

Abstract
Background
The otic placode comprises the progenitors of the inner ear and the neurons that convey hearing and balance information to the brain. Transplantation studies in birds and amphibians demonstrate that when the otic placode is morphologically visible as a thickened patch of ectoderm, it is first committed to an otic fate. Fibroblast Growth Factor (FGF) signaling initiates induction of the otic placode, and levels of FGF signaling are fine-tuned by the Sprouty family of antagonists of receptor tyrosine kinase signaling.
Results
Here, we examined the size of the otic placode and cup by combinatorial inactivation of the Sprouty1 and Sprouty2 genes. Interestingly, in a Sprouty gene dosage series, early enlargement of the otic placode was progressively restored to normal. Restoration of otic size was preceded by normal levels of FGF signaling, reduced cell proliferation and reduced cell death.
Conclusions
Our study demonstrates that excess otic placode cells, which form in response to increased FGF signaling, are not maintained in mammals. This suggests that growth plasticity exists in the mammalian otic placode and cup, and that FGF signaling may not be sufficient to induce the genetic program that maintains otic fate.
Keywords: inner ear, otic placode, Sprouty1, Sprouty2, gene dosage, FGF, induction, cell proliferation, cell death
INTRODUCTION
The otic placode is made up of the progenitors of the membraneous labyrinth of the inner ear and neurons of the cochleovestibular ganglion. Acquisition of otic placode fate appears to occur in steps (for reviews see Ohyama et al., 2007; Ladher et al., 2010; Schlosser, 2010; Groves and Fekete, 2012; Chen and Streit, 2013). The first step occurs at the end of gastrulation when a band of ectoderm, the pre-placodal region, forms adjacent to the developing neural plate and acquires the ability to develop into the cranial placodes. Next, a posterior domain of the pre-placodal region is induced to form the otic-epibranchial progenitor domain (OEPD, also called the posterior placodal area (PPA) or pre-otic field), which comprises the progenitors of the otic placode, epibranchial placodes and posterior cranial epidermis. Finally, morphologically distinct otic, epibranchial and epidermal cells segregate within the OEPD, so that two thickened otic placodes, one on each side of the hindbrain, can first be observed at 8 to 9 somite stages (s).
Transplantation studies in bird and amphibian embryos suggest that cells are first committed to an otic fate at the otic placode stage. Thus, when otic placodes from 9 s embryos are transplanted to a dissimilar tissue environment, such as the lateral trunk, amniocardiac vesicle or limb bud, they are capable of forming otic vesicles and of expressing some, but not all otic markers (Waddington, 1937; Swanson et al., 1990; Ginsburg, 1995; Giraldez, 1998; Herbrand et al., 1998; Groves and Bronner-Fraser, 2000). The ability of transplanted otic tissue to recapitulate aspects of otic vesicle morphogenesis, sensory cell differentiation, and otic marker gene expression is improved with transplantation of older otic tissue (Yntema, 1933; Ginsburg, 1995; Gallagher et al., 1996; Herbrand et al., 1998). These data suggest that although cells may begin commitment to an otic fate at the time of formation of the otic placode, continued signaling from surrounding tissues is required for normal patterning and morphogenesis of otic cells. When cells first become committed to an otic fate in placental mammals, such as mouse and human, is not known.
Studies in multiple organisms support the conclusion that Fibroblast Growth Factor (FGF) signaling initiates otic development by inducing formation of the OEPD from the pre-placodal region (for reviews see Ladher et al., 2010; Groves and Fekete, 2012; Chen and Streit, 2013). In all organisms studied, Fgfs are expressed in mesoderm, endoderm and hindbrain during OEPD induction (for review see Schimmang, 2007). In the mouse, Fgf8, expressed in endoderm, and Fgf3, expressed in hindbrain, maintain expression of Fgf10 in the adjacent mesoderm, thus controlling OEPD induction through an FGF-signaling relay (Ladher et al., 2005; Zelarayan et al., 2007). Loss-of-function studies in multiple organisms further support the conclusion that FGF signaling induces the OEPD. In the mouse, compound inactivation of Fgf3 and Fgf10 (Fgf3−/−; Fgf10−/− embryos) or Fgf3 and Fgf8 (Fgf3−/−; Fgf8hypomorph/− embryos) result in reduction or loss of the OEPD marker Pax2, and in smaller or absent otic vesicles (Alvarez et al., 2003; Wright and Mansour, 2003; Ladher et al., 2005; Zelarayan et al., 2007). In zebrafish, inactivation of fgf3 and fgf8 results in loss of pax2 and pax8 expression and absence of otic vesicle formation (Phillips et al., 2001; Leger and Brand, 2002; Maroon et al., 2002). In the chick, culture of either whole embryos or head explants, containing pre-otic ectoderm, with the FGF signaling antagonist, SU5402, prevents induction of otic markers, including Pax2 (Martin and Groves, 2006; Yang et al., 2013). Furthermore, knockdown of Fgf3 and Fgf19 abolishes Pax2 expression in the OEPD (Freter et al., 2008).
The Sprouty gene family, of which there are four members (Spry1 – 4) in mouse and human, encode intracellular antagonists of receptor tyrosine kinase signaling, including FGF signaling (for reviews see Kim and Bar-Sagi, 2004; Mason et al., 2006; Cabrita and Christofori, 2008; Guy et al., 2009). We have previously shown that compound inactivation of the Spry1 and Spry2 genes (Spry1−/−; Spry2−/− double mutant embryos) results in enlargement of the otic placode due to increased response of tissue to FGF signaling (Mahoney Rogers et al.). Enlargement of the otic placode was due to increased differentiation of otic cells rather than increased cell size, increased cell proliferation, decreased cell death, or enlargement of the entire embryo. Interestingly, the size of the OEPD was unaffected in Spry1−/−; Spry2−/− mutant embryos, suggesting that FGF signaling also regulates otic vs. non-otic cell fate decisions within the OEPD. A study in the chick also suggests that FGF signaling regulates otic cell fate after OEPD formation (Freter et al., 2008). However, rather than resulting in enlargement of the otic placode, sustained overexpression of Fgf3 and Fgf19 inhibited expression of committed otic marker genes, Soho-1 and Hmx3 (previously known as Nkx5.1). The cause of these different outcomes is unclear, but presumably reflects variant response of pre-otic cells to differences in timing, spatial distribution, and levels of FGF overexpression (Padanad et al., 2012).
Here, we analyzed embryos in which the Spry1 and Spry2 genes have been combinatorially inactivated to achieve a Spry gene dosage series. We observed an enlargement of the otic placode in multiple dosage combinations of these two genes. However, we found that enlargement was not maintained. This allowed us to define a window during which otic placode cells can regulate their size in the mouse and to explore cellular processes that contribute to size regulation.
RESULTS
Enlargement of the otic placode in a Sprouty gene dosage series
Over the course of analyzing Spry1−/−;Spry2−/− (or DKO embryos), we found the otic placode also appeared larger in Spry1−/−;Spry2−/+ (or Spry1Null2Het) and Spry1−/+;Spry2−/− (or Spry1Het2Null) littermate embryos at 8 – 11 s. Examination of whole mount expression patterns of Pax8 and Dlx5, two genes expressed in the otic placode, revealed that approximately 90% of Spry1Het2Null embryos had larger Pax8 and Dlx5 expression domains that were indistinguishable from DKO embryos (Fig. 1C, D, G, H, Table 1). In addition, in Spry1Null2Het embryos, 50% had expanded otic expression of Pax8 (Fig. 1B) and Dlx5 (Table 1), whereas the remaining 50% appeared indistinguishable from Spry1−/+;Spry2−/+ (or DHet) embryos (compare Fig. 1E and F). Otic expression of Pax8 and Dlx5 in DHet, Spry1−/− single mutant, and Spry2−/− single mutant embryos were identical to wild-type controls (Fig. 1A, E, data not shown), suggesting that loss of two functional copies of either Spry1 or Spry2 had no detectable effect on otic placode size.
Figure 1. Reduction of Spry1 and Spry2 gene dosage increases otic placode size.
