

Abstract
Expression of the pro-oncogenic mucin MUC1 is elevated by inflammation in airway epithelial cells, but the contributions of MUC1 to the development of lung cancer are uncertain. In this study, we developed our finding that cigarette smoke (CS) increases Muc1 expression in lung macrophages, where we hypothesized it might contribute to CS-induced transformation of bronchial epithelial cells. In human macrophages, CS extract (CSE) strongly induced MUC1 expression through a mechanism involving the nuclear receptor PPAR-γ. CSE-induced ERK activation was also required for MUC1 expression, but it had little effect on MUC1 transcription. RNAi-mediated attenuation of MUC1 suppressed CSE-induced secretion of TNF-α from macrophages, by suppressing the activity of the TNF-α processing enzyme TACE, arguing that MUC1 is required for CSE-induced and TACE-mediated TNF-α secretion. Similarly, MUC1 blockade after CSE induction through suppression of PPAR-γ or ERK inhibited TACE activity and TNF-α secretion. Conditioned media from CSE-treated macrophages induced MUC1 expression and potentiated CSE-induced transformation of human bronchial epithelial cells (HBEC) in a TNF-α-dependent manner. Together, our results identify a signaling pathway involving PPAR-γ, ERK and MUC1 that is used by CSE to trigger TNF-α secretion from macrophages. Further, our results show how that MUC1 contributes to smoking-induced lung cancers that are driven by inflammatory signals driven by macrophages
Keywords: MUC1, PPAR-γ, TACE, TNF-α, transformation
INTRODUCTION
While an inflammatory microenvironment plays an important role in lung cancer development (1, 2), how inflammation promotes lung carcinogenesis has not been clearly elucidated. Cigarette smoke (CS), which elicits chronic pulmonary inflammation, is a major risk factor for lung cancer (3, 4). Carcinogens derived from CS such as benzo(a)pyrene induce lung cancer through DNA damage that results in mutations and epigenetic alterations (4, 5). In the microenvironment, inflammatory cells such as macrophages secrete cytokines that affect epithelial cells to favor carcinogenesis, which may involve suppression of DNA repair and promotion of apoptosis resistance, proliferation, metastasis, and secondary secretion of cytokines and growth factors (4, 6). TNF-α is an important pro-inflammatory cytokine that plays a key role in both inflammation and cancer development (7, 8). Therefore, in the tumor microenvironment, TNF-α produced by inflammatory cells may be a key mediator for inflammation-associated carcinogenesis (8, 9). However, the role and mechanism of TNF-α in lung cancer development are not well defined.
MUC1, a mucin-like glycosylated protein induced by airway inflammation and expressed on the bronchial epithelial cell membrane, plays an important role in the resolution of inflammation during respiratory tract infection (10, 11). MUC1 consists of two subunits derived from a single polypeptide: the N-terminal subunit containing highly conserved repeats of 20 amino acids that are modified by O-glycosylation and the transmembrane C-terminal subunit containing 72 amino acid residues that bind to various proteins involved in signal transduction (12, 13). MUC1 is regarded as a tumor antigen because it is aberrantly overexpressed in various cancers including lung cancer (14–16). In non-small cell lung cancer, MUC1 is overexpressed and correlated with poor patient survival (17). A variety of cellular partners for MUC1 have been identified, which may contribute to the malignancy of cancer cells and their resistance to chemotherapy (13). However, direct evidence for MUC1 in lung carcinogenesis is still lacking. Our previous study showed that chronic CS carcinogen exposure induces persistent MUC1 overexpression in human lung bronchial epithelial cells, which facilitates CS-induced cell transformation through EGFR-mediated cell survival signaling (18). Because MUC1 expression is sustained during chronic inflammation, MUC1 is likely involved in inflammation-associated cancer development (12).
While CS induces chronic pulmonary inflammation and MUC1 expression in airway epithelial cells, it is unclear if inflammation is involved in the upregulation of MUC1 in airway epithelial cells in smokers. Macrophages are the main inflammatory cell types that infiltrate into the lung, where they secrete cytokines such as TNF-α that promote recruitment of other inflammatory cells and production of other cytokines. Although its tumor-promoting effects have been documented, the role and mechanism of TNF-α in lung cancer development has not been well studied. Specifically, while TNF-α plays a role in MUC1 induction in airway epithelial cells during acute bacterial infection (19), whether it is involved in CS-induced and MUC1-mediated lung cancer development is unknown. Furthermore, it is known that MUC1 is expressed in some immune cells (20, 21), but it is unclear if MUC1 is expressed and functions in macrophages.
In this study, we investigated the role and mechanism of CS-induced inflammatory response in MUC1-mediated lung cancer development with a conditioned medium transfer approach and a validated HBEC transformation model. The results identify a novel function for MUC1 in mediating CS-induced macrophage activation that facilitates HBEC transformation. Together with its function in epithelial cells (18), our findings suggest that MUC1 plays a dual role in CS-induced and inflammation-associated lung cancer development: to facilitate TNFα secretion from macrophages and to potentiate transformation of HBECs.
