

Abstract
Point mutations in the calcium-permeable TRPV4 ion channel have been identified as the cause of autosomal-dominant human motor neuropathies, arthropathies, and skeletal malformations of varying severity. The objective of this study was to determine the mechanism by which TRPV4 channelopathy mutations cause skeletal dysplasia. The human TRPV4V620I channelopathy mutation was transfected into primary porcine chondrocytes and caused significant (2.6-fold) up-regulation of follistatin (FST) expression levels. Pore altering mutations that prevent calcium influx through the channel prevented significant FST up-regulation (1.1-fold). We generated a mouse model of theTRPV4V620I mutation, and found significant skeletal deformities (e.g., shortening of tibiae and digits, similar to the human disease brachyolmia) and increases in Fst/TRPV4 mRNA levels (2.8-fold). FST was significantly up-regulated in primary chondrocytes transfected with 3 different dysplasia-causing TRPV4 mutations (2- to 2.3-fold), but was not affected by an arthropathy mutation (1.1-fold). Furthermore, FST-loaded microbeads decreased bone ossification in developing chick femora (6%) and tibiae (11%). FST gene and protein levels were also increased 4-fold in human chondrocytes from an individual natively expressing the TRPV4T89I mutation. Taken together, these data strongly support that up-regulation of FST in chondrocytes by skeletal dysplasia-inducing TRPV4 mutations contributes to disease pathogenesis.—Leddy, H. A., McNulty, A. L., Lee, S. H., Rothfusz, N. E., Gloss, B., Kirby, M. L., Hutson, M. R., Cohn, D. H., Guilak, F., Liedtke, W. Follistatin in chondrocytes: the link between TRPV4 channelopathies and skeletal malformations.
Keywords: cartilage, calcium signaling, growth plate, bone morphogenetic protein
The discovery of the transient receptor potential (TRP) superfamily of ion channels has significantly increased our understanding of how cells can sense and integrate a diverse array of physical cues that are vital to normal physiology (1–3). In particular, transient receptor potential channel vanilloid 4 (TRPV4) is a polymodally activated Ca2+-permeable nonselective cation channel that is activated by a variety of factors, including osmotic and mechanical stimuli (4–9). For some members of the TRP superfamily of ion channels, human diseases are linked to point mutations in coding regions. Of these, TRPV4 channelopathy mutations are both striking and enigmatic, causing a wide range of neurological and musculoskeletal diseases (10–13). To date, >40 skeletal disease-causing mutations have been identified. For example, the V620I mutation occurs in the 5th transmembrane domain, which is adjacent to the pore loop. It causes elevated basal Ca2+ and an elevated response to TRPV4 agonists in heterologous cellular systems (11). Individuals with the V620I mutation exhibit brachyolmia, a skeletal dysplasia characterized by a mild phenotype with short stature, shortened limbs and digits, and scoliosis (11). At the other end of the disease spectrum, the T89I mutation causes neonatal lethal metatrophic dysplasia. This mutation is located in the cytoplasmic domain of TRPV4 near the N terminus (14). T89I causes elevated basal Ca2+ and elevated response to TRPV4 agonists (15). The A716S mutation, on the other hand, causes disease without an in vitro increase of cellular Ca2+ levels (10). This mutation occurs at the cytoplasmic side of the 6th transmembrane domain, adjacent to the pore loop, and causes a more moderate skeletal phenotype, termed spondylometaphyseal dysplasia, Kozlowski type. These skeletal dysplasia-causing mutations are also characterized by irregular chondrocyte growth (14). Another group of TRPV4 mutations that affects chondrocytes causes a premature osteoarthritis phenotype of joints in the hands and feet, but not a generalized skeletal dysplasia. The prototypical F273L mutation is located within the 3rd ankyrin repeat domain and causes altered membrane trafficking of the channel in heterologous cells so that mutant TRPV4 channels do not reach the plasma membrane (16).
In the musculoskeletal system, TRPV4 has been shown to be a critical regulator of the response of chondrocytes to mechanoosmotic stimuli (17, 18); however, our knowledge of TRPV4 function in cartilage is limited to wild-type (WT) TRPV4. In articular cartilage, osmotic signals play a key role in chondrocyte biology (19) and TRPV4 exhibits high osmotic sensitivity and controls cellular volume recovery and other physiological responses to osmotic stress (18). Mice lacking TRPV4 develop osteoarthritis earlier and more severely than their WT counterparts (17), suggesting that TRPV4-mediated mechanotransduction plays a critical role in maintaining cartilage integrity. Furthermore, TRPV4 may play an important role in cartilage development, as TRPV4 is up-regulated during chondrogenesis (20). Activation of TRPV4 can increase growth of tissue engineered cartilage constructs (21, 22) and chondrogenesis of stem cells (20), while adult stem cells lacking TRPV4 exhibit decreased chondrogenesis (23). These studies provide evidence for a critical regulatory role for TRPV4 in cartilage development (7).
Whereas the genetic basis of these disorders is clear, the mechanism by which TRPV4 mutations cause skeletal dysplasia remains to be determined. In this study, we developed model systems using primary chondrocytes and transgenic mice for the expression of mutant channels. We focused on genes in these cells that, on dysregulation by the channelopathy mutation, might alter skeletal development. We initially performed a gene array in a humanized porcine chondrocyte model expressing huTRPV4V620I in order to identify transcriptional reprogramming by the mutation. Follistatin (FST) was robustly up-regulated by TRPV4V620I. FST is a powerful antagonist of bone morphogenetic proteins (BMPs) and other members of the transforming growth factor-β (TGF-β) superfamily. FST acts by directly binding target proteins, similar to a neutralizing antibody (24). By inhibiting BMP activity, FST plays a key role in skeletal patterning, chondrogenesis, and bone formation (25, 26). Thus, we hypothesized that excessive Ca2+ influx via mutant TRPV4 channels caused FST up-regulation, which prevented proper bone formation and resulted in skeletal dysplasia. To test this hypothesis, we used a humanized porcine chondrocyte model to assess skeletal dysplasia-causing TRPV4 channelopathy mutations in an appropriate cellular context. To examine the effect of the V620I mutation at the whole animal level, we generated transgenic mice that expressed huTRPV4WT or huTRPV4V620I, both driven by the human TRPV4 promoter. We demonstrated the effect of excess FST on ossification of developing chick limbs. Finally, we examined FST expression in chondrocytes from a TRPV4T89I patient. Our results suggest that FST overexpression induced by skeletal dysplasia-causing TRPV4 mutations functions as a significant dysregulatory mechanism that contributes to the disease phenotype.
MATERIALS AND METHODS
Humanized porcine chondrocyte model
Chondrocytes were isolated from the femoral condyles of female adult pigs as described previously (27). Briefly, cartilage was minced, removed from the bone, and sequentially digested in Pronase (Calbiochem, San Diego, CA, USA) and type 2 collagenase (Worthington, Lakewood, NJ, USA). Chondrocytes were maintained in feed medium [Dulbecco's modified Eagle's medium–high glucose (DMEM-HG), 15 mM HEPES, 0.1 mM nonessential amino acids, and 10% FBS; Life Technologies, Grand Island, NY, USA] at 380 mOsm (osmolarity was adjusted by adding sucrose) and pH 7.4.
Cells were plated at 1.9 × 105 cells/cm2 for 3 d and then transfected with huTRPV4WT or mutant huTRPV4 using Amaxa's Nucleofector device (Lonza, Basel, Switzerland) and the primary human chondrocyte kit, following the manufacturer's protocol. Cells were transfected with TRPV4 mutations that cause skeletal dysplasias of varying severity: V620I = mild (11), A716S = moderate (10), T89I = severe (14), or an arthropathy-causing mutation (F273L; ref. 16). WT TRPV4 and all mutant constructs were C-terminally tagged with red fluorescent protein (RFP) in pcDNA3.1, and mutagenesis was performed using the Quick-Change site-directed mutagenesis kit (Stratagene, Santa Clara, CA, USA). All constructs were verified by sequencing. TRPV4-RFP expressing cells were selected 48 h after the transfection using FACS DiVa Cell Sorter (BD Biosciences, Franklin Lakes, NJ, USA), thus ensuring similar expression levels of all transgenes. Given the dominant gain-of-function nature of the mutations, the endogenous porcine TRPV4 was unaltered in these cells to reflect the presence of the healthy TRPV4 allele in human patients with a TRPV4 channelopathy.
Ca2+ imaging
Cells were loaded with 2 μM Fura-2-AM (Life Technologies), and imaged using a protocol for ratiometric Ca2+ imaging using 340/380-nm blue light for dual excitation, recording emissions with specific filter sets, and generating ratios. Basal Ca2+ levels (averaged over 10 images prior to stimulus) and peak Ca2+ levels (maximum measurement after stimulus) in response to −100 mOsm stimulation were determined.
