

Abstract
Natural Killer (NK) cells are innate lymphocytes that exhibit many features of adaptive immunity including clonal proliferation and long-lived memory. Here we demonstrate that the BTB-ZF transcription factor Zbtb32 (also known as ROG, FAZF, TZFP, and PLZP) is essential for the proliferative burst and protective capacity of virus-specific NK cells. Signals from proinflammatory cytokines are both necessary and sufficient to induce high Zbtb32 expression in NK cells. Mechanistically, we show that Zbtb32 facilitates NK cell proliferation during infection by antagonizing the anti-proliferative factor Blimp-1 (Prdm1). Taken together, our data support a model in which Zbtb32 acts as a cellular “hub” through which pro-inflammatory signals instruct a “proliferation-permissive” state in NK cells, thereby allowing their prolific expansion in response to viral infection.
Natural killer (NK) cells are critical mediators of host immunity to malignancy and viral infection. NK cell activation is primarily controlled by germline-encoded surface receptors that recognize self- or virally encoded molecules expressed on infected or stressed cells and by proinflammatory cytokines, such as interleukin 12 (IL-12), IL-18, and type I interferons (IFNs). Activated NK cells respond by proliferating and by rapidly secreting cytokines such as interferon-γ (IFN-γ) and tumor necrosis factor (TNF) and cytotoxic molecules (perforin and granzyme B) that lyse infected or malignant target cells1.
Although classically considered cells of the innate immune system, NK cells are now appreciated to share a number of developmental and effector function features with T and B cells of the adaptive immune system. Similarities include development from a common lymphoid progenitor (CLP) cell, a requirement for common γ-chain-dependent cytokines (e.g. IL-15 and IL-7) during development and homeostasis, and an education process in the bone marrow analogous to T cell development in the thymus1. Moreover, much like their B and T cell counterparts, which utilize the B cell receptor (BCR) and T cell receptor (TCR), respectively, to recognize antigens, NK cells also express activating receptors capable of directly binding foreign, often virally derived, antigens. For example, in C57BL/6 mice, the activating receptor Ly49H binds the mouse cytomegalovirus (MCMV)-encoded glycoprotein m157 (refs. 2,3); receptor-ligand engagment drives a proliferative “burst” and effector response that is both necessary and sufficient for resistance to MCMV in this mouse strain4–7. The kinetics of the Ly49H+ NK cell response to MCMV infection are highly reminiscent of antigen-specific B and T cell responses. Most notably, the primary response, which peaks in cell number at around day 7 post-infection7,8, contracts into a long-lived pool of antigen-specific NK cells that exhibit many features of immunological “memory”, including long-term persistence and enhanced functionality upon antigen re-challenge8.
Broad complex, Tramtrack, Bric á brac and Zinc Finger (BTB-ZF) proteins are a large family of transcripton factors with important roles in development, differentiation and oncogenesis9. Family members are characterized by the presence of C-terminal C2H2 Krüppel-type zinc finger domains, which facilitate sequence-specific DNA binding at target loci, and an N-terminal BTB domain that recruits corepressors and histone modification enzymes to the site of regulation9,10. BTB-ZF proteins are currently thought to act primarily as repressors of target gene transcription, in part by modifying chromatin accessibility at target loci.
A growing number of studies have revealed an essential and nonredundant role for BTB-ZF proteins in the regulation of lineage commitment, development and effector function in lymphocytes10. Examples include LRF, which controls T cell versus B cell commitment11; MAZR and ThPOK, which direct CD8+ and CD4+ T cell lineage commitment, respectively12–16; PLZF, which regulates the development and function of NKT and γδ T cells17–20; and Bcl-6, which is required for germinal center B-cell and follicular T helper cell formation21–25. The extent to which BTB-ZF proteins regulate NK cell development and function is largely unknown. Given their prominent role in T and B cell biology, we hypothesized that BTB-ZF proteins may act as important regulators of the adaptive lymphocyte-like features of NK cells. Here, we identify the BTB-ZF transcription factor Zbtb32 as an essential regulator of the proliferative burst of MCMV-specific NK cells responding to viral infection in vivo and show that Zbtb32 is required for NK cell-mediated protection against lethal viral challenge.
Results
MCMV infection induces Zbtb32 expression in NK cells
To identify BTB-ZF genes that might regulate antigen-specific NK cell responses, we first compared expression of 47 BTB-ZF genes in sorted Ly49H+ NK cells from MCMV-infected and uninfected animals by microarray. Remarkably, only three (Zbtb32, Hic1 and Bcl6) were significantly modulated in NK cells on day 1.5 post-infection (p.i.), and of these three, Zbtb32 was the most highly upregulated (Fig. 1a). Indeed, of the >35,000 genes evaluated on the microarray (GEO code: GSE15907)26, Zbtb32 was among the top 30 most highly induced on day 1.5 p.i. (Fig. 1b). The microarray data were confirmed by quantitative reverse-transcription polymerase chain reaction (qRT-PCR), which revealed a >100-fold upregulation of Zbtb32 transcript in Ly49H+ NK cells at day 2 p.i. with MCMV (Fig. 1c), and by flow cytometry, which showed elevated Zbtb32 protein expression on day 2 and day 3 p.i. (Supplementary Fig. 1). Expression of Zbtb32 transcript and protein were transient, and both returned to baseline abundance by day 4 p.i., suggesting an equally rapid down-regulation of this transcription factor following its induction during viral infection.
Figure 1. Zbtb32 is highly upregulated in NK cells during viral infection.

(a) Expression of 47 BTB-ZF genes in splenic Ly49H+ NK cells sorted from uninfected and MCMV-infected animals on day 1.5 p.i., as assessed by microarray (data provided by the Immunological Genome Consortium26). Shown as fold microarray signal intensity for the infected versus uninfected samples (n = 3 biological replicates per group). Solid black bars denote significant upregulation or downregulation. (b) Top 30 most highly induced genes in splenic Ly49H+ NK cells from MCMV-infected versus uninfected control animals. Heat map shows mean microarray signal intensity (n = 3 biological replicates per time point). (c) Zbtb32 mRNA abundance measured by qRT-PCR in splenic Ly49H+ NK cells sorted from MCMV-infected animals on day 2 (n = 6 mice), 4 (n = 3 mice), and 7 (n = 3 mice) shown as fold expression relative to day 0 (n = 5 mice). Data are representative of 3 independent experiments.
Zbtb32 is required for NK cell anti-viral immunity
Given its rapid upregulation following viral infection, we hypothesized that Zbtb32 might regulate the function and/or phenotype of activated antigen-specific NK cells. It has previously been shown that adoptively transferred Ly49H+ NK cells can rescue NK cell-deficient or -impaired animals from otherwise fatal doses of MCMV8. To test the protective capacity of Zbtb32−/− NK cells, we transferred Ly49H+ wild-type or Zbtb32-deficient NK cells into neonate mice, which lack mature B, T and NK cells and are highly susceptible to viral infection, and assessed the ability of the transferred cells to protect against lethal MCMV challenge8,27. As expected, wild-type Ly49H+ NK cells had a significantly protective effect, with 33% of animals surviving past 3 weeks (median survival of 15 days; Fig. 2a). In contrast, neonates receiving Zbtb32−/− NK cells died early after infection (median survival of 8 days), similar to control animals that did not receive NK cells. To further confirm the requirement for Zbtb32 in protective NK cell responses, we compared the ability of wild-type and Zbtb32−/− Ly49H+ NK cells to protect adult Ly49H-deficient mice against lethal doses of recombinant vesicular stomatitis virus expressing the MCMV-derived m157 protein (VSV-m157). In this model, VSV-m157-infected animals rapidly succumb to paralysis and death in the absence of transferred Ly49H+ NK cells. Whereas adoptively transferred wild-type Ly49H+ NK cells protected ∼50% of infected animals from death, all recipients of Zbtb32−/− Ly49H+ NK cells died rapidly after infection, at a time comparable to animals that did not receive NK cells (Fig. 2b). Thus, Zbtb32 is required for protective anti-viral responses by antigen-specific NK cells.
Figure 2. Zbtb32 is required for protective anti-viral NK cell responses.
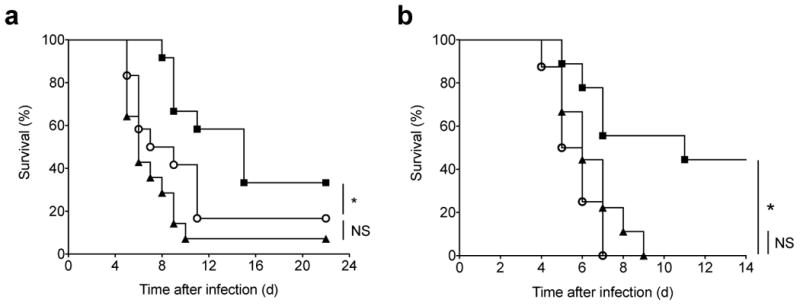
(a) Kaplan-Meier survival curves for neonate Ly49H-deficient hosts receiving splenic Ly49H+ NK cells from WT (n = 12 mice) or Zbtb32−/− (n = 12 mice) donors, or receiving PBS only (n = 14 mice), one day prior to infection with MCMV. Data are pooled from 4 independent experiments. (b) Kaplan-Meier survival curves for adult Ly49H-deficient hosts receiving splenic Ly49H+ NK cells from WT (n = 9 mice) or Zbtb32−/− (n = 8 mice) donors, or receiving PBS only (n = 9 mice), one day prior to infection with VSV-m157. Data are pooled from 3 independent experiments.
