min at 4°C to collect the cells between washes. 1–5 × 107 liver cells were then fixed for a second time in fixation buffer (1% formaldehyde, 750 μg/ml BSA in DMEM/Ham’s F12 [Invitrogen]) for 10–30 min at room temperature. The fixation reaction was quenched using 0.125 M glycine for 5 min at room temperature. Samples were washed twice with 10 ml PBS, pelleted into 1 × 107 cells aliquots, and stored at −80°C. Mouse Hi-C libraries were prepared from fresh liver samples of biological replicates (9-week-old C57/BL6 mouse and the pooled livers from 2- to 4-week-old outbred mice. The libraries for the other three organisms were technical replicates.
Propidium Iodide Staining of Hepatocytes
Formaldehyde-fixed liver cells were lysed on ice in a hypotonic buffer (10 mM Tris-HCl [pH 8], 10 mM NaCl, 0.2% Igepal CA-640, EDTA-free protease inhibitors) for 30 min. Nuclei were stained with a propidium iodide (PI) staining buffer (100 μg/ml PI, 50 μg/ml RNase A, 0.05% Triton X-100) for 60 min on ice. Samples were analyzed on a MoFlo cell sorter (Beckman Coulter).
High-Throughput Mapping of Chromatin Interactions via Hi-C
The Hi-C method previously used (Sofueva et al., 2013) was modified to accommodate primary liver samples. Hepatocytes were lysed in Hi-C lysis buffer (10 mM Tris-HCl [pH 8], 10 mM NaCl, 0.2% Igepal CA-640, EDTA-free protease inhibitors) for 30 min on ice. The sample was transferred to Protein LoBind tubes (Eppendorf) and the nuclei were permeabilized by incubation with 0.1%–0.6% SDS for 1 hr at 37°C with 800 rpm shaking. The reaction was quenched with 0.67%–4% Triton X-100, 1 hr at 37°C, 800 rpm shaking. Nuclei were digested in 500 μl 1X NEBuffer 2 with 1500 U HindIII (New England Biolabs) and monitored for maximal digestion of the chromatin template, thus digestion times ranged from 24-72 hr. All other parts of the Hi-C protocol, including library preparation were performed as previously described. 75 bp paired-end sequencing was performed for each library according to manufacturers conditions using the Illumina Hi-seq platform.
Hi-C Interaction Matrix Generation and Domain Calling
Sequencing reads were aligned to the mouse (mm10), rabbit (oryCun2), macaque (rheMac2), and dog (canFam3) genome assemblies using Bowtie 0.12.8 (Langmead et al., 2009). The parameters used for the alignment allowed a maximum of three mismatches and strictly one alignment per read. Processing of the aligned reads and normalization of the interaction matrices were performed as previously described (Yaffe and Tanay, 2011; Sofueva et al., 2013). The pipeline produced normalized matrices of interactions binning the genome at different resolutions. Interaction matrices for each library were generated displaying seven different resolutions simultaneously (12,500, 25,000, 50,000, 100,000, 250,000, 500,000, and 1,000,000 bp). Domains were identified and clustered as described (Sexton et al., 2012) with the modification that scaling factors were inferred using fends 100–400 kb apart, to account for the lower resolution of the mouse map compared to the Drosophila map. Domain borders were called using the 95% percentile of the scaling track. A domain-level map was partitioned into two clusters, and clusters were assigned as passive/active according to Lamin B mouse embryonic fibroblast (MEF) data, as before. For the rabbit, macaque, and dog genome, the Lamin B MEF track for mouse was lifted over to the corresponding genome to label domain clusters. Domain calls in mouse and dog are available in Table S1.