Gene array
RNA was extracted from porcine chondrocytes that were transfected with huTRPV4WT or huTRPV4V620I and exposed to isoosmotic or hypoosmotic (−100 mOsm) medium for 15 min, using TRIzol reagent (Life Technologies) according to the manufacturer's protocol. This RNA was run on Agilent 4x44k porcine microarrays (representing all known protein-coding mRNAs from Sus scrofa). Microarrays were conducted in triplicate. Hybridization was background subtracted, normalized for a panel of housekeeping genes, and analyzed as described previously (28). Differences between huTRPV4WT and huTRPV4V620I were calculated for genes with significant up-regulation.
Gene expression
FST gene expression was measured in transfected chondrocytes using quantitative reverse transcription polymerase chain reaction (qRT-PCR). RNA was isolated from transfected chondrocytes using TRIzol reagent (Life Technologies) according to the manufacturer's protocol. After purification, 2 μg of total RNA was treated with RNase-free DNase (Qiagen, Valencia, CA, USA). RNA (0.5 μg) was then reverse transcribed using oligo dT primers and the SuperScript first strand synthesis kit (Life Technologies) according to the manufacturer's protocol. Gene expression was assessed by qRT-PCR, using the ABI 7900HT Fast Real Time PCR System and Power SYBR Green PCR Master Mix (Applied Biosystems, Grand Island, NY, USA). Specificity of primers was verified by dissociation/melting curve for the amplicons when using SYBR Green as a detector. mRNA abundance was quantified using the 2−ΔΔCt method with all data normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and the huTRPV4WT sample as the calibrator.
Western blotting
For the detection of FST and actin proteins, huTRPV4-RFP transfected and sorted chondrocytes were lysed in 100 μl of CHAPS buffer containing 30 mM Tris-Cl (pH 7.5), 150 mM NaCl, 1% CHAPS, 1 mM phenylmethylsulfonyl fluoride, 1× protease inhibitor cocktail (Roche Applied Science, Indianapolis, IN, USA), incubated on ice for 30 min and centrifuged at 10,000 rpm for 3 min. Proteins from the cleared supernatant were then quantified using Bradford reagent (Sigma-Aldrich, St. Louis, MO, USA). Equal amounts of total proteins were analyzed by SDS-polyacrylamide gel electrophoresis and transferred onto Immobilon-P membranes (Millipore, Billerica, MA, USA) using a semidry transfer apparatus. Membranes were blocked in phosphate-buffered saline (PBS), 0.1% Tween 20 containing 5% nonfat powdered milk. Proteins were detected by incubating with antibodies at 0.2 μg/ml in blocking solution. Anti-FST antibodies used in this study were from AbCam (Cambridge, MA, USA), and antiactin antibodies were from Santa Cruz Biotechnology (Dallas, TX, USA). Primary antibodies were detected with goat anti-rabbit or anti-mouse secondary antibody conjugated to horseradish peroxidase (Jackson ImmunoResearch, West Grove, PA, USA). Immunocomplexes were detected using ECL Plus Western blotting Detection Reagents (GE Healthcare Life Sciences, Pittsburgh, PA, USA).
Transfection of FST-promoter-driven luciferase reporter into ATDC5 cells
Transient transfections of ATDC5 cells were performed in 12-well plates at 70–85% confluence with Lipofectamine 2000 transfection reagent (Life Technologies) according to the manufacturer's instructions. A 2.4-kb fragment of the pig follistatin promoter was cloned into the pGL3 basic luciferase vector (Promega, Madison, WI, USA), as either a WT isoform or a cAMP-response-element (CRE) mutant that comprises the transcription start site (Supplemental Fig. S1). These two promoter constructs were cotransfected with either huTRPV4WT or huTRPV4V620I expression constructs (pcDNA3.1 backbone). At 6 h after transfection, cells were treated with insulin-transferrin-sodium selenite (ITS) liquid medium supplement (Sigma-Aldrich) for chondrogenic differentiation and harvested on d 3. Lysates were assayed for luciferase activity following manufacturer's instructions (Promega). Results were normalized to Renilla luciferase activity resulting from cotransfection of the pRL Renilla luciferase reporter vector (Promega).
Humanized TRPV4 mouse generation
All animal protocols were approved by the Duke University Institutional Animal Care and Use Committee, in full compliance with U.S. National Institutes of Health guidelines. Transgenic humanized TRPV4 mice were generated using a human bacterial artificial chromosome (BAC; RP11-126K19; Children's Hospital Oakland Research Unit, Oakland, CA, USA). The human BAC was modified by recombineering (29–31) so that the IRES-yellow fluorescent protein (YFP) Venus cassette was inserted immediately following the translational stop signal, into the 3′ untranslated region of the human gene (huTRPV4WT); for the mutant version, a point mutation was introduced in exon 12, resulting in the V620I mutant protein (huTRPV4V620I). All modifications were sequence verified. Modified BAC DNA was injected into pronuclei of B6SJLF1 mice following standard procedures (32). Several transgenic lines were obtained per construct, which transmitted the transgene to the next generation. We detected human TRPV4 cDNA, using specific primers, in similar abundance from the two lines (one each WT, V620I) that were ultimately selected for further experimentation. These lines were backcrossed to C57BL/6N over >5 generations. We also tested for expression of YFP-Venus (30, 33–36). Its expression in cartilage was confirmed by confocal microscopy (LSM 510; Carl Zeiss, Inc., Thornwood, NY, USA; Supplemental Fig. S2). Mouse femurs were dissected from 3 animals/group: huTRPV4WT, huTRPV4V620I, and nontransgenic control. Femurs were soaked in a 1.5 μM solution of Mitotracker Deep Red (Life Technologies) to stain all living cells. YFP expression and Mitotracker were imaged in intact femoral condyles on a confocal microscope. Cells were visible in all mice using the Mitotracker, but YFP was detectable only in chondrocytes from the transgenic mice, with expression found in virtually all articular chondrocytes, similar to native TRPV4.
Skeletal phenotyping
Imaging of the huTRPV4WT and huTRPV4V620I mice was performed after euthanasia on 6-mo-old mice (n=8 mice/genotype) using dual-emission X-ray absorptiometry (DEXA; Lunar PIXImus, Madison, WI, USA) and high resolution digital X-ray (Model MX-20 Digital; Faxitron X-ray Corp., Wheeling, IL, USA). From the DEXA scans, bone mineral density (BMD), bone mineral content (BMC), bone area, tissue area, percent fat, total tissue mass, lean tissue, and fat tissue were measured for the whole body, then specifically for spine, tibia, and femur, using the Lunar PIXImus software. The following dimensions were measured from digital X-ray images: length of the T13–L6 vertebrae and width of the L6 vertebrae (including the transverse process; Supplemental Fig. S3A), length of the femur (Supplemental Fig. S3B), length of the tibia and width of the tibial plateau (Supplemental Fig. S3C), length of the third upper extremity digit (metacarpal plus phalanges of the third digit; Supplemental Fig. S3D), length of the third lower extremity digit (metatarsal plus phalanges of the third digit; Supplemental Fig. S3E), and width of the acetabulum and femoral head (Supplemental Fig. S3F).
Micro-computed tomography (microCT)
The right knee of 2-wk-old and 22- to 24-wk-old (6-mo-old) huTRPV4WT and huTRPV4V620I mice were fixed at 90° flexion in formaldehyde (VWR, Radnor, PA, USA) at 4°C for 5 d and then assessed by microCT and histology. For quantitative 3-D evaluation of the mouse bone parameters, the entire mouse knee joints were scanned by a microCT system (microCT 40, Scanco Medical AG, Bassersdorf, Switzerland). Samples were scanned at 55 kV with a slice increment of 16 μm. Calcified tissues were segmented from soft tissues using a threshold of 182 for the 2-wk-old limbs and 360 for the 6-mo-old limbs. Bone density values (mg HA/cm3) were calibrated with the use of a hydroxyapatite calibration phantom. For the 2-wk-old knees, the entire femoral condyles and tibial plateau above the growth plate were assessed. In the 6-mo-old knees, the following regions of each knee were examined: the femoral condyles, tibial plateau epiphysis, tibial metaphysis, and femoral condyles (17, 37). The condyles were defined by the first transverse slice that contained trabecular bone and consisted of the next 10 slices. The tibial plateau was defined by the first transverse slice that contained the tibia until the first slice where the growth plate appeared. The tibial metaphysis was defined by the first transverse slice that contained the fibula and consisted of the next 20 slices. A 3-D reconstruction was generated of each region and analyzed using the internal software of the microCT system to determine bone volume (mm3), total volume (mm3), calcified bone fraction (bone volume/total volume), and BMD (mg HA/cm3) and trabecular number (mm−1) and trabecular thickness (mm) for the cancellous bone in both huTRPV4WT and huTRPV4V620I mice (n=8/genotype for each time point).