Zbtb32 is dispensable for NK cell activation and effector function
NK cells respond to MCMV infection by rapidly producing cytolytic proteins and pro-inflammatory cytokines, and for those expressing the Ly49H receptor, by proliferating to enlarge the overall pool of effector cells1,7. Given the protective defect of Zbtb32−/− NK cells, we sought to delineate which of these effector functions was controlled by Zbtb32. Zbtb32 has previously been shown to suppress cytokine production in activated T cells28–32. Zbtb32−/− CD4+ T cells expressed more TH2 cytokines, including IL-4, IL-5 and IL-13, following activation in vitro29 or in the context of allergen-induced models of airway and contact hypersensitivity in vivo31,32. Similarly, activated Zbtb32−/− CD8+ T cells produced more IFN-γ than their wild-type counterparts29. In contrast to the findings in T cells, Zbtb32−/− and wild-type NK cells were indistinguishable in their ability to produce IFN-γ in response to MCMV infection in vivo (Fig. 3a), and following stimulation in vitro with pro-inflammatory cytokines or via cross-linking of activating receptors (Supplementary Fig. 2a). Furthermore, wild-type and Zbtb32−/− NK cells from MCMV-infected mice similarly upregulated expression of the potent cytotoxic protein Granzyme B (Fig. 3b) and CD107a, a marker of degranulation (data not shown). In addition, NK cells in Zbtb32−/− animals were similar to those in wild-type animals with respect to their ability to kill adoptively transferred target cells (Supplementary Fig. 2b). Following MCMV infection, wild-type and Zbtb32−/− NK cells were comparable with respect to expression of the activation markers CD69 and KLRG1 (Fig. 3c), and both populations exhibited similar conversion to fully mature CD27loCD11bhi cells by day 7 p.i. (Fig. 3d). Taken together, our data indicate that the protective deficiency of Zbtb32−/− NK cells does not stem from a defect in killing capacity, cytokine production or maturation following viral infection.
Figure 3. Zbtb32 is dispensable for NK cell activation, maturation, and expression of effector molecules.

(a) Intracellular IFN-γ and (b) granzyme B in splenic WT or Zbtb32−/− Ly49H+ NK cells from mixed bone marrow chimeric animals on day 2 p.i. with MCMV. Representative of n = 5 (infected) and n = 2 (uninfected) animals from 2 independent experiments. (c) WT and Zbtb32−/− Ly49H+ NK cells were co-transferred into Ly49H-deficient mice and CD69 and KLRG1 expression was assessed on transferred cells in the spleen on day 2 (for CD69) or day 7 (for KLRG1) p.i. with MCMV. Representative of n = 5 animals per time point from 2 independent experiments. (d) CD27 and CD11b expression were used to assess the relative percentage of immature (CD27hiCD11blo and CD27hiCD11bhi) and mature (CD27loCD11bhi) Ly49H+ NK cells from uninfected (UI) or MCMV-infected (day 7 p.i.) mixed bone marrow chimeric animals. Representative of n = 7 (infected) and n = 2 (uninfected) animals from 2 independent experiments.
Cell-intrinsic requirement for Zbtb32 in NK cell expansion
Given that effector responses were largely intact in Zbtb32-deficient NK cells, we postulated that their protective defect may arise from an impairment in antigen-driven proliferation. To test this hypothesis, we co-transferred equal numbers of Zbtb32−/− and wild-type Ly49H+ NK cells into Ly49H-deficient hosts and evaluated the ability of the transferred Ly49H+ cells to expand after MCMV infection (Supplementary Fig. 3a). Whereas transferred wild-type Ly49H+ NK cell populations rapidly expanded and reached peak numbers and percentages in the spleen by day 7 p.i., transferred Zbtb32−/− Ly49H+ NK cells were barely detectable at this time point (Fig. 4a,b). Similar results were obtained when Zbtb32−/− and wild-type Ly49H+ NK cells were transferred separately (Fig. 4c), indicating that wild-type NK cells were not simply out-competing their Zbtb32-deficient counterparts. The low numbers and percentages of Zbtb32−/− NK cells in the spleen were not due to aberrant trafficking, since a similar deficiency was observed in the liver, lymph node and lung (Fig. 4d). Moreover, the relative percentages of wild-type and Zbtb32−/− NK cells (∼95% vs. ∼5%) remained constant from day 5 through day 45 p.i., ruling out delayed population expansion kinetics and a role for Zbtb32 in memory NK cell formation and maintenance (Fig. 4e). The requirement for Zbtb32 in antigen-specific NK cell expansion was not limited to MCMV infection, since the Zbtb32−/− Ly49H+ NK cell population also exhibited an expansion defect following infection with m157-expressing VSV or m157-expressing VacV, although the magnitude of this defect was directly proportional to number of cellular divisions driven by each virus (Fig. 4f and data not shown).
Figure 4. Zbtb32 is essential for NK cell expansion during viral infection.
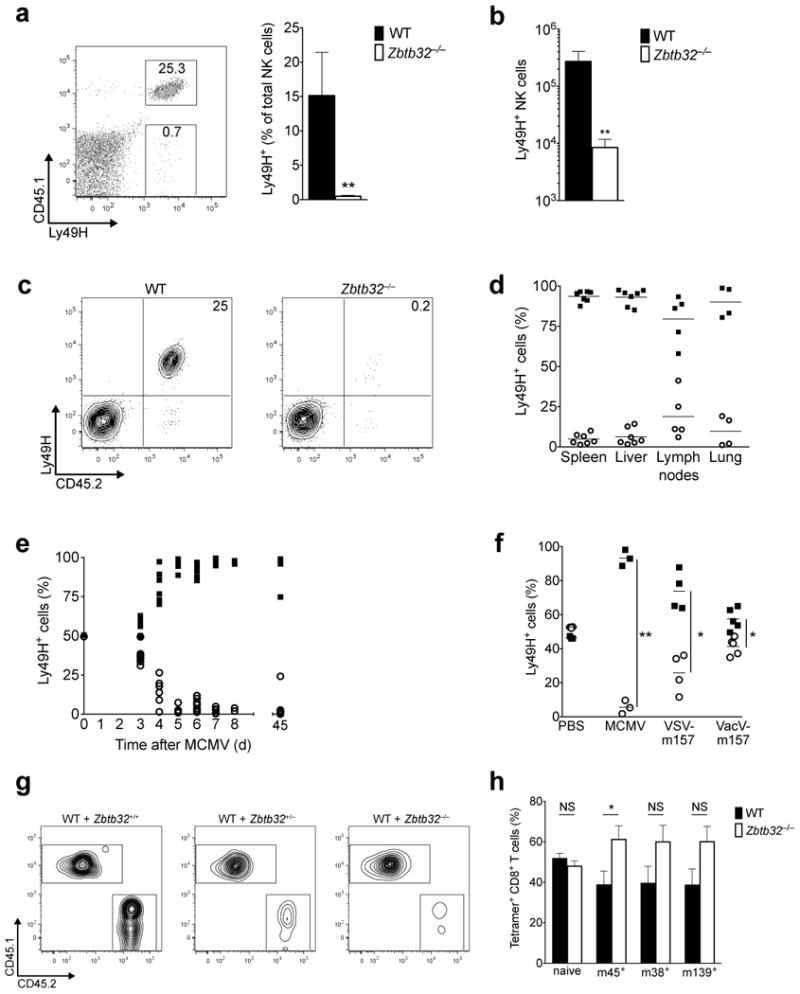
(a – b) and (d – f) Equal numbers of WT (CD45.1+) and Zbtb32−/− (CD45.2+) Ly49H+ NK cells were co-transferred into Ly49H-deficient hosts prior to MCMV infection. (a) The percentage of transferred Ly49H+ NK cells (within the total NK cell gate) in the spleen on day 7 p.i. (n = 3 mice). Representative of 4 independent experiments. P < 0.05 in a ratio paired two-tailed t-test. (b) As in (a), except showing absolute number. Representative of 2 independent experiments. (c) As in (a), except WT or Zbtb32−/− NK cells (CD45.2+) were separately transferred into independent Ly49H-deficient hosts (CD45.1+). Representative of n = 4 mice per group from 2 independent experiments. (d) Relative percentages of co-transferred WT and Zbtb32−/− Ly49H+ NK cells in various organs on day 7 p.i. Symbols represent individual mice from 3 independent experiments. (e) Relative percentages of co-transferred WT and Zbtb32−/− Ly49H+ NK cells in the spleen at various time points. Symbols represent individual mice. (f) Relative percentages of co-transferred Ly49H+ WT and Zbtb32−/− NK cells in the spleen on day 8 p.i. with the indicated viruses. Symbols represent individual mice. Representative of at least 2 independent experiments. (g) Ly49H+ NK cells from Zbtb32−/−, Zbtb32+/−, or Zbtb32+/+ littermates (CD45.2+) were co-transferred with equal numbers of Ly49H+ NK cells from WT animals (CD45.1+). Plots show the relative percentages of transferred Ly49H+ WT and Zbtb32 littermate NK cells in the spleen on day 7 p.i. with MCMV (n = 3 animals per group). Representative of 2 independent experiments. (h) Relative percentage of WT or Zbtb32−/− tetramer+ CD8+ T cells in WT:Zbtb32−/− mixed bone marrow chimeric mice (n = 8 animals) on day 7 p.i. (for M45+) or day 14 p.i. (for m139+ or M38+) with MCMV. Representative of 3 independent experiments.