ChIP-Seq Analysis
We used previously published ChIP-seq data for CTCF from mouse, macaque, and dog livers (Schmidt et al., 2012) and for Rad21 for mouse liver (Faure et al., 2012). Rad21 ChIP-seq data for macaque and dog was prepared as for CTCF. Mouse, macaque, and dog ChIP-seq reads were mapped using bowtie. Alignment was followed by extension of sequenced tags to 300-bp fragments and pileup into 50-bp bins. We normalized ChIP-seq coverage by computing the distribution of pile-up coverage on 50-bp bins and transforming each coverage value v into −log10 (1-quantile(v)). To define binding sites, we used a simple threshold on the sum of values from two biological replicates for each CTCF dataset and for the macaque Rad21 data. Rad21 ChIP data from mouse and dog were done in single, and the data were thresholded. Thresholds used were as follows: mouse CTCF = 2.2, macaque CTCF = 2.4, dog CTCF = 2.2, mouse Rad21 = 2.3, macaque Rad21 = 2.5, dog Rad21 = 3. Different thresholds did not change the results. Binding site width was standardized at 200 bp, and the ChIP-seq intensity for each site was calculated as the maximum value across the 200 bp. The relative distribution of CTCF within topological domains (Figures 1E and S5) was calculated as the distance of each CTCF site from the center of its domain. Half the size of the domain was added to convert it to a measure of distance from the edge of the domain, and this number was then divided by the size of the domain.
Interspecies Comparison of CTCF Sites
Macaque and dog CTCF ChIP-seq libraries were converted to mouse genome coordinates using the liftOver tool from UCSC. To reduce the chance of inaccurate liftOver, a number of filters were implemented: sites within low-mappability regions, repeats, or windows of 100 kb with insufficient synteny were excluded. To estimate mappability, each genome was broken into 50-bp reads and the whole-genome sequence was split into artificial reads and then mapped back to the genome. For each 50-bp bin, the mappability score was then defined to be the portion of artificial reads mapped uniquely to that bin. To estimate the level of synteny in the 100 kb around a CTCF site, the mappability tracks for macaque and dog were converted to the mouse genome using liftOver and all bins for which liftOver was not possible were converted to zeroes. The converted tracks were subsequently smoothed over 100 kb, and CTCF sites falling in regions below the top quartile of such smoothed tracks were excluded from all subsequent analysis. Divergent CTCF sites in mouse and dog are available in Table S1.
CTCF Binding Energy Function
A CTCF DNA-binding energy function from the Cortex CTCF binding sites (ENCODE Cortex CTCF mouse, GSM769019; Shen et al., 2012) was used to profile all genomes for their similarity to the CTCF consensus motif. The consensus motif is very highly conserved across all species (Schmidt et al., 2012). Given a set of genomic sites, we compute for each site the maximal energy value within a 200-bp window centered on the point.
Motif Orientation Analysis
Orientation of the motifs underneath conserved CTCF peaks was obtained using MEME (http://meme.nbcr.net/meme/), (Bailey and Elkan, 1994) with the parameters -revcomp -dna -nmotifs 1 -w 20 -mod zoops -maxsize 100,000.
Crossover Analysis
Crossover analysis was performed as described previously (Sofueva et al., 2013). The bands used were 5–7.5, 7.5–11.25, 10–15, 15–22.5, 20–30, 30–45, 40–60, 60–90, and 80–120 kb.
Distal Contact Analysis
To calculate the average interaction profiles for a group of genomic landmarks, HindIII fragment ends were grouped into classes by associating each end with a genomic element located within 5 kb and then grouping all fragment ends associated with an element of the same class. For the mouse, macaque, and dog genomes, three classes of CTCF sites (conserved, divergent present, divergent absent) and TSS sites were defined. These classes were further divided to sites within active or passive Hi-C domains. The remaining fragment end (not classified given other landmarks) was defined as the background.
4C-Seq
Preparation of 4C-seq samples, libraries, sequencing analysis, and normalization were all performed as previously described (Sofueva et al., 2013). Primer sequences were chosen to viewpoint sites that were as close as possible to CTCF ChIP-seq peaks (Table 1). Mouse primers were designed according to the genome-wide 4C-seq primer database from (van de Werken et al., 2012). For dog primers, a similar database was generated for the regions of interest.
