

Skeletal muscle is the main site of insulin-dependent glucose uptake. Insulin inactivation of the Akt substrate Rab-GAP AS160 leads to Rab8A activation in myocytes. The molecular motor myosin Va is a Rab8A effector in this pathway, leading to GLUT4 translocation to the myocyte surface, linking signal transduction to vesicle traffic.
Abstract
Rab-GTPases are important molecular switches regulating intracellular vesicle traffic, and we recently showed that Rab8A and Rab13 are activated by insulin in muscle to mobilize GLUT4-containing vesicles to the muscle cell surface. Here we show that the unconventional motor protein myosin Va (MyoVa) is an effector of Rab8A in this process. In CHO-IR cell lysates, a glutathione S-transferase chimera of the cargo-binding COOH tail (CT) of MyoVa binds Rab8A and the related Rab10, but not Rab13. Binding to Rab8A is stimulated by insulin in a phosphatidylinositol 3-kinase–dependent manner, whereas Rab10 binding is insulin insensitive. MyoVa-CT preferentially binds GTP-locked Rab8A. Full-length green fluorescent protein (GFP)–MyoVa colocalizes with mCherry-Rab8A in perinuclear small puncta, whereas GFP–MyoVa-CT collapses the GTPase into enlarged perinuclear depots. Further, GFP–MyoVa-CT blocks insulin-stimulated translocation of exofacially myc-tagged GLUT4 to the surface of muscle cells. Mutation of amino acids in MyoVa-CT predicted to bind Rab8A abrogates both interaction with Rab8A (not Rab10) and inhibition of insulin-stimulated GLUT4myc translocation. Of importance, small interfering RNA–mediated MyoVa silencing reduces insulin-stimulated GLUT4myc translocation. Rab8A colocalizes with GLUT4 in perinuclear but not submembrane regions visualized by confocal total internal reflection fluorescence microscopy. Hence insulin signaling to the molecular switch Rab8A connects with the motor protein MyoVa to mobilize GLUT4 vesicles toward the muscle cell plasma membrane.
INTRODUCTION
Skeletal muscle is the primary tissue disposing of dietary glucose, a response regulated by insulin and necessary to maintain whole-body glucose homeostasis. Insulin also stimulates glucose uptake into adipocytes, where glucose is converted into triglycerides, whereas in muscle it is stored as glycogen. In both cell types, glucose entry is rate limiting and mediated by the transmembrane facilitative glucose transporter protein GLUT4. GLUT4 dynamically cycles to and from the plasma membrane in vesicles, with fast endocytic and slower exocytic rates creating a larger intracellular pool of GLUT4. The molecular basis for insulin-stimulated glucose uptake is a gain in surface GLUT4 brought about by a surge in the exocytic rate of GLUT4-containing vesicles. Myoblasts and preadipocytes in culture have been used to study the mechanisms of insulin action upon GLUT4 traffic, successfully identifying the fusion machinery involved in GLUT4 vesicle fusion with the plasma membrane, as well as with insulin receptor–derived signals triggering GLUT4 translocation. However, it is unknown how signal transduction interacts with molecules enacting mechanical mobilization of GLUT4 vesicles.
Insulin signals leading to GLUT4 translocation include activation of phosphatidylinositol-3-kinase (PI3K) to produce phosphatidylinositol 3,4,5-trisphosphate, responsible for the recruitment to and activation of Akt/PKB on the plasma membrane. Akt then phosphorylates and thereby inactivates the Rab-GAP AS160 (Akt substrate of 160 kDa, TBC1D4). As a result, the Rab GTPase targets of AS160 can prevail in their active, GTP-bound form. Indeed, we showed that insulin leads to GTP loading of Rab8A and Rab13 (but not Rab10) in muscle cells, and these Rab GTPases lie downstream of AS160 insofar as their overexpression rescues GLUT4 translocation from inhibition by constitutively active AS160 (AS160-4A; Ishikura and Klip, 2008; Sun et al., 2010; Chen et al., 2012). Rab8A, Rab13, and Rab10 are closely related evolutionarily (Pereira-Leal and Seabra, 2000) and show a high degree of homology, although they differ in their C-terminal region. Rab10 appears to be the target of AS160 in 3T3-L1 adipocytes, since silencing its expression restores basal surface GLUT4 levels that are elevated upon AS160 depletion (Sano et al., 2007). Conversely, this restoration of surface GLUT4 levels in muscle cells is achieved by silencing Rab8A (Ishikura and Klip, 2008). Consistent with Rab8A and Rab13 participating in GLUT4 traffic in muscle cells and Rab10 in adipocytes, silencing these GTPases in the respective cells reduces insulin-dependent GLUT4 translocation (Ishikura et al., 2007; Ishikura and Klip, 2008; Sano et al., 2007; Sun et al., 2010). In muscle cells, insulin-dependent Rab8A GTP loading precedes that of Rab13, and whereas Rab8A localization is predominantly perinuclear, Rab13 is both perinuclear and at/near the cell surface (Sun et al., 2010). These Rabs are not redundant, since the inhibition of insulin-dependent GLUT4 translocation brought about by Rab13 knockdown was rescued by overexpression of a Rab13 orthologue but not by Rab8A overexpression.
As molecular switches, Rab molecules signal to effector molecules through their C-terminal region, and hence a key challenge is to identify effectors that are engaged by Rab GTPases in an insulin-dependent manner. In particular, linking Rab signaling to proteins with mechanical traffic function would point to a convergence of signal transduction and the vesicle traffic machinery. Of note, Rab8A, Rab10, and Rab11 bind MyoV motors directly, and Rab8A can bind to all three MyoV isoforms (Hume et al., 2001; Rodriguez and Cheney, 2002; Roland et al., 2009). MyoVa is expressed as two spliced variants—the brain (–D exon) and melanocyte (+D exon) isoforms—and both Rab8A and Rab10 bind exclusively to the melanocyte isoform of MyoVa (Hume et al., 2001; Wu et al., 2002; Roland et al., 2009).
We previously showed that a construct encoding the cargo-binding COOH tail of MyoVb mislocalizes Rab8A and Rab10 and inhibits GLUT4 translocation, suggesting that a Rab-regulated step of GLUT4 traffic may signal through its association with myosin V motors in muscle cells (Ishikura and Klip, 2008). Conversely, silencing MyoVa reduces GLUT4 translocation in 3T3-L1 adipocytes (Yoshizaki et al., 2007), and it was reported that MyoVa interacts with and governs the localization of Rab10 in those cells (Chen et al., 2012). Given the differences in Rab GTPase engagement with the GLUT4 system existing between muscle cells and adipocytes, it becomes paramount to identify the myosin V that operates in muscle cells and whether it is engaged through Rab8A or other GTPases.
Here we show that 1) rat muscle and muscle cells express MyoVa but not MyoVb or MyoVc; 2) MyoVa does not bind Rab13, but it does bind Rab8A in its GTP-bound form, and only the binding to Rab8A (and not that of Rab10) is stimulated by insulin, in a PI3K-dependent manner; 3) a C-terminal fragment of MyoVa that binds Rab8A mislocalizes the GTPase and inhibits GLUT4 translocation to the plasma membrane; 4) mutations in the predicted Rab8A-binding sites at the C-terminus of MyoVa prevent Rab8A binding (but not Rab10 binding) and eliminate the effects on Rab8A mislocalization and impairment of GLUT4 translocation; 5) silencing MyoVa reduces GLUT4 translocation; and 6) MyoVa and Rab8A colocalize at perinuclear and cytosolic regions, as do Rab8A and GLUT4, although Rab8A does not reach the submembrane region visualized through total internal reflection fluorescence (TIRF) microscopy, whereas GLUT4 does. We propose that insulin-activated Rab8A binds to MyoVa to mobilize GLUT4 out of the perinuclear region toward the muscle cell periphery. Other Rabs, such as Rab13, may promote traffic beyond this step.
RESULTS
Interaction of MyoVa with Rab8A, Rab10, and Rab13
Of the MyoV isoforms, only MyoVa was detected in rat L6 myoblast or myotube cDNA by semiquantitative PCR, with no detectable signals for either myosin Vb or Vc (Supplemental Figure S1). Both the –D and +D mRNA splice variants are expressed in both myoblasts and myotube stages of the culture. MyoVa is also expressed in mouse C2C12 muscle cells, 3T3-L1 adipocytes, and mouse and rat skeletal muscles (Supplemental Figure S1). MyoVb was not detectable in any of the foregoing cells or tissues, and MyoVc was only observed in mouse muscle but not in rat muscle or the rat muscle cell line; a trace amount of MyoVc was observed in 3T3-L1 adipocytes and C2C12 myoblasts (Supplemental Figure S1).
MyoVa is an actin-based motor, composed of an N-terminal (head) motor domain and a C-terminal (tail) Rab-binding domain (Figure 1A). Using the +D splice variant cDNA, we created a truncated MyoVa–glutathione S-transferase (GST) fusion protein containing the Rab-binding domain (amino acids 1260–1854; GST–MyoVa-CT) and tested its ability to interact with GFP fusion proteins of Rabs 8A, 10, and 13 transfected into CHO cells stably expressing the insulin receptor (CHO-IR cells) in basal and insulin-stimulated conditions (100 nM, 15 min). CHO-IR cells were chosen for these biochemical experiments, given their proven sensitivity to insulin and their high yield of transfectability necessary to perform pull-down experiments. GST–MyoVa-CT pulled down Rab8A and Rab10 but not Rab13, and only the interaction of Rab8A with MyoVa-CT was elevated (greater than twofold) in an insulin-dependent manner (Figure 1B). Insulin stimulates GTP loading of Rab8A but not Rab10 (Sun et al., 2010), and therefore we tested the ability of GST–MyoVa-CT to interact with wild-type (wt), activated (GTP-locked) or inactivated (GDP-locked) mutants of Rab8A expressed in CHO-IR cells. Figure 1C shows that GST–MyoVa-CT bound wt and GTP-locked mutant Rab8A but not GDP-locked mutant Rab8A. Although a nonspecific band appeared in all input samples that barely separated from GFP-Rab8Awt and more clearly separated from the GTP- or GDP-locked mutants (Figure 1C gel), this band was not present in the pull-down material (Figure 1C, bar graph). Binding of Rab8A-GTP was 2.5-fold higher than that of wt Rab8A when normalized for total GFP-Rab fusion protein expression (Figure 1C), consistent with the hypothesis that MyoVa is an effector of active Rab8A.