MATERIALS AND METHODS
Reagents
Bisphenol A diglycidyl ether (BADGE) was obtained from Enzo Life Science. The inhibitors U0126 for ERK, SB203580 for p38, SC-514 for IKK, and Necrostatin-1 for RIP1 kinase were purchased from Calbiochem. GW9662 and U0124, the negative control for U0126, were obtained from Millipore. Small interfering RNA (siRNA; SiGenome SMARTpool) for MUC1, PPAR-γ, and TNFR1 and negative control siRNA were purchased from Dharmacon. The primary antibodies against Mucin 1 (GP1.4) and GAPDH were from Santa Cruz Biotechnology. MUC1Ab-5, a hamster monoclonal antibody that recognizes the MUC1 CT domain was purchased from Lab Vision (Fremont, CA). Antibodies against β-Tubulin and β-Actin were from Sigma-Aldrich. Antibodies against PPAR-γ and phospho-PPAR-γ (S112) were purchased from Abcam. Antibodies against ERK, and phospho-ERK (Y185/187) were from Invitrogen. The human TNF-α detection ELISA kit was purchased from eBioscience (San Diego, CA). Filters collected from the AMESA Type 1300 smoking machine, which generates mainstream CS, were used to prepare cigarette smoke extract (CSE) by sequential extraction of material from CS filter with DMSO (for dissolving water-insoluble components) and culture medium (for dissolving water-soluble components). Before use, the water-soluble and -insoluble fractions were proportionally mixed to make total CSE. Total particulate material (TPM) was determined by weighing the filter before and after extraction (22).
Cell Culture
THP-1 and U937 macrophages were cultured in RPMI 1640 supplemented with 10% fetal bovine serum (FBS), 2 mM of L-glutamine, 100 U/ml of penicillin, and 100μg/ml of streptomycin. These cell lines, obtained from ATCC (Manassas, VA), were used within 6 months of receipt. The immortalized human bronchial epithelial cells, HBEC-13, were generously provided by Drs. Shay and Minna, Southwestern Medical Center, Dallas, TX (23). HBEC cells were maintained in Keratinocyte serum free medium (KSFM) (Invitrogen), supplemented with 5 μg/L of human recombinant EGF and 50 mg/L of bovine pituitary extract in plates coated with fibronectin (Athena ES).
Transfections
THP-1 and U937 macrophages were cultured in the presence of 50 nM PMA for 24 h. The differentiated cells were transfected with MUC1 and control small interfering RNA (siRNA) with INTERFERin™ siRNA Transfection Reagent (Polyplus-transfection) according to manufacturer’s instructions. MUC1 protein level was determined by Western blot at 48 h after transfection.
Western Blot
Cells were washed twice with cold PBS, collected, and lysed with M2 buffer (20 mM Tris-HCl, pH 7.6, 0.5 % Nonidet P-40, 250 mM NaCl, 3 mM EDTA, 3 mM EGTA, 2 mM dithiothreitol, 0.5 mM phenylmethylsulfonyl fluoride, 20 mM β-glycerophosphate, 1 mM sodium vanadate, and 1 μg/ml leupeptin). Concentration of protein was measured by Bradford assay (Bio-Rad). Equal amounts of proteins were separated by 12 % SDS-polyacrylamide gels and then transferred to PVDF membranes. The proteins were detected by enhanced chemiluminescence (Immobilon Western Chemiluminescent HRP Substrate, Millipore). The intensity of the individual bands was quantified by densitometry (ImageJ) and normalized to the corresponding input control (β-Actin) bands. Fold changes were calculated with the control taken as 1.
Detecting TNF-α by ELISA
Cells were plated at 70–80% confluence in 12-well plates. After differentiation, cells were treated as described in the figure legends. After CSE treatment for 1 h, treated culture media was removed and cells were maintained in fresh RPMI 1640 without serum. Twenty-four hours later, the culture media was collected and following the manufacturer’s instructions the concentration of TNF-α was detected by ELISA analysis with the Human TNF alpha ELISA Ready-SET-Go!® (eBioscience).
Cell Viability Assay
Cell Viability was assessed using a 3-(4,5-dimethylthiazolyl-2-)-2,5-diphenyltetrazolium bromide (MTT) cell proliferation assay as previously described (24). Briefly, cells were seeded in 12-well plates at ~70% confluence, differentiated for 24h in the presence of PMA (50 nM), and then treated as indicated in the figure legends. After treatment, cells were washed twice with PBS, and were incubated in MTT solution for 2–3 h. The percentage of viable cells was calculated using the following formula: Cell viability (%) = (Absorbance of treated sample/Absorbance of control) ×100. The experiments were performed in triplicate.
TACE activity assay
Following the manufacturer’s instructions, TACE activity was assessed using SensoLyte® 520 TACE (α - Secretase) Activity Assay Kit (ANASPEC). After treatment, cells were washed with PBS, collected, and lysed with assay buffer. TACE enzymatic activity was quantified by continuous measurement of fluorescence intensity in a microplate fluorometer (λex 485 nm and λem 535 nm). TACE activity was normalized with the total protein level of each sample as determined by Bradford protein assay (Bio-Rad, Hercules, CA, USA).
Reverse transcription-polymerase chain reaction (RT-PCR)
Total RNA was extracted from each sample using TRIzol® Reagent (Life Technologies). Two micrograms of RNA were used as a template for cDNA synthesis with a reverse transcription kit (Promega). An equal volume of cDNA product was subjected to PCR analysis with some modifications according to ref. 18. The following primers were used in the PCR reactions: MUC1, forward primer 5′-ACAATTGACTCTGGCCTTCCG-3′ and reverse primer 5′-TGGGTTTGTGTAAGAGAGGCT-3′; β-actin, forward primer: 5′-CCAGCCTTCCTTCCTGGGCAT-3′ and reverse primer 5′-AGGAGCAATGATCT TGATCTTCATT-3′; TNF-α, forward primer 5′-AATCGGCCCGACTATCTCGACTTT-3′ and reverse primer 5′-TTTGAGCCAGAAGAGGGT-3′. For MUC1 and TNF-α, the PCR cycles were 32 and 31, respective, while for β-actin the cycles were 23. The amplified PCR products were resolved on 2% agarose gels with 0.5 μg/ml ethidium bromide, visualized, and photographed.