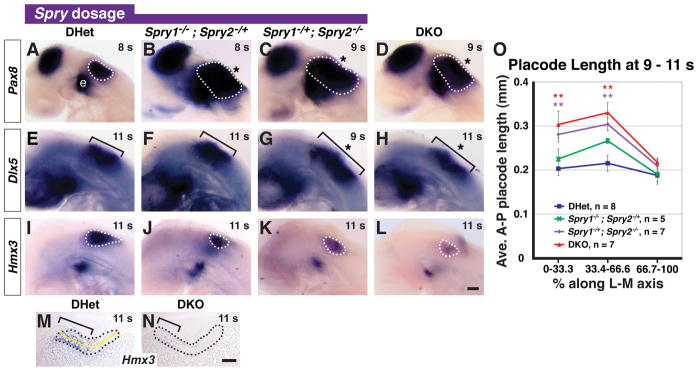
In situ hybridization analysis of Pax8, Dlx5, and Hmx3 in 8 – 11 s embryos with the genotypes indicated (A – L). Lateral views are shown with anterior to the left. Otic placode staining of Pax8 (A – D) and Hmx3 (I – L) is outlined (white dashed line) and of Dlx5 (E – H) is bracketed. Epibranchial placode (“e”) staining adjacent to the otic domain is indicated. Expansions of gene expression domains are highlighted (asterisk). (M, N) Mid-placodal, sagittal sections of 11 s DHet and DKO embryos stained for Hmx3 (brackets). Otic placode is outlined (black dashed line). (O) Average anterior to posterior (A-P) lengths of the otic placode in 9 – 11 s embryos with the genotypes indicated. Yellow line in (M) depicts where the A-P length of the otic placode was measured. **, p < 0.01 by ANOVA in comparisons between DHet and the genotype indicated by color coding; error bars represent standard error of sample means. Scale bars (A – L), 50 μm and (M, N), 100 μm.
Table 1.
Gene expression analysis in a Sprouty gene dosage series
| Percent with enlarged or reduced otic expression (no./total)
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| 8 – 11 s | 12 – 16 s | ||||||||
|
| |||||||||
| Enlarged | Reduced | Enlarged | |||||||
|
| |||||||||
| Dlx5 | Pax8 | Hmx3 | Dlx5 | Foxi2a | Pax8 | Hmx3 | Etv4 | Etv5 | |
| DHet | 25% (1/4) | 0% (0/11) | 17% (1/6) | 0% (0/7) | 0% (0/7) | 0% (0/6) | 0% (0/9) | 0% (0/8) | 14% (1/7) |
| Spry1−/−; Spry2−/+ | 50% (3/6) | 50% (3/6) | 33% (2/6) | 0% (0/7) | 0% (0/5) | 0% (0/6) | 14% (1/7) | n/db | n/db |
| Spry1−/+; Spry2−/− | 88% (7/8) | 89% (8/9) | 71% (5/7) | 83% (5/6) | 75% (3/4) | 60% (3/5) | 20% (1/5) | 67% (4/6) | 62% (5/8) |
| DKO | 100% (5/5) | 100% (8/8) | 83% (5/6) | 100% (7/7) | 100% (7/7) | 83% (5/6) | 90% (9/10) | 100% (7/7) | 100% (5/5) |
Percentage of embryos in which the Foxi2-negative otic domain was larger
Expression was not determined
In contrast, examination of a third marker of the otic placode, Hmx3, revealed that the intensity and domain of Hmx3 expression was reduced in Spry1Het2Null and DKO embryos compared to DHet embryos (compare Fig. 1K and L to Fig. 1I, Table 1). In Spry1Null2Het embryos, the Hmx3 domain appeared either slightly reduced in size (Fig. 1J) or indistinguishable from DHet embryos (Table 1). To compare the Hmx3 expression domain with the morphology of the otic placode, we sagittally sectioned DHet and DKO embryos after whole mount in situ hybridization for Hmx3 transcript. We found that in DHet embryos, a posterior-lateral region of the otic placode did not stain for Hmx3 (Fig. 1M and data not shown). In DKO embryos, the otic epithelium was thickened and appeared expanded, but the Hmx3 expression domain was reduced (Fig. 1N). These data indicate that loss of Spry1 and Spry2 regulate Hmx3 expression independently of pseudostratification of the otic ectoderm.
To quantify the size of the otic placode in the Sprouty gene dosage series, we sagittally sectioned 9 – 11 s embryos and measured the anterior to posterior (A-P) length of the otic placode in each section, following the curvature of the epithelium (diagramed with a yellow line, Fig. 1M). Consistent with our previous analysis (Mahoney Rogers et al.), in DKO embryos the average cross-sectional area of the otic placode, a global measurement of size, was 0.028 ± 0.004 mm2 (average area ± st. dev.). This was a 1.8-fold enlargement over the average cross-sectional area in DHet embryos (0.016 ± 0.005 mm2). We found that the average cross-sectional area of the otic placode in Spry1Het2Null embryos was 0.023 ± 0.008 mm2, a 1.4-fold (p = 0.04) enlargement over the average cross-sectional area in DHet embryos. No statistical difference in average cross-sectional area was detected between Spry1Null2Het (0.017 ± 0.003 mm2) and DHet embryos (p = 0.57). Thus, although in approximately 50% of Spry1Null2Het embryos the expression domains of Pax8 and Dlx5 in the otic placode appeared bigger (see Fig. 1B and Table 1), the average size of the otic placode was not significantly different from the size in DHet embryos. This may reflect either discordance between otic gene expression domains with the morphology of the otic placode or the obscuring of incompletely penetrant increases in otic placode size when measurements from all Spry1Null2Het embryos were averaged.
To detect regional size differences, sagittal sections were binned along the lateral to medial axis. In the lateral (0 – 33.3%) and mid-placode (33.4 – 66.6%) bins, the average AP lengths of the otic epithelium in DKO and Spry1Het2Null embryos were significantly longer than DHet controls (Fig. 1O; for Spry1Het2Null embryos, 0 – 33.3%: p = 0.006, 33.4 – 66.6%: p = 0.002; for DKO embryos, 0 – 33.3%: p = 0.009, 33.4 – 66.6%: p = 0.002). In Spry1Null2Het embryos, average A-P otic length in the mid-placode bin (33.4 – 66%) was larger than in DHet embryos, but was not significant (p = 0.056). No difference in size was detected among all genotypes in the medial bin (66.7 – 100%), containing sections closest to the hindbrain. Combined, these data suggest that with respect to otic placode size, progressive inactivation of Spry1 and Spry2 result in a gene dosage series in which wild type = DHet ≤ Spry1Null2Het < Spry1Het2Null < DKO.
Progressive reduction of otic placode enlargements in a Sprouty gene dosage series
To perform a longitudinal analysis, we examined size of the otic placode several somite stages later (12 – 16 s), during invagination. For all Spry1Null2Het embryos examined, otic expression of Pax8, Dlx5 and Hmx3 was comparable to DHet controls (compare Fig. 2A, E, I with Fig. 2B, F, J and Table 1). Foxi2 is expressed in cranial epidermal and epibranchial progenitor cells, but is excluded from otic placode (Ohyama and Groves, 2004). No difference in size of the Foxi2-negative otic region was detected in Spry1Null2Het embryos compared to DHet controls (compare Fig. 2M and Fig. 2N, Table 1). Thus, whereas at 8 – 11 s, 50% of Spry1Null2Het embryos had expanded Pax8 and Dlx5 expression domains, at 12 – 16 s, no expansions of Pax8, Dlx5, Hmx3 or Foxi2-negative domains were detected. We conclude that in the percentage of Spry1Null2Het embryos with enlarged Pax8 and Dlx5 otic expression domains at 8 – 11 s, expansion was restored to normal by 12 – 16 s.
Figure 2. The otic placode remains larger in Spry1−/+; Spry2−/− and DKO embryos during early invagination.

In situ hybridization analysis of Pax8, Dlx5, Hmx3, and Foxi2 in 12 – 14 s embryos with the genotypes indicated. Otic placode staining of Pax8 (A – D), Dlx5 (E – H), and Hmx3 (I – L) is bracketed. Epibranchial placode (“e”) staining of Pax8, adjacent to the otic domain, is labeled (A). Absence of Foxi2 staining in the otic placode (M – P) is outlined (white dashed line). *, expansion of the otic domain. Lateral views are shown with anterior to the left. Scale bar, 50 μm.
Examination of expression of Pax8, Dlx5, and Foxi2 at 12 – 16 s in Spry1Het2Null and DKO embryos revealed that the otic placode remained larger in a majority of these embryos (compare Fig. 2C, G, O and Fig. 2D, H, P with Fig. 2A, E, M, Table 1). Interestingly, Hmx3 staining intensity was no longer reduced in all Spry1Het2Null and DKO embryos examined (Fig. 2I, K, L). In particular, in DKO embryos, the Hmx3 expression domain appeared larger (Fig. 2L).
To determine whether enlargement was maintained in Spry1Het2Null and DKO embryos, we sagittally sectioned embryos at a late invagination stage (17 – 20 s). At this stage, the morphology of the otic cup was indistinguishable between DHet, Spry1Null2Het, and Spry1Het2Null embryos. In mid-placodal sections, the rim of the otic cup was curved in the direction of closure (Fig. 3A – C), and in medial sections close to the hindbrain, the otic vesicle was closed (data not shown). In DKO embryos, formation of the otic cup was delayed: in all sections the otic epithelium was wide-open, with no indication of closure (Fig. 3D).