FIGURE 1:

MyoVa-CT binds Rab8A and Rab10 but not Rab13. Binding to Rab8A is insulin, GTP, and PI3K dependent. (A) Schematic representation of MyoVa and the GST fusion protein with the C-terminal amino acids 1260–1854 of MyoVa (GST–MyoVa-CT). (B) Cell lysates prepared from CHO-IR cells expressing GFP-Rab8A, -Rab10, -Rab13, or GFP and treated with/without insulin for 15 min were incubated with glutathione–Sepharose (GSHS) beads loaded with GST–MyoVa-CT. The beads were sedimented, and complexes in the GST–MyoVa-CT pull down or aliquots of lysates (input) were subjected to SDS–PAGE and immunoblotted with anti-GFP antibody, as described in Materials and Methods. The pixel intensity of the pulled-down bands (upper gel) was expressed as the ratio of the pixel intensity of the corresponding band in the input lysates (lower gel) and is presented as the ratio of the insulin to basal samples in the graph bars to the right of the gel (mean ± SE, ***p < 0.001). Phospho-Akt S473 (p-Akt) was measured in lysates to confirm insulin effectiveness. Data are the mean of three independent experiments. (C) Cell lysates prepared from CHO-IR cells expressing GFP-Rab8Awt, the constitutively GTP-bound Rab8AQ67L mutant (GFP–Rab8A-GTP), the constitutively GDP-bound Rab8AT22N mutant (GFP–Rab8A-GDP), or GFP were incubated with GSHS beads loaded with MyoVa-CT. The complexes were pulled down and analyzed as in B. The graph beneath the gels show the quantified results (n = 3, mean ± SE, **p < 0.01). #, Nonspecific band detected by anti-GFP in the input lysates. For the quantification shown in the bar graph, we zoomed in on the image, which allowed us to outline the band separation and select the specific Rab8A band without including the nonspecific one in the input gel. (D) CHO-IR cells were transfected with GFP-Rab8A or GFP for 48 h, followed by pretreatment with 100 nM wortmannin in DMSO (or DMSO alone) for 30 min, followed by treatment with/without insulin (100 nM) for 15 min and with/without wortmannin (or DMSO). Cells were lysed and lysates subjected to GST–MyoVa-CT pull down using GSHS beads, and then complexes or lysate aliquots (input) were separated by SDS–PAGE and immunoblotted with anti-GFP antibody. Lysates were also blotted for p-Akt to confirm insulin and wortmannin effectiveness. The pixel intensity of each binding reaction was quantified as in B and expressed relative to GFP–Rab8Awt-DMSO (mean ± SE, **p < 0.01: insulin vs. insulin + wortmannin). Data are the mean of three independent experiments.
Insulin increases Rab8A–MyoVa interaction downstream of PI3K
To examine the MyoVa–Rab8A interaction dependence on insulin signaling in muscle cells, we tested the effect of wortmannin, a PI3K inhibitor that blocks activation of Akt, on GFP-Rab8A pull down by GST–MyoVa-CT. GFP-Rab8A was expressed in CHO-IR cells to equivalent levels in all conditions before stimulation with insulin (100 nM, 15 min) in the absence or presence of wortmannin (100 nM, with 30-min pretreatment). Binding of GFP-Rab8A to GST–MyoVa-CT was 1.9 times greater in lysates of cells treated with insulin than with those of unstimulated, dimethyl sulfoxide (DMSO)–treated control cells, and wortmannin effectively eliminated the insulin-induced response (Figure 1D). These results are consistent with a role of the PI3K axis in regulating the GTP-loaded levels of Rab8A (Sano et al., 2003; Miinea et al., 2005; Ishikura et al., 2007; Zaid et al., 2008).
MyoVa colocalizes with Rab8A in L6 myoblasts
To study the reciprocal localization of MyoVa and Rab8A in muscle cells, we expressed them with individual fluorescent tags into L6 muscle cells. We are aware of the caveat that the information obtained pertains to overexpressed proteins rather than the endogenous ones, but this is a common practice, as regrettably there are no useful antibodies to detect the latter. By confocal fluorescence microscopy of L6 myoblasts cotransfected with full-length (FL) GFP-MyoVa and mCherry-Rab8A (MC-Rab8A), the two proteins showed a very similar punctate perinuclear distribution in untreated (basal) cells (Figure 2A), which suggested a high degree of colocalization. Some GFP-MyoVa fluorescence spread into the cell periphery, where very little MC-Rab8A was detected and barely overlapped with GFP-MyoVa. The ratio of peripheral to total signal of MC-Rab8A fluorescence intensity was quantified in 10–15 cells for basal and insulin-stimulated states and compared with that in control cells expressing GFP plus MC-Rab8A (Figure 2C). Insulin stimulation significantly promoted the distribution of Rab8A to the cell periphery in cells overexpressing GFP-MyoVa (but not in those expressing only GFP), suggesting that the excess motor protein sufficed to mobilize the GTPase toward the cell periphery under the influence of insulin. This resulted in increased colocalization of GFP-MyoVa with MC-Rab8A in the peripheral cytoplasmic region (region of interest excluded the area of intense perinuclear fluorescence of GFP–MyoVa-FL; Figure 2, A, B, and D).
FIGURE 2:
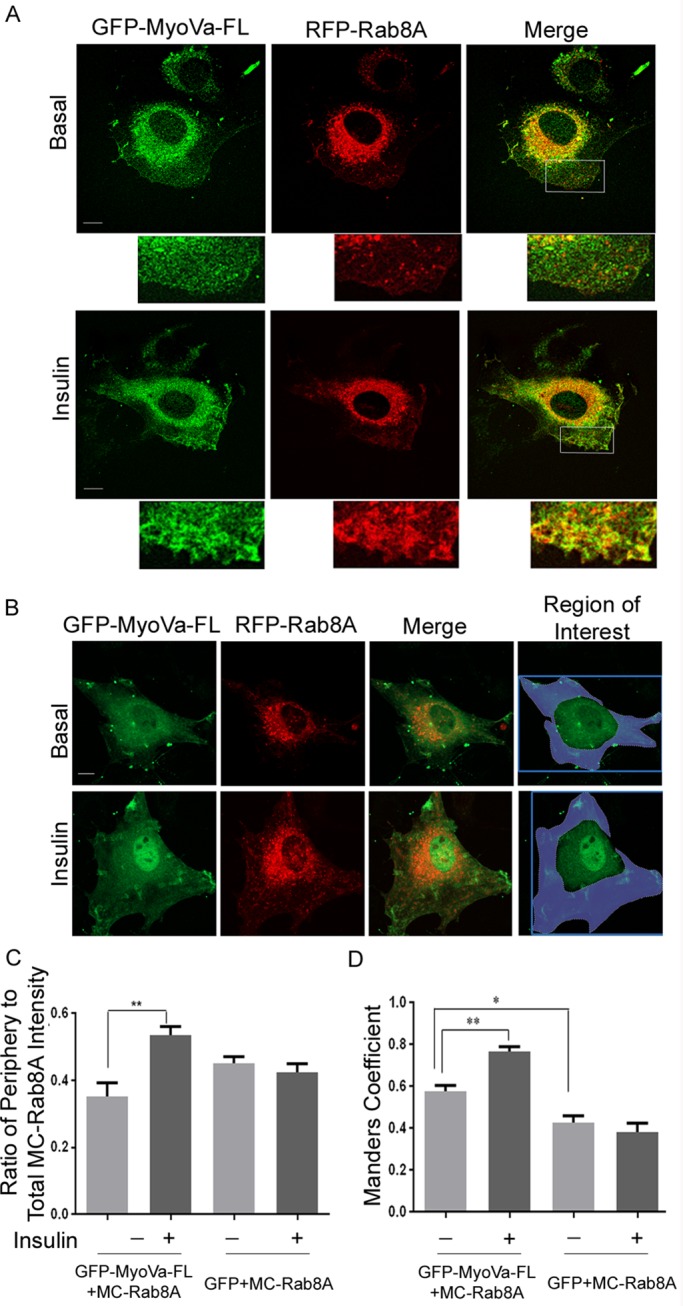
Full-length MyoVa colocalizes with Rab8A in L6 myoblasts, and insulin increases this colocalization in cytoplasmic peripheral regions. L6 cells grown on glass coverslips were cotransfected with GFP–MyoVa-FL and MC-Rab8A for 24 h, followed by no further treatment (basal) or 100 nM insulin for 15 min (insulin), and then processed for spinning-disk fluorescence microscopy. (A) Fluorescence of GFP–MyoVa-FL (green) and MC-Rab8A (red) in representative cells. Excerpts are magnified regions from the original image represented by the outlined region of interest indicated in the merges. Representative images collected from three independent experiments in which >25 cells/experiment were analyzed. (B) Similar experiment as in A but with the area of interest indicated in blue including all regions of the cell except the nuclear and perinuclear regions. (C) Quantification of the ratio of peripheral MC-Rab8A to total MC-Rab8A fluorescence intensity and its response to insulin. (D) Quantification of the Manders coefficient of colocalization of MC-Rab8A relative to GFP–MyoVa-FL.
The MyoVa-CT–Rab8A interaction mislocalizes Rab8A and reduces insulin-dependent GLUT4 translocation
Because full-length MyoVa and Rab8A showed partial colocalization and the GST–MyoVa-CT fragment was able to pull down Rab8A, we next examined whether MyoVa-CT and Rab8A can interact in situ. GFP–MyoVa-CT was coexpressed with MC–Rab8A-wt, -GTP, or -GDP constructs in CHO-IR cells (used here again due to their high rate of transfection), and complexes were immunoprecipitated with anti-mCherry antibody. MC-Rab8A coprecipitated with GFP–MyoVa-CT, and, of note, the magnitude of this response was higher in the insulin-stimulated state (Figure 3A). Further, GFP–MyoVa-CT coprecipitated MC–Rab8A-GTP, whereas there was only minimal coprecipitation of MC–Rab8A-GDP above the background level observed when using control immunoglobulin G instead of anti-mCherry antibody (Figure 3B). These findings are consistent with the GST pull-down results in Figure 1.