Detection of MUC1 in mouse lung by immunohistostaining
Lungs from A/J mice exposed to CS or filtered air (FA) for 20 weeks were paraformaldehyde-fixed and stained with hematoxylin and eosin (H&E). Immunohistochemistry (IHC) was carried out using the VECTASTAIN® ABC Kit with peroxidase labeling and DAB (3, 3′-diaminobenzidine) Peroxidase Substrate Kit (Vector laboratories, Burlingame, CA). Briefly, the slides were deparaffinized in xylene and rehydrated in gradient ethanol. Antigen retrieval was done by boiling the slide in 0.1 % citrate buffer for 15 min. Then, the slide was treated with 3% H2O2 for 20 min followed by blocking with 5% normal rabbit serum in PBS for 2 h at room temperature. After blocking the slide was incubated with primary antibody against MUC1 (rabbit antibody, Santa Cruz) overnight at 4°C. The ABC kit was then applied and the slide was developed with DAB according to instructions from the manufacturer.
Soft agar assay
HBEC-13 cells were exposed to CSE (20 μg/ml TPM) with or without TNF-α every three days for a total of 4 treatments. For each treatment, the cells were pre-treated with TNF-α (100 pg/ml) for 30 min followed by CSE treatment for 1 h. After CSE treatment, cells were cultured in fresh KSFM. A total of 1×104 cells in each well (12-well plate) were seeded in soft agar for colony formation. To study the influence of conditioned medium from CSE-treated macrophages on cell transformation, HBEC-13 cells were exposed to conditioned medium with or without anti-TNF-α neutralizing antibody for 24 h prior to CSE (20 μg/ml TPM) treatment. Cells were then treated similarly as mentioned above. After 3-week incubation, colonies in the agar were photographed and counted. The average number of colonies ± S.D. was determined using 6 randomly selected fields (18). All experiments were run in triplicate.
Chromatin immunoprecipitation assay (CHIP)
Antibodies specific to PPAR (Abcam) were used to capture protein–DNA complexes. Mouse immunoglobulins were used for isotype controls. qPCR was carried out using Power SYBR Green PCR Master Mix (Applied Biosystems) with the ABI PRISM 7900HT. Results were generated in triplicate independent experiments. Primer sequences are: forward primer: 5′-GACCGGTATAAAGCGGTAGG-3′ and reverse primer: 5′-GTCATGGTGGTGGTGAAATG-3′. Results were quantified as fold enrichment using a 2−DDCT method.
Statistics
All data are expressed as mean ± S.D. Statistical significance was examined by one-way analysis of variance. In all analyses, p<0.05 was considered statistically significant.
RESULTS
Increased MUC1 expression in CS exposed mouse lung macrophages and human macrophage cell lines
To investigate the effect of CS on MUC1 expression in lung cells, we first compared Muc1 expression in lung tissues from A/J mice chronically exposed to CS or filtered air (FA) for 20 weeks by immunohistostaining (25). Compared to Muc1 expression in FA exposed mice, Muc1 expression in airway and alveolar cells of CS exposed mice was significantly increased (Fig. S1). Interestingly, while CS exposure increased infiltration of inflammatory cells into the lung, a clear signal for Muc1 was also detected in lung macrophages. Both the number of Muc1-positive macrophages and the extent of Muc1 expression were promoted by CS exposure (Figs 1A and S1).
Figure 1. CS increases Muc1 expression in lung macrophages in vivo and human macrophage cell lines.

A), Quantitative representation of the macrophages expressing Muc1 after CS or FA treatment. Bars show the averages of macrophage numbers of 10 randomly selected fields. Data shown are mean ± S.D; * p<0.05. B), THP-1 cells were differentiated for 24 h in the presence of PMA (50 ng/ml) before the cells were treated with CSE (20 μg/ml TPM) at different time points. MUC1 expression was detected by Western blot with antibody Muc1 GP1.4 against the extracellular domain (MUC1-N) and antibody MUC1Ab-5 recognizing the C-terminal domain (MUC1-CT). GAPDH was detected as an input control. C), U937 cells were differentiated for 72 h in the presence of PMA (50ng/ml) before the cells were treated with CSE for 24h. MUC1 expression was detected by Western blot. β-Actin was detected as an input control. D), Differentiated THP-1 and U937 cells were transfected with MUC1 siRNA or negative control siRNA for 48 h. MUC1 expression was detected by Western blot. β-Actin was detected as an input control.
In human macrophage cells (THP-1 and U937), MUC1 expression was confirmed by Western blot with antibodies against both the extracellular domain (MUC1-N) and the C-terminal domain (MUC1-CT) (Fig. 1B, 1C). To validate the specificity of the Western blot, MUC1 siRNA was used to specifically block MUC1 expression. The MUC1 signal was effectively eliminated with MUC1 siRNA transfection (Fig. 1D). These results strongly suggest that MUC1 is expressed in macrophages. Furthermore, treating the human macrophage cells with CS extract (CSE) strikingly induced the expression of MUC1 (Fig. 1B, 1C). Collectively, these results suggest expression of MUC1 in lung macrophages is strongly induced by CS. Thus, we proceeded to examine if MUC1 plays a role in CS-induced inflammatory response in macrophages.