To test whether there exists a gene dosage effect for Zbtb32 in antiviral NK cell responses, NK cells from Zbtb32−/−, Zbtb32+/−, or Zbtb32+/+ littermates were individually co-transferred along with congenic wild-type NK cells into Ly49H-deficient hosts. As expected, Ly49H+ NK cells from Zbtb32+/+ animals increased as well as those from wild-type donors, whereas Ly49H+ NK cells from Zbtb32−/− littermates barely increased at all, confirming that Zbtb32-deficiency, and not background-related effects, was responsible for the expansion defect (Fig. 4g). Surprisingly, Ly49H+ NK cells containing only one Zbtb32 allele (Zbtb32+/−) were almost as dysfunctional as fully-deficient Zbtb32−/− NK cells, underscoring that maximal Zbtb32 expression is required for NK cell population expansion after infection.
Because Zbtb32 is also upregulated in antigen-specific CD8+ T cells following viral or bacterial infection (data not shown), and has previously been reported to regulate T cell proliferation in vitro (although these studies suggested a restrictive role for Zbtb32 in TCR-driven proliferation)29,33, we measured the expansion of antigen-specific CD8+ T cells in wild-type:Zbtb32−/− mixed bone marrow chimeras during MCMV infection (Fig. 4h). In contrast to our findings in NK cells, Zbtb32-deficient antigen-specific CD8+ T cell populations expanded similarly or better than their wild-type counterparts. Thus, taken together, our data indicate an essential cell-intrinsic and lineage-specific function for Zbtb32 in promoting NK cell expansion after infection.
Zbtb32 controls NK cell proliferation, but not survival
We next investigated the extent to which Zbtb32 controlled the various cellular processes of proliferation, survival and apoptosis in activated NK cells, any one of which could explain the observed population expansion defect. Transfer of CFSE-labeled Ly49H+ NK cells confirmed that Zbtb32-deficiency significantly impaired MCMV-driven proliferation in vivo (Fig. 5a). The impaired proliferative capacity of Zbtb32−/− Ly49H+ NK cells was further corroborated by their decreased expression of the proliferation marker Ki67 (Fig. 5b) and by their inability to incorporate BrdU after short-term pulses on day 4 and day 6 p.i. (Fig. 5c). This defect was mirrored at a molecular level by a failure of Zbtb32-deficient NK cells to upregulate a suite of genes involved in cell cycle progression (Fig. 5d).
Figure 5. Zbtb32 regulates NK cell proliferation following MCMV infection.
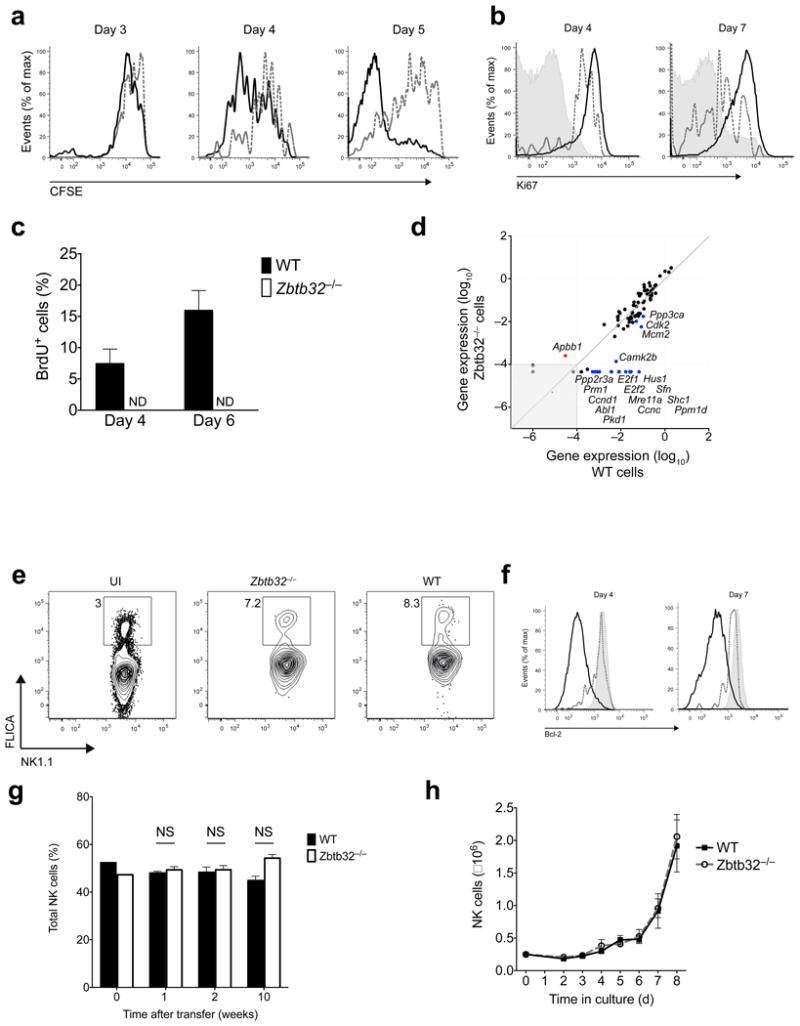
(a–f) Equal numbers of WT (CD45.1+; black lines in histograms) and Zbtb32−/− (CD45.2+; red lines in histograms) Ly49H+ NK cells were co-transferred into Ly49H-deficient hosts one day prior to MCMV infection. (a) CFSE dilution of transferred Ly49H+ NK cell populations in the spleen at indicated time points p.i. Representative of n = 6 mice per time point from 3 independent experiments. (b) Intracellular Ki67 expression in transferred Ly49H+ NK cells on day 4 and 7 p.i. (shaded histograms show uninfected controls). Representative of n = 4 mice per time point from 2 independent experiments. (c) Percentage of BrdU incorporation in transferred Ly49H+ NK cells in the spleen at indicated time points p.i. with MCMV (n = 2 mice per time point). Representative of 2 independent experiments. (d) Expression of 84 genes involved in cell cycle regulation on day 4 p.i. as measured by Cell Cycle PCR Array™ (Qiagen). Genes up- (red) or down-regulated (blue) >4-fold in Zbtb32−/− versus WT Ly49H+ splenic NK cells are annotated. (e) Pan-caspase activation in transferred Ly49H+ NK cell populations in the spleens day 4 p.i. or ininfected (UI) mice. Representative of n = 2 animals per group from 2 independent experiments. (f) Intracellular Bcl-2 expression in transferred Ly49H+ NK cells in the spleen on day 4 and 7 p.i. (shaded histogram shows uninfected controls). Representative of n = 2 – 3 animals per group from 3 independent experiments. (g) WT and Zbtb32−/− Ly49H+ NK cells were co-transferred into Rag2−/− × Il2rg−/− hosts and the relative percentage of each population in the peripheral blood (0, 1, and 2 weeks) or spleen (10 weeks) was determined (n = 3 mice per time point). Representative of 2 independent experiments. (h) Sorted splenic NK cells from WT and Zbtb32−/− mice were incubated with IL-2 and IL-15 and the absolute number of NK cells was determined daily. Data are representative of n = 3 biological replicates per group from 2 independent experiments.
Because Zbtb32 was found to be upregulated to a similar degree in both Ly49H+ and Ly49H− NK cells after infection (Supplementary Fig. 3b), we tested whether Zbtb32 might also regulate the low-level NK cell proliferation that occurs independently of Ly49H-m157 engagement. Indeed, even Ly49H− NK cells showed impaired proliferation in the absence of Zbtb32, as did Ly49H+ NK cells responding to infection with MCMV Δm157 (Supplementary Fig. 3c,d), although this defect was subtle given that all NK cells proliferated very little in these settings.
In contrast to their proliferation defect, Zbtb32−/− NK cells exhibited no evidence of enhanced apoptosis by staining for Annexin V, the pro-apoptotic molecule Bim, or activated caspases (Fig. 5e and data not shown). In T cells, the pro-survival protein Bcl-2 is downregulated in effector cells with the highest proliferative capacity, and subsequent apoptosis of these highly proliferative Bcl-2lo cells marks the contraction phase of the effector T cell response34,35. Consistent with a defect in proliferation but not survival, Zbtb32−/− NK cells failed to undergo activation-induced downregulation of Bcl-2 (Fig. 5f). Thus, these findings highlight a specific requirement for Zbtb32 in the proliferation but not survival of NK cells following viral infection.