FIGURE 3:
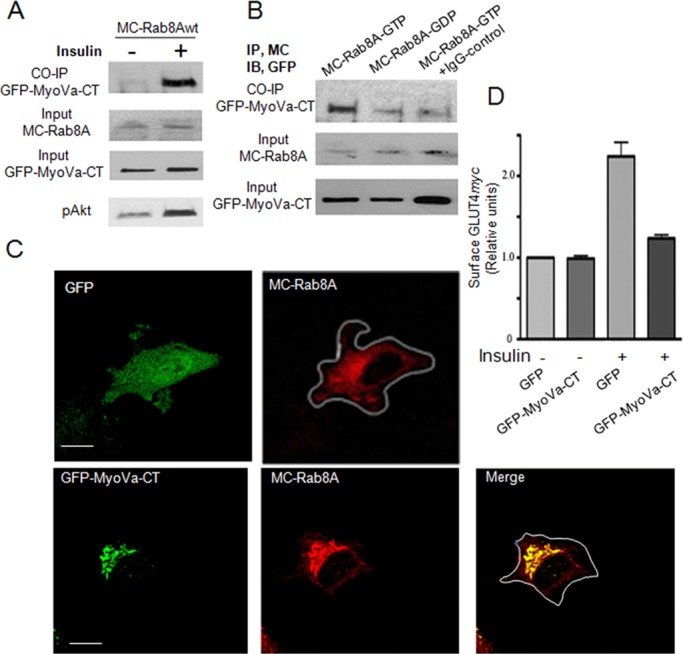
MyoVa–CT-Rab8A interaction occurs in situ and leads to mislocalization of Rab8A into large puncta and impairs GLUT4 translocation to the cell surface in muscle cells. (A) CHO-IR cells were cotransfected with GFP–MyoVa-CT and mCherry-Rab8A (MC-Rab8A) for 48 h, followed by treatment with the cell-permeant cross-linker DSP (1.5 mM for 30 min) before stimulation with or without insulin (100 nM, 15 min). Cells were lysed in 50 mM Tris, pH 7.4, with 2% SDS. Coimmunoprecipitation (CO-IP) was with mouse anti-mCherry antibody. Immunoprecipitates were immunoblotted with rabbit anti-GFP antibody to detect GFP–MyoVa-CT. Aliquots of lysates were immunoblotted directly for MC-Rab8a (anti-mCherry) or GFP–MyoVa-CT (anti-GFP) (input). (B) CHO-IR cells were cotransfected with GFP–MyoVa-CT and Q67L MC-Rab8A (MC–Rab8A-GTP) or with T22N MC-Rab8A (MC–Rab8A-GDP) for 48 h, followed by treatment with DSP as in A and coimmunoprecipitation with mouse anti-mCherry or mouse immunoglobulin G. (C) L6-GLUT4myc muscle cells were cotransfected overnight with GFP and MC-Rab8A or GFP–MyoVa-CT with MC-Rab8A in 12-well plates. Cells were suspended and plated on glass coverslips for 24 h and then imaged by confocal fluorescence microscopy. Representative collapsed images of two independent experiments with >25 cells quantified/experiment. (D) L6-GLUT4myc muscle cells transfected as described were also stimulated with or without insulin (100 nM, 15 min), processed for measurement of cell surface GLUT4myc, and imaged using a LSM510 Zeiss confocal microscope and quantified as described in Materials and Methods. Bar graph, mean responses from two independent experiments of >25 cells/experiment.
The interaction of GFP–MyoVa-CT with Rab8A was next verified in L6 muscle cells. On confocal fluorescence microscopy of L6 myoblasts, MC–Rab8A-wt displayed a typical, dispersed vesicle–like, perinuclear distribution when coexpressed with GFP (Figure 3C). This disperse, small punctate morphology was observed in all cells transfected with GFP and MC-Rab8A (50 cells imaged). When cotransfected with GFP–MyoVa-CT, however, MC-Rab8A was considerably mislocalized into vastly enlarged perinuclear depots that colocalized with GFP–MyoVa-CT (Figure 3C). This pattern was observed in 76% of cells imaged (38 of 50 cells), and similarly collapsed depots but of somewhat smaller size were observed in the remaining 12 of 50 cells. Because MyoVa-CT lacks its motor domain and cannot interact with actin filaments, it probably interacts with MC-Rab8A on vesicles or compartments but fails to connect them with actin filaments (Wu et al., 2001). In contrast, there was no obvious change in the distribution of MC-Rab13 when coexpressed with either GFP or GFP–MyoVa-CT in muscle cells (Supplemental Figure S2), consistent with the inability of GFP-Rab13 to bind GST–MyoVa-CT (Figure 1B).
Given that Rab8A is required for insulin-dependent GLUT4 translocation in muscle cells (Ishikura et al., 2007; Ishikura and Klip, 2008; Sun et al., 2010) a prediction of the mislocalization of Rab8A by GFP–MyoVa-CT is that the latter would impair insulin-dependent GLUT4 translocation. Indeed, the insulin-induced gain in GLUT4myc at the muscle cell surface was vastly reduced in cells expressing GFP–MyoVa-CT (Figure 3D). Thus Rab8A and MyoVa-CT can interact in situ, and this leads to abnormal localization of Rab8A and inhibition of insulin-stimulated translocation of GLUT4 to the muscle cell surface.
Mutations on MyoVa-CT largely reduce the interaction with Rab8A and restore insulin-stimulated GLUT4myc translocation
We hypothesized that the inhibition of insulin-stimulated GLUT4myc translocation by MyoVa-CT is related to its ability to interact with and mislocalize Rab8A. Thus we sought to identify the binding sites for Rab8A on MyoVa-CT. Goldenring and coworkers (Roland et al., 2011) showed that binding of Rab8A to another MyoV isoform, MyoVb, requires Q1300 and Y1307, but the sites on MyoVa are unknown. Aligning the C-terminal sequences of MyoVa and MyoVb revealed potential equivalence of DVQ1300 in MyoVa to VDQ1300 in MyoVb and of AY1312IG in MyoVa with AY1307HG in MyoVb. Moreover, Q1300 and Y1312 in MyoVa were conserved in mice, rats, and humans (Supplemental Figure S3); therefore, we introduced the mutations Q1300L (M1) and Y1312C (M2) on MyoVa-CT to generate the double mutant (2M) in both GST and GFP expression vectors of MyoVa-CT. In pull-down assays with CHO-IR cell lysates, very little transfected GFP-Rab8A associated with GST–MyoVa-CT(2M) compared with GST–MyoVa-CT (Figure 4A), suggesting the Rab8A-MyoVa interaction requires Q1300 and Y1312 in MyoVa. In contrast, GST–MyoVa-CT(2M) bound transfected GFP-Rab10 to a similar extent as did GST–MyoVa-CT. These results indicate that Q1300 and Y1312 are required to bind Rab8A but not Rab10 to MyoVa (Supplemental Figure S4). In line with the inability of GST–MyoVa-CT(2M) to bind Rab8A in CHO-IR cell lysates, coexpression of GFP-MyoVa(2M) with MC-Rab8A in muscle cells did not cause MC-Rab8A proteins to aggregate as did GFP–MyoVa-CT (Figure 4B).
FIGURE 4:

Mutation of putative binding sites for Rab8A on MyoVa-CT abolished the interaction between Rab8A and MyoVa-CT, allowing GLUT4 translocation to proceed. (A) GST–MyoVa-CT or GST–MyoVa-CT(2M) was bound to GSHS beads and incubated with cell lysates from CHO-IR transfected with GFP-Rab8A or GFP, and then the pull-down complexes were subjected to SDS–PAGE. The gel was cut horizontally, and the upper section was stained with Coomassie blue to reveal the amount of GST–MyoVa-CT or GST–MyoVa-CT(2M) on the GSHS beads (input), and the lower section was immunoblotted with anti-GFP antibody (middle and top), as described in Materials and Methods. The binding of GFP-Rab8A to GST–MyoVa-CT (2M) was calculated (as in Figure 1) and is represented as change relative to that of GST–MyoVa-CT with GFP-Rab8A (mean ± SE, ***p < 0.001); data are the summary of three independent experiments. (B) L6-GLUT4myc muscle cells were cotransfected GFP–MyoVa-CT or GFP–MyoVa-CT(2M) with MC-Rab8A, followed by imaging with confocal fluorescence microscopy, presented as collapsed images. MC-Rab8A mislocalized into GFP–MyoVa-CT–positive enlarged puncta; however, GFP–MyoVa-CT(2M) did not colocalize with or induce enlarged MC-Rab8A puncta. Representative images of three independent experiments. (C) L6-GLUT4myc cells were transfected with GFP, GFP–MyoVa-CT, or GFP–MyoVa-CT(2M) in 12-well plates overnight. Cells were split and plated on coverslips the next day, and after 24 h they were treated with or without insulin (100 nM, 15 min) and then processed for measurement of cell-surface GLUT4myc and imaged by confocal microscopy and quantified as described in Materials and Methods. Mean responses from three independent experiments of >25 cells/experiment (mean ± SE, **p < 0.01, insulin GFP vs. insulin GFP–MyoVa-CT; *p < 0.05 insulin GFP–MyoVa-CT vs. insulin GFP–MyoVa-CT(2M).
The influence of GST–MyoVa-CT(2M) on GLUT4 traffic was then tested. As before, insulin promoted a GLUT4myc translocation to the cell surface (by 2.7-fold) in control L6 muscle cells expressing GFP that was markedly impaired by transfected GFP–MyoVa-CT, whereas GFP–MyoVa-CT(2M) allowed an almost complete (88%) insulin-stimulated GLUT4myc response (Figure 4C). These experiments strongly suggest that preventing Rab8A binding to the MyoVa-CT fragment in situ eliminates the ability of MyoVa-CT to interfere with GLUT4 traffic to the cell surface. Of note, MyoVa-CT(2M) could still bind Rab10 like the nonmutated MyoVa-CT fragment, suggesting that this GTPase is not responsible for the differential effect of these fragments on GLUT4 translocation.
Given that the binding of MyoVa-CT to Rab8A correlated with the ability of MyoVa-CT to interfere with GLUT4myc translocation to the cell surface, these findings implicate MyoVa as an effector of Rab8A required for insulin-stimulated GLUT4 traffic.
Rab8A colocalizes with GLUT4 in perinuclear regions but not in the TIRF zone of muscle cells
The foregoing results indicate that MyoVa interacts with Rab8A and that this interaction is required for GLUT4 translocation to the plasma membrane. To begin to address the cellular locus where the input of Rab8A:MyoVa takes place during the itinerary of GLUT4, we examined the subcellular localization of Rab8A vis-à-vis GLUT4. Using spinning-disk confocal microscopy, we found GFP-GLUT4 and MC-Rab8A to partially colocalize at the perinuclear region in both basal and insulin pretreated states (Figure 5A). By this approach, Rab8A is not detected near the cell surface; however, L6 myoblasts are relatively flat cells, and hence we examined the localization of GFP-GLUT4 and MC-Rab8A within ∼100 nm of the plasma membrane using two-color TIRF microscopy (Mattheyses et al., 2010). Imaging fixed muscle cells, we readily detected GFP-GLUT4 in the TIRF zone (Figure 5B), where we previously characterized its mobility and response to insulin (Boguslavsky et al., 2012). Although molecules on GLUT4 vesicles can be detected in the TIRF region, such as the nonprocessive myosin 1c (Boguslavsky et al., 2012), Rab8A was not present (Figure 5B), despite prominent overlap with GLUT4 in perinuclear regions. Based on the predominantly perinuclear colocalization of Rab8A with MyoVa-FL (Figure 2A), the higher overlap of Rab8A with MyoVa at the cell periphery after insulin stimulation (Figure 2B and C), and the perinuclear colocalization of Rab8A with GLUT4 (Figure 5A; Sun et al., 2010), we envisage that the Rab8A:MyoVa complex directs GLUT4 traffic before it reaches the cell periphery.