Histology
The 2-wk-old mouse knee joints were decalcified using Cal-Ex (Fisher Scientific, Hampton, NH, USA) followed by sequential dehydration and paraffin embedding. Sections (8 μm thick) were taken in the coronal plane at the A/P location of normal loading and then stained with Harris hematoxylin (Poly Scientific, Bay Shore, NY, USA), Safranin O (Sigma-Aldrich), and aqueous fast green (Sigma-Aldrich) to label cell nuclei, proteoglycans, and collagens, respectively. Images were acquired and measurements were made of the width of the tibial plateau cartilage and the thickness of the proliferative and hypertrophic zones in the growth plate, and the thickness of the total growth plate in the huTRPV4WT (n=7) and huTRPV4V620I (n=6) mouse knees.
In situ Ca2+ signaling
In situ Ca2+ signaling was carried out as described previously (17) in three 4-mo-old huTRPV4WT and huTRPV4V620I mice. Briefly, femora were isolated from freshly euthanized mice and incubated with fura red (30 μM, Life Technologies) and fluo-4 (15.5 μM, Life Technologies) for 40 min at 37°C. With the cartilage fully intact, the femora were imaged in a 37°C perfusion chamber on a confocal microscope. All experiments were performed in medium (DMEM-HG without phenol red, 15 mM HEPES, 4 mM l-glutamine, and 1 mM Na pyruvate; Life Technologies) at 300 mOsm. From each mouse, the control femur was perfused with 0.7 μM dimethyl sulfoxide (DMSO; Sigma-Aldrich) and the experimental femur was perfused with 10 μM 4α-phorbol 12,13-didecanoate (4αPDD) dissolved in 0.7 μM DMSO (EMD Millipore, Germany) to specifically activate TRPV4. The ratio of fluo-4 (increases fluorescence with Ca2+ concentration) to fura red (decreases fluorescence with Ca2+ concentration) was recorded for each cell in the field of view. Cells in which this ratio increased to >3 sd above baseline were considered positive for Ca2+ signaling.
Gene expression
Knee and hip cartilage from 2-wk-old huTRPV4WT and huTRPV4V620I mice (n=3 samples/genotype; each sample contained pooled tissue from 6 mice) were collected and stored in RNA later (Life Technologies) until RNA extraction. Cartilage was then transferred to Trizol (Life Technologies) and homogenized using a Mini-Beadbeater (Biospec, Bartlesville, OK, USA). The RNA was extracted according the Trizol protocol through the precipitation step and then was processed according to the manufacturer's protocol for the RNeasy Plus Kit (Qiagen). RNA (0.5 μg) was then reverse transcribed using Superscript VILO (Life Technologies), according to the manufacturer's protocol. Gene expression was assessed by qRT-PCR as described above. Using the 2−ΔΔCt method, data were normalized for Gapdh, and a huTRPV4WT sample was set as the calibrator. Fst mRNA abundance was normalized for TRPV4 mRNA.
Chick limb ossification
Fertilized Ross Hubert chick eggs (Gallus gallus domesticus, Pilgrim's Pride Hatchery, Siler City, NC, USA) were incubated for a total of 9 d at 37°C and 70% humidity. Embryos were staged according to Hamburger and Hamilton (HH; ref. 38). Eggs were windowed on d 2 (HH14), sealed with tape, and incubated until d 7. Affi-gel blue gel 150- to 300-μm-diameter beads (Bio-Rad, Hercules, CA, USA) were incubated overnight with either PBS (Mediatech, Manassas, VA, USA) or 100 μg/ml carrier free recombinant human FST 288 (R&D Systems, Minneapolis, MN, USA). On d 7 (HH30), a single PBS- or FST-soaked bead was placed in the right leg of each chick near the tibial growth plate. The window chambers were resealed and incubated for an additional 2 d. On d 9 (HH34), embryos were harvested (n=14 for PBS treated and n=18 for FST treated), both legs were removed and stained with alcian blue (Acros Organics, Morris Plains, NJ, USA) and alizarin red (Electron Microscopy Sciences, Hatfield, PA, USA) to identify cartilage and bone, respectively, using a previously published protocol (39). The stained legs were imaged, and the bone length and ossification regions were measured for the tibia and femur using the LSM Image 5 browser (Zeiss). The percentage ossification of the tibia and femur were calculated as follows: (length of ossification region/total bone length) × 100.
Human chondrocytes
Primary human chondrocytes were isolated from the femoral cartilage of control (n=7) or International Skeletal Dysplasia Registry case R94-316, who has the neonatally lethal TRPV4T89I mutation (14). Human TRPV4, human FST, and human GAPDH gene expression in the chondrocytes was determined by isolating RNA from the cells using the Trizol (Life Technologies) protocol and reverse transcription using Superscript VILO (Life Technologies), followed by qRT-PCR as described above. Using the 2−ΔΔCt method, data were normalized for GAPDH, and a control sample was set as the calibrator. FST mRNA abundance was normalized for TRPV4 mRNA. The levels of FST secreted into the culture medium were determined using a human FST ELISA kit (R&D Systems) according to the manufacturer's instructions. Total FST secreted into the medium was normalized for total micrograms of RNA in the cell lysate.
Statistical analyses
All data are presented as means ± se. Data were tested for normality with Kolmogorov-Smirnov test. Non-normal data were log transformed to achieve normality, and statistics were performed on log transformed data. Student's t test or ANOVA was used to compare means (α=0.05). The percentage of murine chondrocytes responding with a Ca2+ signal was compared between conditions and genotypes by χ2 test of proportions. Paired 2-tailed t tests were used to determine significant differences between the right and left legs in the chick experiments.
RESULTS
Humanized TRPV4 porcine chondrocyte model: assessing huTRPV4V620I mutant channels
Ca2+ signaling is increased by the V620I mutation
To examine the effect of TRPV4V620I in an appropriate cellular context, adult porcine articular chondrocytes were isolated and transfected with WT (huTRPV4WT) or V620I (huTRPV4V620I) human TRPV4 with C-terminally fused RFP. Chondrocyte transfection was confirmed by imaging RFP fluorescence (Supplemental Fig. S4). Chondrocytes responded to a 100 mOsm decrease with increased intracellular Ca2+ (Fig. 1A). Chondrocytes expressing huTRPV4WT, however, responded largely with Ca2+ oscillations, whereas chondrocytes expressing huTRPV4V620I tended to respond with long-lasting Ca2+ increases. The basal Ca2+ level in huTRPV4V620I expressing chondrocytes was significantly greater than that of huTRPV4WT cells (Fig. 1B). The peak Ca2+ level in response to hypoosmotic stimuli was also significantly greater in huTRPV4V620I than in huTRPV4WT cells (Fig. 1C). Thus, the brachyolmia mutation TRPV4V620I is responsible for excess Ca2+ influx into primary chondrocytes.
Figure 1.

Ca2 signaling is increased and FST is up-regulated in huTRPV4V620I porcine chondrocytes. A) Representative Ca2+ traces (each trace is an individual cell) showing chondrocytes with huTRPV4V620I (V620I) or huTRPV4WT (huWT) channels responding to −100 mOsm stimulus. B) Basal Ca2+ levels of chondrocytes with directed expression of V620I or huWT (means±sem; n=19). *P = 0.003; t test on log-transformed data. C) Peak Ca2+ levels of chondrocytes with directed expression of V620I or huWT in response to a −100 mOsm change (means±sem; n=10). *P = 0.0002; t test on log-transformed data. D) Fold change in FST mRNA of chondrocytes expressing huWT and V620I (means±sem; n=7). V620I showed >2-fold up-regulation vs. huWT. E) Western blot showing FST and actin protein levels for chondrocytes with directed expression of V620I or huWT.
FST is up-regulated by the V620I mutation in a Ca2+-dependent manner
A microarray was performed to identify potential genes altered by TRPV4V620I. FST was among the most highly regulated genes in response to the V620I mutation (Supplemental Fig. S5 and Supplemental Table S1). This microarray finding was confirmed by measuring FST levels in the humanized chondrocyte model. FST mRNA was significantly up-regulated in chondrocytes expressing huTRPV4V620I vs. huTRPV4WT (Fig. 1D). This FST increase was confirmed at the protein level by Western blotting (Fig. 1E). Furthermore, the increase in FST was eliminated when the V620I mutation was combined with pore mutations that alter channel permeability. The pore-blocking mutation, M680K (40), or a mutation that renders the channel impermeable to Ca2+ but permeable to monovalent cations, M680D (ref. 41 and Fig. 2A), both prevented the V620I-induced FST increase. This suggests that influx of Ca2+ through the channel is necessary for FST up-regulation. Western blotting confirmed that the presence of the M680D mutation also prevented FST up-regulation at the protein level (Fig. 2B).