Figure 3. Restoration of otic size in Spry1−/+; Spry2−/− and DKO embryos.
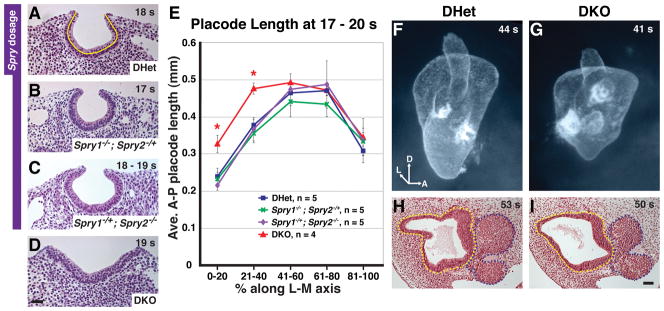
(A – D) Mid-placodal, sagittal sections of 17 – 19 s embryos with the genotypes indicated. Sections are located between 41% and 60% along the medial to lateral axis. Anterior is to the left. (E) Average anterior to posterior lengths of the otic placode in 17 – 20 s embryos with the genotypes indicated. Yellow line in (A) depicts where the A-P length of the otic placode was measured. *, p < 0.05 by ANOVA in comparisons between DHet and DKO embryos; error bars represent standard error of sample means. (F, G) Paint fills of DHet and DKO otic vesicles to visualize the shape and size of the lumenal space. Axial orientation is indicated: D, dorsal; A, anterior; L, lateral. (H, I) Mid-otic, transverse sections of DHet and DKO embryos. Basal surface of the otic epithelium and outline of the nuclei in the cochleovestibular ganglion are traced with yellow and blue dashed lines respectively. Scale bar (A – D, H, I), 50 μm.
We measured the A-P length of otic epithelium, following the curvature of the otic cup, in sagittal sections of 17 – 20 s embryos and binned sections into five groups along the lateral to medial axis. Consistent with the observed normal otic cup morphology, the otic epithelium in Spry1Het2Null embryos was comparable in length to DHet and Spry1Null2Het embryos in all bins (Fig. 3E). Only DKO embryos had a statistically significant increase in A-P length in the two most lateral bins (Fig. 3E; 0 – 20% along the lateral to medial axis, p = 0.0005; 21 – 40%, p = 0.03). Thus, by 17 – 20 s, size of the otic epithelium was restored to normal in Spry1Het2Null embryos. In DKO embryos, although the lateral portion of the otic placode was expanded, mid-placode and medial regions (41 – 100%) were not significantly enlarged (Fig. 3E). In contrast, at 9 – 11 s, a larger region of the otic placode was expanded: enlargement of the otic placode extended from lateral regions, past the mid-placode and into the medial portion (see Fig. 1O). This indicates that there was partial restoration of otic placode size in DKO embryos prior to 17 – 20 s.
To determine whether enlargement of the otic epithelium was ever completely restored to normal in DKO embryos, we compared size of otic vesicles in DKO and DHet embryos. In DKO embryos, although closure of otic cups is delayed, otic vesicles form by E11.5 (Mahoney Rogers et al., 2011). Paint fills of the lumen of otic vesicles at E11.5 revealed that although DKO otic vesicles were not as elongated as DHet vesicles, the lumenal space was not obviously larger (Fig. 3F, G; for DKO, n = 3 embryos, 4 inner ears; for DHet, n = 2 embryos, 4 inner ears). To examine the otic epithelium directly, we sectioned E11.5 embryos transversely and obtained volume measurements of the otic epithelium. No significant difference in average volume of the otic epithelium in DKO vs. DHet embryos was detected (p = 0.89, Fig. 3H, I). In DHet embryos the average volume of the otic epithelium was 0.014 ± 0.004 mm3 (average volume ± st. dev., n = 4 embryos) and in DKO embryos, the average otic epithelial volume was 0.014 ± 0.005 mm3 (n = 4 embryos).
To determine whether the restoration of normal size of the otic epithelium in DKO embryos was due to increased neuroblast delamination, we measured the volume of the cell bodies of the cochleovestibular ganglion (cvg). No statistically significant difference in average volumes of the cell bodies of the cvg was detected in DKO compared to DHet embryos (p = 0.62, Fig. 3H, I). Average volumes of the cell bodies of the cochleovestibular ganglion were 0.013 ± 0.003 mm3 (average volume ± st. dev., n = 3) in DHet embryos and 0.015 ± 0.003 mm3 (n = 3) in DKO embryos. Crown-rump measurements in DKO embryos were identical to somite-matched DHet embryos, suggesting that overall body size was unaffected. We conclude that although defects in otic vesicle morphogenesis were never restored in DKO embryos, enlargement of the otic epithelium were not maintained. Thus, for each genotype in a Spry1 and Spry2 gene dosage series, the increased size of the otic placode was progressively restored to normal, with genotypes with less dramatic placode expansions undergoing earlier restoration.
Expression of feedback inhibitors of FGF signaling in Spry1−/+; Spry2−/− embryos
To investigate cellular mechanisms by which enlargement of the otic placode was restored to normal, we chose Spry1Het2Null embryos for more detailed study. At the otic placode stage (8 – 11 s), Spry1Het2Null embryos had an enlarged otic placode, which was restored to normal by the late invagination stage (17 – 20 s, compare Fig. 1 and 3). This established a defined window from 12 - 16 s, during which otic placode enlargement was restored to normal in Spry1Het2Null embryos. We first determined whether size restoration correlated with increased transcription of the remaining functional Spry1 allele. In situ hybridization revealed no change in Spry1 expression in Spry1Het2Null (n = 7) compared to DHet embryos (n = 6, Fig. 4A, B). Furthermore, quantitative real-time PCR analysis of RNA extracted from microdissected, otic cup-containing tissue revealed no difference in Spry1 transcript levels in Spry1Het2Null compared to DHet controls at 13 – 16 s (Fig. 4C, p = 0.15, n = 6 embryos for each genotype). The Spry4 gene is expressed predominantly in the mesoderm adjacent to the otic placode (A. A. Mahoney Rogers and J. Zhang, unpublished observations). We detected no change in Spry4 expression levels in 12 – 16 s Spry1Het2Null or DKO embryos compared to DHet controls by in situ hybridization (n = 3 embryos for each genotype, data not shown).
Figure 4. Transcriptional readouts of FGF signaling in Spry1−/+; Spry2−/− and DKO embryos.

(A, B) In situ hybridization for Spry1 transcript in 14 – 15 s embryos. Otic region is bracketed. Lateral views are shown, with anterior to the left. (C) Real-time PCR analysis of Spry1 and Dusp6 transcript levels in RNA collected from 13 – 16 s otic cup-containing tissue. (D – L) In situ hybridization analysis of Etv4 expression. Embryos are oriented laterally, with anterior to the left. The OEPD (D – F), otic cup during size restoration in Spry1−/+;Spry2−/− embryos (G – I), and otic cup after size restoration in Spry1−/+;Spry2−/− embryos (J – L) are bracketed. Expanded Etv4 expression domains are highlighted (asterisk). (M) Real-time PCR analysis of Etv4 and Etv5 transcript levels in RNA collected from 13 – 16 s otic cup-containing tissue. (C, M) Tissue from two embryos was pooled into one sample; error bars represent standard error of sample means. *, p < 0.05; **, p < 0.001. Scale bar, (A, B, D – L), 50 μm.
We examined other negative feedback inhibitors of the FGF signaling pathway to determine whether their expression was increased in Spry1Het2Null and DKO embryos compared to DHet controls. Members of the dual-specificity phosphatase (Dusp) gene family negatively regulate mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK) signaling by dephosphorylating activated ERK (for review see Patterson et al., 2009). In particular, Dusp6 and Dusp9, are expressed in the otic cup (Urness et al., 2008), and Dusp6 expression is FGF-dependent (Eblaghie et al., 2003; Kawakami et al., 2003; Tsang et al., 2004; Echevarria et al., 2005; Gomez et al., 2005; Smith et al., 2005; Vieira and Martinez, 2005; Li et al., 2007). In addition, Sef encodes an antagonist of FGF signaling, whose expression is FGF-dependent (Furthauer et al., 2002; Tsang et al., 2002; Kovalenko et al., 2003; Yang et al., 2003; Torii et al., 2004; Tsang and Dawid, 2004; Abraira et al., 2007). RNA was extracted from microdissected, otic-cup-containing tissues from Spry1Het2Null and DHet embryos at 13 – 16 s (n = 6 embryos for each genotype). Real-time PCR revealed no significant difference in Dusp6, Dusp9, or Sef expression levels in Spry1Het2Null compared to DHet embryos (p = 0.18 for Dusp6 (Fig. 4C), p = 0.41 for Dusp9, p = 0.82 for Sef). Furthermore, whole mount in situ hybridization for Dusp6, Dusp9, and Sef transcript revealed no difference in staining intensity in the otic cup in Spry1Het2Null compared to DHet embryos at 12 – 16 s (n = 3 embryos for each genotype, data not shown).