FIGURE 5:
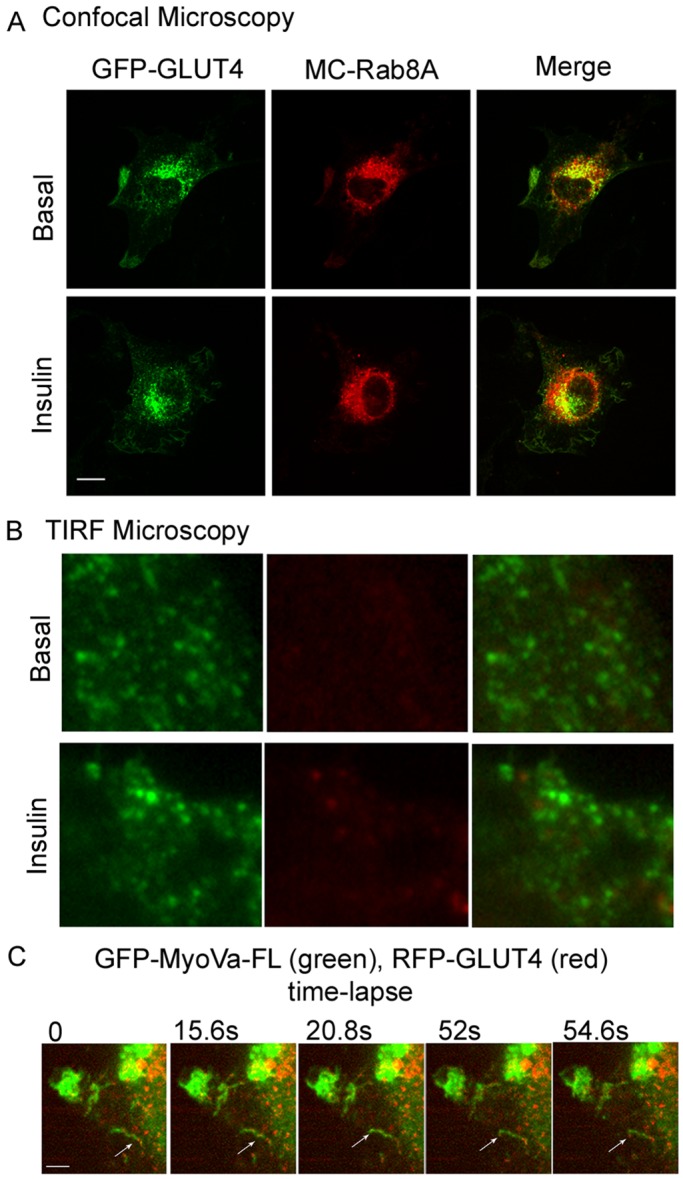
GLUT4 vesicles colocalizes with Rab8A in perinuclear regions but not in the submembrane “TIRF” zone and position along MyoVa structures. L6 muscle cells were cotransfected with GFP-GLUT4 and MC-Rab8A for 48 h, followed by treatment with or without insulin (100 nM, 15 min). (A) The cells were imaged by spinning-disk confocal microscopy as described in Materials and Methods. Representative images of focal planes through the center of the cells from three independent experiments. (B) Paraformaldehyde-fixed myoblasts were imaged with an Olympus 1XR1 TIRF microscope to examine GFP-GLUT4– and MC-Rab8A–positive vesicles beneath the cell surface as described in Materials and Methods. Results are representative of two independent experiments of 15 cells/experiment. (C) Time-lapse images of the position of GLUT4 vesicles along MyoVa structures. L6 muscle cells were cotransfected with GFP–MyoVa-FL and RFP-GLUT4 and then treated with 100 nM insulin for 10 min; live cells were imaged for the subsequent 100 s. Shown are selected time-lapse frames taken at the indicated time points during video recording. Selected time-lapse frames at the indicated time points during the video recording. The arrow points to a representative GLUT4 vesicle on the MyoVa structure.
A prediction of this scenario is that GLUT4 vesicles would be mobilized along with MyoVa, possibly positioned along actin filaments. We therefore determined the position of red fluorescent protein (RFP)–GLUT4 vis-à-vis full-length GFP-MyoVa in insulin-stimulated L6 myoblasts over time, using real-time imaging. Figure 5C shows several time-lapse images of the localization of the transporter and the motor protein and illustrates that RFP-GLUT4 puncta—likely vesicles—organize sequentially along cytosolic GFP-MyoVa structures.
Knockdown of MyoVa reduces insulin-stimulated GLUT4myc translocation
To ascertain the functional contribution of MyoVa in enabling insulin-dependent GLUT4 traffic, we reduced the expression of endogenous MyoVa via small interfering RNA (siRNA)–mediated knockdown. With an achieved knockdown efficiency of ∼60% (Figure 6A and B), the net insulin-stimulated GLUT4myc gain at the cell surface was reduced by ∼47% compared with the response in cells expressing a nonrelated (NR) control siRNA. Under these conditions, there was no reduction in insulin-dependent phosphorylation of Akt (Figure 6A). These results demonstrate the participation of endogenous MyoVa in insulin-mediated GLUT4 exocytic movement.
FIGURE 6:
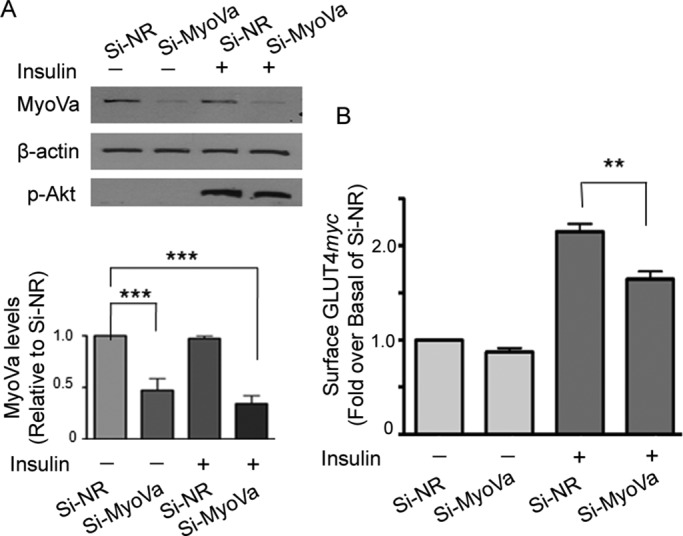
Knockdown of MyoVa reduces insulin-stimulated GLUT4 translocation to the cell surface of muscle cells. L6-GLUT4myc cells were transfected with siRNA to rat MyoVa (si-MyoVa) or scrambled nonrelated siRNA (si-NR) (300 nM for 72 h), followed by treatment with or without insulin (100 nM, 15 min). (A) Cell lysates were immunoblotted for MyoVa, β-actin, and phospho-Akt S473. The intensities of MyoVa and β-actin were quantified and expressed as ratio of MyoVa/β-actin in si-MyoVa–treated cells relative to that of si-NR (mean ± SE, ***p < 0.001). (B) Si-MyoVa– or si-NR–transfected L6-GLUT4myc cells were processed for cell-surface GLUT4myc levels using a colorimetric assay as described in Materials and Methods. Fold changes relative to si-NR. Mean responses from three independent experiments (mean ± SE, **p < 0.01).
To determine the effect that silencing MyoVa had on GLUT4 intracellular localization, we cotransfected sh-GFP-MyoVa (or control sh-expression vector encoding GFP) along with RFP-GLUT4 in L6 myoblasts. Silencing MyoVa resulted in the collapse of RFP-GLUT4 into a tight and dense location at the perinuclear region, which contrasted with the punctate perinuclear distribution observed in the control condition (Figure 7). This finding suggests that MyoVa normally allows proper positioning of GLUT4 in the perinuclear region, from where it is dynamically recruited to the cell periphery. In the absence of MyoVa, GLUT4 is unable to leave this region and instead coalesces in a tight perinuclear locale. The fact that insulin no longer allows its translocation to the membrane in cells depleted of MyoVa (Figure 6A and B) suggests that MyoVa serves the function of allowing GLUT4 departure (possibly motored by MyoVa) from its normally punctate perinuclear compartment.
FIGURE 7:
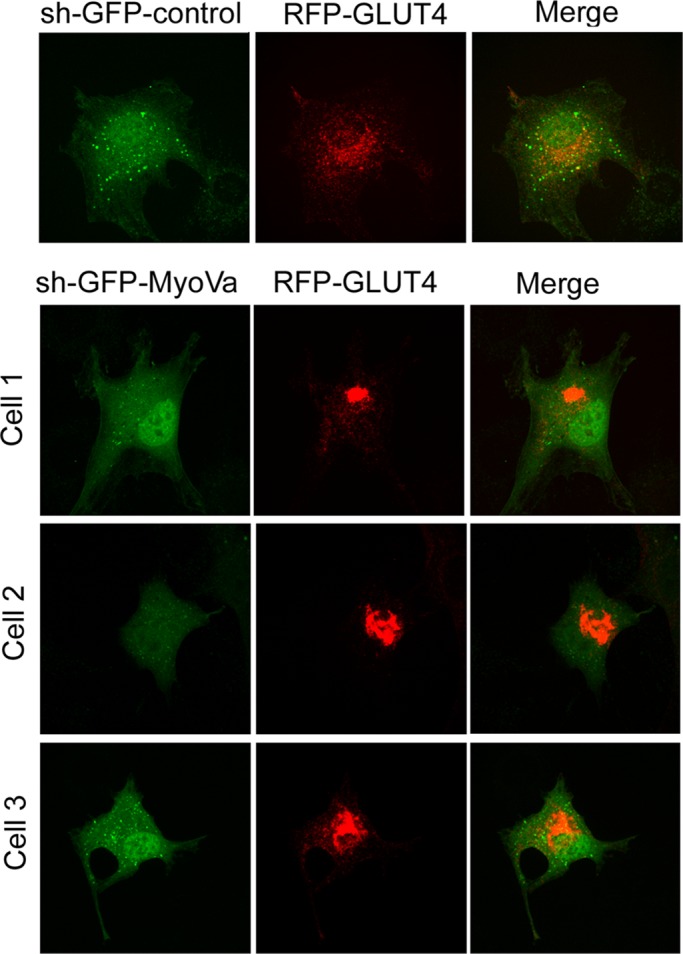
Silencing the expression of MyoVa causes GLUT4 collapse in the perinuclear region. L6 muscle cells were cotransfected with sh-GFP-MyoVa or sh-GFP control vector, along with RFP-GLUT4 for 48 h, and then processed for confocal spinning-disk fluorescence microscopy. Representative images from a sh-GFP control–transfected cell or three separate sh-GFP-MyoVa–transfected cells and their relative RFP-GLUT4 distribution. Results are representative from three independent experiments.
Finally, we assessed the specificity of MyoVa toward GLUT4 vis-à-vis its possible effects on the traffic or distribution of other membrane proteins. Unlike the mislocalization of GLUT4 caused by silencing MyoVa shown earlier, sh-GFP-MyoVa did not alter the localization of the transferrin receptor, furin, or syntaxin-6 (Figure 8). Similarly, their localization was not altered by expression of the C-terminal-fragment GFP–MyoVa-CT (Figure 8). Moreover, unlike inhibition of GLUT4 translocation caused by silencing MyoVa or expressing GFP-MyoVa-CT, these two strategies failed to appreciably affect transferrin recycling (Supplemental Figure S5).