Figure 2.
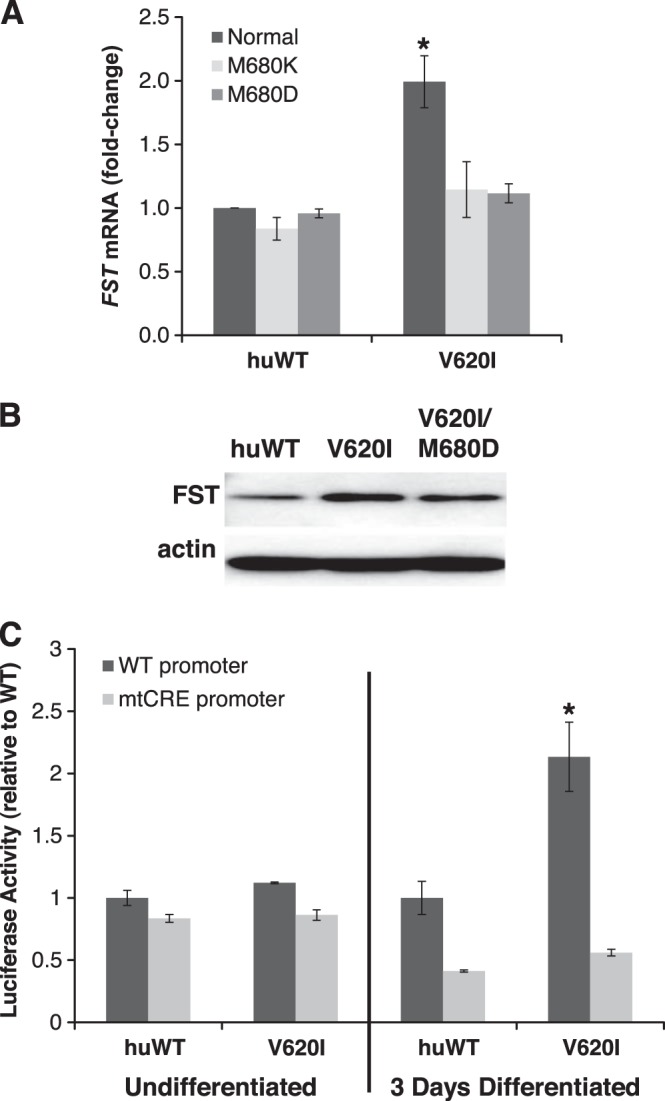
FST up-regulation requires a TRPV4 channelopathy mutation with a Ca2+-permeable pore and requires the CRE in the FST promoter. A) Fold change in FST mRNA abundance of chondrocytes with directed expression of huTRPV4V620I (V620I) or huTRPV4WT (huWT) with pore blocking (M680K) or monovalent cation permeable but Ca2+ impermeable (M680D) mutations (means±sem; huWT, n=6; V620I, n=6; V620I/M680K, n=4; M680K, n=2; M680D, n=3; V620I/M680D, n=3). *P = 0.0003 vs. all other groups; ANOVA on log-transformed data. B) Western blot showing FST and actin levels for chondrocytes with directed expression of V620I, V620I/M680D, or huWT. Relative luciferase activity of ATDC5 cells that were either undifferentiated (left panel) or chondrogenically differentiated for 3 d (right panel) expressing V620I or huWT, coexpressing a luciferase reporter gene driven by the porcine FST promoter (WT) or the FST promoter containing a mutation in the CRE (mtCRE). Only in differentiated cells was luciferase activity significantly increased in the presence of the V620I mutation as compared to huWT. This increase was completely eliminated when the FST promoter was mutated (means±sem; n=3/group). *P < 0.003; ANOVA.
ATDC5 chondrocyte cell line model: assessment of transcriptional regulation of FST by huTRPV4V620I
To better understand how excess Ca2+ entering the cell through huTRPV4V620I up-regulates FST transcription, we conducted reporter gene experiments in a chondrogenic cell line. We transfected ATDC5 cells with huTRPV4V620I, huTRPV4WT, and a FST promoter construct that drives luciferase expression. Two versions of the FST promoter were used, 2.4 kb of the WT porcine FST proximal promoter (FSTCRE-WT) and the same DNA sequence with an engineered mutation in the CRE that is part of the FST transcriptional start site (FSTCRE-mut; ref. 42 and Supplemental Fig. S1). Undifferentiated ATDC5 cells expressing huTRPV4V620I did not exhibit altered luciferase activity. Notably, ATDC5 cells that were differentiated toward a chondrogenic phenotype showed a significant increase in FST reporter activity in the presence of the V620I mutation, which was abolished when the FSTCRE-mut promoter was present (Fig. 2C). These findings demonstrate that huTRPV4V620I channels, compared with huTRPV4WT, increase transcriptional drive on a 2.4-kb proximal promoter fragment of porcine FST, but only once cells are driven into the chondrogenic lineage. These data are likely relevant for all mammals because of the highly conserved nature of the CRE sequence and relative position in the FST promoter in different mammalian species.
huTRPV4V620I transgenic mice: humanized mouse model of brachyolmia caused by TRPV4V620I
With evidence for Ca2+ influx through huTRPV4V620I activating FST transcription and leading to its overexpression in primary chondrocytes, we examined the in vivo function of huTRPV4V620I in a mouse model. huTRPV4V620I transgenic mice were generated to recapitulate the skeletal features of brachyolmia in these mice, and to probe Ca2+ signaling and Fst expression abnormalities in chondrocytes of these mice. For controls, huTRPV4WT mice were generated.
Phenotyping revealed altered bone and cartilage shape in huTRPV4V620I mice
We measured huTRPV4V620I-induced changes in skeletal morphology to determine whether our mouse model accurately reflected the phenotype of the human disease. In 6-mo-old mice, we recorded confirmatory evidence; namely, huTRPV4V620I mice had significantly shorter tibia and digits than huTRPV4WT mice (Fig. 3A). The L6 vertebra was longer and wider in the huTRPV4V620I mice, as compared to the huTRPV4WT mice (Fig. 3A, B). However, there was no significant difference in the length of the femur (Fig. 3A) or the T13–L5 vertebrae between huTRPV4WT and huTRPV4V620I mice. In addition to the altered bone length in the huTRPV4V620I mice, the acetabulum, femoral head, and tibial plateau were all wider in the huTRPV4V620I mice than the huTRPV4WT mice (Fig. 3C). Additional biometrics between huTRPV4V620I and huTRPV4WT transgenic mice (whole body, spine-, tibia-, or femur-specific measurements of bone mineral density, bone mineral content, bone area, tissue area, percentage fat, total tissue mass, lean tissue, or fat tissue) revealed no difference between the huTRPV4V620I and huTRPV4WT mice. In addition, there were no differences between the huTRPV4WT and huTRPV4V620I mice in the bone parameters in 2-wk-old mice (Table 1) or the epiphysis and metaphysis of the tibial plateau in 6-mo-old mice (Table 2). However, the calcified bone fraction in the femoral condyles of the older huTRPV4V620I mice was significantly decreased, as compared to the huTRPV4WT mice (P<0.001).
Figure 3.

Humanized huTRPV4V620I mice exhibited skeletal changes, increased Ca2+ signaling, and increased Fst expression. A) Length of the femur, tibia, third lower extremity digit (metatarsal plus phalanges of the third digit), third upper extremity digit (metacarpal plus phalanges of the third digit), and L6 vertebrae, as well as the width of the L6 vertebrae (including the transverse process) of the huTRPV4WT (huWT; darker bars) and huTRPV4V620I mice (V620I; lighter bars) (means±sem; n=8). *P < 0.05; t test. B) Faxitron images of the vertebrae. C) Width of the acetabulum, femoral head, and tibial plateau of the huWT and V620I mice (means±sem; n=8). *P < 0.05; t test. D) Percentage of cells in intact femoral cartilage responding with a Ca2+ signal to vehicle control or 10 μM 4αPDD differs between huWT and V620I mice. Bars with different letters differ significantly from one another (P<0.009, χ2; huWT control, n=118; huWT 4αPDD, n=147; V620I control, n=116; V620I 4αPDD, n=119; cells from 3 mice/group). E) Fst mRNA, which was isolated directly from mouse cartilage, was normalized for TRPV4 mRNA in V620I vs. huWT mice. Fst mRNA levels prior to normalization for TRPV4 mRNA were 1.07 ± 0.21 for huWT and 1.38 ± 0.02 for V620I mice (P=0.21; means±sem; n=3). *P = 0.009; t test.