Table 1.
4C-Seq Primers Used in This Study
| Viewpoint | CTCF Peak | Reading Primer | Non-reading Primer |
|---|---|---|---|
| Mouse 4C-seq primers (mm10) | |||
| Mmus+ Cfam+ 1 (Figure 4) | chr10:94609250 | 5′-CCATCTGTTTGAACAAGATC-3′ | 5′-CAAGAGAGAGTGGAAACAGG-3′ |
| Mmus+ Cfam+ 2 (Figure 4) | chr10:94623583 | 5′-AGTCAGATGGAATGCAGATC-3′ | 5′-CTAGATACAGCAATCAGCCC-3′ |
| Mmus+ Cfam+ 3 (Figure 4) | chr10:94958324 | 5′-ATTGCTTTCTCTGGTTGATC-3′ | 5′-AGTCACTCCTGCTCCTGTAA-3′ |
| Mmus+ Cfam+ 4 (Figure 4) | chr10:94991353 | 5′-GTTTCTGTTGGTTCACGATC-3′ | 5′-AAGCATTGTCCTACGTGATT-3′ |
| Mmus+ Cfam− (Figure 4) | chr10:95218005 | 5′-CTACTCTGGCTTCTATGATC-3′ | 5′-CCCTTCCCTTCTATGTTTCT-3′ |
| Dog 4C-seq primers (canFam3) | |||
| Mmus+ Cfam+ 1 (Figure 4) | chr15:34606369 | 5′-GCTCTTGCTCTAAACTGATC-3′ | 5′-TGGACCTCACCTCTCCTA-3′ |
| Mmus+ Cfam+ 2 (Figure 4) | chr15:34596229 | 5′-TGAGGTCCAGCAGAGATC-3′ | 5′-GTCGCATCACTTACTGGG-3′ |
| Mmus+ Cfam+ 3 (Figure 4) | chr15:34269944 | 5′-CTCCACTGAGCATTAAGATC-3′ | 5′-GCGGGATAGTTCTTTTCTCT-3′ |
| Mmus+ Cfam+ 4 (Figure 4) | chr15:34244336 | 5′-CTTATGTGCTCCTCCAGATC-3′ | 5′-AATCATATGCCTCCTCCTCT-3′ |
| Mmus+ Cfam− (Figure 4) | chr15:33989305 | 5′-AAAGTAATCCCACCCAGATC-3′ | 5′-CTGAAGGAAACAACAATGTCA-3′ |
Author Contributions
M.V.R., D.T.O., and S.H. initiated the project. M.V.R. performed the Hi-C and 4C-Seq experiments, including library preparations. D.T.O. and C.E. provided the liver samples for all species (except mouse) and sequenced the Hi-C libraries. M.V.R., C.B., and A.T. processed and statistically analyzed the data. M.V.R., A.T., and S.H. wrote the manuscript, with contributions from all authors.
Acknowledgments
The authors wish to thank Sevil Sofueva for help with Hi-C and 4C-seq library preparations; Wen-Ching Chan for analysis; Christine Feig for Rad21 ChIP-seq data in macaque and dog; Bianca Schmidt and the Cancer Research UK CI Genomic Core facility for technical assistance with Hi-C sequencing and Pedro Olivares for advice on analysis and data manipulation. We would also like to acknowledge all members of the Hadjur group for discussions. This work was supported by the Medical Research Council UK (G0900491/1 and G1001649) (S.H.), the EPIGENESYS EU NoE (S.H. and A.T.), and Cancer Research UK (studentship to M.V.R.). D.T.O. is supported by Cancer Research UK.
Published: February 26, 2015
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/3.0/).
Supplemental Information includes eight figures and one table and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2015.02.004.