Zbtb32 is dispensible for development and homeostasis
The NK cell compartment of Zbtb32−/− animals was phenotypically indistinguishable from that of wild-type animals, suggesting that Zbtb32-deficiency did not impair NK cell development or abrogate proliferation in that context. The percentage and number of mature NK cells in peripheral organs and of NK cell developmental subsets in the bone marrow of Zbtb32-deficient animals was comparable to that observed in wild-type animals (Supplementary Fig. 4a,b and data not shown). Similarly, NK cells in Zbtb32−/− animals expressed normal amounts of markers typically associated with NK cell identity and function, including Ly49H, Ly49D, NK1.1, DX5, Ly49C or Ly49I and Ly49A (Supplementary Fig. 4c,d).
In addition, wild-type and Zbtb32-deficient NK cells repopulated the hematopoietic compartment of mixed bone marrow chimeric animals with similar efficiencies (Supplementary Fig. 5, left panel), further confirming that Zbtb32 was dispensible for NK development. However, when wild-type and Zbtb32−/− Ly49H+ NK cells from mixed bone marrow chimeric animals were adoptively transferred into naive Ly49H-deficient recipients, only the wild-type population was able to expand following MCMV challenge, reaffirming that even Zbtb32−/− NK cells that developed in a Zbtb32-sufficient environment exhibited an expansion defect after infection (Supplementary Fig. 5, middle and right panels). Consistent with a dispensable role in NK cell proliferation during development, Zbtb32−/− NK cells exhibited normal homeostatic proliferation when transferred into lymphocyte-deficient hosts (Fig. 5g) and when expanded ex vivo with IL-2 and IL-15, cytokines important for NK cell proliferation and survival during development and at steady-state (Fig. 5h). Thus, Zbtb32 does not appear to be necessary for NK cell proliferation in a non-inflammatory environment. Collectively, our data indicate that Zbtb32 is dispensible for development and homeostasis-associated proliferation, but rather specifically regulates NK cell proliferation in the context of an infectious or inflammatory setting.
Inflammation drives Zbtb32 expression in NK cells
Because maximal levels of Zbtb32 mRNA in NK cells were observed at early time points in vivo (by ∼48 h p.i.), when inflammation is high but viral dissemination (and thus antigen availability) is still lagging, and because both Ly49H+ and Ly49H− NK cells were able to upregulate Zbtb32 after infection, we hypothesized that signals from proinflammatory cytokines, rather than interactions between the Ly49H receptor and the m157 viral antigen, might control Zbtb32 expression in activated NK cells. Indeed, resting NK cells incubated ex vivo with the proinflammatory cytokines IL-12, IL-18, or IFN-α/β induced high expression of Zbtb32 mRNA, with IL-12 and IL-18 co-treatment exerting a strongly synergistic effect (Fig. 6a). In contrast, Zbtb32 mRNA was barely detectable following crosslinking of the activating receptors Ly49H, Ly49D, NKG2D or NKp46. To test whether signals from pro-inflammatory cytokines were required for Zbtb32 induction in vivo, we utilized mixed bone marrow chimeric animals harboring both wild-type NK cells and NK cells lacking receptors for either type I IFNs, IL-12, or IL-18, or for both type I IFNs and IL-12. Compared to their wild-type counterparts, cytokine receptor-deficient NK cells were markedly impaired in their ability to upregulate Zbtb32 mRNA following MCMV infection in vivo, with NK cells lacking both the IL-12 and the type I interferon receptors showing the greatest defect (Fig. 6b).
Figure 6. Inflammatory cytokines are necessary and sufficient for maximal Zbtb32 expression in NK cells.
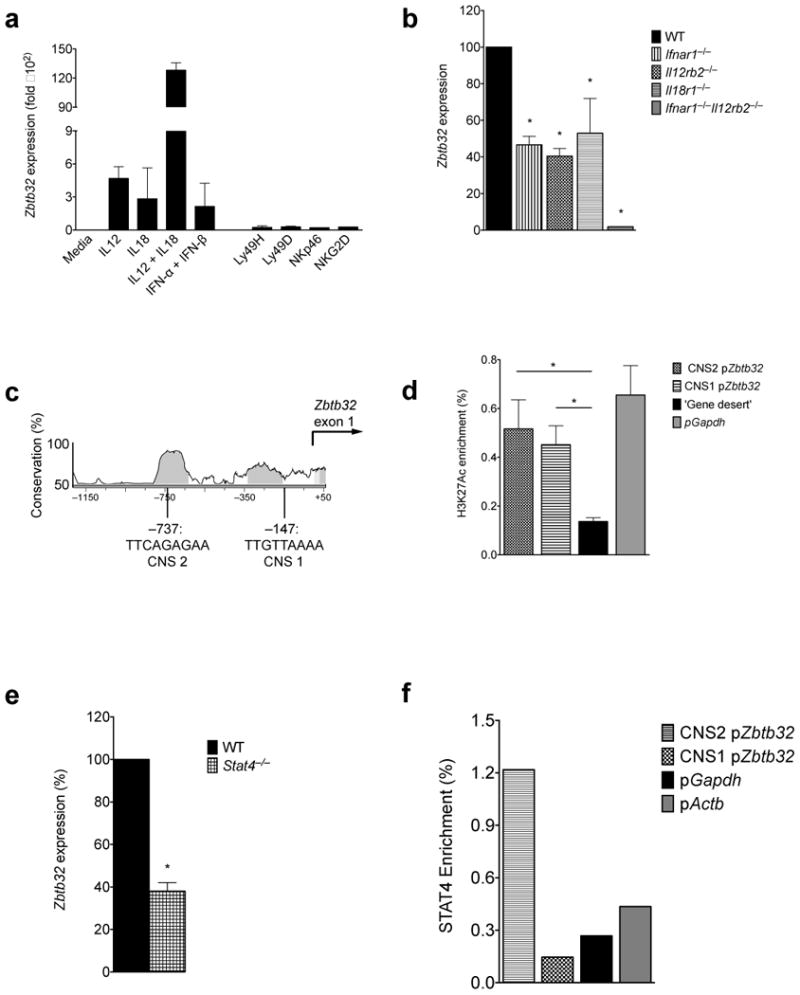
(a) Expression of Zbtb32 mRNA in stimulated NK cells, expressed as fold expression after treatment with medium only, as assessed by qRT-PCR (n = 3 biological replicates per condition). Data are representative of 3 independent experiments. (b) WT or mutant Ly49H+ NK cells were sorted from the spleens of mixed bone marrow chimeric mice on day 2 p.i. with MCMV. Zbtb32 mRNA abundance, as a percentage of expression in WT cells, was assessed by qRT-PCR (n = 2 – 8 animals) from 3 pooled experiments. (c) Vista browser image of conserved noncoding sequences (CNS; gray shaded regions) and 2 predicted STAT4 binding sites within the mouse Zbtb32 promoter. (d) Enrichment of acetylated (lysine 27) histone 3 at the Zbtb32 promoter, as assessed by ChIP followed by qPCR on sorted WT NK cells (n = 3 biological replicates). Enrichment of target (Zbtb32 promoter) or control DNA (Gapdh promoter or ‘gene desert’ ∼50 kb upstream of Foxp3 gene) is expressed as percentage of input. Dotted line denotes “background-level” enrichment at the gene desert locus. Representative of 3 independent experiments. (e) As in (b), except using WT:Stat4−/− mixed bone marrow chimeric mice (n = 4 mice from 2 independent experiments). (f) Purified WT NK cells were stimulated for 24 h with IL-12 and IL-18, and STAT4 binding at the Zbtb32 promoter was assessed by ChIP followed by qPCR. Target (Zbtb32) or control (Actb and Gapdh) promoter DNA levels are expressed as a percentage of input. Dotted line denotes “background-level” enrichment, set as the mean enrichment of the pGapdh and pActb control loci. Representative of 2 independent experiments.
Given that pro-inflammatory cytokines induce Zbtb32, we postulated that specific transcriptional activators downstream of cytokine receptors might directly regulate Zbtb32 expression in NK cells. Analysis of the Zbtb32 promoter revealed several conserved non-coding sites (CNS) within a DNase I hypersensitivity region previously identified in human NK cells36 (Fig. 6c). Chromatin immunoprecipitation (ChIP) experiments showed enrichment of acetylated histone marks at these sites (specifically, H3K27Ac, which marks transcriptionally active promoters), consistent with a likely role in transcriptional activation of Zbtb32 (Fig. 6d). In support of direct regulation by cytokine receptor signaling, the CNS regions of the Zbtb32 promoter were found to harbor putative binding sites for the transcription factor STAT4, which acts downstream of the IL-12 receptor to induce target gene expression in lymphocytes (Fig. 6c). Similar to IL-12R-deficient NK cells, NK cells lacking STAT4 were also significantly impaired in MCMV-induced Zbtb32 expression (Fig. 6e). Using STAT4 ChIP, we observed substantial binding of STAT4 at a CNS in the Zbtb32 promoter approximately 750 base pairs upstream of the transcriptional start site, indicating that Zbtb32 may be a direct target of STAT4, acting as a transmitter of IL-12 signaling (Fig. 6f). Decreased Zbtb32 expression correlates with the impaired proliferation recently observed in IL-12R- or STAT4-deficient NK cells37, consistent with a central role for Zbtb32 in transmitting pro-proliferative signals downstream of inflammatory cues. Thus, signals from proinflammatory cytokines, but not activating receptors, are sufficient to induce and are required for maximal Zbtb32 expression in primary NK cells.