We also compared Dusp6, Dusp9, and Sef expression levels in DKO and DHet otic cup-containing tissue microdissected from 13 – 16 s embryos. Real-time PCR analysis revealed an increase in Dusp6 expression levels in DKO (n = 8 embryos) compared to DHet tissue (n = 6 embryos, Fig. 4C, p = 0.02), but no significant difference in Dusp9 and Sef expression levels (p = 0.81 for Dusp9, p = 0.07 for Sef, average relative 1.15-fold increase in Sef expression levels in DKO embryos). In summary, none of the negative regulators of FGF signaling tested, Spry1, Spry4, Dusp6, Dusp9, or Sef, were significantly upregulated in Spry1Het2Null embryos to account for size restoration at 12 – 16 s. Consistent with elevated levels of FGF signaling in DKO embryos, expression of the FGF-responsive negative regulator, Dusp6, was increased.
FGF signaling activity in Spry1−/+; Spry2−/− and DKO embryos
Increased expression levels of Dusp6 in DKO, but not Spry1Het2Null embryos suggested that FGF signaling activity was high in DKO embryos, but was not elevated in Spry1Het2Null embryos at 12 – 16 s. Like Dusp6, the ETS-domain containing transcription factors, Etv4 and Etv5, are transcriptionally induced by FGF signaling in multiple organs (Raible and Brand, 2001; Roehl and Nusslein-Volhard, 2001; Firnberg and Neubuser, 2002; Liu et al., 2003; Brent and Tabin, 2004). To examine FGF signaling activity over a timecourse from just prior to otic placode formation to the time when otic placode expansion was restored to normal, we performed in situ hybridization on 5 – 20 s embryos using probes for Etv4 and Etv5. We found that sizes of Etv4 and Etv5 expression domains were expanded in Spry1Het2Null and DKO embryos at 5 – 7 s (Fig. 4D – F and data not shown; Etv4, n = 6 embryos for each genotype; Etv5, n = 6 embryos for each genotype). This suggests that as in DKO embryos (Mahoney Rogers et al.), the enlargement of the otic placode in Spry1Het2Null embryos was due to the presence of more FGF-responsive cells in the OEPD. When the otic placode was morphologically distinct, both Etv4 and Etv5 were expressed in the otic placode epithelium and their expression domains reflected the size of otic placode. Thus, at 12 – 16 s, whereas no DHet embryos had expanded Etv4 (0/5) or Etv5 (0/4) expression domains, 67% and 62% of Spry1Het2Null embryos had expanded Etv4 and Etv5 expression domains respectively (Fig. 4H and Table 1). This is consistent with the observed percentage of Spry1Het2Null embryos with expanded Pax8, Dlx5, or Foxi2-negative stained otic domains at this stage (see Table 1). By 17 – 20 s, 20% (1/5) and 0% (0/4) of Spry1Het2Null embryos had expanded Etv4 and Etv5 expression domains respectively (compare Fig. 4J and 4K, data not shown), consistent with the observed restoration of otic placode size in this genotype at this stage (see Fig. 3E). Expression of both Etv4 and Etv5 in DKO embryos continued to appear expanded at 17 – 20 s (Fig. 4L and data not shown; 5/5 embryos for Etv4; 4/4 embryos for Etv5), presumably reflecting a combination of both increased size of the otic placode and delay in otic cup formation (see Fig. 3D, E).
During the stage prior to restoration of otic cup size in Spry1Het2Null embryos, the intensity of both Etv4 and Etv5 staining appeared more intense in DKO embryos compared to Spry1Het2Null and DHet embryos (Fig. 4G – I, data not shown). This is consistent with increased levels of FGF signaling in DKO, but not Spry1Het2Null, embryos at 12 – 16 s. To quantify Etv4 and Etv5 transcript levels, we performed real-time PCR on microdissected otic-cup-containing tissue from 13 – 16 s embryos. Whereas Etv4 and Etv5 transcript levels were increased in DKO embryos at 13 – 16 s (n = 8 embryos, p = 0.03 for Etv4, p = 0.001 for Etv5), no significant differences in Etv4 and Etv5 transcript levels were detected in Spry1Het2Null (n = 6, p = 0.21 for Etv4, p = 0.87 for Etv5) compared to DHet embryos (n = 6, Fig. 4M). Combined, these data suggest that prior to otic placode formation, in both Spry1Het2Null and DKO embryos, more cells respond to FGF signaling, resulting in larger Etv4 and Etv5 expression domains in the OEPD and subsequently a larger otic placode. However, at 13 – 16 s, prior to restoration of otic placode size, FGF signaling in Spry1Het2Null embryos is lower than in DKO embryos, as indicated by relative expression levels of FGF-induced genes, Etv4, Etv5, and Dusp6. Lower levels of FGF signaling may explain why otic placode morphology and size are completely restored to normal in Spry1Het2Null embryos, but not in DKO embryos by 17 – 20 s.
Cell proliferation and cell death in a Sprouty gene dosage series
We next determined whether changes in cell proliferation or cell death contributed to the restoration of otic placode size in Spry1Het2Null and DKO embryos. Transverse sections of embryos in the Spry gene dosage series were stained using an antibody that detects the activated form of caspase-3 protease, an initiator of apoptotic cell death, and an antibody that detects the mitosis marker, phosphorylated Histone H3 (pH3). To account for differences in otic cup size among genotypes, total numbers of caspase-3+ or pH3+ cells were normalized by total volume of otic tissue measured (number/mm3).
Consistent with previous reports (Washausen and Knabe, 2012), apoptotic cells were predominantly found in the most ventral-lateral portion of the otic placode at 12 – 16 s (Fig. 5A–D). We detected a statistically significant reduction in cell death in Spry1Het2Null and DKO embryos at 12 – 16 s, compared to DHet controls (Fig. 5E; for DKO embryos, p = 0.02; for Spry1Het2Null embryos, p = 0.02). Thus, the amount of cell death in lateral regions of the otic cup is directly or indirectly controlled by Spry gene dosage. However, the reduction in lateral cell death in Spry1Het2Null and DKO embryos cannot explain how the larger size of the otic placodes in these embryos is restored to normal.
Figure 5. Cell proliferation and death in a Sprouty gene dosage series.
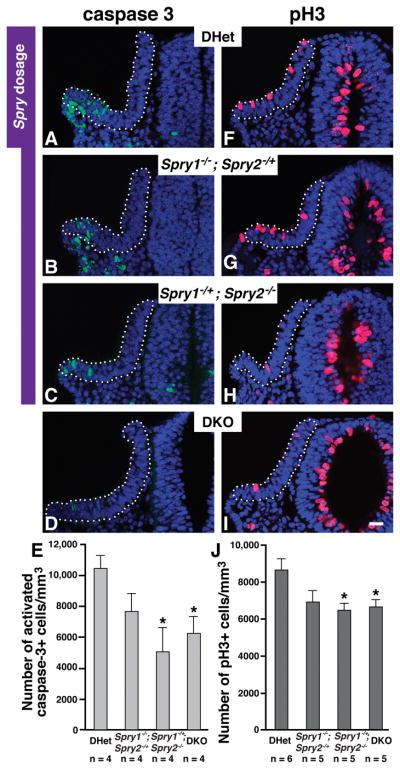
Immunolabeling to detect activated caspase 3 (A – D) and phosphorylated Histone H3 (F – I) in transverse sections of 13 – 16 s embryos with the genotypes indicated. (E) Average number of activated-caspase-3+ cells normalized by volume of the otic epithelium. (J) Average number of phosphorylated-Histone-H3+ cells normalized by volume of the otic epithelium. Decreases in average numbers of caspase-3+ and pH3+ cells were observed in Spry1−/−;Spry2−/+ embryos, but were not statistically significant (caspase-3+ cells, p = 0.11; pH3+ cells, p = 0.09). *, p < 0.05 by ANOVA in comparisons with DHet measurements; error bars represent standard error of sample means. Scale bar, 50 μm.
In contrast to the pattern of apoptotic cells in the otic placode, mitotic cells were evenly distributed throughout the otic placode (Fig. 5F–I). Quantification of the total number of pH3+ cells, revealed that both Spry1Het2Null and DKO embryos had a statistically significant reduction in mitotic index compared to DHet controls (Fig. 5J; for DKO embryos, p = 0.03; for Spry1Het2Null embryos, p = 0.02). As we discuss below, this reduction in cell proliferation in 12 – 16 s embryos likely explains how the size of the otic cup is completed restored in Spry1Het2Null embryos and is partially restored in DKO embryos by 17 – 20 s.