FIGURE 8:
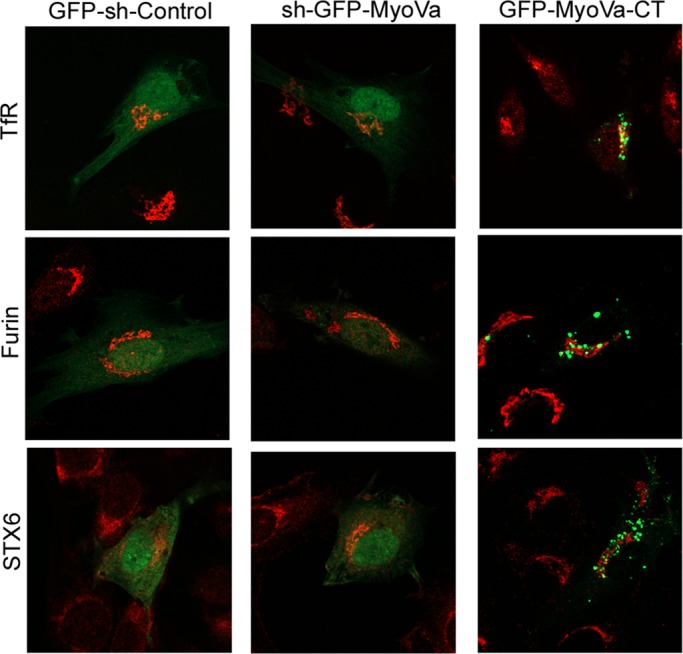
Silencing the expression of MyoVa or expression of MyoVa-CT does not alter the localization of the transferrin receptor, furin, or syntaxin-6. L6-GLUT4myc cells were transfected with sh-GFP-MyoVa, GFP–MyoVa-CT, or sh-GFP control vector for 45 h, followed by fixation with 3% PFA. Transferrin receptor, furin, or syntaxin-6 (STX6) were then detected with cognate antibodies coupled with appropriate Alexa 555 (red)–conjugated secondary antibodies. Results are representative from three independent experiments.
Overall these results support participation of MyoVa in the mobilization of GLUT4 toward the muscle cell surface promoted by Rab8A binding.
DISCUSSION
The regulation of GLUT4 translocation by insulin is central to the ability of skeletal muscle to retrieve dietary glucose from the circulation, and as such is a major element in the maintenance of glucose homeostasis. Here we focused on the fundamental question of how insulin-derived intracellular signals engage molecules that control vesicle traffic, essentially identifying a step that links signal transduction to molecular motors delivering mechanical input to GLUT4. Specifically, we show that Rab8A activation, which occurs downstream of the Akt substrate Rab-GAP AS160 (Sun et al., 2010), leads to the recruitment of the processive molecular motor MyoVa. This interaction is mapped to the C-terminal region of MyoVa, involving residues Q1300 and Y1312. Binding is preferential to the GTP-bound state of Rab8A and is potentiated by insulin in a PI3 kinase–dependent manner. This interaction is shown by both pull down from lysates and coimmunoprecipitation and found to be required for effective insulin-dependent GLUT4 translocation to the muscle cell membrane. Accordingly, lowering MyoVa expression through siRNA-mediated gene silencing proportionally reduces GLUT4 translocation. Finally, Rab8A and MyoVa colocalize on endomembranes not reaching the TIRF-imaged zone, whereas GLUT4 reaches those regions before fusion with the membrane (Boguslavsky et al., 2012), consistent with a role of Rab8A in mobilizing GLUT4 out of perinuclear compartments in the muscle cell periphery. Accordingly, GLUT4 vesicles were visualized along cytosolic MyoVa structures.
MyoVa thus fits the requirements of being a bona fide effector of Rab8A in the context of GLUT4 mobilization, according to the criteria defined for Rab-effectors (Khan, 2013): “(i) recognition of the GTP-bound conformation of a Rab with higher affinity than the GDP-bound conformation; (ii) regulation of biological activity through its binding to an active Rab, and (iii) loss of Rab recruitment and subsequent biological activity following GAP-mediated hydrolysis of GTP to GDP.”
Function of Rab8A in defined intracellular traffic events
Rab8A has gained interest in recent years, as it has been increasingly shown to participate in directional endosomal and exocytic events, as summarized in Table 1. These collective functions are extended here to the regulation of GLUT4 traffic in muscle cells. Of note, Rab8A associates with tubular membranes that are connected to kinesin and dynein motors promoted by Rab35, EHD1, and MICAL-L1 (Roland et al., 2007; Giridharan et al., 2012). This finding suggests the possibility that Rab8A may act at a point of transference of vesicular cargo from microtubules (mobilized by kinesin or dynein) to actin filaments. This scenario may also play out in GLUT4 vesicle mobilization, as Rab35, EHD1, and kinesin have all been implicated in this process (Guilherme et al., 2004; Davey et al., 2012). We entertain a model by which Rab8A would “hand over” GLUT4 vesicles from microtubules to actin filaments, where they would be propelled by MyoVa, as this molecular motor has been shown to functionally couple microtubules to actin filaments (Cao et al., 2004), and insulin promotes MyoVa association with actin filaments (Yoshizaki et al., 2007). This model should be the subject of future experimental enquiry.
TABLE 1:
Participation of Rab8A in intracellular traffic events.
| Traffic event | Reference |
|---|---|
| Endosomal recycling | Gerges et al. (2004), Esseltine et al. (2012) |
| Functions downstream of Rab11 | Westlake et al. (2011) |
| Synaptic traffic | Esseltine et al. (2012) |
| Docking/fusion of exocytic carriers | Grigoriev et al. (2011) |
| Glutamate receptor traffic | Esseltine et al. (2012) |
| Dense granule release | Hampson et al. (2013) |
| Traffic of basolateral proteins in epithelial cells | Henry and Sheff (2008) |
| Cholesterol traffic from late endosomes to periphery | Linder et al. (2007) |
| Apical morphogenesis and sorting of apical proteins | Kato et al. (2009) |
| Formation of primary cilia | Knodler et al. (2010) |
Rab GTPases mobilize GLUT4 in muscle cells and adipocytes
Rab8A and Rab10 are both inactivated by the Rab-GTPase activating protein AS160, which is inhibited by phosphorylation in response to insulin (Sano et al., 2007). Whereas in muscle cells overexpression of Rab8A rescues GLUT4 translocation from the inhibition imposed by the constitutively active AS160-4A (Sun et al., 2010), in 3T3-L1 adipocytes Rab10 is the more relevant AS160-regulated GTPase (Sano et al., 2007; Chen et al., 2012; Sadacca et al., 2013). It is therefore important to explore whether there are similarities and differences in the functions of these GTPases, as both are expressed in each cell type (Sun et al., 2010).
To begin with, the subcellular localizations of Rab8A and Rab10 are markedly different. Whereas Rab8A in muscle cells is largely located in perinuclear/cytoplasmic endomembranes and it cannot be detected in the TIRF-imaged zone (Figure 5B), Rab10 in adipocytes is readily detected in this region, where it contributes to GLUT4 vesicle docking with the membrane (Chen et al., 2012). Of note, in muscle cells, Rab13 is the insulin-stimulated GTPase that reaches the TIRF-imaged zone, where it interacts with proteins that may enact GLUT4 vesicle docking (Y.S. and A.K., unpublished data).
Although it has been difficult to detect the activity status of Rab10 in response to insulin in adipocytes (Sano et al., 2008), silencing DENND4C (a Rab10 GEF) reduces the insulin-dependent gain in surface GLUT4, and DENND4C overexpression promotes a gain in surface GLUT4 (Sano et al., 2011; Sadacca et al., 2013). DENND4C is tonically active in adipose cells (Sadacca et al., 2013), and in addition, GDI-1 may control Rab10 functional availability (Chen et al., 2009). In muscle cells, the activated state of Rab8A has been directly measured upon binding of a photoactivatable GTP (Sun et al., 2010), and its activation is supported here by our GST-dependent pull-down approach by which GTP-bound Rab8A binds to the CT domain of MyoVa (Figure 1C). This finding further suggests that MyoVa-CT may be used in pull-down experiments as an indicator of Rab8A activity, akin to the use of domain effectors of Rho-family GTPases (e.g., the Cdc42/Rac interactive binding domain of PAK for Rac and the Rho-binding domain of rhotekin for Rho). Of note, binding of Rab8A to this CT domain rose in response to insulin, whereas Rab10 binding did not (Figure 1B). Finally, of importance, we identified two amino acids required for Rab8A binding to MyoVa-CT (Q1300 and Y1312) that were dispensable for binding Rab10 (Supplemental Figure S4). Because mutating these residues in MyoVa-CT abrogated the inhibitory action of the transfected MyoVa fragment on GLUT4 translocation in muscle cells, we conclude that Rab8A but not Rab10 binding to MyoVa is important for insulin-regulated GLUT4 traffic in muscle cells.
Additional differences have been reported in the function of Rab8A and Rab10 in GLUT4 traffic. In muscle cells, silencing Rab8A largely reduces the insulin-dependent gain in surface GLUT4, and this is not observed upon silencing Rab10 expression (Ishikura et al., 2007). In contrast, the reverse is observed in 3T3-L1 adipocytes (Sano et al., 2007), although a partial contribution for Rab8A in these cells has also been reported (Brewer et al., 2011). In keeping with AS160 acting as a retaining signal, silencing this Rab-GAP elevates surface GLUT4 levels in both muscle cells and adipocytes, an effect selectively counteracted by concomitantly silencing Rab8A in muscle cells (Ishikura and Klip, 2008) and Rab10 in adipose cells (Sano et al., 2007).
Finally, in both cell types, Rab14 silencing reduces GLUT4 traffic, and emerging findings place this GTPase at a step before GLUT4 sorting into the insulin-sensitive compartment (Reed et al., 2013; Sadacca et al., 2013), potentially controlled more by TBC1D1 than by AS160 (Ishikura and Klip, 2008).
Based on the foregoing analysis, the function of Rab8A in GLUT4 traffic in muscle cells is likely not the same as that of Rab10 in adipocytes, and more studies are required to elucidate the cohort of Rab GTPases functioning downstream of AS160 that contribute to GLUT4 traffic in each cell type.