Table 1.
Bone morphology in 2-wk-old mice
| Bone parameter | huTRPV4WT | huTRPV4V620I | P |
|---|---|---|---|
| Femoral condyles | |||
| TV (mm3) | 2.683 ± 0.257 | 2.677 ± 0.150 | 0.983 |
| BV (mm3) | 1.373 ± 0.170 | 1.40 ± 0.131 | 0.899 |
| BV/TV | 0.502 ± 0.030 | 0.516 ± 0.020 | 0.692 |
| BD (mg/cm3) | 422 ± 11 | 430 ± 9 | 0.593 |
| Tibial plateau | |||
| TV (mm3) | 1.074 ± 0.123 | 1.052 ± 0.088 | 0.889 |
| BV (mm3) | 0.611 ± 0.083 | 0.598 ± 0.077 | 0.916 |
| BV/TV | 0.562 ± 0.029 | 0.556 ± 0.028 | 0.888 |
| BD (mg/cm3) | 414 ± 17 | 429 ± 11 | 0.499 |
TV, total volume; BV, bone volume; BV/TV, bone fraction; BD, bone density. Data are presented as means ± sem with statistical analysis by 2-tail unpaired t test.
Table 2.
Bone morphology in 6-mo-old mice
| Bone parameter | huTRPV4WT | huTRPV4V620I | P |
|---|---|---|---|
| Femoral condyles–cancellous bone | |||
| TV (mm3) | 0.046 ± 0.003 | 0.052 ± 0.004 | 0.209 |
| BV (mm3) | 0.035 ± 0.002 | 0.035 ± 0.003 | 0.907 |
| BV/TV | 0.760 ± 0.010 | 0.668 ± 0.019 | 0.001 |
| BD (mg/cm3) | 1208 ± 17 | 1209 ± 16 | 0.960 |
| Tb.N (mm−1) | 49.275 ± 1.760 | 50.511 ± 1.000 | 0.551 |
| Tb.Th (mm) | 0.045 ± 0.001 | 0.0440 ± 0.001 | 0.397 |
| Tibial plateau | |||
| Epiphysis | |||
| TV (mm3) | 1.250 ± 0.099 | 1.357 ± 0.071 | 0.393 |
| BV (mm3) | 0.877 ± 0.051 | 0.897 ± 0.034 | 0.744 |
| BV/TV | 0.710 ± 0.019 | 0.665 ± 0.020 | 0.132 |
| BD (mg/cm3) | 1204 ± 8 | 1180 ± 9 | 0.070 |
| Tibial plateau | |||
| Metaphysis | |||
| TV (mm3) | 1.515 ± 0.073 | 1.692 ± 0.114 | 0.213 |
| BV (mm3) | 0.573 ± 0.041 | 0.611 ± 0.068 | 0.639 |
| BV/TV | 0.379 ± 0.024 | 0.342 ± 0.025 | 0.304 |
| BD (mg/cm3) | 1100 ± 18 | 1115 ± 22 | 0.621 |
TV, total volume; BV, bone volume; BV/TV, bone fraction; BD, bone density; Tb.N, trabecular number; Tb.Th, trabecular thickness. Data are presented as means ± sem with statistical analysis by 2-tail unpaired t test.
The cartilage and growth plate of 2-wk-old huTRPV4WT and huTRPV4V620I mice were assessed by histological staining and then measurement of the cartilage width and thickness of the growth plate zones (Table 3). The huTRPV4V620I mice have significantly wider cartilage on the tibial plateau than the huTRPV4WT mice (P<0.01). This wider cartilage template is consistent with the wider tibial plateau in the older huTRPV4V620I mice. However, the growth plate thickness in these mice was not different between the two genotypes (Supplemental Fig. S6). These findings in the mouse skeleton and growth plate cartilage are consistent with the generalized defects in endochondral ossification and mild short stature observed clinically in brachyolmia, which affect both the spine and the long bones.
Table 3.
Measurements of tibial plateau cartilage and growth plate in 2-wk-old mice
| Parameter | huTRPV4WT | huTRPV4V620I | P |
|---|---|---|---|
| Cartilage width (mm) | 2.33 ± 0.05 | 2.51 ± 0.02 | 0.01 |
| Total growth plate (mm) | 0.46 ± 0.02 | 0.44 ± 0.03 | 0.43 |
| Proliferative zone (mm) | 0.25 ± 0.01 | 0.23 ± 0.02 | 0.51 |
| Hypertrophic zone (mm) | 0.21 ± 0.02 | 0.20 ± 0.02 | 0.66 |
Data are presented as mean ± sem with statistical analysis by 2-tail unpaired t test.
In situ Ca2+ signaling was elevated in huTRPV4V620I mice
To confirm altered Ca2+ signaling in chondrocytes of huTRPV4V620I transgenic mice, we used a novel organ culture model to measure Ca2+ signaling in response to a selective TRPV4 agonist in intact femoral condyles (17). Significantly more chondrocytes in cartilage from huTRPV4V620I transgenic mice signaled without stimulation and in response to specific chemical activation of TRPV4 with 4αPDD than the huTRPV4WT controls (Fig. 3D). The relative amount of 4αPDD-induced signaling (normalized to control response), however, was lower in huTRPV4V620I mice (0.5) than huTRPV4WT mice (1.6).
Fst gene expression was increased in huTRPV4V620I transgenic mouse chondrocytes
To address whether increased Ca2+ signaling in chondrocytes derived from huTRPV4V620I transgenic mice evoke increased Fst gene expression, Fst mRNA abundance was measured in mouse chondrocytes by qRT-PCR. We detected a significant, almost 3-fold up-regulation of Fst for huTRPV4V620I vs. huTRPV4WT when using huTRPV4 mRNA abundance for normalization (Fig. 3E). huTRPV4 mRNA was expressed by both huTRPV4WT (1.30±0.25; fold change±sem) and huTRPV4V620I (0.61±0.27) mice, using qRT-PCR. In view of these findings, we deemed it necessary to normalize for huTRPV4 mRNA abundance in chondrocytes. Because expression levels of the huTRPV4 transgene were moderately more abundant in the huTRPV4WT control, the differences between huTRPV4WT and huTRPV4V620I might be slightly underrepresented. However, given the dominant effect of TRPV4V620I and other dysplasia-inducing mutations, we are confident that results derived from our humanized mouse model were reflective of in vivo pathogenesis.
Humanized TRPV4 porcine chondrocyte model: assessing a wider range of TRPV4 mutations
To address whether other skeletal dysplasia-inducing TRPV4 mutations would also up-regulate FST, we expanded our humanized porcine chondrocyte model to include examples of a clinical severity spectrum of skeletal dysplasia mutations [V620I=mild (11), A716S=moderate (10), T89I=severe (14)], and used an arthropathy-causing mutation, F273L (16), which is not associated with skeletal dysplasia to test the specificity of our proposed FST channelopathy mechanism.
Basal and peak Ca2+ levels varied with different TRPV4 mutations
The effects of the mutations on basal Ca2+ and Ca2+ response to a 100-mOsm decrease were measured. Overall, we found that Ca2+ levels (either baseline or peak) did not correlate with disease severity among the dysplasia-causing mutations (Fig. 4A, B). Furthermore, huTRPV4F273L, the nondysplasia causing mutation, had significantly elevated baseline Ca2+. These data suggest that, although Ca2+ levels may be altered by these mutations, the Ca2+ levels may not be directly associated with a disease mechanism.
Figure 4.

Basal and peak Ca2+ levels varied with different TRPV4 mutations, but FST is up-regulated in a TRPV4-dependent manner by humanized porcine chondrocytes expressing skeletal dysplasia causing mutations. A) Basal Ca2+ levels in porcine chondrocytes expressing TRPV4 mutations (means±sem; huWT, n=38; V620I, n=50; A716S, n=36; T89I, n=16; F273L, n=20). *P < 0.00001 vs. huWT; ANOVA on log-transformed data. B) Peak Ca2+ levels in response to −100 mOsm change in porcine chondrocytes expressing TRPV4 mutations (means±sem; huWT, n=28; V620I, n=40; A716S, n=26; T89I, n=6; F273L, n=15). *P < 0.0001 vs. huWT; ANOVA on log-transformed data. C) Fold change of FST mRNA with different TRPV4 mutations without (darker bars) and with (lighter bars) selective TRPV4 antagonist, GSK205 (25 μM) (means±sem; huWT, n=10; V620I, n=9; A716S, n=7; T89I, n=4; F273L, n=3). All dysplasia mutations showed >2-fold up-regulation. D) Fold change of FST mRNA abundance for huWT, A716S, and A716S/M680D (means±sem; n=3). *P = 0.003 vs. other groups; ANOVA.