MUC1 is required for CSE-induced TNF-α production from macrophages
CS induces pulmonary macrophage infiltration and MUC1 was reported to regulate an inflammatory response in epithelial cells (26, 27). Therefore, we examined the role of MUC1 in CSE-induced inflammatory response in macrophages in an in vitro cell culture system. CSE strongly induced TNF-α secretion from both THP-1 and U937 cells, starting at 4h post-treatment and lasting for over 24h (Fig. 2A). Strikingly, knockdown of MUC1 expression by RNA interference effectively reduced CSE-induced TNF-α secretion from both THP-1 and U937 cells (Figs. 2B and 2C). MUC1 knockdown was confirmed by Western blot (Figs. 2B and 2C, insert). Remarkably, knockdown of MUC1 had no effect on cell viability with or without CSE treatment in both THP-1 and U937 cells (Figs. 2B and 2C). These results strongly suggest that MUC1 is required for TNF-α secretion in CSE-treated human macrophages.
Figure 2. MUC1 is required for CSE-induced TNF-α secretion from human macrophages.

A), THP-1 and U937 cells were differentiated for 24 h and 72 h, respectively, in the presence of PMA (50 ng/ml). THP-1 and U937 cells were treated with CSE (20 μg/ml TPM) at the indicated time points. Conditioned media were collected for detection of TNF-α secretion by ELISA assay. B) and C), Inserts are the confirmations of MUC1 knockdown by Western blot. THP-1 cells B) and U937 cells C) were transfected with MUC1 siRNA or negative control siRNA for 24 h. The cells were then treated with CSE (20 μg/ml TPM) for 1h. At 24h post-treatment, conditioned media were collected for detection of TNF-α by ELISA assay. Meanwhile, cell viability was detected by MTT assay. Data shown are mean ± S.D; ** p< 0.01, * p<0.05.
CSE-induced MUC1 expression and TNF-α secretion involve PPAR-γ
To investigate how CSE induces MUC1 expression in macrophages, MUC1 mRNA level was detected by RT-PCR. In THP-1 cells, CSE treatment robustly increased MUC1 transcript beginning at 30 min and persisting for 2 h (Fig. 3A). Consistently, CSE also induced MUC1 mRNA expression in U937 cells (Fig. 3B). These results indicate that CSE activates MUC1 transcription.
Figure 3. CSE induces MUC1 expression in macrophages depending on PPAR-γ.

A), THP-1 cells were differentiated for 24 h in the presence of PMA (50 ng/ml). THP-1 cells were treated with CSE (20 μg/ml TPM) at the indicated time points. MUC1 mRNA level was detected by RT-PCR. β-Actin was detected as an input control. B), U937 cells were differentiated for 72 h in the presence of PMA (50 ng/ml). U937 cells were treated with CSE (20 μg/ml TPM) for 1h. MUC1 mRNA level was detected by RT-PCR. β-Actin was detected as an input control. C), THP-1 cells were treated with CSE (20 μg/ml TPM) with or without different inhibitors (U0126, SP, SB, SC-514, and BHA) for 30 min. MUC1 mRNA level was detected by RT-PCR. β-Actin was detected as an input control. D), THP-1 cells were pre-treated with BADGE (10 μM) for 30 min, followed by CSE treatment (20 μg/ml TPM) for 24h for Western blot or 30 min for RT-PCR. E), U937 cells were transfected with PPARγ siRNA or negative control siRNA for 24 h. After transfection, cells were exposed to CSE (40 μg/ml TPM) for 24h. MUC1 and PPARγ expression were detected by Western blot. β-Actin was detected as an input control. F), ChIP assay shows the enrichment of PPARγ at the hMUC1 promoter. U937 cells were exposure to CSE (40 μg/ml TPM) for 1h. Data shown are mean ± S.D. G), THP-1 cells were pre-treated with BADGE (10 μM) for 30 min before the cells were treated with CSE (20 μg/ml TPM) for 1h. At 24h post-treatment, conditioned media were collected for detection of TNF-α by ELISA assay. BHA and R031 served as negative controls. Cell viability was detected by MTT assay. Data shown are mean ± S.D; ** p< 0.01.
The MUC1 promoter is under control of several transcription factors such as NF-κB, SP1 and PPAR-γ (27–29). To explore which transcription factor is involved in CSE-induced MUC1 expression, THP-1 cells were pre-exposed to a number of inhibitors before CSE treatment. The PPAR-γ inhibitors BADGE and GW9662, but not inhibitors for NF-κB, MAPKs (JNK, ERK, and p38) or ROS, remarkably attenuated CSE-induced MUC1 mRNA expression (Figs. 3C, 3D and S2A). Further, CSE caused an early activation of PPAR-γ in THP-1 cells (S2B). These results are in agreement with reports that PPAR-γ activates MUC1 gene transcription in human lung and gastric epithelial cells, and mouse placenta (27–29). Consistently, suppressing PPAR-γ with either BADGE or PPAR-γ siRNA dramatically suppressed CSE-induced MUC1 protein expression (Fig. 3D, 3E). Further, in a CHIP assay, binding of PPAR-γ to the MUC1 promoter was robustly induced by CSE (Fig. 3F). Thus, these results suggest that CSE induces MUC1 expression in human macrophages through PPAR-γ mediated transcription.