Function of MyoVa in vesicle traffic as an effector of Rab GTPases
The interaction of noncontractile myosin motors with Rab GTPases has emerged as a canonical pathway for vesicle traffic (Hume et al., 2001), as illustrated for the MyoV family in Table 2. Here we report that muscle cells only express MyoVa (Supplemental Figure S1). Of note, pigment granule transport propelled by MyoVa requires actin filament dynamics (Semenova et al., 2008), and insulin triggers actin filament dynamics (branching and severing) in muscle cells (Chiu et al., 2013). MyoVa was first linked to GLUT4 traffic in 3T3-L1 adipocytes upon demonstration that insulin promotes its association with actin filaments and silencing the protein expression reduces insulin-dependent GLUT4 translocation (Yoshizaki et al., 2007). Here we provide mechanistic insight into this role by showing that MyoVa and Rab8A colocalize in perinuclear/cytosolic endomembranes (Figure 2) and that the C-terminal fragment of MyoVa can coimmunoprecipitate with or pull down insulin-activated Rab8A (Figures 1 and 3). Further, upon expression of this truncated fragment in muscle cells, Rab8A becomes mislocalized, and insulin-dependent GLUT4 translocation is aborted (Figure 3D). We take these collective findings as evidence of the participation of MyoVa in GLUT4 traffic in muscle cells and link it to the upstream activation of Rab8A in response to insulin. Buttressing this conclusion, we find that a MyoVa-CT altered by site-directed mutagenesis to eliminate its binding to Rab8A is no longer inhibitory of insulin-dependent GLUT4 translocation. We surmise that normally the endogenous MyoVa links Rab8A-containing GLUT4 vesicles to actin filaments and that the overexpressed MyoVa-CT displaces the endogenous motor protein from Rab8A, preventing the link of GLUT4 vesicles to actin filaments. Whether these actin filaments are stress fibers or the more cortically located branched filaments induced by insulin remains to be determined. It is tantalizing to propose that, by increasing actin filament dynamics (Chiu et al., 2013), insulin may promote binding of MyoVa to actin filaments. Based on literature reports, the Rab8A-MyoVa interaction may allow its vesicular cargo to switch from microtubules to actin filaments (Cao et al., 2004) and/or be mobilized along the latter (Semenova et al., 2008).
TABLE 2:
MyoV interactions with Rab GTPases and effect on cargo mobilization.
| MyoV–Rab interaction | Cargo mobilized | Reference |
|---|---|---|
| MyoVa, b, c with Rab8A, 10, 11 | – | Rodriguez and Cheney (2002) |
| Yeast Myo2 (MyoV) with Ypt31/31 (Rab11) | – | Lipatova et al. (2008) |
| MyoVa | Synaptic vesicles | Rudolf et al. (2011) |
| Melanosomes and insulin granules | Hume et al. (2001), Varadi et al. (2005) | |
| Na/K-ATPase | Lecuona et al. (2009) | |
| MyoVb | Epithelial membrane recycling, lumen formation, cell polarity (via Rab8A) | Bryant et al. (2010), Roland et al. (2011) |
| Apical exocytic vesicles (via Rab8A) | Khandelwal et al. (2013) | |
| MyoVc | Secretory granules (adenocarcinoma cells) | Jacobs et al. (2009) |
| Exocrine granules (pancreas, colon) | Rodriguez and Cheney (2002) |
We previously used the C-terminal fragment of the MyoVb isoform to similarly mislocalize Rab8A and impair GLUT4 translocation (Ishikura and Klip, 2008). The fragment also mislocalized Rab10, but only silencing the expression of Rab8A and not Rab10 prevented insulin-dependent GLUT4 translocation (Ishikura et al., 2007). Because MyoVb is not expressed in either mature muscle or muscle cells (Supplemental Figure S1), the previous result using C-terminal fragment cannot be taken to indicate participation of MyoVb in GLUT4 traffic, and instead the fragment must have acted by sequestering away Rab8A. This suggests that both the MyoVa-CT and MyoVb-CT fragments act as scavengers that mislocalize Rab GTPases and thereby prevent their cargo from achieving its destination within the cell. Consistent with this view, Chen et al. (2012) transfected a MyoVa short-tail fragment (virtually equivalent to MyoVa-CT) into 3T3-L1 adipocytes and found that it delayed and reduced the maximal insertion into the membrane of the GLUT4 surrogate protein IRAP-phluorin. On the basis of colocalization of the MyoVa fragment with Rab14 and Rab10 at endosomal compartments and the TIRF-imaged zone, respectively, these authors proposed that the endogenous MyoVa may have a dual role, promoting GLUT4/IRAP endosomal traffic and vesicle positioning for docking/fusion (Chen et al., 2012). Hence, in adipocytes, MyoVa is engaged by Rabs 10 and 14, whereas in muscle cells it is engaged by Rab8A.
In summary, we provide evidence of a functional link between a molecular switch, Rab8A, and a molecular motor, MyoVa, in the signaling relay triggered by insulin to mobilize GLUT4 to the muscle cell membrane. The results are consistent with the following model. Insulin-activated Akt phosphorylates/inactivates AS160, allowing GTP-loaded Rab8A on perinuclear/cytosolic GLUT4 vesicles to prevail and engage with MyoVa, effectively linking the vesicles to actin filaments as part of the mechanism that relays the vesicles to the plasma membrane.
MATERIALS AND METHODS
Reagents and DNA constructs
DMSO and cocktail proteinase inhibitor were from Sigma-Aldrich (St. Louis, MO). Isopropyl-β-d-thiogalactoside (IPTG) was from Fermentas (Glen Burnie, MD), DSP PI22585 cross-linker was purchased from Thermo Scientific (Waltham, MA). Wortmannin was from Enzo Life Science (Farmingdale, NY). Humulin human insulin was from Eli Lilly (Indianapolis, IN). Mouse anti-myc (9E1) antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Rabbit anti-pAkt (473) and anti-myosin Va were from Cell Signaling Technology (Danvers, MA). Polyclonal anti-GFP antibodies were from Abcam (Cambridge, MA) and Clontech (Mountain View, CA). Monoclonal anti-mCherry antibody was from Clontech. Alexa 555–conjugated goat anti-mouse antibody was from Life Technologies (Carlsbad, CA). Horseradish-peroxidase (HRP)–conjugated goat anti-rabbit and goat anti-mouse secondary antibodies were from Jackson ImmunoResearch (West Grove, PA). Glutathione–Sepharose 4B and protein G beads were from GE Healthcare (Uppsala, Sweden).
GFP-MyoVa-FL (full-length) and GFP-MyoVa-CT (melanocyte specific) were kind gifts from M. C. Seabra (Imperial College, London, United Kingdom). The constitutively active, GTP-bound Rab8AQ67L and the dominant-negative, GDP-bound Rab8AT22N mutant cDNA constructs were kind gifts from I. Mellman (Genentec, South San Francisco, CA); the mCherry-Rab8A (MC-Rab8A), as well as its mutants (MC-Rab8A-GTP, MC-Rab8A-GDP), were kind gifts from J. Goldenring (Vanderbilt University, Nashville, TN). The C-terminal melanocyte-specific MyoVa C-terminal codons (1260–1854) were PCR amplified from the GFP–MyoVa-FL construct and subcloned into the pGEX-6p-2 (Clontech) vector by BamHI/EcoRI to generate GST–MyoVa-CT. Double mutagenesis (Q1300L and Y1312C) was performed on GFP/GST–MyoVa-CT by using the QuikChange Site-Directed Mutagenesis Kit (Stratagene, Santa Clara, CA) to generate GFP–MyoVa-CT(2M) and GST–MyoVa-CT(2M). Predesigned siRNA oligomers targeting rat MyoVa or a nontargeting control siRNA (siNR) were from Qiagen (Mississauga, Canada). A pSuper-basic vector encoding a GFP-tagged short hairpin RNA to the same sequence as the siRNA oligomer targeting rat MyoVa (sh-GFP-MyoVa) was a kind gift from A. El-Husseini (University of British Columbia, Vancouver, Canada). A pSuper-basic vector tagged with GFP but without the short hairpin sequence was used as control.
Cell culture and transfection
Rat L6 muscle cells stably expressing GLUT4 with an exofacial myc epitope (L6-GLUT4myc) were cultured in α-MEM supplemented with 10% fetal bovine serum and antibiotics as described previously (Wang et al., 1998; Foster et al., 2001). Transfection of cDNA constructs was performed with FuGENE HD (Promega, Madison, WI) or X-treme GENE HP (Roche, Mannheim, Germany) according to the manufacturer's protocol. Briefly, cDNA/reagent mixture was applied to the cells in serum-free medium for 4 h without antibiotics, medium was changed, and cells were grown for 24, 48, or 72 h, as indicated.
Reverse Transcriptase-PCR
Total RNA was isolated from L6-GLUT4myc muscle cells, 3T3-L1 adipocytes, C2C12 myoblasts, and mouse and rat skeletal muscles using RNeasy Mini Kit (Qiagen). cDNAs were prepared using Superscript III Reverse Transcriptase (Life Technologies). Specific primer pairs were designed to detect rat/mouse MyoVa, Vb, or Vc expression in L6 cells, using glyceraldehyde-3-phosphate dehydrogenase expression as a reaction control.
Transferrin recycling
L6 GLUT4myc myoblasts were transfected with sh-GFP-MyoVa-FL, GFP-MyoVa-CT, or sh-GFP-control plasmids for 48 h, followed by serum deprivation for 1.5 h. Cells were incubated with 50 μg/ml Alexa 546–conjugated transferrin in serum-free medium containing 1% bovine serum albumin (BSA) for 10 min at 37°C. Some cells were then chased with 500 μg/ml holotransferrin (Sigma-Aldrich) for 10 min at 37°C (intensity without chasing was the initial total signal). Cells were placed on ice, washed two times with ice-cold phosphate-buffered saline (PBS), acid buffer (0.15 M NaCl, 0.1 M glycine, pH 3.0), and PBS, then fixed with 4% paraformaldehyde in PBS at room temperature for 90 min. Samples were quenched with 50 mM NH4Cl for 10 min, followed by washing with PBS, and then processed for fluorescence microscopy of Alexa 546–conjugated transferrin using a Zeiss LSM 510 laser-scanning confocal microscope (Carl Zeiss, Jena, Germany). Cells were scanned along the z-axis, and collapsed xy-projections were assembled from optical stacks taken at 1-μm intervals. The pixel intensity of each cell (≥15 cells per condition) was quantified by ImageJ software (National Institutes of Health, Bethesda, MD).
Cell lysis, immunoprecipitation, and immunoblotting
After experimental treatments, cells were washed and lysed in lysis buffer (1% Triton-X100, 50 mM Tris, 140 mM NaCl, 1:500 dilution of Sigma-Aldrich cocktail proteinase inhibitor, 20 mM NaF, 2 mM Na3VO4, 5 mM MgCl2, pH 7.4). For coimmunoprecipitation, CHO-IR cells were grown in 10-cm-diameter dishes and transfected for 48 h, followed by serum deprivation for 3 h. During the last 30 min, cells were treated with 1.5 mM dithiobis succinimidyl propionate (DSP), a cell-permeant thiol-cleavable and amine-reactive cross-linking agent, followed by treatment with or without insulin. Lysates were passed through a 19-gauge syringe needle 10 times, and supernatants were collected after centrifugation at 11,000 × g at 4°C for 10 min. Supernatants were incubated with protein G beads conjugated to 1 μg of antibody at 4°C for 4 h, followed by three washes with washing buffer (0.4% Triton, 10 mM Tris-HCl, 140 mM NaCl, pH 7.4). Coimmunoprecipitated complexes were eluted with 2× Laemmli buffer, then separated by SDS–PAGE. Immunoblotting was conducted on polyvinylidene difluoride (PVDF) membrane (Bio-Rad, Richmond, CA) with respective primary antibodies and visualized by HRP-conjugated secondary antibodies by enhanced chemiluminescence (GE Life Sciences).