FST is up-regulated in a TRPV4-dependent manner by chondrocytes expressing skeletal dysplasia causing mutations but not by an arthropathy-causing mutation
We examined FST expression levels to determine whether FST is up-regulated by different skeletal dysplasia causing TRPV4 mutations. Chondrocytes expressing TRPV4 with each of the dysplasia causing mutations had ≥2-fold FST up-regulation relative to huTRPV4WT (Fig. 4C). In contrast, the arthropathy causing mutation, huTRPV4F273L, did not alter FST mRNA levels relative to huTRPV4WT. All FST up-regulation was virtually eliminated by adding the TRPV4-selective inhibitor, GSK205. This indicates mutant TRPV4 channel activity as the likely cause of FST up-regulation. GSK205 did not significantly affect basal calcium levels in chondrocytes (control normalized to 1; GSK205=0.99±0.03; n>20 cells/3 independent experiments).
We examined the A716S mutation more closely because it up-regulates FST expression, but it does not alter Ca2+ dynamics in chondrocytes. When the A716S mutation was combined with the M680D monovalent cation pore mutation, it prevented FST up-regulation (Fig. 4D). This finding indicates the critical importance of Ca2+ entering through huTRPV4A716S to drive FST up-regulation.
FST decreases bone ossification in a chick embryo model system
To show that FST up-regulation could cause alterations in the developing skeleton, we measured the effects of excess FST on bone ossification in chick legs (Fig. 5A). The treatment of chick legs with PBS soaked beads did not alter the percent ossification of either the tibia or the femur (Fig. 5B, C), as compared to the contralateral untreated limb. However, FST bead implantation significantly decreased the percentage ossification of both the tibia (Fig. 5B) and the femur (Fig. 5C) when compared to the contralateral untreated leg. Thus, excess FST significantly decreases ossification of long bones in the developing skeleton of a vertebrate embryo.
Figure 5.

FST decreases ossification of developing chick limbs. A) Representative images of chick tibia stained with alizarin red (bone) and alcian blue (cartilage). Red circles indicate location of PBS- or FST-soaked beads near the tibia of the right legs. Scale bar = 500 μm. B) Percentage ossification of untreated (darker bars) and treated (with a PBS- or FST-soaked bead; lighter bars) tibiae (means±sem; PBS, n=14; FST, n=18). *P = 0.01 vs untreated bead; paired t test. C) Percentage ossification of untreated (darker bars) and treated (with a PBS- or FST-soaked bead; lighter bars) femurs. means±sem; PBS, n=14; FST, n=18). *P = 0.02 vs. untreated bead; paired t test.
Native TRPV4T89I in human chondrocytes increases FST
In a human case study, we measured TRPV4 and FST mRNA and secreted FST protein levels in human chondrocytes from a neonatally deceased TRPV4T89I individual, compared to a cohort of control human chondrocytes. Human TRPV4 was expressed by both control human chondrocytes (0.92±0.21; fold change) and chondrocytes from the TRPV4T89I patient (0.5). The TRPV4 expression levels were 1.8-fold higher in the control chondrocytes, perhaps indicative of an adaptive response by the T89I chondrocytes to the cellular injury that a channel that gates excess Ca2+ can inflict. On normalization to TRPV4 expression levels, FST gene expression was elevated nearly 4-fold in T89I chondrocytes (Fig. 6A). Notably, FST protein secretion from TRPV4T89I chondrocytes was 3.8-fold higher than from control chondrocytes (Fig. 6B). These findings indicate that FST is overexpressed in chondrocytes in a case of a rare but lethal human skeletal dysplasia caused by a TRPV4 mutation, TRPV4T89I.
Figure 6.

Case study of primary human chondrocytes from a TRPV4T89I individual demonstrates increased FST expression; model to explain how TRPV4 mutations may cause skeletal dysplasia. A) Relative FST mRNA levels corrected for relative TRPV4 mRNA levels in human control chondrocytes and chondrocytes from a lethal TRPV4T89I (T89I) case. Prior to normalization for TRPV4 mRNA, FST mRNA levels were 2.4-fold higher in the T89I chondrocytes (5.79), as compared to control chondrocytes (2.41±0.69). Bars represent means ± sem. B) FST protein secreted by control chondrocytes or chondrocytes from a patient with the TRPV4 T89I mutation. Data are expressed as picograms FST per microgram RNA. Control, n = 7; T89I, n = 1. Bars represent means ± sem. C) Our proposed model of how TRPV4 mutations cause skeletal dysplasia. The skeletal dysplasia mutant TRPV4 channels allow altered Ca2+ influx into chondrocytes. Ca2+ signaling facilitates phosphorylation and binding of CREB to the CRE in the FST promoter (45, 46), thus up-regulating FST expression. In turn, FST functions as a BMP inhibitor in the extracellular milieu, thus inhibiting BMP activity (25, 26), and this attenuation of BMP signaling prevents proper bone formation, resulting in skeletal dysplasias.
DISCUSSION
TRPV4 mutations that cause skeletal dysplasias occur at different sites throughout the channel protein, but may share a common disease-causing mechanism involving the Ca2+-mediated up-regulation of FST expression in chondrocytes. Our findings are consistent with this hypothesis and show FST up-regulation in 3 different dysplasia-inducing TRPV4 mutations, but not with an arthropathy-causing TRPV4 mutation. In a humanized mouse model where huTRPV4V620I was expressed under the control of the human TRPV4 promoter, we observed similar skeletal features as in human TRPV4V620I brachyolmia. Moreover, we demonstrated elevated FST levels in chondrocytes from these mice. We showed in chick embryos that excess FST will impair proper bone development. We further confirmed that FST was elevated in human chondrocytes derived from an individual with a lethal TRPV4T89I mutation. Taken together, these data strongly support that up-regulation of FST in chondrocytes by skeletal dysplasia-inducing TRPV4 mutations contributes to disease pathogenesis. We are proposing here a transcriptional mechanism that partially accounts for the skeletal phenotype by gene regulation of FST, which in excess, neutralizes BMP signaling in the developing skeleton.
Ca2+ entry through the mutated TRPV4 channel clearly plays a role in mediating the effects of channelopathy mutations that cause skeletal dysplasias, but some inconsistencies need to be resolved further. We found that neither basal nor peak Ca2+ was strongly associated with disease presence or severity. Nonetheless, both chemical antagonists and pore blocking mutations of the TRPV4 channel prevented the increase in FST. Furthermore, mutation of the pore to allow transit of only monovalent cations also prevented the FST increase. This finding suggests that Ca2+ must pass through the channel to cause this effect. Importantly, we observed up-regulation of FST expression in cells where the channels have not been specifically activated, which suggests involvement of basal channel activity. In the developing skeleton, this could possibly be modified by growth-associated mechanical cues and/or presence of a growth factor milieu that renders TRPV4 channels particularly sensitive.
In previous studies, heterologous expression in frog oocytes was used to examine a wide range of TRPV4 channelopathies (15). In this study, disease severity was found to be inversely correlated to relative channel activity, largely driven by an increase in basal channel activity (15). Our mouse data are consistent with this concept, as the relative amount of Ca2+ signaling in the huTRPV4V620I mice was lower than in the huTRPV4WT mice, likely due to the elevated basal Ca2+ signaling in the huTRPV4V620I mice.
FST up-regulation also clearly plays a role in these TRPV4 channelopathies, but the FST levels that we measured did not correlate directly with disease severity. As our measurements represent a single time point in isolated cells with directed expression of huTRPV4, it is possible that FST protein levels in cartilage in situ measured at a specific developmental stage would correlate better with disease severity. Consistent with our results, previous studies have shown that FST soaked beads prevent digit formation in chick embryos (43). In addition, FST up-regulation has been shown to cause smaller, weaker bones in mice (44), supporting our concept that FST is driving skeletal changes, and also suggesting that the mechanical properties of the bones from TRPV4 channelopathy patients might be compromised. Our data suggest that the Ca2+ sensitivity of FST up-regulation may be mediated by the CRE in the FST promoter. Ca2+ signaling via mutant TRPV4 channels may activate Ca2+/calmodulin-activated kinases that in turn promote the phosphorylation of the CRE binding protein (CREB), which binds to the CRE (45) and promotes transcription of FST.