DISCUSSION
In this study, we observed that the otic placode was enlarged in a Sprouty gene dosage series at 8 – 11 s, the stage at which cells are first committed to an otic fate in birds and amphibians. By 17 – 20 s, size and morphogenesis of the otic cup in Spry1Het2Null embryos was restored to normal. Even in DKO embryos, there was partial restoration of the size of the otic cup by this stage. This suggests that there is sufficient growth plasticity in the otic placode and cup from 8 – 20 s to correct size abnormalities. The window of regulative growth may extend later than 20 s, since in DKO embryos, the size of the otic vesicle and cochleovestibular ganglion was restored by E11.5.
At 12 – 16 s, prior to complete restoration of otic size in Spry1Het2Null embryos and partial size restoration in DKO embryos, both cell proliferation and cell death in the otic cup were reduced. The reduction in cell proliferation could contribute to the restoration of normal otic cup size. However, lateral cell death was also significantly reduced in both Spry1Het2Null and DKO embryos, which should increase otic cup size. If reduced cell proliferation is the primary driver for size restoration, then the duration of the division cycle must outpace the time it takes for a cell to die by apoptosis. We propose that cell proliferation may be the primary determinant of otic size at this stage. There is significant growth of the otic placode between 8 – 11 s and 17 – 20 s. In DHet embryos, the average cross-sectional area of the otic placode/cup increases 3.5-fold from 0.016 ± 0.005 mm2 (average area ± st. dev.) to 0.056 ± 0.011 mm2, indicating that cell proliferation does outpace cell death. Thus, we infer that as the otic cup grows from 12 – 16 s, fewer cells are added in Spry1Het2Null and DKO than in DHet embryos due to reduced cell proliferation. Furthermore, we infer that the amount of cells that are retained in Spry1Het2Null and DKO embryos due to reduced cell death is outweighed by the fast pace of cell division.
Also during restoration of otic cup size at 12 – 16 s, levels of Etv4, Etv5, and Dusp6 expression were similar in Spry1Het2Null and DHet embryos, suggesting that FGF signaling was not elevated in Spry1Het2Null embryos at this stage. Normal levels of FGF signaling may contribute to restoration of otic cup size and the absence of morphogenetic defects in Spry1Het2Null embryos. In DKO embryos, expression levels of Etv4, Etv5, and Dusp6 were increased (Fig. 4C, M), however partial restoration of otic cup size still occurred. Elevated levels of FGF signaling in DKO embryos may explain why the lateral placode was enlarged and morphogenesis of the otic cup and vesicle was abnormal. However, our data that partial restoration of otic size can occur despite elevated levels of FGF signaling, suggests that increased FGF signaling was not sufficient to upregulate the genetic program required to maintain otic fate.
A recent study by Yang et al. using chick embryos demonstrates that although FGF signaling is necessary for expression of all otic genes tested, it is not sufficient to induce the full otic gene expression program (Yang et al., 2013). Microarray analysis indicates that only 15% of otic genes are induced when FGF is added to cultures of non-otic, pre-placodal ectoderm. Furthermore, ectopic expression of FGF in vivo can induce the expression of some, but not all otic genes (Adamska et al., 2001; Alvarez et al., 2003). We found that expression of one gene, Hmx3, was not expanded in the otic placode (9 – 11 s) in the Sprouty gene dosage series, but instead was transiently reduced. It has been demonstrated that some cellular responses do not change in proportion to increasing levels of FGF signaling (Storm et al., 2003). Our data suggest that in the otic placode, Hmx3 expression does not change proportionately with FGF signaling activity. When FGF activity is low (Fgf3−/−; Fgf10−/− mice, (Urness et al., 2010) or when FGF activity is high, Hmx3 expression is reduced (Spry1Het2Null and DKO embryos). Intermediate levels of FGF signaling (DHet and Spry1Null2Het embryos) result in the highest level of Hmx3 expression. In zebrafish, sox3 expression in the otic placode reflects a similar disproportionate response to FGF dosage (Padanad and Riley, 2011; Padanad et al., 2012). However, other responses to FGF signaling, such as pseudostratification of the otic epithelium and induction of Pax8 and Dlx5 gene expression, increase proportionately with increasing FGF activity in the Sprouty gene dosage series. This highlights the importance of regulation of FGF dose to elicit appropriate cellular responses in the otic placode.
Ectopic overexpression of Fgf in multiple organisms has resulted in enlargement of the otic placode or the formation of ectopic vesicles that express otic marker genes (for review see Schimmang, 2007). Regression of ectopic vesicles has been reported in chick (Adamska et al., 2001), and restoration of the size of an enlarged otic vesicle has been reported in zebrafish (Hans et al., 2007). In Adamska et al., FGF2-soaked beads were placed either anterior or posterior to the developing chick otic placode at 10 – 14 s. Occasionally, bead implantation just anterior or posterior to the normal otic placode resulted in the induction of small, ectopic vesicles with epithelial morphology that express Hmx3, SOHo-1, Pax2, but not Dlx5. These vesicles are not maintained past 3 days after bead implantation. In Hans et al., overexpression of fgf8 during gastrulation or early segmentation resulted in enlarged endogenous otic vesicles. Despite their increased size, axial patterning in these vesicles was normal, and by three days of development, size was restored.
In non-mammalian animals, such as insects, amphibians, fish and birds, embryonic organs possess the ability to compensate for experimental reduction or increase in their size (Summerbell, 1981; Bryant and Simpson, 1984; Raff, 1996; Martin and Morata, 2006). This growth plasticity is particularly robust in urodeles (such as salamanders), which retain their ability to regenerate resected body parts as adults (Mescher, 1996; Nye et al., 2003; Song et al., 2010). In other vertebrates, such as mammals, this regenerative ability is progressively lost for most organs during embryogenesis. For example, in mouse, the embryonic pancreas is unable to accelerate growth in response to a reduction in progenitors, even at the initial stages of its development (Stanger et al., 2007). Like salamanders, zebrafish are a highly regenerative organism, in which regenerative ability of many organs including the inner ear, is maintained in adults (for reviews see Meyers and Corwin, 2008; Brigande and Heller, 2009; Warchol, 2011). The mouse is not a highly regenerative organism, and the adult mammalian inner ear does not possess a robust ability to regenerate after injury. We provide evidence that compensatory regulation of otic size is conserved from fish to chick to mammals: in multiple species FGF-induced enlargement of the otic placode is not maintained. Furthermore, our data point to reduced cell proliferation and failure to upregulate otic cell fate maintenance programs as potential drivers of this size restoration.
EXPERIMENTAL PROCEDURES
Mouse Lines
Mouse lines carrying null or floxed alleles of Spry1 (Basson et al., 2005), Spry2 (Shim et al., 2005), and β-actin cre (Lewandoski et al., 1997) were maintained and genotyped as described. Embryos were generated by crossing β-actin cre/β-actin cre; Spry1−/+; Spry2−/+ to Spry1flox/flox; Spry2flox/flox animals. All mouse procedures were approved by the Institutional Animal Care and Use Committee of the Medical College of Wisconsin.
In Situ Hybridization
Embryos were staged so that noon on the day of vaginal plug detection was designated as embryonic (E) day 0.5. Embryos were dissected in phosphate buffered saline, 0.1% Tween-20 and fixed by immersion in 4% paraformaldehyde for 1 hour at 4°C. Whole-mount in situ hybridization using digoxigenin-labeled probes was performed according to standard protocols, using the following probes: Pax8, Dlx5, Foxi2, Spry1, Spry4, Hmx3, Etv4 and Etv5, Dusp6, Dusp9, and Sef. Scoring of enlargement or reduction in otic gene expression domains (Table 1) was based upon the observation of a substantial difference in expression domain size compared to the size in the vast majority of DHet control embryos. Photography of embryos was done at the same exposure, using a Zeiss Discovery V.12 microscope.
Morphometric Analysis and Paint Filling
For measurement of otic placode size, embryos that had been stained for Pax8 or Dlx5 expression by whole-mount in situ hybridization were embedded in plastic (JB-4, Polysciences, Warrington PA). Serial sagittal sections were cut at 6 μm thickness and then stained with hematoxylin and eosin. Embryos in which no more than 3 sections were lost per otic placode were saved for measurement. Anterior-to-posterior (A-P) length of the otic placode was measured using Image J software. The otic placode was identified based upon its pseudostratified epithelial morphology and by presence of Pax8 or Dlx5 expression (9 – 11 s embryos). Using Image J, a freehand line was drawn, midway along the apical-to-basal axis of the otic epithelium, which followed the entire A-P curvature of the otic placode or otic cup. Example locations of these freehand lines are diagramed in Fig. 1M and Fig. 3A. To ensure uniformity of the plane of section among embryos, only right-side placodes were measured in 9 – 11 s embryos, and left-side placodes were measured in 17 – 20 s embryos.