Accession Numbers
The data analyzed in this study have been deposited in the GEO database with the accession number GSE65126.
Supplemental Information
References
- Bailey T.L., Elkan C. Fitting a mixture model by expectation maximization to discover motifs in bipolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1994;2:28–36. [PubMed] [Google Scholar]
- Bell A.C., West A.G., Felsenfeld G. The protein CTCF is required for the enhancer blocking activity of vertebrate insulators. Cell. 1999;98:387–396. doi: 10.1016/s0092-8674(00)81967-4. [DOI] [PubMed] [Google Scholar]
- Birney E., Stamatoyannopoulos J.A., Dutta A., Guigó R., Gingeras T.R., Margulies E.H., Weng Z., Snyder M., Dermitzakis E.T., Stamatoyannopoulos J.A. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borneman A.R., Gianoulis T.A., Zhang Z.D., Yu H., Rozowsky J., Seringhaus M.R., Wang L.Y., Gerstein M., Snyder M. Divergence of transcription factor binding sites across related yeast species. Science. 2007;317:815–819. doi: 10.1126/science.1140748. [DOI] [PubMed] [Google Scholar]
- Boyden L.M., Lewis J.M., Barbee S.D., Bas A., Girardi M., Hayday A.C., Tigelaar R.E., Lifton R.P. Skint1, the prototype of a newly identified immunoglobulin superfamily gene cluster, positively selects epidermal gammadelta T cells. Nat. Genet. 2008;40:656–662. doi: 10.1038/ng.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dermitzakis E.T., Clark A.G. Differential selection after duplication in mammalian developmental genes. Mol. Biol. Evol. 2001;18:557–562. doi: 10.1093/oxfordjournals.molbev.a003835. [DOI] [PubMed] [Google Scholar]
- Dixon J.R., Selvaraj S., Yue F., Kim A., Li Y., Shen Y., Hu M., Liu J.S., Ren B. Topological domains in Mamm. Genomes identified by analysis of chromatin interactions. Nature. 2012;485:376–380. doi: 10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong X., Han S., Zylka M.J., Simon M.I., Anderson D.J. A diverse family of GPCRs expressed in specific subsets of nociceptive sensory neurons. Cell. 2001;106:619–632. doi: 10.1016/s0092-8674(01)00483-4. [DOI] [PubMed] [Google Scholar]
- Dowen J.M., Fan Z.P., Hnisz D., Ren G., Abraham B.J., Zhang L.N., Weintraub A.S., Schuijers J., Lee T.I., Zhao K., Young R.A. Control of cell identity genes occurs in insulated neighborhoods in mammalian chromosomes. Cell. 2014;159:374–387. doi: 10.1016/j.cell.2014.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faure A.J., Schmidt D., Watt S., Schwalie P.C., Wilson M.D., Xu H., Ramsay R.G., Odom D.T., Flicek P. Cohesin regulates tissue-specific expression by stabilizing highly occupied cis-regulatory modules. Genome Res. 2012;22:2163–2175. doi: 10.1101/gr.136507.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippova G.N., Fagerlie S., Klenova E.M., Myers C., Dehner Y., Goodwin G., Neiman P.E., Collins S.J., Lobanenkov V.V. An exceptionally conserved transcriptional repressor, CTCF, employs different combinations of zinc fingers to bind diverged promoter sequences of avian and mammalian c-myc oncogenes. Mol. Cell. Biol. 1996;16:2802–2813. doi: 10.1128/mcb.16.6.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guacci V., Koshland D., Strunnikov A. A direct link between sister chromatid cohesion and chromosome condensation revealed through the analysis of MCD1 in S. cerevisiae. Cell. 1997;91:47–57. doi: 10.1016/s0092-8674(01)80008-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadjur S., Williams L.M., Ryan N.K., Cobb B.S., Sexton T., Fraser P., Fisher A.G., Merkenschlager M. Cohesins form chromosomal cis-interactions at the developmentally regulated IFNG locus. Nature. 