Skeletal dysplasia causing TRPV4 mutations appear to mostly affect endochondral ossification. The shorter, wider bones and wider cartilage template in the huTRPV4V620I mice suggest an abnormality in endochondral ossification. Chondrocytes express high levels of TRPV4 (11, 18), which may be why the TRPV4 channelopathies seem to have large effects on cartilage and bone with little effects elsewhere. Robust expression of TRPV4 in the chondrocytic lineage justifies directing expression of huTRPV4 and its mutant isoforms in porcine chondrocytes. The use of our humanized porcine chondrocyte model allowed us to examine these mutations in an appropriate cellular context. Furthermore, in the ATDC5 cells, the effect of the mutant TRPV4 was only present when the cells were induced toward a chondrogenic phenotype. This finding illustrates the powerful influence of differentiation state in determining normal and pathological activities of TRPV4. TRPV4 expression levels have been shown to increase as ATDC5 cells are differentiated (20), and this increase in expression or an increase in the presence of chondrocyte-specific factors may explain why FST promoter activity only increased in the differentiated cells.
We have shown directly that FST decreased bone ossification in developing chick limbs, highlighting the crucial role that FST plays in proper endochondral ossification during development (43). Based on our results and the literature, we propose the following model to explain how TRPV4 mutations cause skeletal dysplasias (Fig. 6C): the skeletal dysplasia mutant TRPV4 channels allow abnormal Ca2+ influx into chondrocytes; Ca2+ signaling promotes phosphorylation and binding of CREB to the CRE in the FST promoter (45, 46); FST gene expression is up-regulated and FST protein is secreted; FST inhibits activities of BMP-type growth factors and morphogens (25, 26); and inhibition of BMP activity prevents proper bone formation, resulting in skeletal dysplasias. Thus, we have built a case for the role of FST in mediating the effects of skeletal dysplasia causing TRPV4 channelopathies. Despite the likely scenario that other effector mechanisms also are operative, both FST and TRPV4 now appear therapeutic targets. In particular, blocking mutant TRPV4 channels with specific TRPV4 inhibitors (47) to down-regulate FST expression may be a promising and rational therapeutic tool to prevent or at least mitigate skeletal dysplasias in patients with TRPV4 gain-of-function mutations.
Supplementary Material
Acknowledgments
The authors thank Stephen Johnson and Daniel Stapor for technical assistance, and Michele Yeo for assistance with Fig. 6C. The authors also thank Dr. Brian Johnstone and his laboratory for providing the ATDC5 cells.
This work is supported by U.S. National Institutes of Health (NIH) grants AR48182 (F.G.), AR48852 (F.G.), AG15768 (F.G.), AR50245 (F.G.), AG46927 (F.G.), DE018549 (W.L.), HD022657 (D.C.), and AR062651 (D.C.), and by the Arthritis Foundation. Venus plasmid (pBES2) was obtained from the Yeast Resource Center (University of Washington, Seattle, WA. USA) funded under NIH grant P41RR11823-03. Recombineering and zygote injections were performed by the Duke Neurotransgenic Laboratory, supported with funding from NIH–National Institute of Neurological Disorders and Stroke Center Core grant 5P30NS061789.
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
- 4αPDD
- 4α-phorbol 12,13-didecanoate
- BAC
- bacterial artificial chromosome
- BMC
- bone mineral content
- BMD
- bone mineral density
- BMP
- bone morphogenetic protein
- CRE
- cAMP-response-element
- DMEM-HG
- Dulbecco's modified essential medium-high glucose
- FST
- follistatin
- GAPDH
- glyceraldehyde 3-phosphate dehydrogenase
- HH
- Hamburger and Hamilton
- microCT
- micro-computed tomography
- PBS
- phosphate-buffered saline
- qRT-PCR
- quantitative reverse transcription polymerase chain reaction
- RFP
- red fluorescent protein
- YFP
- yellow fluorescent protein
- TRP
- transient receptor potential
- TRPV4
- transient receptor potential vanilloid 4
- WT
- wild type
REFERENCES
- 1. Nilius B., Voets T. (2004) Diversity of TRP channel activation. Novartis Found. Symp. 258, 140–149; discussion 149–159, 263–266 [PubMed] [Google Scholar]
- 2. Liedtke W. (2006) TRPV channels' function in sensory transduction and cellular signaling cascades. In TRP Ion Channel Function in Sensory Transduction and Cellular Signaling Cascades (Liedtke W., Heller S., eds) pp. 303–318, CRC Press/Taylor & Francis, Boca Raton, FL, USA: [PubMed] [Google Scholar]
- 3. Montell C. (2001) Physiology, phylogeny, and functions of the TRP superfamily of cation channels. Sci. STKE. 2001, RE 1. [DOI] [PubMed] [Google Scholar]
- 4. Liedtke W., Choe Y., Marti-Renom M. A., Bell A. M., Denis C. S., Sali A., Hudspeth A. J., Friedman J. M., Heller S. (2000) Vanilloid receptor-related osmotically activated channel (VR-OAC), a candidate vertebrate osmoreceptor. Cell 103, 525–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nilius B., Watanabe H., Vriens J. (2003) The TRPV4 channel: structure-function relationship and promiscuous gating behaviour. Pflügers Arch. 446, 298–303 [DOI] [PubMed] [Google Scholar]
- 6. Strotmann R., Harteneck C., Nunnenmacher K., Schultz G., Plant T. D. (2000) OTRPC4, a nonselective cation channel that confers sensitivity to extracellular osmolarity. Nat. Cell Biol. 2, 695–702 [DOI] [PubMed] [Google Scholar]
- 7. Guilak F., Leddy H. A., Liedtke W. (2010) Transient receptor potential vanilloid 4: The sixth sense of the musculoskeletal system? Ann. N.Y. Acad. Sci. 1192, 404–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Liedtke W., Kim C. (2005) Functionality of the TRPV subfamily of TRP ion channels: add mechano-TRP and osmo-TRP to the lexicon! Cell. Mol. Life Sci. 62, 2985–3001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mutai H., Heller S. (2003) Vertebrate and invertebrate TRPV-like mechanoreceptors. Cell Calcium 33, 471–478 [DOI] [PubMed] [Google Scholar]
- 10. Krakow D., Vriens J., Camacho N., Luong P., Deixler H., Funari T. L., Bacino C. A., Irons M. B., Holm I. A., Sadler L., Okenfuss E. B., Janssens A., Voets T., Rimoin D. L., Lachman R. S., Nilius B., Cohn D. H. (2009) Mutations in the gene encoding the calcium-permeable ion channel TRPV4 produce spondylometaphyseal dysplasia, Kozlowski type and metatropic dysplasia. Am. J. Hum. Genet. 84, 307–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rock M. J., Prenen J., Funari V. A., Funari T. L., Merriman B., Nelson S. F., Lachman R. S., Wilcox W. R., Reyno S., Quadrelli R., Vaglio A., Owsianik G., Janssens A., Voets T., Ikegawa S., Nagai T., Rimoin D. L., Nilius B., Cohn D. H. (2008) Gain-of-function mutations in TRPV4 cause autosomal dominant brachyolmia. Nat. Genet. 40, 999–1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nilius B., Voets T. (2013) The puzzle of TRPV4 channelopathies. EMBO Rep. 14, 152–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hurd L. M., Boggs M. B., Kirwin S. M., Vinette K. M., Bober M. B., Mackenzie W. G., Funanage V. L., Duncan R. L. (2013) Indentification and characterization of novel transient receptor potential vanilloid 4 (TRPV4) mutations associated with skeletal dysplasia. Trans. Orthop. Res. Soc. 59, 0036 [Google Scholar]
- 14. Camacho N., Krakow D., Johnykutty S., Katzman P. J., Pepkowitz S., Vriens J., Nilius B., Boyce B. F., Cohn D. H. (2010) Dominant TRPV4 mutations in nonlethal and lethal metatropic dysplasia. Am. J. Med. Genet. A 152A, 1169–1177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Loukin S., Su Z., Kung C. (2011) Increased basal activity is a key determinant in the severity of human skeletal dysplasia caused by TRPV4 mutations. PLoS One 6, e19533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lamande S. R., Yuan Y., Gresshoff I. L., Rowley L., Belluoccio D., Kaluarachchi K., Little C. B., Botzenhart E., Zerres K., Amor D. J., Cole W. G., Savarirayan R., McIntyre P., Bateman J. F. (2011) Mutations in TRPV4 cause an inherited arthropathy of hands and feet. Nat. Genet. 43, 1142–1146 [DOI] [PubMed] [Google Scholar]