Zbtb32 promotes proliferation by antagonizing Blimp-1
We next sought to identify the specific pathway(s) regulated by Zbtb32 in proliferating NK cells. Zbtb32-deficiency did not affect expression or phosphorylation of STAT1, STAT3, STAT4 or T-bet in activated NK cells, transcription factors with proven or suspected roles in NK cell effector function37–39, nor did it influence the expression of Gata3, a known target of Zbtb32 in T cells28,30 (Supplementary Fig. 6a). These data are consistent with our findings that the non-proliferative features of NK cell activation, including production of IFN-γ and cytotoxic molecules, are intact in the absence of Zbtb32.
Because cell cycle genes are dysregulated in Zbtb32−/− NK cells during MCMV infection (Fig. 5d), we investigated the mechanism(s) by which Zbtb32 may be promoting cell cycle progression in NK cells. In T and B cells, proliferation of effector lymphocytes (for example, germinal center B cells) depends upon suppression of the anti-proliferative/pro-terminal differentiation factor, Blimp-1 (encoded by Prdm1)40. Blimp-1 is upregulated in NK cells during development, where it functions to restrict proliferation, and remains highly expressed in mature resting NK cells. Although the BTB-ZF protein, Bcl-6, is the main antagonist of Blimp-1 in B and T cells40, it does not play this role in developing NK cells41 and is unlikely to do so in activated NK cells, since Bcl-6 is actually down-regulated after infection (Fig. 1b and Supplementary Fig. 6b). Given the high degree of homology between Bcl-6 and Zbtb32, we hypothesized that Zbtb32 rather than Bcl-6 may function in activated NK cells to suppress Blimp-1 expression or function and thereby facilitate antigen-driven proliferation. This idea was supported by a recent report showing a physical interaction between Zbtb32 and Blimp-1 in B cells42 and our own observation that Prdm1 expression was elevated in Zbtb32−/− Ly49H+ NK cells (Fig. 7a). However, to formally test this hypothesis, we generated mice harboring NK cells in which both Zbtb32 and Prdm1 have been genetically deleted (Zbtb32−/−Prdm1fl/flNkp46-Cre+). We predicted that if the primary function of Zbtb32 was to suppress or antagonize Blimp-1 function, then deletion of Blimp-1 in Zbtb32-deficient NK cells should result in a partial or complete restoration of Ly49H+ NK cell expansion during MCMV infection. Indeed, we found that whereas NK cells from Zbtb32−/−Prdm1fl/flNkp46-Cre- (Zbtb32-deficient only) donors were severely impaired in their ability to expand after infection, NK cells from Zbtb32−/−Prdm1fl/flNkp46-Cre+ (Zbtb32 and Prdm1 double-deficient) littermates were fully rescued from this defect and proliferated as well as wild-type cells (Fig. 7b,c). Thus, consistent with our hypothesis, Zbtb32 was no longer required for MCMV-driven NK cell proliferation when Blimp-1 was not present. These findings provide a molecular explanation for how Zbtb32 acts downstream of pro-inflammatory signals to promote a pro-proliferative state in activated NK cells, and reveal a novel antagonistic interaction between Zbtb32 and Blimp-1 that regulates NK cell expansion during infection (Supplementary Fig. 7).
Figure 7. Zbtb32 promotes NK cell proliferation by antagonizing Blimp-1.

(a) WT and Zbtb32−/− Ly49H+ NK cells were co-transferred into Ly49H-deficient hosts and then recovered from the spleen by sorting on day 4 p.i. with MCMV. Prdm1 mRNA abundance, as a percentage of expression in WT cells, was quantified by qRT-PCR in each population (n = 4 mice). Data are representative of 2 independent experiments. (b,c) Ly49H+ NK cells from Prdm1fl/flNkp46-Cre−, Prdm1fl/flNkp46-Cre+, Zbtb32−/−Prdm1fl/flNkp46-Cre−, or Zbtb32−/−Prdm1fl/flNkp46-Cre+ littermate animals (CD45.2+) were co-transferred with equal numbers of Ly49H+ NK cells from WT animals (CD45.1+) into Ly49H-deficient hosts one day prior to infection with MCMV. The relative percentages of transferred Ly49H+ WT and CD45.2+ littermate NK cells in the spleen on day 7 p.i. are shown. Depicted are (b) representative responses in individual animals (except the Prdm1fl/flNkp46-Cre+ cohort) and (c) summary data (symbols represent individual mice). Data are representative of 3 independent experiments.
Discussion
A growing number of recent studies in mice and humans have led to the unexpected discovery that NK cells can mount antigen-specific responses with features of adaptive immunity, including robust clonal proliferation, long-term persistence, and enhanced secondary responses of virus-specific NK cells1. Before now, the molecular events that regulate these antigen-specific responses were almost completely unknown. Here, we demonstrate that the transcription factor Zbtb32 is essential for the proliferation and protective functionality of MCMV-specific NK cells responding to infection. Zbtb32-deficiency ablates the proliferative burst of antigen-specific NK cells and impairs the low-level proliferation driven by antigen-independent signals in “bystander” NK cells. We further show that signals from pro-inflammatory cytokines were necessary and sufficient to induce Zbtb32 expression in activated NK cells. At a mechanistic level, Zbtb32 was found to promote a pro-proliferative state in activated NK cells by antagonizing the tumor suppressor factor, Blimp-1. In summary, this study is the first to elucidate the molecular pathway behind the proliferative burst of antigen-specific NK cells, demonstrating not only that Zbtb32 is the molecular “link” between inflammation and proliferation in NK cells, but also revealing a previously uncharacterized antagonistic interaction between Zbtb32 and Blimp-1.
The finding that proinflammatory cytokines are essential for maximal Zbtb32 expression provides a mechanistic explanation for how and why inflammatory signals are required for the robust proliferation of antigen-specific NK cells during MCMV infection, even when high amounts of viral antigen are available37. This pathway may be analgous to “Signal 3” in the widely-accepted model of T cell activation, which holds that full efffector function relies on three independent and contemporal signals from the TCR (Signal 1), co-stimulatory receptors such as CD28 (Signal 2), and cytokine receptors such as IFN-αR and IL12R (Signal 3). In a similar manner, inflammation-driven upregulation of Zbtb32 may act as an important checkpoint for effector responses by NK cells, only allowing robust antigen-driven proliferation when clear signals of infection are present. Indeed, the intimate connection between inflammation and Zbtb32 expression is consistent with our finding that Zbtb32 is dispensible for NK cell proliferation in non-inflammatory settings, such as during development, homeostatic turn-over, and long-term memory cell maintenance.
During infection, both IL-12 signaling (e.g. as reflected by STAT4 phosphorylation) and Zbtb32 expression peak early after infection, yet this pathway has a critical, long-lasting impact on the proliferative response that continues for days after these initiating signals have returned to baseline. These findings are consistent with a model in which Zbtb32 acts as a “priming” signal in inflammation-activated NK cells, suppressing Blimp-1 function and thereby unleashing a long-lasting proliferative potential in antigen-specific and non-specific NK cells responding to infection. We speculate that this may be achieved, at least in part, by the well-established function of both Blimp-1 and BTB-ZF proteins (including Zbtb32) in remodeling the chromatin environment at target loci by recruiting histone modifying enzymes, including histone acetylases (HATs), histone methyltransferases, and histone deacetylases (HDACs)10,43,44. Perhaps Zbtb32 promotes proliferation in NK cells by suppressing or subverting the ability of Blimp-1 to alter chromatin structure at certain loci such as pro- and anti-proliferative target genes, and in this way permits the proliferative responses that continue for days after Zbtb32 expression peaks. In support of this idea, a recent study in B cells demonstrated that Zbtb32 and Blimp-1 could physically interact at a protein level and could co-localize at certain loci targeted by Blimp-1 (ref. 42). It will be interesting to determine whether Zbtb32 also complexes with Blimp-1 in NK cells, and whether such an interaction might explain the antagonistic relationship between Zbtb32 and Blimp-1 in NK cell proliferation.