Average cross-sectional area (mm2) of the otic placode was calculated as the sum of the A-P lengths of the otic placode in all sections multiplied by 0.006 mm (the thickness of each section). For regional comparisons along the lateral-to-medial axis of the otic placode, sections were grouped into three (9 – 11 s embryos) or five (17 – 20 s embryos) bins, consisting of 5 – 6 sections of each bin, in which 0% represented the most lateral section and 100% represented the most medial section closest to the hindbrain. Significance of differences between genotypes was measured by one-way analysis of variance (ANOVA).
For measurement of the volume of otic vesicles and the cochleovestibular ganglion, E11.5 embryos were fixed in 4% paraformaldehyde, 0.1% Tween-20 overnight, then embedded in paraffin, sectioned, and stained with hematoxylin and eosin using standard procedures. Sections were imaged using a Zeiss AxioImager Z1. The area of the otic epithelium or of the cell bodies of the cochleovestibular ganglion was traced freehand and measured using ImageJ. Volume was calculated as the total area multiplied by 0.012 mm (the distance between each section in which area measurements were taken). Significance of differences between genotypes was measured by unpaired Student’s t-test.
Paint filling of inner ears from E11.5 embryos was performed as described (Kiernan, 2006), except that a 1% solution of Correction Fluid in methyl salicylate was used instead of paint.
Quantification of cell proliferation and cell death
Embryos were fixed in 4% paraformaldehyde, 0.1% Tween-20 overnight; washed with phosphate-buffered saline, 0.1% Tween-20; embedded in 3.5% low melting point agarose; and cut into 40 μm-thick transverse sections by vibratome. Sections were processed for immunohistochemistry as described (Shim et al., 2005). Primary antibodies were: anti-phospho-histone-H3 (Millipore 05-806, 1:1,000) and anti-activated-caspase-3 (Cell Signaling 9661, 1:200). Secondary antibodies were labeled with either Alexa-488 or Alexa-568 (Life Technologies, 1:1,000). DAPI (Sigma) was used to counter-stain the nuclei.
Vibratome sections containing the otic placode were imaged using a Zeiss LSM510 laser scanning confocal microscope. In each vibratome section, the total number of otic cells from both placodes, staining for either phospho-histone-H3 or activated caspase-3 was counted. The volume of the otic tissue in each vibratome section was calculated as the area of the otic region in the central image from the confocal z-stack (mm2, measured in ImageJ), multiplied by 0.04 mm (the thickness of each vibratome section). For each embryo, the total number of pH3+ or activated caspase-3+ cells was normalized by the total volume of otic tissue measured (number/mm3). Significance of differences between genotypes was measured by ANOVA.
Otic Placode RNA Isolation and Quantitative PCR Analysis
The otic region, including the ectoderm, adjacent neural ectoderm, and underlying mesenchyme was dissected from 13 – 16 s embryos using tungsten needles and fine forceps (Urness et al., 2010). Tissue from each embryo was stored separately in TRIzol (Life Technologies) at −80°C prior to genotyping. RNA from tissue pooled from two embryos of the same genotype was extracted using TRIzol reagent, followed by reverse transcription using the Superscript III First-Strand Synthesis kit and oligo-dT primers (Life Technologies). Quantitative, real-time PCR was performed on a Bio-Rad iQ5 thermocycler using the Power SYBR Green PCR kit (Life Technologies). All PCRs were performed in duplicate on three or four biological replicates (total of 6 – 8 embryos per genotype). Quantification was performed using Gapdh as a reference gene, and normalized by Pax8 expression to account for variability in otic cup dissection. Significance was determined using the unpaired Student’s t-test.
Primers were validated for efficiency, and primer specificity was validated by inclusion of “no reverse transcriptase” controls, dissociation curve analysis, and gel electrophoresis of products. Primer sequences were: Spry1 forward 5′-AGGCCGAGGATTTCAGATG-3′ and reverse 5′-TCACCACTAGCGAAGTGTGG-3′; Dusp6 forward 5′-ATGCGGGCGAGTTCAAATA-3′ and reverse 5′-CACCAGGACACCACAGTTT-3′; Dusp9 forward 5′-AGAAGGCTACCCAGCATACTA-3′ and reverse 5′-GCACTGGGCTAGACATTGAG-3′; Sef forward 5′-TCACCTTCAGATACGACAACTG-3′ and reverse 5′-CTGGCTGATGGTGATGTTCT-3′; Etv4 forward 5′ATGAAAGGCGGATACTTGGACCAG-3′ and reverse 5′-ATGAGCTTTCCCTGCGGGACCAT-3′; Etv5 forward 5′-TAGCGGAGACTTTGGAAGCACCAT-3′ and reverse 5′-GTCGACAGTCCTCTGATCGAGAT-3′; Pax8 forward 5′-GGTCCCACCCTTCAATGCCTTT-3′and reverse 5′-ATAGCAGAAGAGGCATAGCTGCCC-3′; Gapdh forward 5′-GGGCTGGCATTGCTCTCAATGACAACTTT-3′ and reverse 5′-CACCCTGTTGCTGTAGCCGTATTCAT-3′.
KEY FINDINGS.
The otic placode is enlarged in a Sprouty gene dosage series, but size is progressively restored to normal.
Although the otic placode is larger in mutant embryos, Hmx3 gene expression is transiently reduced.
Analysis of the Spry1−/+; Spry2−/− mutant reveals that normal levels of FGF signaling, decreased cell proliferation, and decreased cell death precede restoration of otic placode size.
In Spry1−/−; Spry2−/− mutant embryos, partial restoration of otic cup size can occur despite elevated FGF signaling levels.
Acknowledgments
We wish to thank Drs. Suzi Mansour and Lisa Urness for providing a detailed protocol for the microdissection of otic cup-containing tissue and for the Hmx3 plasmid; Dr. Ramani Ramchandran and members of his laboratory for sharing equipment; Dr. Pippa Simpson in the Quantitative Health Sciences division of the Children’s Research Institute for expert statistical advice; Dr. Suresh Kumar at the Children’s Research Institute imaging core for advice on confocal microscopy; Dr. Bill Jackson and members of the Shim lab for critical reading of the manuscript; and the anonymous reviewers of this manuscript, whose astute comments helped to improve its quality. This work was supported by N.I.H. grant DC010387 (to K. S.).