To corroborate the role of PPAR-γ in MUC1 expression and function, BADGE was used to treat macrophages before CSE treatment for TNF-α secretion. BADGE strongly inhibited CSE-induced TNF-α secretion, while it had no detectable cell cytotoxicity in macrophages (Fig. 3G). These results demonstrate that PPAR-γ-mediated MUC1 expression plays a critical role for CSE-induced TNF-α secretion from macrophages.
ERK is involved in CSE-induced MUC1 expression and TNF-α secretion in macrophages
It has been reported that the ERK pathway is involved in MUC1 expression (28). Thus, we also examined if this MAPK is involved in CSE-induced MUC1 expression. The ERK inhibitor U0126, but not the negative control U0124, effectively suppressed CSE-induced MUC1 protein expression (Fig. 4A). Consistent with this observation, CSE effectively activated ERK, beginning at 1 h and persisting for more than 4h after treatment (Fig. 4B). Interestingly, ERK inhibition had no detectable effect on CSE-induced MUC1 mRNA expression (Fig. 3C). These results suggest that the ERK pathway is likely involved in CSE-induced MUC1 expression in macrophages through a posttranscriptional mechanism. Indeed, the stability of MUC1 protein was reduced in ERK-inhibited and CSE-treated cells. The half-life of MUC1 was 2h in ERK-inhibited cells compared to 4.5h in control cells (Fig. 4C). In addition, U0126 also significantly blocked CSE-induced TNF-α secretion from macrophages with no cytotoxicity to the cells (Fig. 4D). Altogether, these results suggested that ERK is involved in CSE-induced MUC1 expression and TNF-α secretion in macrophages.
Figure 4. ERK pathway is involved in CSE-induced MUC1 expression and TNF-α secretion in macrophages.

A), THP-1 cells were pre-treated with U0126 (5 μM) for 30 min, followed by CSE treatment (20 μg/ml TPM) for 24 h. U0124 served as the negative control. MUC1expression was detected by Western blot. β-Actin was detected as an input control. B), THP-1 cells were treated with CSE (20 μg/ml TPM) for the indicated time points. ERK activation was detected by Western blot. β-Tubulin was detected as an input control. C), U937 cells were treated with cycloheximide (CHX, 10 μM) and CSE (40 μg/ml TPM) with or without U0126 (10 μM) for the indicated time periods. MUC1 expression was detected by Western blot. β-Actin was detected as an input control. The intensity of the individual bands was quantified by densitometry (Image J) and normalized to the corresponding input control bands. MUC1 expression changes were calculated with the control taken as 100%. D), THP-1 cells were pre-treated with U0126 (5 μM) for 30 min before the cells were treated with CSE (20 μg/ml TPM) for 1h. At 24h post-treatment, conditioned media were collected for detection of TNF-α by ELISA assay. SB and SC-514 served as negative controls. Cell viability was detected by MTT assay. Data shown are mean ± S.D, ** p< 0.01.
MUC1 mediates CSE-induced activation of TNF-α-converting enzyme (TACE)
TNF-α secretion is mainly mediated by shedding of the cell membrane-bound pro-TNF-α by TNF-α-converting enzyme (TACE) (30). To explore the underlying mechanism by which CSE induces TNF-α secretion, we examined if MUC1 is involved in regulation of TACE activity. Throughout the time period assessed, CSE induced TACE activity in a time-dependent manner (Fig. 5A). CSE-induced TNF-α secretion was effectively blocked by TACE inhibitor TAPI-1, confirming that CSE activates TACE activity to induce TNF-α secretion (Fig. S3). Interestingly, knockdown of MUC1 significantly attenuated CSE-induced TACE activity (Fig. 5B), suggesting that the induction of TACE activity by CSE was MUC1 dependent. CSE treatment or MUC1 knockdown had no effect on TACE expression (Fig. 5C), implying that CSE modulates TACE protease activity but not expression through MUC1. Similarly, suppression of ERK by U0126, which suppresses MUC1 expression, had no effect on TACE expression (Fig. 5C). CSE-induced MUC1 expression inhibited by treatment with BADGE or U0126 (Fig. 3D, 4A) effectively blocked CSE-induced but not basal TACE activity (Fig. 5D). These results are consistent with the important role of PPAR-γ and ERK in CSE-induced MUC1 expression. Altogether, these results demonstrate that CSE induces TNF-α secretion from macrophages through MUC1-dependent activation of TACE activity.
Figure 5. MUC1 mediates CSE-induced TACE activation.