- 17. Clark A. L., Votta B. J., Kumar S., Liedtke W., Guilak F. (2010) Chondroprotective role of the osmotically sensitive ion channel transient receptor potential vanilloid 4: age- and sex-dependent progression of osteoarthritis in Trpv4-deficient mice. Arthritis Rheum. 62, 2973–2983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Phan M. N., Leddy H. A., Votta B. J., Kumar S., Levy D. S., Lipshutz D. B., Lee S. H., Liedtke W., Guilak F. (2009) Functional characterization of TRPV4 as an osmotically sensitive ion channel in porcine articular chondrocytes. Arthritis Rheum. 60, 3028–3037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chao P. H., West A. C., Hung C. T. (2006) Chondrocyte intracellular calcium, cytoskeletal organization, and gene expression responses to dynamic osmotic loading. Am. J. Physiol. Cell Physiol. 291, C718–C725 [DOI] [PubMed] [Google Scholar]
- 20. Muramatsu S., Wakabayashi M., Ohno T., Amano K., Ooishi R., Sugahara T., Shiojiri S., Tashiro K., Suzuki Y., Nishimura R., Kuhara S., Sugano S., Yoneda T., Matsuda A. (2007) Functional gene screening system identified TRPV4 as a regulator of chondrogenic differentiation. J. Biol. Chem. 282, 32158–32167 [DOI] [PubMed] [Google Scholar]
- 21. O'Conor C. J., Leddy H. A., Benefield H. C., Liedtke W. B., Guilak F. (2014) TRPV4-mediated mechanotransduction regulates the metabolic response of chondrocytes to dynamic loading. Proc. Natl. Acad. Sci. U.S.A. 111, 1316–1321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Eleswarapu S. V., Athanasiou K. A. (2013) TRPV4 channel activation improves the tensile properties of self-assembled articular cartilage constructs. Acta Biomater. 9, 5554–5561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. O'Conor C. J., Griffin T. M., Liedtke W., Guilak F. (2013) Increased susceptibility of Trpv4-deficient mice to obesity and obesity-induced osteoarthritis with very high-fat diet. Ann. Rheum. Dis. 72, 300–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Abe Y., Abe T., Aida Y., Hara Y., Maeda K. (2004) Follistatin restricts bone morphogenetic protein (BMP)-2 action on the differentiation of osteoblasts in fetal rat mandibular cells. J. Bone Miner. Res. 19, 1302–1307 [DOI] [PubMed] [Google Scholar]
- 25. Funaba M., Ogawa K., Murata T., Fujimura H., Murata E., Abe M., Takahashi M., Torii K. (1996) Follistatin and activin in bone: expression and localization during endochondral bone development. Endocrinology 137, 4250–4259 [DOI] [PubMed] [Google Scholar]
- 26. Rosen V. (2006) BMP and BMP inhibitors in bone. Ann. N.Y. Acad. Sci. 1068, 19–25 [DOI] [PubMed] [Google Scholar]
- 27. Kuettner K. E., Pauli B. U., Gall G., Memoli V. A., Schenk R. K. (1982) Synthesis of cartilage matrix by mammalian chondrocytes in vitro. I. Isolation, culture characteristics, and morphology. J. Cell Biol. 93, 743–750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liedtke W. B., McKinley M. J., Walker L. L., Zhang H., Pfenning A. R., Drago J., Hochendoner S. J., Hilton D. L., Lawrence A. J., Denton D. A. (2011) Relation of addiction genes to hypothalamic gene changes subserving genesis and gratification of a classic instinct, sodium appetite. Proc. Natl. Acad. Sci. U.S.A. 108, 12509–12514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Murphy K. C. (1998) Use of bacteriophage lambda recombination functions to promote gene replacement in Escherichia coli. J. Bacteriol. 180, 2063–2071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang Y., Buchholz F., Muyrers J. P., Stewart A. F. (1998) A new logic for DNA engineering using recombination in Escherichia coli. Nat. Genet. 20, 123–128 [DOI] [PubMed] [Google Scholar]
- 31. Yu D., Ellis H. M., Lee E. C., Jenkins N. A., Copeland N. G., Court D. L. (2000) An efficient recombination system for chromosome engineering in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 97, 5978–5983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nagy A. (2003) Manipulating the Mouse Embryo: a Laboratory Manual, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, USA [Google Scholar]
- 33. Gong S., Yang X. W., Li C., Heintz N. (2002) Highly efficient modification of bacterial artificial chromosomes (BACs) using novel shuttle vectors containing the R6Kgamma origin of replication. Genome Res. 12, 1992–1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Muyrers J. P., Zhang Y., Stewart A. F. (2000) ET-cloning: think recombination first. Genet. Eng. (N. Y). 22, 77–98 [DOI] [PubMed] [Google Scholar]
- 35. Lakso M., Pichel J. G., Gorman J. R., Sauer B., Okamoto Y., Lee E., Alt F. W., Westphal H. (1996) Efficient in vivo manipulation of mouse genomic sequences at the zygote stage. Proc. Natl. Acad. Sci. U.S.A. 93, 5860–5865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liu H., Kishi T., Roseberry A. G., Cai X., Lee C. E., Montez J. M., Friedman J. M., Elmquist J. K. (2003) Transgenic mice expressing green fluorescent protein under the control of the melanocortin-4 receptor promoter. J. Neurosci. 23, 7143–7154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Furman B. D., Strand J., Hembree W. C., Ward B. D., Guilak F., Olson S. A. (2007) Joint degeneration following closed intraarticular fracture in the mouse knee: a model of posttraumatic arthritis. J. Orthop. Res. 25, 578–592 [DOI] [PubMed] [Google Scholar]
- 38. Hamburger V., Hamilton H. L. (1951) A series of normal stages in the development of the chick embryo. J. Morphol. 88, 49–92 [PubMed] [Google Scholar]
- 39. Yamazaki Y., Yuguchi M., Kubota S., Isokawa K. (2011) Whole-mount bone and cartilage staining of chick embryos with minimal decalcification. Biotech. Histochem. 86, 351–358 [DOI] [PubMed] [Google Scholar]
- 40. Liedtke W., Tobin D. M., Bargmann C. I., Friedman J. M. (2003) Mammalian TRPV4 (VR-OAC) directs behavioral responses to osmotic and mechanical stimuli in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U.S.A. 100(Suppl. 2), 14531–14536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Voets T., Prenen J., Vriens J., Watanabe H., Janssens A., Wissenbach U., Bodding M., Droogmans G., Nilius B. (2002) Molecular determinants of permeation through the cation channel TRPV4. J. Biol. Chem. 277, 33704–33710 [DOI] [PubMed] [Google Scholar]
- 42. Boss V., Roback J. D., Young A. N., Roback L. J., Weisenhorn D. M., Medina-Flores R., Wainer B. H. (2001) Nerve growth factor, but not epidermal growth factor, increases Fra-2 expression and alters Fra-2/JunD binding to AP-1 and CREB binding elements in pheochromocytoma (PC12) cells. J. Neurosci. 21, 18–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Merino R., Macias D., Ganan Y., Rodriguez-Leon J., Economides A. N., Rodriguez-Esteban C., Izpisua-Belmonte J. C., Hurle J. M. (1999) Control of digit formation by activin signalling. Development 126, 2161–2170 [DOI] [PubMed] [Google Scholar]
- 44. Gajos-Michniewicz A., Pawlowska E., Ochedalski T., Piastowska-Ciesielska A. (2012) The influence of follistatin on mechanical properties of bone tissue in growing mice with overexpression of follistatin. J. Bone Miner. Metab. 30, 426–433 [DOI] [PubMed] [Google Scholar]
- 45. West A. E., Chen W. G., Dalva M. B., Dolmetsch R. E., Kornhauser J. M., Shaywitz A. J., Takasu M. A., Tao X., Greenberg M. E. (2001) Calcium regulation of neuronal gene expression. Proc. Natl. Acad. Sci. U.S.A. 98, 11024–11031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Blount A. L., Vaughan J. M., Vale W. W., Bilezikjian L. M. (2008) A Smad-binding element in intron 1 participates in activin-dependent regulation of the follistatin gene. J. Biol. Chem. 283, 7016–7026 [DOI] [PubMed] [Google Scholar]
- 47. Thorneloe K. S., Cheung M., Bao W., Alsaid H., Lenhard S., Jian M. Y., Costell M., Maniscalco-Hauk K., Krawiec J. A., Olzinski A., Gordon E., Lozinskaya I., Elefante L., Qin P., Matasic D. S., James C., Tunstead J., Donovan B., Kallal L., Waszkiewicz A., Vaidya K., Davenport E. A., Larkin J., Burgert M., Casillas L. N., Marquis R. W., Ye G., Eidam H. S., Goodman K. B., Toomey J. R., Roethke T. J., Jucker B. M., Schnackenberg C. G., Townsley M. I., Lepore J. J., Willette R. N. (2012) An orally active TRPV4 channel blocker prevents and resolves pulmonary edema induced by heart failure. Sci. Transl. Med. 4, 159ra148. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.