2009;460:410–413. doi: 10.1038/nature08079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou C., Li L., Qin Z.S., Corces V.G. Gene density, transcription, and insulators contribute to the partition of the Drosophila genome into physical domains. Mol. Cell. 2012;48:471–484. doi: 10.1016/j.molcel.2012.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T.H., Abdullaev Z.K., Smith A.D., Ching K.A., Loukinov D.I., Green R.D., Zhang M.Q., Lobanenkov V.V., Ren B. Analysis of the vertebrate insulator protein CTCF-binding sites in the human genome. Cell. 2007;128:1231–1245. doi: 10.1016/j.cell.2006.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., Trapnell C., Pop M., Salzberg S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelis C., Ciosk R., Nasmyth K. Cohesins: chromosomal proteins that prevent premature separation of sister chromatids. Cell. 1997;91:35–45. doi: 10.1016/s0092-8674(01)80007-6. [DOI] [PubMed] [Google Scholar]
- Mishiro T., Ishihara K., Hino S., Tsutsumi S., Aburatani H., Shirahige K., Kinoshita Y., Nakao M. Architectural roles of multiple chromatin insulators at the human apolipoprotein gene cluster. EMBO J. 2009;28:1234–1245. doi: 10.1038/emboj.2009.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nativio R., Wendt K.S., Ito Y., Huddleston J.E., Uribe-Lewis S., Woodfine K., Krueger C., Reik W., Peters J.-M., Murrell A. Cohesin is required for higher-order chromatin conformation at the imprinted IGF2-H19 locus. PLoS Genet. 2009;5:e1000739. doi: 10.1371/journal.pgen.1000739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nora E.P., Lajoie B.R., Schulz E.G., Giorgetti L., Okamoto I., Servant N., Piolot T., van Berkum N.L., Meisig J., Sedat J. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature. 2012;485:381–385. doi: 10.1038/nature11049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parelho V., Hadjur S., Spivakov M., Leleu M., Sauer S., Gregson H.C., Jarmuz A., Canzonetta C., Webster Z., Nesterova T. Cohesins functionally associate with CTCF on mammalian chromosome arms. Cell. 2008;132:422–433. doi: 10.1016/j.cell.2008.01.011. [DOI] [PubMed] [Google Scholar]
- Pauli A., Althoff F., Oliveira R.A., Heidmann S., Schuldiner O., Lehner C.F., Dickson B.J., Nasmyth K. Cell-type-specific TEV protease cleavage reveals cohesin functions in Drosophila neurons. Dev. Cell. 2008;14:239–251. doi: 10.1016/j.devcel.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips-Cremins J.E., Sauria M.E.G., Sanyal A., Gerasimova T.I., Lajoie B.R., Bell J.S.K., Ong C.-T., Hookway T.A., Guo C., Sun Y. Architectural protein subclasses shape 3D organization of genomes during lineage commitment. Cell. 2013;153:1281–1295. doi: 10.1016/j.cell.2013.04.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rollins R.A., Morcillo P., Dorsett D. Nipped-B, a Drosophila homologue of chromosomal adherins, participates in activation by remote enhancers in the cut and Ultrabithorax genes. Genetics. 1999;152:577–593. doi: 10.1093/genetics/152.2.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt D., Wilson M.D., Ballester B., Schwalie P.C., Brown G.D., Marshall A., Kutter C., Watt S., Martinez-Jimenez C.P., Mackay S. Five-vertebrate ChIP-seq reveals the evolutionary dynamics of transcription factor binding. Science. 2010;328:1036–1040. doi: 10.1126/science.1186176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt D., Schwalie P.C., Wilson M.D., Ballester B., Gonçalves A., Kutter C., Brown G.D., Marshall A., Flicek P., Odom D.T. Waves of retrotransposon expansion remodel genome organization and CTCF binding in multiple mammalian lineages. Cell. 2012;148:335–348. doi: 10.1016/j.cell.2011.11.