The role of Zbtb32 in regulating tumor suppressor and cell cycle pathways in NK cells is consistent with the reported functions of other BTB-ZF family members, both in lymphocytes and in non-hematopoietic cells9,10. For example, LRF, has been shown to repress Cdkn1a/p21 and Cdkn2a/p19Arf in germinal center B cells45. Similarly, Bcl-6 facilitates the survival and proliferation of germinal center B cells undergoing class switch recombination and somatic hypermutation by repressing transcription of the tumor suppressor genes, TP53 and Prdm1, and the cell cycle arrest gene, CDNK1A/P21 (refs. 40, 46–49). Our finding that Zbtb32 regulates NK cell proliferation principally by antagonizing Blimp-1 sheds light on the previously unexplained observation that NK cells could robustly proliferate during infection despite expressing consistently high amounts of Blimp-1 (ref. 41), a factor generally recognized to restrain lymphocyte proliferation40.
Similar to our findings in MCMV-infected mice, recent studies have described clonal-like NK cell proliferation and long-lived memory-like NK cells in humans infected with human CMV50–52. In as much as pro-inflammatory cytokines can drive certain adaptive immune properties in human NK cells in vitro53, it will be important to determine whether the human Zbtb32 homolog also controls antigen-specific NK cell responses in humans during pathogen infection. Nonetheless, our current findings uncover novel molecular events that control antigen-specific NK cell responses and may have potential utility in informing therapeutic approaches that harness NK cells in the treatment of human disease.
Methods
Mice
Mice were bred at MSKCC in accordance with the guidelines of the Institutional Animal Care and Use Committee (IACUC). The following strains were used in this study: C57BL/6 (CD45.2+; The Jackson Laboratory), B6.SJL (CD45.1+; Taconic), Rag2−/− × Il2rg−/− (Taconic), Il18r1−/−54, Il12rb2−/−55, Stat4−/−56, Ifnar1−/−57, Klra8−/− (Ly49H-deficient)58, B2m−/− (Taconic), m157-transgenic59, Nkp46iCre (herein called Nkp46-Cre)60, Prdm1fl/fl61, and Zbtb32−/−31 mice. Femurs from Il12rb2−/− × Ifnar1−/− mice were provided by S. Way (University of Minnesota) for making mixed bone marrow chimeras. Zbtb32−/−Prdm1fl/flNkp46iCre/wt animals and littermate controls were generated by breeding at MSKCC. Unblinded experiments were conducted using age- and gender-matched animals in accordance with approved institutional protocols.
Mixed bone marrow chimeric mice were generated as described8. Briefly, host C57BL/6 × B6.SJL animals (CD45.1+CD45.2+) were lethally irradiated with 900 grays of radiation, and reconstituted with a 1:1 mixture of bone marrow cells from B6.SJL WT (CD45.1+) and knock-out donor animals (CD45.2+), co-injected with anti-NK1.1 (clone PK136) to deplete any residual donor or host mature NK cells. Host NK cells were excluded from all analyses.
Virus infections
MCMV (Smith Strain) and MCMVΔm157 were obtained from L. Lanier (UCSF) and U. Koszinowski (Max von Pettenkofer-Institute), respectively. MCMV was passaged serially through BALB/c hosts two times, then viral stocks were prepared by douncing the salivary glands of infected animals 3 weeks p.i. For neonate protection studies, 4-day old Ly49H-deficient neonates received 1 × 106 WT or Zbtb32−/− Ly49H+ NK cells by intraperitoneal (i.p.) injection and were infected by i.p. injection of 2 × 103 PFU of MCMV on the following day. WT, Zbtb32−/− and mixed bone marrow chimera mice were infected by i.p. injection of 7.5 × 103 PFU of MCMV. For the adoptive transfer studies, animals were infected by i.p. injection of 7.5 × 102 PFU of MCMV one day after receiving approximately 1 × 106 Ly49H+ NK cells.
VSV-Indiana and recombinant VSV-m157, made by cloning the coding sequence for the MCMV glycoprotein m157 into the parental VSV-Indiana strain62 were provided by K. Schluns (MD Anderson). For the VSV protection studies, adult Ly49H-deficient mice were infected by intravenous (i.v.) injection of 1.5 × 109 PFU of VSV-m157 one day after receiving 1 × 106 WT or Zbtb32−/− Ly49H+ NK cells. In other adoptive transfer experiments, mice were infected by i.v. injection of 1 × 107 PFU of VSV-m157 or VSV-Indiana one day after receiving approximately 1 × 106 Ly49H+ NK cells.
VacV-m157 was provided by G. Gasteiger (Memorial Sloan-Kettering Cancer Center) and animals were infected by i.p. injection with 1 × 107 PFU.
Flow cytometry and cell sorting
Cell surface staining was performed using the following fluorophore-conjugated antibodies (purchased from BD Biosciences, eBioscience, BioLegend, and R&D Systems): NK1.1 (PK136), CD11b (M1/70), CD27 (LG.3A10), CD49b/DX5 (DX5), KLRG1 (2F1), NKp46 (29A1.4), CD69 (H1.2F3), Ly49H (3D10) Granzyme B (16G6), CD107a (1D4B), CD45.1 (A20), CD45.2 (104), CD8α (53-6.7), CD122 (TM-β1), TCRβ (H57-597), CD3ε (145-2C11), IFN-γ (XMG1.2), Ly49D (4E5), Ly49A (A1/Ly49A), Ly49C/I (5E6), T-bet (4B10), Bcl-2 (3F11), Ki67 (B56), Gata3 (L50-823), Stat1-pY701 (4a), Stat3-pY705 (4/P-STAT3), Stat4-pY693 (38/p-Stat4) and peptide-loaded MHC class I tetramers (MCMV peptides m45, m38, or m139; NIH Tetramer Facility). Unless otherwise indicated, NK cells were defined as TCRβ-NK1.1+ or CD3-NK1.1+ cells. Intracellular staining was performed by fixing and permeabilizing with the Foxp3/Transcription Factor Staining Kit (eBioscience) for staining intranuclear proteins, including Zbtb32; with Cytofix/Cytoperm™ Plus (BD) for staining cytokines; or with formaldehyde and methanol for staining phosphorylated STAT proteins. For the detection of intracellular Zbtb32, permeabilized cells were stained with 4 μg/ml of a rat anti-Zbtb32 primary antibody (#853111, generously provided by R&D Systems) for 45 min at 4 °C, then with a FITC-conjugated anti-rat secondary antibody for 10 min at 4 °C. Pan-caspase staining was carried out using the FAM FLICA™ in vitro Poly Caspase Kit (Immunochemistry Technologies) per the manufacturer's instructions. Positive control cells were first incubated with 20 μg/ml of anti-mouse CD95 (clone Jo-2; BD Pharmingen) for 2.5 – 4 h at 37 °C (data not shown). For the BrdU labeling experiments, animals were injected i.p. with 1.5 mg of BrdU in PBS on days 3.5 and 5.5 p.i. with MCMV, and BrdU labeling was determined approximately 16 hours later using the APC BrdU Flow Kit (BD Biosciences).
Flow cytometry and cell sorting were performed on the LSR II and Aria II cytometers (BD Biosciences), respectively. For experiments involving qRT-PCR, cell populations were sorted to >95% purity. Data were analyzed with FlowJo software (Tree Star).
NK cell enrichment and adoptive transfer
T, B, and red blood cells were labeled with 10 μg per spleen of rat monoclonal antibodies against CD4 (GK1.5), CD8 (53.6.72), CD19 (1D3) and Ter119 (obtained from the UCSF Core Facility) and magnetically depleted from total splenocyte suspensions using anti-rat IgG-coupled magnetic beads (Qiagen)37. Approximately 1 × 106 enriched NK cells (typically 25-70% of the total cells) were injected i.v. into adult or i.p into neonate recipients one day before infection. In some experiments, splenocytes were labeled prior to transfer with 5 μM Vybrant™ CFDA SE (Molecular Probes).
Ex vivo stimulation of NK cells
Approximately 1 × 105 sorted NK cells were stimulated for 18 hours in RPMI containing 5% fetal bovine serum with recombinant mouse IL-12 (20 ng/ml; R&D Systems), IL-18 (10 ng/ml; R&D Systems), or IFN-α/β (400 U/mL) or with plate-bound antibodies (10 μg/ml; eBioscience) against the activating NK cell receptors Ly49H (3D10), Ly49D (4E5), NKp46 (29A1.4), or NK1.1 (PK136).
For ex vivo NK cell expansion experiments, 2.5 × 105 sorted NK cells (>98% purity) were stimulated in RPMI supplemented with 10% fetal bovine serum, penicillin/streptomycin, L-glutamine and β-mercaptoethanol, and containing recombinant mouse IL-2 (20 ng/ml; R&D Systems) and IL-15 (40 ng/ml; R&D Systems). Fresh media and cytokines were added on alternating days of culture.
Quantitative RT-PCR
Cells were lysed in Tri-Reagent (Ambion). RNA purification and cDNA synthesis were carried out with the Qiagen RNeasy kit (with on-column DNase I treatment), and MuLV reverse transcriptase and Oligo dT(16) primers (Applied Biosystems). iQ Sybr Green SuperMix (BioRad) was used for qRT-PCR. Data were normalized to Actb and expressed as relative target abundance via the ΔΔCT method. Supplementary Table 1 lists relevant primer sequences. For Fig. 5d, the Mouse Cell Cycle PCR Array, RT2 SYBR Green qPCR Master Mix, and Mouse Cell Cycle RT2 PreAMP cDNA synthesis kits (Qiagen) were used.