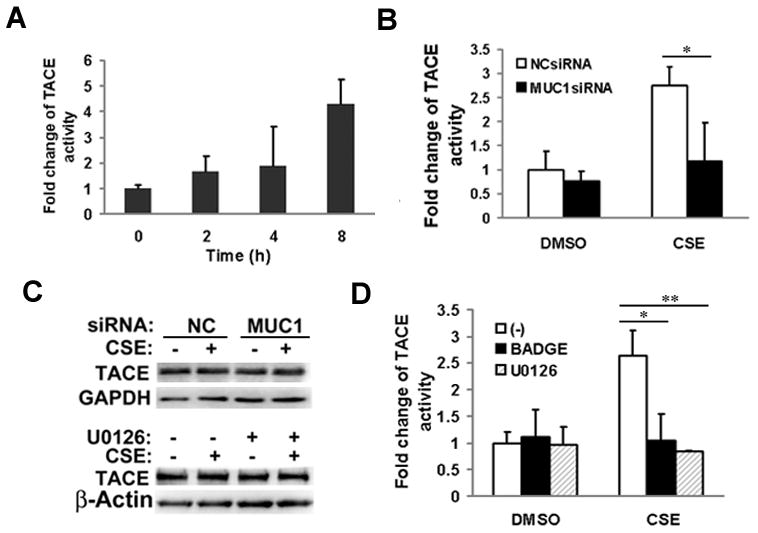
A), THP-1 were treated with CSE (20 μg/ml TPM) for the indicated time points. TACE activity was detected using SensoLyte® 520 TACE (α-Secretase) Activity Assay Kit. B), THP-1 cells were transfected with MUC1 siRNA or negative control siRNA for 24 h. After transfection, the cells were treated with CSE (20 μg/ml TPM) for 8 h for TACE activity assay. C), Upper, TACE expression was detected by Western blot. GAPDH was detected as a loading control. Lower, THP-1 cells were exposed to CSE (20 μg/ml TPM) for 8 h with or without U0126 (10 μM). TACE expression was detected by Western blot. β-Actin was detected as a loading control. D), THP-1 cells were pre-treated with BADGE (10 μM) or U0126 (5 μM) for 30 min before CSE treatment for 8 h. Cells were lysed for TACE activity assay. Data shown are mean ± S.D; ** p< 0.01, * P<0.05.
TNF-α secreted from macrophages significantly induces MUC1 expression in human bronchial epithelial cells (HBECs)
Because our previous study showed that MUC1 in HBECs plays an important role in CS carcinogen-induced HBEC transformation and TNF-α is a potential cytokine involved in MUC1 induction (18, 19), we investigated if TNF-α secreted from macrophages induces MUC1 expression in HBECs. Conditioned media from CSE-treated macrophages significantly increased MUC1 expression in HBEC-13 cells, while the control medium from untreated macrophages showed little effect on MUC1 expression (Fig. 6A). It is interesting that CSE by itself had a minor induction of MUC1 expression whereas conditioned medium from CSE-treated macrophages had a much stronger effect. On the other hand, the combination of CSE and conditioned medium from CSE-treated macrophages had no additive effect in MUC1 expression (Fig. 6B). These results suggest that the inflammatory response plays a major role in MUC1 expression in HBECs, although the direct induction of MUC1 by CSE in HBECs may have some minor contribution to MUC1 expression. Importantly, a TNF-α neutralizing antibody, but not a negative control antibody effectively blocked MUC1 induction by conditioned medium from CSE-exposed macrophages, suggesting that the MUC1 induction is TNF-α-dependent (Fig. 6C). Further, recombinant TNF-α was used to substantiate the role of TNF-α in MUC1 induction in HBECs. Indeed, MUC1 expression was strongly induced by the recombinant TNF-α (Fig. 6D). Taken together, these results establish the role of TNF-α secreted from macrophages in stimulating MUC1 expression in HBECs.
Figure 6. TNF-α secreted from macrophages induces MUC1 expression in human bronchial epithelial cells.

A), HBEC-13 cells were treated with conditioned medium from CSE or DMSO-treated macrophages for the indicated time points. B), HBEC-13 cells were treated as described in the figure legend. MUC1 expression was detected by Western blot. The intensity of the individual bands was quantified by densitometry (Image J) and normalized to the corresponding input control bands. C), HBEC-13 cells were treated with conditioned medium from CSE-treated macrophages with or without a TNF-α neutralizing antibody (50 ng/ml) for 24 h. D), HBEC-13 cells were exposed to recombinant TNF-α (1 ng/ml) for the indicated time points. The expression of MUC1 was detected by Western blot. β-Tubulin and GAPDH were detected as loading controls.
TNF-α secreted from macrophages facilitates CSE-induced transformation of HBECs
We next investigated the influence of conditioned media from CSE-treated macrophages on CSE-induced HBEC transformation. For induction of MUC1 expression, HBEC-13 cells were exposed to conditioned medium for 24 h, followed by exposure to CSE (Fig. 6A). Cell transformation was assayed by colony formation in soft agar assay. While CSE modestly induced colony formation, the conditioned medium from CSE-treated macrophages robustly enhanced CSE-induced cell transformation (Fig. 7A, 7B). Importantly, the effect of the conditioned medium was completely abolished by incubation with a TNF-α neutralizing antibody, but not the negative control antibody (Figs. 7A and 7B). In HBEC-13, recombinant TNF-α effectively and consistently augmented CSE-induced colony formation (Fig. S4). Together with our previous finding that MUC1 plays an important role in CS carcinogen-induced HBEC transformation (18), these results suggest that TNF-α secreted from macrophages potentiates CSE-induced transformation through the induction of MUC1 expression in HBECs.
Figure 7. TNF-α in conditioned medium from CSE-treated macrophages facilitates CSE-induced HBEC-13 cell transformation.

A) Representative images of and B) quantitative representation of colony formation of HBEC-13 cells in soft agar. HBEC-13 cells were pre-treated with the conditioned medium from CSE or DMSO-treated macrophages with or without a TNF-α neutralizing antibody (50 ng/ml) for 24 h and followed by CSE treatment (10 μg/ml TPM) for 1h. The cells were treated identically twice a week for 2 continuous weeks before they were seeded in soft agar. Colonies were allowed to develop for 3 weeks before they were photographed and counted. Bars show the average colony numbers of 6 randomly selected fields. Data shown are mean ± S.D; ** p< 0.01. C), A model of MUC1 in CS-induced and inflammation-associated lung cancer development. Cigarette smoke (CS) triggers MUC1 expression in macrophages, which facilitates TNF-α secretion from macrophages. TNF-α secreted from macrophages enhances MUC1 expression in bronchial epithelial cells, potentiating transformation of HBECs.