References
- Abraira VE, Hyun N, Tucker AF, Coling DE, Brown MC, Lu C, Hoffman GR, Goodrich LV. Changes in Sef levels influence auditory brainstem development and function. J Neurosci. 2007;27:4273–4282. doi: 10.1523/JNEUROSCI.3477-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adamska M, Herbrand H, Adamski M, Kruger M, Braun T, Bober E. FGFs control the patterning of the inner ear but are not able to induce the full ear program. Mech Dev. 2001;109:303–313. doi: 10.1016/s0925-4773(01)00550-0. [DOI] [PubMed] [Google Scholar]
- Alvarez Y, Alonso MT, Vendrell V, Zelarayan LC, Chamero P, Theil T, Bosl MR, Kato S, Maconochie M, Riethmacher D, Schimmang T. Requirements for FGF3 and FGF10 during inner ear formation. Development. 2003;130:6329–6338. doi: 10.1242/dev.00881. [DOI] [PubMed] [Google Scholar]
- Basson MA, Akbulut S, Watson-Johnson J, Simon R, Carroll TJ, Shakya R, Gross I, Martin GR, Lufkin T, McMahon AP, Wilson PD, Costantini FD, Mason IJ, Licht JD. Sprouty1 Is a Critical Regulator of GDNF/RET-Mediated Kidney Induction. Dev Cell. 2005;8:229–239. doi: 10.1016/j.devcel.2004.12.004. [DOI] [PubMed] [Google Scholar]
- Brent AE, Tabin CJ. FGF acts directly on the somitic tendon progenitors through the Ets transcription factors Pea3 and Erm to regulate scleraxis expression. Development. 2004;131:3885–3896. doi: 10.1242/dev.01275. [DOI] [PubMed] [Google Scholar]
- Brigande JV, Heller S. Quo vadis, hair cell regeneration? Nat Neurosci. 2009;12:679–685. doi: 10.1038/nn.2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant PJ, Simpson P. Intrinsic and extrinsic control of growth in developing organs. Q Rev Biol. 1984;59:387–415. doi: 10.1086/414040. [DOI] [PubMed] [Google Scholar]
- Cabrita MA, Christofori G. Sprouty proteins, masterminds of receptor tyrosine kinase signaling. Angiogenesis. 2008;11:53–62. doi: 10.1007/s10456-008-9089-1. [DOI] [PubMed] [Google Scholar]
- Chen J, Streit A. Induction of the inner ear: stepwise specification of otic fate from multipotent progenitors. Hear Res. 2013;297:3–12. doi: 10.1016/j.heares.2012.11.018. [DOI] [PubMed] [Google Scholar]
- Eblaghie MC, Lunn JS, Dickinson RJ, Munsterberg AE, Sanz-Ezquerro JJ, Farrell ER, Mathers J, Keyse SM, Storey K, Tickle C. Negative feedback regulation of FGF signaling levels by Pyst1/MKP3 in chick embryos. Curr Biol. 2003;13:1009–1018. doi: 10.1016/s0960-9822(03)00381-6. [DOI] [PubMed] [Google Scholar]
- Echevarria D, Martinez S, Marques S, Lucas-Teixeira V, Belo JA. Mkp3 is a negative feedback modulator of Fgf8 signaling in the mammalian isthmic organizer. Dev Biol. 2005;277:114–128. doi: 10.1016/j.ydbio.2004.09.011. [DOI] [PubMed] [Google Scholar]
- Firnberg N, Neubuser A. FGF signaling regulates expression of Tbx2, Erm, Pea3, and Pax3 in the early nasal region. Dev Biol. 2002;247:237–250. doi: 10.1006/dbio.2002.0696. [DOI] [PubMed] [Google Scholar]
- Freter S, Muta Y, Mak SS, Rinkwitz S, Ladher RK. Progressive restriction of otic fate: the role of FGF and Wnt in resolving inner ear potential. Development. 2008;135:3415–3424. doi: 10.1242/dev.026674. [DOI] [PubMed] [Google Scholar]
- Furthauer M, Lin W, Ang SL, Thisse B, Thisse C. Sef is a feedback-induced antagonist of Ras/MAPK-mediated FGF signalling. Nat Cell Biol. 2002;4:170–174. doi: 10.1038/ncb750. [DOI] [PubMed] [Google Scholar]
- Gallagher BC, Henry JJ, Grainger RM. Inductive processes leading to inner ear formation during Xenopus development. Dev Biol. 1996;175:95–107. doi: 10.1006/dbio.1996.0098. [DOI] [PubMed] [Google Scholar]
- Ginsburg AS. Determination of the labyrinth in different amphibian species and its correlation with determination of the other ectoderm derivatives. Roux’s Archives in Developmental Biology. 1995;204:351–358. doi: 10.1007/BF00360480. [DOI] [PubMed] [Google Scholar]
- Giraldez F. Regionalized organizing activity of the neural tube revealed by the regulation of lmx1 in the otic vesicle. Dev Biol. 1998;203:189–200. doi: 10.1006/dbio.1998.9023. [DOI] [PubMed] [Google Scholar]
- Gomez AR, Lopez-Varea A, Molnar C, de la Calle-Mustienes E, Ruiz-Gomez M, Gomez-Skarmeta JL, de Celis JF. Conserved cross-interactions in Drosophila and Xenopus between Ras/MAPK signaling and the dual-specificity phosphatase MKP3. Dev Dyn. 2005;232:695–708. doi: 10.1002/dvdy.20227. [DOI] [PubMed] [Google Scholar]
- Groves AK, Bronner-Fraser M. Competence, specification and commitment in otic placode induction. Development. 2000;127:3489–3499. doi: 10.1242/dev.127.16.3489. [DOI] [PubMed] [Google Scholar]
- Groves AK, Fekete DM. Shaping sound in space: the regulation of inner ear patterning. Development. 2012;139:245–257. doi: 10.1242/dev.067074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy GR, Jackson RA, Yusoff P, Chow SY. Sprouty proteins: modified modulators, matchmakers or missing links? J Endocrinol. 2009;203:191–202. doi: 10.1677/JOE-09-0110. [DOI] [PubMed] [Google Scholar]
- Hans S, Christison J, Liu D, Westerfield M. Fgf-dependent otic induction requires competence provided by Foxi1 and Dlx3b. BMC Dev Biol. 2007;7:5. doi: 10.1186/1471-213X-7-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbrand H, Guthrie S, Hadrys T, Hoffmann S, Arnold HH, Rinkwitz-Brandt S, Bober E. Two regulatory genes, cNkx5-1 and cPax2, show different responses to local signals during otic placode and vesicle formation in the chick embryo. Development. 1998;125:645–654. doi: 10.1242/dev.125.4.645. [DOI] [PubMed] [Google Scholar]
- Kawakami Y, Rodriguez-Leon J, Koth CM, Buscher D, Itoh T, Raya A, Ng JK, Esteban CR, Takahashi S, Henrique D, Schwarz MF, Asahara H, Izpisua Belmonte JC. MKP3 mediates the cellular response to FGF8 signalling in the vertebrate limb. Nat Cell Biol. 2003;5:513–519. doi: 10.1038/ncb989. [DOI] [PubMed] [Google Scholar]
- Kiernan AE. The paintfill method as a tool for analyzing the three-dimensional structure of the inner ear. Brain Res. 2006;1091:270–276. doi: 10.1016/j.brainres.2006.02.037. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Bar-Sagi D. Modulation of signalling by Sprouty: a developing story. Nat Rev Mol Cell Biol. 2004;5:441–450. doi: 10.1038/nrm1400. [DOI] [PubMed] [Google Scholar]
- Kovalenko D, Yang X, Nadeau RJ, Harkins LK, Friesel R. Sef inhibits fibroblast growth factor signaling by inhibiting FGFR1 tyrosine phosphorylation and subsequent ERK activation. J Biol Chem. 2003;278:14087–14091. doi: 10.1074/jbc.C200606200. [DOI] [PubMed] [Google Scholar]
- Ladher RK, O’Neill P, Begbie J. From shared lineage to distinct functions: the development of the inner ear and epibranchial placodes. Development. 2010;137:1777–1785. doi: 10.1242/dev.040055. [DOI] [PubMed] [Google Scholar]
- Ladher RK, Wright TJ, Moon AM, Mansour SL, Schoenwolf GC. FGF8 initiates inner ear induction in chick and mouse. Genes Dev. 2005;19:603–613. doi: 10.1101/gad.1273605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leger S, Brand M. Fgf8 and Fgf3 are required for zebrafish ear placode induction, maintenance and inner ear patterning. Mech Dev. 2002;119:91–108. doi: 10.1016/s0925-4773(02)00343-x. [DOI] [PubMed] [Google Scholar]
- Lewandoski M, Meyers EN, Martin GR. Analysis of Fgf8 gene function in vertebrate development. Cold Spring Harb Symp Quant Biol. 1997;62:159–168. [PubMed] [Google Scholar]
- Li C, Scott DA, Hatch E, Tian X, Mansour SL. Dusp6 (Mkp3) is a negative feedback regulator of FGF-stimulated ERK signaling during mouse development. Development. 2007;134:167–176. doi: 10.1242/dev.02701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Jiang H, Crawford HC, Hogan BL. Role for ETS domain transcription factors Pea3/Erm in mouse lung development. Dev Biol. 2003;261:10–24. doi: 10.1016/s0012-1606(03)00359-2. [DOI] [PubMed] [Google Scholar]
- Mahoney Rogers AA, Zhang J, Shim K. Sprouty1 and Sprouty2 limit both the size of the otic placode and hindbrain Wnt8a by antagonizing FGF signaling. Dev Biol. 2011;353:94–104. doi: 10.1016/j.ydbio.2011.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroon H, Walshe J, Mahmood R, Kiefer P, Dickson C, Mason I. Fgf3 and Fgf8 are required together for formation of the otic placode and vesicle. Development. 2002;129:2099–2108. doi: 10.1242/dev.129.9.2099. [DOI] [PubMed] [Google Scholar]