Chromatin Immunoprecipitation (ChIP)
Approximately 5 × 106 enriched splenic NK cells were purified by sorting on TCRβ−NK1.1+ cells. Proteins were cross-linked to chromatinized DNA for 8 min at 25 °C by addition of 1% formaldehyde to the medium. ChIP was carried out as previously described63 using 10 μg of rabbit polyclonal anti-STAT4 (Santa Cruz, sc-468, clone C-20) or 10 μg of rabbit serum IgG antibody as a control (data not shown; Sigma, I5006), or 0.5 μg of anti-H3K27Ac (Abcam, 4729). Relative abundance of regulatory sequences in the Zbtb32 promoter or, as controls, in the Actb and Gapdh promoters or a gene desert ∼50 kb upstream of the Foxp3 gene was measured by qPCR in the antibody-precipitated DNA. Percent input was calculated by 100 × 2̂(CTadjusted input – CTtarget), where the CTinput was adjusted from 5% to 100% by subtracting Log2 20 CTs. Primer sequences are listed in Supplementary Table 2.
Statistical Analyses
For graphs, data are shown as mean ± SEM and, unless otherwise indicated, statistical differences were evaluated using a two-tailed unpaired Student's t-test, assuming equal sample variance. P values < 0.05 were considered significant and are denoted by an asterisk (*). Statistical differences in survival were determined by Gehan-Breslow-Wilcoxon Test analysis. Graphs were produced and statistical analyses were performed using GraphPad Prism.
Supplementary Material
Acknowledgments
We thank members of the Sun lab for technical support and experimental assistance, and members of the MSKCC NK club for insightful comments and helpful discussions. We thank A. Rudensky, M. van den Brink, M. Li, L. Lanier (UCSF), D. Sant'Angelo (Rutgers University), and L. Denzin (Rutgers University) for sharing antibodies and flow cytometry resources, and for providing expertise critical to this study and manuscript. We thank G. Gasteiger, K. Schluns (MD Anderson), and U. Koszinowski (Max von Pettenkofer-Institute) for generously providing many of the parent and recombinant viruses used in our study. We thank S. Way (University of Minnesota) for providing femurs from Il12rb2−/− × Ifnar1−/− mice for use in making bone marrow chimeras. We thank the Immunological Genome Consortium for providing the microarray data used in this study26. We thank J.P. Houchins and his team at R&D Systems for generously providing the experimental anti-Zbtb32 flow cytometry antibody used in this study. A.M.B. was supported by an NIH T32 award (CA009149). J.C.S. was supported by the Searle Scholars Program, the Cancer Research Institute, and grants from the NIH (AI085034 and AI100874).
Footnotes
Author Contributions: A.B. and C.Z. performed the experiments; T.N. provided the Zbtb32−/− mice and feedback on the manuscript; A.B. and J.S. designed the study and wrote the manuscript.
Competing Financial Interests: The authors declare no financial conflicts of interest.
References
- 1.Sun JC, Lanier LL. NK cell development, homeostasis and function: parallels with CD8(+) T cells. Nat Rev Immunol. 2011;11:645–657. doi: 10.1038/nri3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Smith HR, et al. Recognition of a virus-encoded ligand by a natural killer cell activation receptor. Proc Natl Acad Sci U S A. 2002;99:8826–8831. doi: 10.1073/pnas.092258599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arase H, Mocarski ES, Campbell AE, Hill AB, Lanier LL. Direct recognition of cytomegalovirus by activating and inhibitory NK cell receptors. Science. 2002;296:1323–1326. doi: 10.1126/science.1070884. [DOI] [PubMed] [Google Scholar]
- 4.Daniels KA, et al. Murine cytomegalovirus is regulated by a discrete subset of natural killer cells reactive with monoclonal antibody to Ly49H. J Exp Med. 2001;194:29–44. doi: 10.1084/jem.194.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brown MG, et al. Vital involvement of a natural killer cell activation receptor in resistance to viral infection. Science. 2001;292:934–937. doi: 10.1126/science.1060042. [DOI] [PubMed] [Google Scholar]
- 6.Bubic I, et al. Gain of virulence caused by loss of a gene in murine cytomegalovirus. J Virol. 2004;78:7536–7544. doi: 10.1128/JVI.78.14.7536-7544.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dokun AO, et al. Specific and nonspecific NK cell activation during virus infection. Nat Immunol. 2001;2:951–956. doi: 10.1038/ni714. [DOI] [PubMed] [Google Scholar]
- 8.Sun JC, Beilke JN, Lanier LL. Adaptive immune features of natural killer cells. Nature. 2009;457:557–561. doi: 10.1038/nature07665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kelly KF, Daniel JM. POZ for effect--POZ-ZF transcription factors in cancer and development. Trends Cell Biol. 2006;16:578–587. doi: 10.1016/j.tcb.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 10.Beaulieu AM, Sant'Angelo DB. The BTB-ZF family of transcription factors: key regulators of lineage commitment and effector function development in the immune system. J Immunol. 2011;187:2841–2847. doi: 10.4049/jimmunol.1004006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maeda T, et al. Regulation of B versus T lymphoid lineage fate decision by the proto-oncogene LRF. Science. 2007;316:860–866. doi: 10.1126/science.1140881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bilic I, et al. Negative regulation of CD8 expression via Cd8 enhancer-mediated recruitment of the zinc finger protein MAZR. Nat Immunol. 2006;7:392–400. doi: 10.1038/ni1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sakaguchi S, et al. The zinc-finger protein MAZR is part of the transcription factor network that controls the CD4 versus CD8 lineage fate of double-positive thymocytes. Nat Immunol. 2010;11:442–448. doi: 10.1038/ni.1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun G, et al. The zinc finger protein cKrox directs CD4 lineage differentiation during intrathymic T cell positive selection. Nat Immunol. 2005;6:373–381. doi: 10.1038/ni1183. [DOI] [PubMed] [Google Scholar]
- 15.He X, et al. The zinc finger transcription factor Th-POK regulates CD4 versus CD8 T-cell lineage commitment. Nature. 2005;433:826–833. doi: 10.1038/nature03338. [DOI] [PubMed] [Google Scholar]
- 16.Muroi S, et al. Cascading suppression of transcriptional silencers by ThPOK seals helper T cell fate. Nat Immunol. 2008;9:1113–1121. doi: 10.1038/ni.1650. [DOI] [PubMed] [Google Scholar]
- 17.Kovalovsky D, et al. The BTB-zinc finger transcriptional regulator PLZF controls the development of invariant natural killer T cell effector functions. Nat Immunol. 2008;9:1055–1064. doi: 10.1038/ni.1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alonzo ES, et al. Development of promyelocytic zinc finger and ThPOK-expressing innate gamma delta T cells is controlled by strength of TCR signaling and Id3. J Immunol. 2010;184:1268–1279. doi: 10.4049/jimmunol.0903218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Savage AK, et al. The transcription factor PLZF directs the effector program of the NKT cell lineage. Immunity. 2008;29:391–403. doi: 10.1016/j.immuni.2008.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kreslavsky T, et al. TCR-inducible PLZF transcription factor required for innate phenotype of a subset of gammadelta T cells with restricted TCR diversity. Proc Natl Acad Sci U S A. 2009;106:12453–12458. doi: 10.1073/pnas.0903895106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dent AL, Shaffer AL, Yu X, Allman D, Staudt LM. Control of inflammation, cytokine expression, and germinal center formation by BCL-6. Science. 1997;276:589–592. doi: 10.1126/science.276.5312.589. [DOI] [PubMed] [Google Scholar]
- 22.Ye BH, et al. The BCL-6 proto-oncogene controls germinal-centre formation and Th2-type inflammation. Nat Genet. 1997;16:161–170. doi: 10.1038/ng0697-161. [DOI] [PubMed] [Google Scholar]
- 23.Yu D, et al. The transcriptional repressor Bcl-6 directs T follicular helper cell lineage commitment. Immunity. 2009;31:457–468. doi: 10.1016/j.immuni.2009.07.002. [DOI] [PubMed] [Google Scholar]
- 24.Johnston RJ, et al. Bcl6 and Blimp-1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science. 2009;325:1006–1010. doi: 10.1126/science.1175870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nurieva RI, et al. Bcl6 mediates the development of T follicular helper cells. Science. 2009;325:1001–1005. doi: 10.1126/science.1176676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bezman NA, et al. Molecular definition of the identity and activation of natural killer cells. Nat Immunol. 2012 doi: 10.1038/ni.2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bukowski JF, Warner JF, Dennert G, Welsh RM. Adoptive transfer studies demonstrating the antiviral effect of natural killer cells in vivo. J Exp Med. 1985;161:40–52. doi: 10.1084/jem.161.1.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Omori M, et al. CD8 T cell-specific downregulation of histone hyperacetylation and gene activation of the IL-4 gene locus by ROG, repressor of GATA. Immunity. 2003;19:281–294. doi: 10.1016/s1074-7613(03)00210-3. [DOI] [PubMed] [Google Scholar]