DISCUSSION
In this study, we provide evidence showing a novel function for MUC1 in a CS-induced inflammatory response in macrophages. MUC1 was modestly expressed in mouse lung macrophages and was robustly induced by chronic CS exposure. The expression of MUC1 in human macrophages was also up-regulated by CSE in vitro. Suppressing MUC1 expression significantly attenuated CSE-induced TACE activation and TNF-α secretion from macrophages. Furthermore, we found that conditioned medium from CSE-treated macrophages significantly induced MUC1 expression in HBECs and potentiated CSE-induced HBEC transformation in a TNF-α dependent manner. Therefore, together with our previous finding that MUC1 in HBECs plays an important role in CS carcinogen-induced transformation (18), our observations establish a dual role for MUC1 in CS-induced and inflammation-associated lung cancer development: to facilitate TNF-α secretion in macrophages and to potentiate transformation in HBECs (Fig. 7C).
We found the induction of MUC1 in macrophages by CSE is dependent on PPAR-γ mediated transcription and ERK-mediated protein stabilization. The expression and function of MUC1 in respiratory mucosal epithelial cells are well documented (12). MUC1 expression was also found in immune cells such as T lymphocytes and dendritic cells (20, 21). However, whether MUC1 is functionally expressed in macrophages has not been determined. In this report, we carefully determined the expression of MUC1 in vivo in mouse macrophages by immunohistochemistry and in vitro in human macrophages by Western blot and RT-PCR. The detection of MUC1 was validated by RNA interference. Furthermore, we determined that MUC1 is inducible by CSE and is functional in macrophages. Knockdown of MUC1 effectively attenuated CSE-induced TNF-α secretion. Thus, our results for the first time establish the expression and function of MUC1 in macrophages. The pro-inflammatory function of MUC1 in macrophages is contradictory to reports showing an anti-inflammatory function for MUC1 in epithelial cells (27). The reason for this discrepancy is currently unknown but it is likely that MUC1 may function distinctively in different cell types.
We further identified the main pathways for CS-induced MUC1 expression in macrophages. The direct binding of PPAR-γ to the MUC1 promoter induced by CSE was detected by CHIP assay. The blockage of PPAR-γ effectively inhibited CSE-induced MUC1 mRNA and protein expression in macrophages. This is consistent with reports that PPAR-γ directly binds and activates MUC1 transcription (27–29). Also, suppressing PPAR-γ blocked CSE-induced TNF-α secretion from macrophages, further substantiating the role of PPAR-γ-mediated MUC1 expression as a pro-inflammatory response in macrophages. Supporting our observation, it was reported that the PPAR-γ inhibitor, BADGE, suppressed TNF-α production in the murine macrophage cell line RAW 264.7 (31). In addition, expression of PPAR-γ was increased in monocytes/macrophages in smokers (32). We further found that ERK is also required for CSE-induced MUC1 expression in macrophages at a posttranscriptional level. Thus, MUC1 expression in macrophages is cooperatively controlled by PPAR-γ and ERK at multiple levels.
The anti-inflammatory role of MUC1 in epithelial cells is likely through suppression of Toll-like receptor signaling (11, 33). We show here that MUC1 plays a pro-inflammatory role in macrophages involving TACE activation. It would be interesting to determine if the contradictory roles of MUC1 in different cell types are due to different molecular partners or different isoforms of MUC1. Similarly, the role of PPAR-γ in the CS-induced inflammatory response seems to also be complex, being either anti- or pro-inflammatory (27, 31, 32, 34). The complexity of the response may be due to different cell types (i.e., epithelial cells vs. macrophages), different stimulations (i.e., CS vs. PMA) or different PPAR-γ modulating agents used (27, 31, 32, 34, 35). Nevertheless, our results clearly show a pro-inflammatory role for PPAR-γ in macrophages in response to CSE through activation of MUC1 expression. Elucidation of the mechanisms for the functional interaction of PPAR-γ and MUC1 in a cell’s response to inflammatory stimuli would help us to understand the complex roles of these molecules in inflammation. Since TNFα is able to induce MUC1 expression in HBECs, we examined if TNFα functions in an autocrine manner to induce MUC1 expression in macrophages. Knockdown of TNFR1, the major TNFα receptor, had no effect on CSE-induced MUC1 expression and TNFα secretion (Fig. S5A, S5B). A similar result was obtained when TNFR2 was knocked down (data not shown). Consistently, knockdown of TNFR1 had no effect on CSE-induced ERK activation in macrophages (Fig. S5C), suggesting the CSE-induced ERK activation is independent of TNFα-induced signaling. Therefore, it is unlikely MUC1 expression in macrophages is triggered by autocrine TNFα. Finally, we determined that MUC1-mediated TNF-α secretion from macrophages potentiates CS-induced transformation of HBECs. This process occurs at least in part through the induction of MUC1 expression in HBECs. Previously, we have shown that increased MUC1 in HBECs potentiates EGFR-mediated cell signaling for CS carcinogen-induced HBEC transformation (18). Therefore, our results suggest that by functioning in different cell types in the tumor-prone microenvironment, MUC1 substantially contributes to lung carcinogenesis. Specifically, MUC1 plays a dual role: to facilitate TNF-α secretion in macrophages and to potentiate transformation in HBECs. Further studies are warranted to determine if these MUC1-mediated mechanisms can be targeted for prevention against lung cancer development.