- Martin FA, Morata G. Compartments and the control of growth in the Drosophila wing imaginal disc. Development. 2006;133:4421–4426. doi: 10.1242/dev.02618. [DOI] [PubMed] [Google Scholar]
- Martin K, Groves AK. Competence of cranial ectoderm to respond to Fgf signaling suggests a two-step model of otic placode induction. Development. 2006;133:877–887. doi: 10.1242/dev.02267. [DOI] [PubMed] [Google Scholar]
- Mason JM, Morrison DJ, Basson MA, Licht JD. Sprouty proteins: multifaceted negative-feedback regulators of receptor tyrosine kinase signaling. Trends Cell Biol. 2006;16:45–54. doi: 10.1016/j.tcb.2005.11.004. [DOI] [PubMed] [Google Scholar]
- Mescher AL. The cellular basis of limb regeneration in urodeles. Int J Dev Biol. 1996;40:785–795. [PubMed] [Google Scholar]
- Meyers JR, Corwin JT. Morphological correlates of regeneration and repair. In: Salvi RJ, Fay RR, Popper AN, editors. Hair cell regeneration, repair, and protection. New York: Springer; 2008. pp. 39–75. [Google Scholar]
- Nye HL, Cameron JA, Chernoff EA, Stocum DL. Regeneration of the urodele limb: a review. Dev Dyn. 2003;226:280–294. doi: 10.1002/dvdy.10236. [DOI] [PubMed] [Google Scholar]
- Ohyama T, Groves AK. Expression of mouse Foxi class genes in early craniofacial development. Dev Dyn. 2004;231:640–646. doi: 10.1002/dvdy.20160. [DOI] [PubMed] [Google Scholar]
- Ohyama T, Groves AK, Martin K. The first steps towards hearing: mechanisms of otic placode induction. Int J Dev Biol. 2007;51:463–472. doi: 10.1387/ijdb.072320to. [DOI] [PubMed] [Google Scholar]
- Padanad MS, Bhat N, Guo B, Riley BB. Conditions that influence the response to Fgf during otic placode induction. Dev Biol. 2012;364:1–10. doi: 10.1016/j.ydbio.2012.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padanad MS, Riley BB. Pax2/8 proteins coordinate sequential induction of otic and epibranchial placodes through differential regulation of foxi1, sox3 and fgf24. Dev Biol. 2011;351:90–98. doi: 10.1016/j.ydbio.2010.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson KI, Brummer T, O’Brien PM, Daly RJ. Dual-specificity phosphatases: critical regulators with diverse cellular targets. Biochem J. 2009;418:475–489. doi: 10.1042/bj20082234. [DOI] [PubMed] [Google Scholar]
- Phillips BT, Bolding K, Riley BB. Zebrafish fgf3 and fgf8 encode redundant functions required for otic placode induction. Dev Biol. 2001;235:351–365. doi: 10.1006/dbio.2001.0297. [DOI] [PubMed] [Google Scholar]
- Raff MC. Size control: the regulation of cell numbers in animal development. Cell. 1996;86:173–175. doi: 10.1016/s0092-8674(00)80087-2. [DOI] [PubMed] [Google Scholar]
- Raible F, Brand M. Tight transcriptional control of the ETS domain factors Erm and Pea3 by Fgf signaling during early zebrafish development. Mech Dev. 2001;107:105–117. doi: 10.1016/s0925-4773(01)00456-7. [DOI] [PubMed] [Google Scholar]
- Roehl H, Nusslein-Volhard C. Zebrafish pea3 and erm are general targets of FGF8 signaling. Curr Biol. 2001;11:503–507. doi: 10.1016/s0960-9822(01)00143-9. [DOI] [PubMed] [Google Scholar]
- Schimmang T. Expression and functions of FGF ligands during early otic development. Int J Dev Biol. 2007;51:473–481. doi: 10.1387/ijdb.072334ts. [DOI] [PubMed] [Google Scholar]
- Schlosser G. Making senses development of vertebrate cranial placodes. Int Rev Cell Mol Biol. 2010;283:129–234. doi: 10.1016/S1937-6448(10)83004-7. [DOI] [PubMed] [Google Scholar]
- Shim K, Minowada G, Coling DE, Martin GR. Sprouty2, a mouse deafness gene, regulates cell fate decisions in the auditory sensory epithelium by antagonizing FGF signaling. Dev Cell. 2005;8:553–564. doi: 10.1016/j.devcel.2005.02.009. [DOI] [PubMed] [Google Scholar]
- Smith TG, Sweetman D, Patterson M, Keyse SM, Munsterberg A. Feedback interactions between MKP3 and ERK MAP kinase control scleraxis expression and the specification of rib progenitors in the developing chick somite. Development. 2005;132:1305–1314. doi: 10.1242/dev.01699. [DOI] [PubMed] [Google Scholar]
- Song F, Li B, Stocum DL. Amphibians as research models for regenerative medicine. Organogenesis. 2010;6:141–150. doi: 10.4161/org.6.3.12039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanger BZ, Tanaka AJ, Melton DA. Organ size is limited by the number of embryonic progenitor cells in the pancreas but not the liver. Nature. 2007;445:886–891. doi: 10.1038/nature05537. [DOI] [PubMed] [Google Scholar]
- Storm EE, Rubenstein JL, Martin GR. Dosage of Fgf8 determines whether cell survival is positively or negatively regulated in the developing forebrain. Proc Natl Acad Sci U S A. 2003;100:1757–1762. doi: 10.1073/pnas.0337736100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summerbell D. Evidence for regulation of growth, size and pattern in the developing chick limb bud. J Embryol Exp Morphol. 1981;65(Suppl):129–150. [PubMed] [Google Scholar]
- Swanson GJ, Howard M, Lewis J. Epithelial autonomy in the development of the inner ear of a bird embryo. Dev Biol. 1990;137:243–257. doi: 10.1016/0012-1606(90)90251-d. [DOI] [PubMed] [Google Scholar]
- Torii S, Kusakabe M, Yamamoto T, Maekawa M, Nishida E. Sef is a spatial regulator for Ras/MAP kinase signaling. Dev Cell. 2004;7:33–44. doi: 10.1016/j.devcel.2004.05.019. [DOI] [PubMed] [Google Scholar]
- Tsang M, Dawid IB. Promotion and attenuation of FGF signaling through the Ras-MAPK pathway. Sci STKE. 2004:pe17. doi: 10.1126/stke.2282004pe17. [DOI] [PubMed]
- Tsang M, Friesel R, Kudoh T, Dawid IB. Identification of Sef, a novel modulator of FGF signalling. Nat Cell Biol. 2002;4:165–169. doi: 10.1038/ncb749. [DOI] [PubMed] [Google Scholar]
- Tsang M, Maegawa S, Kiang A, Habas R, Weinberg E, Dawid IB. A role for MKP3 in axial patterning of the zebrafish embryo. Development. 2004;131:2769–2779. doi: 10.1242/dev.01157. [DOI] [PubMed] [Google Scholar]
- Urness LD, Li C, Wang X, Mansour SL. Expression of ERK signaling inhibitors Dusp6, Dusp7, and Dusp9 during mouse ear development. Dev Dyn. 2008;237:163–169. doi: 10.1002/dvdy.21380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urness LD, Paxton CN, Wang X, Schoenwolf GC, Mansour SL. FGF signaling regulates otic placode induction and refinement by controlling both ectodermal target genes and hindbrain Wnt8a. Dev Biol. 2010;340:595–604. doi: 10.1016/j.ydbio.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira C, Martinez S. Experimental study of MAP kinase phosphatase-3 (Mkp3) expression in the chick neural tube in relation to Fgf8 activity. Brain Res Brain Res Rev. 2005;49:158–166. doi: 10.1016/j.brainresrev.2004.12.038. [DOI] [PubMed] [Google Scholar]
- Waddington CH. The determination of the auditory placode in the chick. Journal of Experimental Biology. 1937;14:232–239. [Google Scholar]
- Warchol ME. Sensory regeneration in the vertebrate inner ear: differences at the levels of cells and species. Hear Res. 2011;273:72–79. doi: 10.1016/j.heares.2010.05.004. [DOI] [PubMed] [Google Scholar]
- Washausen S, Knabe W. Apoptosis contributes to placode morphogenesis in the posterior placodal area of mice. Brain Struct Funct. 2012;218:789–803. doi: 10.1007/s00429-012-0429-y. [DOI] [PubMed] [Google Scholar]
- Wright TJ, Mansour SL. Fgf3 and Fgf10 are required for mouse otic placode induction. Development. 2003;130:3379–3390. doi: 10.1242/dev.00555. [DOI] [PubMed] [Google Scholar]
- Yang L, O’Neill P, Martin K, Maass JC, Vassilev V, Ladher R, Groves AK. Analysis of FGF-dependent and FGF-independent pathways in otic placode induction. PLoS One. 2013;8:e55011. doi: 10.1371/journal.pone.0055011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang RB, Ng CK, Wasserman SM, Komuves LG, Gerritsen ME, Topper JN. A novel interleukin-17 receptor-like protein identified in human umbilical vein endothelial cells antagonizes basic fibroblast growth factor-induced signaling. J Biol Chem. 2003;278:33232–33238. doi: 10.1074/jbc.M305022200. [DOI] [PubMed] [Google Scholar]
- Yntema CL. Experiments on the Determination of the Ear Ectoderm in the Embryo of Amblystoma Punctatum. Journal of Experimental Zoology. 1933;65:317–357. [Google Scholar]
- Zelarayan LC, Vendrell V, Alvarez Y, Dominguez-Frutos E, Theil T, Alonso MT, Maconochie M, Schimmang T. Differential requirements for FGF3, FGF8 and FGF10 during inner ear development. Dev Biol. 2007;308:379–391. doi: 10.1016/j.ydbio.2007.05.033. [DOI] [PubMed] [Google Scholar]