- 29.Piazza F, Costoya JA, Merghoub T, Hobbs RM, Pandolfi PP. Disruption of PLZP in mice leads to increased T-lymphocyte proliferation, cytokine production, and altered hematopoietic stem cell homeostasis. Mol Cell Biol. 2004;24:10456–10469. doi: 10.1128/MCB.24.23.10456-10469.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miaw SC, Choi A, Yu E, Kishikawa H, Ho IC. ROG, repressor of GATA, regulates the expression of cytokine genes. Immunity. 2000;12:323–333. doi: 10.1016/s1074-7613(00)80185-5. [DOI] [PubMed] [Google Scholar]
- 31.Hirahara K, et al. Repressor of GATA regulates TH2-driven allergic airway inflammation and airway hyperresponsiveness. J Allergy Clin Immunol. 2008;122:512–520. e511. doi: 10.1016/j.jaci.2008.06.004. [DOI] [PubMed] [Google Scholar]
- 32.Hirasaki Y, et al. Repressor of GATA negatively regulates murine contact hypersensitivity through the inhibition of type-2 allergic responses. Clin Immunol. 2011;139:267–276. doi: 10.1016/j.clim.2011.02.009. [DOI] [PubMed] [Google Scholar]
- 33.Miaw SC, Kang BY, White IA, Ho IC. A repressor of GATA-mediated negative feedback mechanism of T cell activation. J Immunol. 2004;172:170–177. doi: 10.4049/jimmunol.172.1.170. [DOI] [PubMed] [Google Scholar]
- 34.Marrack P, Kappler J. Control of T cell viability. Annu Rev Immunol. 2004;22:765–787. doi: 10.1146/annurev.immunol.22.012703.104554. [DOI] [PubMed] [Google Scholar]
- 35.Dunkle A, Dzhagalov I, Gordy C, He YW. Transfer of CD8+ T cell memory using Bcl-2 as a marker. J Immunol. 2013;190:940–947. doi: 10.4049/jimmunol.1103481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stergachis AB, et al. Developmental fate and cellular maturity encoded in human regulatory DNA landscapes. Cell. 2013;154:888–903. doi: 10.1016/j.cell.2013.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sun JC, et al. Proinflammatory cytokine signaling required for the generation of natural killer cell memory. J Exp Med. 2012;209:947–954. doi: 10.1084/jem.20111760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miyagi T, et al. High basal STAT4 balanced by STAT1 induction to control type 1 interferon effects in natural killer cells. J Exp Med. 2007;204:2383–2396. doi: 10.1084/jem.20070401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hesslein DG, Lanier LL. Transcriptional control of natural killer cell development and function. Adv Immunol. 2011;109:45–85. doi: 10.1016/B978-0-12-387664-5.00002-9. [DOI] [PubMed] [Google Scholar]
- 40.Crotty S, Johnston RJ, Schoenberger SP. Effectors and memories: Bcl-6 and Blimp-1 in T and B lymphocyte differentiation. Nat Immunol. 2010;11:114–120. doi: 10.1038/ni.1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kallies A, et al. A role for Blimp1 in the transcriptional network controlling natural killer cell maturation. Blood. 2011;117:1869–1879. doi: 10.1182/blood-2010-08-303123. [DOI] [PubMed] [Google Scholar]
- 42.Yoon HS, et al. ZBTB32 Is an Early Repressor of the CIITA and MHC Class II Gene Expression during B Cell Differentiation to Plasma Cells. J Immunol. 2012;189:2393–2403. doi: 10.4049/jimmunol.1103371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shin HM, et al. Epigenetic modifications induced by Blimp-1 Regulate CD8(+) T cell memory progression during acute virus infection. Immunity. 2013;39:661–675. doi: 10.1016/j.immuni.2013.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bikoff EK, Morgan MA, Robertson EJ. An expanding job description for Blimp-1/PRDM1. Curr Opin Genet Dev. 2009;19:379–385. doi: 10.1016/j.gde.2009.05.005. [DOI] [PubMed] [Google Scholar]
- 45.Sakurai N, et al. The LRF transcription factor regulates mature B cell development and the germinal center response in mice. J Clin Invest. 2011;121:2583–2598. doi: 10.1172/JCI45682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Phan RT, Dalla-Favera R. The BCL6 proto-oncogene suppresses p53 expression in germinal-centre B cells. Nature. 2004;432:635–639. doi: 10.1038/nature03147. [DOI] [PubMed] [Google Scholar]
- 47.Phan RT, Saito M, Basso K, Niu H, Dalla-Favera R. BCL6 interacts with the transcription factor Miz-1 to suppress the cyclin-dependent kinase inhibitor p21 and cell cycle arrest in germinal center B cells. Nat Immunol. 2005;6:1054–1060. doi: 10.1038/ni1245. [DOI] [PubMed] [Google Scholar]
- 48.Tunyaplin C, et al. Direct repression of prdm1 by Bcl-6 inhibits plasmacytic differentiation. J Immunol. 2004;173:1158–1165. doi: 10.4049/jimmunol.173.2.1158. [DOI] [PubMed] [Google Scholar]
- 49.Vasanwala FH, Kusam S, Toney LM, Dent AL. Repression of AP-1 function: a mechanism for the regulation of Blimp-1 expression and B lymphocyte differentiation by the B cell lymphoma-6 protooncogene. J Immunol. 2002;169:1922–1929. doi: 10.4049/jimmunol.169.4.1922. [DOI] [PubMed] [Google Scholar]
- 50.Lopez-Verges S, et al. Expansion of a unique CD57(+)NKG2Chi natural killer cell subset during acute human cytomegalovirus infection. Proc Natl Acad Sci U S A. 2011;108:14725–14732. doi: 10.1073/pnas.1110900108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Foley B, et al. Cytomegalovirus reactivation after allogeneic transplantation promotes a lasting increase in educated NKG2C+ natural killer cells with potent function. Blood. 2012;119:2665–2674. doi: 10.1182/blood-2011-10-386995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Foley B, et al. Human cytomegalovirus (CMV)-induced memory-like NKG2C(+) NK cells are transplantable and expand in vivo in response to recipient CMV antigen. J Immunol. 2012;189:5082–5088. doi: 10.4049/jimmunol.1201964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Romee R, et al. Cytokine activation induces human memory-like NK cells. Blood. 2012;120:4751–4760. doi: 10.1182/blood-2012-04-419283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hoshino K, et al. Cutting edge: generation of IL-18 receptor-deficient mice: evidence for IL-1 receptor-related protein as an essential IL-18 binding receptor. J Immunol. 1999;162:5041–5044. [PubMed] [Google Scholar]
- 55.Wu C, et al. IL-12 receptor beta 2 (IL-12R beta 2)-deficient mice are defective in IL-12-mediated signaling despite the presence of high affinity IL-12 binding sites. J Immunol. 2000;165:6221–6228. doi: 10.4049/jimmunol.165.11.6221. [DOI] [PubMed] [Google Scholar]
- 56.Kaplan MH, Sun YL, Hoey T, Grusby MJ. Impaired IL-12 responses and enhanced development of Th2 cells in Stat4-deficient mice. Nature. 1996;382:174–177. doi: 10.1038/382174a0. [DOI] [PubMed] [Google Scholar]
- 57.Muller U, et al. Functional role of type I and type II interferons in antiviral defense. Science. 1994;264:1918–1921. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- 58.Fodil-Cornu N, et al. Ly49h-deficient C57BL/6 mice: a new mouse cytomegalovirus-susceptible model remains resistant to unrelated pathogens controlled by the NK gene complex. J Immunol. 2008;181:6394–6405. doi: 10.4049/jimmunol.181.9.6394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tripathy SK, et al. Continuous engagement of a self-specific activation receptor induces NK cell tolerance. J Exp Med. 2008;205:1829–1841. doi: 10.1084/jem.20072446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Narni-Mancinelli E, et al. Fate mapping analysis of lymphoid cells expressing the NKp46 cell surface receptor. Proc Natl Acad Sci U S A. 2011;108:18324–18329. doi: 10.1073/pnas.1112064108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shapiro-Shelef M, et al. Blimp-1 is required for the formation of immunoglobulin secreting plasma cells and pre-plasma memory B cells. Immunity. 2003;19:607–620. doi: 10.1016/s1074-7613(03)00267-x. [DOI] [PubMed] [Google Scholar]
- 62.Kim SK, et al. Generation of mucosal cytotoxic T cells against soluble protein by tissue-specific environmental and costimulatory signals. Proc Natl Acad Sci U S A. 1998;95:10814–10819. doi: 10.1073/pnas.95.18.10814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zheng Y, et al. Genome-wide analysis of Foxp3 target genes in developing and mature regulatory T cells. Nature. 2007;445:936–940. doi: 10.1038/nature05563. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.