Expression of GST constructs and pull-down experiments
GST–MyoVa-CT or GST–MyoVa-CT(2M) was transformed in BL21 bacteria, and a single clone was picked to grow overnight in 3.5 ml of LB medium containing ampicillin. The culture was expanded into 15 ml during the day and then diluted into 300 ml, followed by induction with 0.3 mM IPTG overnight at room temperature under agitation (100 rpm). For pull-down assays, bacteria expressing GST–MyoVa-CT pellets were lysed by sonication in 1% Triton-X100/TBS buffer (20 mM Tris, 140 mM NaCl, pH 7.4), followed by binding with 35 μl of glutathione–Sepharose beads (GSHS, 60% slurry) at 4°C for 1 h. Lysates expressing different GFP-Rabs or GFP from CHO-IR cells were incubated with similar amounts of GST–MyoVa-CT- or GST–MyoVa-CT(2M) bound to GSHS beads at 4°C for 4 h, and after washes, bound complexes were eluted with 2× Laemmli buffer. Eluates were separated by SDS–PAGE, and the upper section of the gels was stained with Coomassie blue to reveal loading density of GST-MyoVa (95 kDa) and the lower sections transferred to PVDF for immunoblotting of GFP-Rabs using anti-GFP antibody.
Cell-surface GLUT4myc detection
Cell-surface GLUT4myc detection was as described previously (Wang et al., 1998; Ishikura and Klip, 2008). Briefly, myoblasts were grown in 24-well plates and serum starved for 3 h and treated with or without 100 nM insulin for 15 min, followed by rinsing with cold PBS, fixation with 3% (vol/vol) paraformaldehyde (PFA), quenching with 50 mM NH4Cl, and blocking with 3% (vol/vol) goat serum for 30 min on ice. Cells were next incubated with polyclonal anti-myc antibody (1:200) for 1 h on ice, followed by three washes of 5 min with ice-cold PBS. Cells were incubated with HRP-conjugated goat anti-rabbit secondary antibody (1:1000) for 1 h on ice. After three washes with ice-cold PBS, the cells were incubated with o-phenylenediamine (1.0 ml/well of 0.4 mg of o-phenylenediamine/ml in 50 mM NaH2PO4, 25 mM citric acid, pH 5.0) and allowed to develop for 20–30 min at room temperature in dark. The reaction was stopped with 3 M HCl (0.25 ml/well). Supernatants were collected, and absorbance was measured at 490 nm, with background values lacking anti-myc antibody subtracted from all values.
Surface GLUT4myc was also measured by immunofluorescence in L6 myoblasts plated on glass coverslips transiently transfected with cDNA constructs, as previously described (Khayat et al., 2000; Ishikura and Klip, 2008). Briefly, cells were serum starved for 3 h and then treated with or without 100 nM insulin for 15 min, rinsed with PBS, fixed with 3% PFA, quenched with 50 mM NH4Cl, and blocked with 2% (vol/vol) BSA in PBS. Surface GLUT4myc was detected by monoclonal anti-myc (1:100) for 1 h. After three washes, cells were incubated with Alexa 555–conjugated anti-mouse secondary antibody in the dark for 1 h, rinsed with PBS, and mounted on slides. Fluorescence images were taken with a Zeiss LSM 510 laser-scanning confocal microscope. Cells were scanned along the z-axis, and single collapsed images (collapsed xy-projection) were assembled from optical stacks taken at 1-μm intervals. The pixel intensity of each cell (≥25 cells per condition) was quantified by ImageJ software.
Spinning-disk confocal fluorescence microscopy
GFP-GLUT4 and MC-Rab8A, or GFP-MyoVa-FL and MC-Rab8A, were cotransfected in L6 muscle cells for 24 h, followed by serum deprivation for 3 h. Cells were then treated with or without insulin for 15 min and fixed with 3% PFA in PBS. Cells expressing MyoVa-FL and GFP-GLUT4 were also subjected to real-time imaging. In some experiments, fixed cells were incubated with antibodies to transferrin receptor (1:500), furin (1:300), or syntaxin-6 (1:300), followed by reaction with respective secondary antibodies conjugated to Alexa 555 (1:1000). Fluorescence images were acquired with a Leica spinning-disk confocal microscope equipped with Volocity 5.4.1 software (PerkinElmer, Waltham, MA). Cells were scanned along the z-axis from top to bottom at 0.25-μm stack intervals. Images of single optical planes from stacks were exported into Photoshop 7.0 (Adobe, San Jose, CA) for processing. The intensity of the MC-Rab8A fluorescence signal in the cell periphery and the colocalization of GFP-MyoVa-FL (green) with MC-Rab8A (red) were analyzed in raw images of spinning-disk fluorescence microscopy using Volocity 6.1.2 software. The periphery cytoplasmic area signal (avoiding the perinuclear area) of GFP–MyoVa-FL was selected as shown and quantified relative to the total cell signal or for its colocalization with MC-Rab8A. The degree of colocalization was assessed from the Manders coefficient (MC-Rab8A to GFP-MyoVa-FL). Mean values were analyzed by Prism software (GraphPad, La Jolla, CA) and are presented as mean ± SE.
TIRF microscopy
After 24 h of transfection with GFP-GLUT4 and MC-Rab8A, myoblasts were serum deprived for 3 h in 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid–containing RPMI-1640 medium and then treated with or without insulin (100 nM) for 15 min and examined by TIRF microscopy using an Olympus IX81 microscope (Olympus, Tokyo, Japan) equipped with a dual-laser (488, 561 nm using a 150×/1.45 numerical aperture objective), and Volocity 4.0 software was used to visualize GFP-GLUT4 vesicles with possible red fluorescent MC-Rab8A within 100 nm of the cell membrane.
Supplementary Material
Acknowledgments
We thank M. Seabra, J. Goldenring, I. Mellman, and the lab of the late A. El-Husseini for generously providing constructs. This work was supported by Canadian Institutes of Health Research Grant 7307 to A.K. Y.S. was supported by subsequent postdoctoral fellowships from the Banting and Best Diabetes Center of the University of Toronto and the Canadian Diabetes Association. T.C. was supported by a Banting doctoral award from the Canadian Institutes of Health Research.
Abbreviations used:
- Akt/PKB
protein kinase B
- AS160/TBC1D4
Akt substrate of 160kDa/TBC1 domain family member 4
- CHO-IR cells
CHO cells stably expressing the human insulin receptor
- L6 GLUT4myc cells
L6 rat muscle cells stably expressing GLUT4 with an exofacial myc-epitope in the first extracellular loop
- MyoV
myosin V
- PI3K
phosphatidylinositol-3-kinase
- shRNA
short hairpin RNA
- siRNA
small interfering RNA
- TIRF
total internal reflection fluorescence
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E13-08-0493) on January 29, 2014.
REFERENCES
- Boguslavsky S, Chiu T, Foley KP, Osorio-Fuentealba C, Antonescu CN, Bayer KU, Bilan PJ, Klip A. Myo1c binding to submembrane actin mediates insulin-induced tethering of GLUT4 vesicles. Mol Biol Cell. 2012;23:4065–4078. doi: 10.1091/mbc.E12-04-0263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer PD, Romenskaia I, Kanow MA, Mastick CC. Loss of AS160 Akt substrate causes Glut4 protein to accumulate in compartments that are primed for fusion in basal adipocytes. J Biol Chem. 2011;286:26287–26297. doi: 10.1074/jbc.M111.253880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant DM, Datta A, Rodriguez-Fraticelli AE, Peranen J, Martin-Belmonte F, Mostov KE. A molecular network for de novo generation of the apical surface and lumen. Nat Cell Biol. 2010;12:1035–1045. doi: 10.1038/ncb2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao TT, Chang W, Masters SE, Mooseker MS. Myosin-Va binds to and mechanochemically couples microtubules to actin filaments. Mol Biol Cell. 2004;15:151–161. doi: 10.1091/mbc.E03-07-0504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Deng Y, Zhang J, Yang L, Xie X, Xu T. GDI-1 preferably interacts with Rab10 in insulin-stimulated GLUT4 translocation. Biochem J. 2009;422:229–235. doi: 10.1042/BJ20090624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Wang Y, Zhang J, Deng Y, Jiang L, Song E, Wu XS, Hammer JA, Xu T, Lippincott-Schwartz J. Rab10 and myosin-Va mediate insulin-stimulated GLUT4 storage vesicle translocation in adipocytes. J Cell Biol. 2012;198:545–560. doi: 10.1083/jcb.201111091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu TT, Sun Y, Koshkina A, Klip A. Rac-1 superactivation triggers insulin-independent glucose transporter 4 (GLUT4) translocation that bypasses signaling defects exerted by c-Jun N-terminal kinase (JNK)- and ceramide-induced insulin resistance. J Biol Chem. 2013;288:17520–17531. doi: 10.1074/jbc.M113.467647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey JR, Humphrey SJ, Junutula JR, Mishra AK, Lambright DG, James DE, Stockli J. TBC1D13 is a RAB35 specific GAP that plays an important role in GLUT4 trafficking in adipocytes. Traffic. 2012;13:1429–1441. doi: 10.1111/j.1600-0854.2012.01397.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esseltine JL, Ribeiro FM, Ferguson SS. Rab8 modulates metabotropic glutamate receptor subtype 1 intracellular trafficking and signaling in a protein kinase C-dependent manner. J Neurosci. 2012;32:16933–16942a. doi: 10.1523/JNEUROSCI.0625-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster LJ, Li D, Randhawa VK, Klip A. Insulin accelerates inter-endosomal GLUT4 traffic via phosphatidylinositol 3-kinase and protein kinase B. J Biol Chem. 2001;276:44212–44221. doi: 10.1074/jbc.M102964200. [DOI] [PubMed] [Google Scholar]
- Gerges NZ, Backos DS, Esteban JA. Local control of AMPA receptor trafficking at the postsynaptic terminal by a small GTPase of the Rab family. J Biol Chem. 2004;279:43870–43878. doi: 10.1074/jbc.M404982200. [DOI] [PubMed] [Google Scholar]
- Giridharan SS, Cai B, Naslavsky N, Caplan S. Trafficking cascades mediated by Rab35 and its membrane hub effector, MICAL-L1. Commun Integr Biol. 2012;5:384–387. doi: 10.4161/cib.20064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigoriev I, et al. Rab6, Rab8, and MICAL3 cooperate in controlling docking and fusion of exocytotic carriers. Curr Biol. 2011;21:967–974. doi: 10.1016/j.cub.2011.04.030. [DOI] [PubMed] [Google Scholar]