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seitan V.C., Hao B., Tachibana-Konwalski K., Lavagnolli T., Mira-Bontenbal H., Brown K.E., Teng G., Carroll T., Terry A., Horan K. A role for cohesin in T-cell-receptor rearrangement and thymocyte differentiation. Nature. 2011;476:467–471. doi: 10.1038/nature10312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seitan V.C., Faure A.J., Zhan Y., McCord R.P., Lajoie B.R., Ing-Simmons E., Lenhard B., Giorgetti L., Heard E., Fisher A.G. Cohesin-based chromatin interactions enable regulated gene expression within preexisting architectural compartments. Genome Res. 2013;23:2066–2077. doi: 10.1101/gr.161620.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sexton T., Yaffe E., Kenigsberg E., Bantignies F., Leblanc B., Hoichman M., Parrinello H., Tanay A., Cavalli G. Three-dimensional folding and functional organization principles of the Drosophila genome. Cell. 2012;148:458–472. doi: 10.1016/j.cell.2012.01.010. [DOI] [PubMed] [Google Scholar]
- Shen Y., Yue F., McCleary D.F., Ye Z., Edsall L., Kuan S., Wagner U., Dixon J., Lee L., Lobanenkov V.V., Ren B. A map of the cis-regulatory sequences in the mouse genome. Nature. 2012;488:116–120. doi: 10.1038/nature11243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofueva S., Yaffe E., Chan W.-C., Georgopoulou D., Vietri Rudan M., Mira-Bontenbal H., Pollard S.M., Schroth G.P., Tanay A., Hadjur S. Cohesin-mediated interactions organize chromosomal domain architecture. EMBO J. 2013;32:3119–3129. doi: 10.1038/emboj.2013.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Werken H.J.G., Landan G., Holwerda S.J.B., Hoichman M., Klous P., Chachik R., Splinter E., Valdes-Quezada C., Öz Y., Bouwman B.A.M. Robust 4C-seq data analysis to screen for regulatory DNA interactions. Nat. Methods. 2012;9:969–972. doi: 10.1038/nmeth.2173. [DOI] [PubMed] [Google Scholar]
- Wendt K.S., Yoshida K., Itoh T., Bando M., Koch B., Schirghuber E., Tsutsumi S., Nagae G., Ishihara K., Mishiro T. Cohesin mediates transcriptional insulation by CCCTC-binding factor. Nature. 2008;451:796–801. doi: 10.1038/nature06634. [DOI] [PubMed] [Google Scholar]
- Xiao T., Wallace J., Felsenfeld G. Specific sites in the C terminus of CTCF interact with the SA2 subunit of the cohesin complex and are required for cohesin-dependent insulation activity. Mol. Cell. Biol. 2011;31:2174–2183. doi: 10.1128/MCB.05093-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaffe E., Tanay A. Probabilistic modeling of Hi-C contact maps eliminates systematic biases to characterize global chromosomal architecture. Nat. Genet. 2011;43:1059–1065. doi: 10.1038/ng.947. [DOI] [PubMed] [Google Scholar]
- Yaffe E., Farkash-Amar S., Polten A., Yakhini Z., Tanay A., Simon I. Comparative analysis of DNA replication timing reveals conserved large-scale chromosomal architecture. PLoS Genet. 2010;6:e1001011. doi: 10.1371/journal.pgen.1001011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuin J., Dixon J.R., van der Reijden M.I.J.A., Ye Z., Kolovos P., Brouwer R.W.W., van de Corput M.P.C., van de Werken H.J.G., Knoch T.A., van IJcken W.F.J. Cohesin and CTCF differentially affect chromatin architecture and gene expression in human cells. Proc. Natl. Acad. Sci. USA. 2014;111:996–1001. doi: 10.1073/pnas.1317788111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.