- Guilherme A, Soriano NA, Furcinitti PS, Czech MP. Role of EHD1 and EHBP1 in perinuclear sorting and insulin-regulated GLUT4 recycling in 3T3-L1 adipocytes. J Biol Chem. 2004;279:40062–40075. doi: 10.1074/jbc.M401918200. [DOI] [PubMed] [Google Scholar]
- Hampson A, O'Connor A, Smolenski A. Synaptotagmin-like protein 4 and Rab8 interact and increase dense granule release in platelets. J Thromb Haemost. 2013;11:161–168. doi: 10.1111/jth.12068. [DOI] [PubMed] [Google Scholar]
- Henry L, Sheff DR. Rab8 regulates basolateral secretory, but not recycling, traffic at the recycling endosome. Mol Biol Cell. 2008;19:2059–2068. doi: 10.1091/mbc.E07-09-0902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hume AN, Collinson LM, Rapak A, Gomes AQ, Hopkins CR, Seabra MC. Rab27a regulates the peripheral distribution of melanosomes in melanocytes. J Cell Biol. 2001;152:795–808. doi: 10.1083/jcb.152.4.795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikura S, Bilan PJ, Klip A. Rabs 8A and 14 are targets of the insulin-regulated Rab-GAP AS160 regulating GLUT4 traffic in muscle cells. Biochem Biophys Res Commun. 2007;353:1074–1079. doi: 10.1016/j.bbrc.2006.12.140. [DOI] [PubMed] [Google Scholar]
- Ishikura S, Klip A. Muscle cells engage Rab8A and myosin Vb in insulin-dependent GLUT4 translocation. Am J Physiol Cell Physiol. 2008;295:C1016–C1025. doi: 10.1152/ajpcell.00277.2008. [DOI] [PubMed] [Google Scholar]
- Jacobs DT, Weigert R, Grode KD, Donaldson JG, Cheney RE. Myosin Vc is a molecular motor that functions in secretory granule trafficking. Mol Biol Cell. 2009;20:4471–4488. doi: 10.1091/mbc.E08-08-0865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato Y, Sugiura T, Nakadera Y, Sugiura M, Kubo Y, Sato T, Harada A, Tsuji A. Investigation of the role of oligopeptide transporter PEPT1 and sodium/glucose cotransporter SGLT1 in intestinal absorption of their substrates using small GTP-binding protein Rab8-null mice. Drug Metab Dispos. 2009;37:602–607. doi: 10.1124/dmd.108.023689. [DOI] [PubMed] [Google Scholar]
- Khan AR. Oligomerization of Rab/effector complexes in the regulation of vesicle trafficking. Prog Mol Biol Transl Sci. 2013;117:579–614. doi: 10.1016/B978-0-12-386931-9.00021-0. [DOI] [PubMed] [Google Scholar]
- Khandelwal P, Prakasam HS, Clayton DR, Ruiz WG, Gallo LI, van Roekel D, Lukianov S, Peranen J, Goldenring JR, Apodaca G. A Rab11a-Rab8a-Myo5B network promotes stretch-regulated exocytosis in bladder umbrella cells. Mol Biol Cell. 2013;24:1007–1019. doi: 10.1091/mbc.E12-08-0568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khayat ZA, Tong P, Yaworsky K, Bloch RJ, Klip A. Insulin-induced actin filament remodeling colocalizes actin with phosphatidylinositol 3-kinase and GLUT4 in L6 myotubes. J Cell Sci. 2000;113:279–290. doi: 10.1242/jcs.113.2.279. [DOI] [PubMed] [Google Scholar]
- Knodler A, Feng S, Zhang J, Zhang X, Das A, Peranen J, Guo W. Coordination of Rab8 and Rab11 in primary ciliogenesis. Proc Natl Acad Sci USA. 2010;107:6346–6351. doi: 10.1073/pnas.1002401107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecuona E, et al. Myosin-Va restrains the trafficking of Na+/K+-ATPase-containing vesicles in alveolar epithelial cells. J Cell Sci. 2009;122:3915–3922. doi: 10.1242/jcs.046953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linder MD, Uronen RL, Holtta-Vuori M, van der Sluijs P, Peranen J, Ikonen E. Rab8-dependent recycling promotes endosomal cholesterol removal in normal and sphingolipidosis cells. Mol Biol Cell. 2007;18:47–56. doi: 10.1091/mbc.E06-07-0575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipatova Z, Tokarev AA, Jin Y, Mulholland J, Weisman LS, Segev N. Direct interaction between a myosin V motor and the Rab GTPases Ypt31/32 is required for polarized secretion. Mol Biol Cell. 2008;19:4177–4187. doi: 10.1091/mbc.E08-02-0220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattheyses AL, Simon SM, Rappoport JZ. Imaging with total internal reflection fluorescence microscopy for the cell biologist. J Cell Sci. 2010;123:3621–3628. doi: 10.1242/jcs.056218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miinea CP, Sano H, Kane S, Sano E, Fukuda M, Peranen J, Lane WS, Lienhard GE. AS160, the Akt substrate regulating GLUT4 translocation, has a functional Rab GTPase-activating protein domain. Biochem J. 2005;391:87–93. doi: 10.1042/BJ20050887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira-Leal JB, Seabra MC. The mammalian Rab family of small GTPases: definition of family and subfamily sequence motifs suggests a mechanism for functional specificity in the Ras superfamily. J Mol Biol. 2000;301:1077–1087. doi: 10.1006/jmbi.2000.4010. [DOI] [PubMed] [Google Scholar]
- Reed SE, Hodgson LR, Song S, May MT, Kelly EE, McCaffrey MW, Mastick CC, Verkade P, Tavare JM. A role for Rab14 in the endocytic trafficking of GLUT4 in 3T3-L1 adipocytes. J Cell Sci. 2013;126:1931–1941. doi: 10.1242/jcs.104307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez OC, Cheney RE. Human myosin-Vc is a novel class V myosin expressed in epithelial cells. J Cell Sci. 2002;115:991–1004. doi: 10.1242/jcs.115.5.991. [DOI] [PubMed] [Google Scholar]
- Roland JT, Bryant DM, Datta A, Itzen A, Mostov KE, Goldenring JR. Rab GTPase-Myo5B complexes control membrane recycling and epithelial polarization. Proc Natl Acad Sci USA. 2011;108:2789–2794. doi: 10.1073/pnas.1010754108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roland JT, Kenworthy AK, Peranen J, Caplan S, Goldenring JR. Myosin Vb interacts with Rab8a on a tubular network containing EHD1 and EHD3. Mol Biol Cell. 2007;18:2828–2837. doi: 10.1091/mbc.E07-02-0169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roland JT, Lapierre LA, Goldenring JR. Alternative splicing in class V myosins determines association with Rab10. J Biol Chem. 2009;284:1213–1223. doi: 10.1074/jbc.M805957200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolf R, Bittins CM, Gerdes HH. The role of myosin V in exocytosis and synaptic plasticity. J Neurochem. 2011;116:177–191. doi: 10.1111/j.1471-4159.2010.07110.x. [DOI] [PubMed] [Google Scholar]
- Sadacca LA, Bruno J, Wen J, Xiong W, McGraw TE. Specialized sorting of GLUT4 and its recruitment to the cell surface are independently regulated by distinct Rabs. Mol Biol Cell. 2013;24:2544–2557. doi: 10.1091/mbc.E13-02-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano H, Eguez L, Teruel MN, Fukuda M, Chuang TD, Chavez JA, Lienhard GE, McGraw TE. Rab10, a target of the AS160 Rab GAP, is required for insulin-stimulated translocation of GLUT4 to the adipocyte plasma membrane. Cell Metab. 2007;5:293–303. doi: 10.1016/j.cmet.2007.03.001. [DOI] [PubMed] [Google Scholar]
- Sano H, Kane S, Sano E, Miinea CP, Asara JM, Lane WS, Garner CW, Lienhard GE. Insulin-stimulated phosphorylation of a Rab GTPase-activating protein regulates GLUT4 translocation. J Biol Chem. 2003;278:14599–14602. doi: 10.1074/jbc.C300063200. [DOI] [PubMed] [Google Scholar]
- Sano H, Peck GR, Kettenbach AN, Gerber SA, Lienhard GE. Insulin-stimulated GLUT4 protein translocation in adipocytes requires the Rab10 guanine nucleotide exchange factor Dennd4C. J Biol Chem. 2011;286:16541–16545. doi: 10.1074/jbc.C111.228908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano H, Roach WG, Peck GR, Fukuda M, Lienhard GE. Rab10 in insulin-stimulated GLUT4 translocation. Biochem J. 2008;411:89–95. doi: 10.1042/BJ20071318. [DOI] [PubMed] [Google Scholar]
- Semenova I, Burakov A, Berardone N, Zaliapin I, Slepchenko B, Svitkina T, Kashina A, Rodionov V. Actin dynamics is essential for myosin-based transport of membrane organelles. Curr Biol. 2008;18:1581–1586. doi: 10.1016/j.cub.2008.08.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Bilan PJ, Liu Z, Klip A. Rab8A and Rab13 are activated by insulin and regulate GLUT4 translocation in muscle cells. Proc Natl Acad Sci USA. 2010;107:19909–19914. doi: 10.1073/pnas.1009523107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varadi A, Tsuboi T, Rutter GA. Myosin Va transports dense core secretory vesicles in pancreatic MIN6 beta-cells. Mol Biol Cell. 2005;16:2670–2680. doi: 10.1091/mbc.E04-11-1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Khayat Z, Kishi K, Ebina Y, Klip A. GLUT4 translocation by insulin in intact muscle cells: detection by a fast and quantitative assay. FEBS Lett. 1998;427:193–197. doi: 10.1016/s0014-5793(98)00423-2. [DOI] [PubMed] [Google Scholar]
- Westlake CJ, et al. Primary cilia membrane assembly is initiated by Rab11 and transport protein particle II (TRAPPII) complex-dependent trafficking of Rabin8 to the centrosome. Proc Natl Acad Sci USA. 2011;108:2759–2764. doi: 10.1073/pnas.1018823108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Rao K, Bowers MB, Copeland NG, Jenkins NA, Hammer JA 3rd. Rab27a enables myosin Va-dependent melanosome capture by recruiting the myosin to the organelle. J Cell Sci. 2001;114:1091–1100. doi: 10.1242/jcs.114.6.1091. [DOI] [PubMed] [Google Scholar]
- Wu XS, Rao K, Zhang H, Wang F, Sellers JR, Matesic LE, Copeland NG, Jenkins NA, Hammer JA., 3rd Identification of an organelle receptor for myosin-Va. Nat Cell Biol. 2002;4:271–278. doi: 10.1038/ncb760. [DOI] [PubMed] [Google Scholar]
- Yoshizaki T, Imamura T, Babendure JL, Lu JC, Sonoda N, Olefsky JM. Myosin 5a is an insulin-stimulated Akt2 (protein kinase Bbeta) substrate modulating GLUT4 vesicle translocation. Mol Cell Biol. 2007;27:5172–5183. doi: 10.1128/MCB.02298-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaid H, Antonescu CN, Randhawa VK, Klip A. Insulin action on glucose transporters through molecular switches, tracks and tethers. Biochem J. 2008;413:201–215. doi: 10.1042/BJ20080723. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.