Supplementary Material
Acknowledgments
This study was supported by a grant from NIEHS/NIH (R01ES017328). We thank Kieu Do for her help with the Chromatin immunoprecipitation assay.
Abbreviations
- CS
cigarette smoke
- CSE
cigarette smoke extract
- PPAR-γ
peroxisome proliferator-activated receptor-gamma
- TNF-α
tumor necrosis factor-alpha
- BADGE
bisphenol A diglycidyl ether
- MAPK
mitogen-activated protein kinase
- MUC1
Mucin 1
Footnotes
Note: The authors have no conflict of interest to declare.
References
- 1.Lin WW, Karin M. A cytokine-mediated link between innate immunity, inflammation, and cancer. The Journal of clinical investigation. 2007;117:1175–83. doi: 10.1172/JCI31537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cho WC, Kwan CK, Yau S, So PP, Poon PC, Au JS. The role of inflammation in the pathogenesis of lung cancer. Expert opinion on therapeutic targets. 2011;15:1127–37. doi: 10.1517/14728222.2011.599801. [DOI] [PubMed] [Google Scholar]
- 3.Milara J, Cortijo J. Tobacco, inflammation, and respiratory tract cancer. Current pharmaceutical design. 2012;18:3901–38. doi: 10.2174/138161212802083743. [DOI] [PubMed] [Google Scholar]
- 4.Hecht SS. Tobacco smoke carcinogens and lung cancer. Journal of the National Cancer Institute. 1999;91:1194–210. doi: 10.1093/jnci/91.14.1194. [DOI] [PubMed] [Google Scholar]
- 5.Belinsky SA. Gene-promoter hypermethylation as a biomarker in lung cancer. Nat Rev Cancer. 2004;4:707–17. doi: 10.1038/nrc1432. [DOI] [PubMed] [Google Scholar]
- 6.Pfeifer GP, Denissenko MF, Olivier M, Tretyakova N, Hecht SS, Hainaut P. Tobacco smoke carcinogens, DNA damage and p53 mutations in smoking-associated cancers. Oncogene. 2002;21:7435–51. doi: 10.1038/sj.onc.1205803. [DOI] [PubMed] [Google Scholar]
- 7.Aggarwal BB. Signalling pathways of the TNF superfamily: a double-edged sword. Nat Rev Immunol. 2003;3:745–56. doi: 10.1038/nri1184. [DOI] [PubMed] [Google Scholar]
- 8.Wang X, Lin Y. Tumor necrosis factor and cancer, buddies or foes? Acta pharmacologica Sinica. 2008;29:1275–88. doi: 10.1111/j.1745-7254.2008.00889.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gonda TA, Tu S, Wang TC. Chronic inflammation, the tumor microenvironment and carcinogenesis. Cell cycle (Georgetown, Tex. 2009;8:2005–13. doi: 10.4161/cc.8.13.8985. [DOI] [PubMed] [Google Scholar]
- 10.Lu W, Hisatsune A, Koga T, Kato K, Kuwahara I, Lillehoj EP, et al. Cutting edge: enhanced pulmonary clearance of Pseudomonas aeruginosa by Muc1 knockout mice. J Immunol. 2006;176:3890–4. doi: 10.4049/jimmunol.176.7.3890. [DOI] [PubMed] [Google Scholar]
- 11.Kyo Y, Kato K, Park YS, Gajghate S, Umehara T, Lillehoj EP, et al. Antiinflammatory role of MUC1 mucin during infection with nontypeable Haemophilus influenzae. American journal of respiratory cell and molecular biology. 2012;46:149–56. doi: 10.1165/rcmb.2011-0142OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hattrup CL, Gendler SJ. Structure and function of the cell surface (tethered) mucins. Annu Rev Physiol. 2008;70:431–57. doi: 10.1146/annurev.physiol.70.113006.100659. [DOI] [PubMed] [Google Scholar]
- 13.Kufe DW. MUC1-C oncoprotein as a target in breast cancer: activation of signaling pathways and therapeutic approaches. Oncogene. 2012;32:1073–81. doi: 10.1038/onc.2012.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baldus SE, Engelmann K, Hanisch FG. MUC1 and the MUCs: a family of human mucins with impact in cancer biology. Crit Rev Clin Lab Sci. 2004;41:189–231. doi: 10.1080/10408360490452040. [DOI] [PubMed] [Google Scholar]
- 15.Woenckhaus M, Merk J, Stoehr R, Schaeper F, Gaumann A, Wiebe K, et al. Prognostic value of FHIT, CTNNB1, and MUC1 expression in non-small cell lung cancer. Hum Pathol. 2008;39:126–36. doi: 10.1016/j.humpath.2007.05.027. [DOI] [PubMed] [Google Scholar]
- 16.Schroeder JA, Masri AA, Adriance MC, Tessier JC, Kotlarczyk KL, Thompson MC, et al. MUC1 overexpression results in mammary gland tumorigenesis and prolonged alveolar differentiation. Oncogene. 2004;23:5739–47. doi: 10.1038/sj.onc.1207713. [DOI] [PubMed] [Google Scholar]
- 17.Guddo F, Giatromanolaki A, Koukourakis MI, Reina C, Vignola AM, Chlouverakis G, et al. MUC1 (episialin) expression in non-small cell lung cancer is independent of EGFR and c-erbB-2 expression and correlates with poor survival in node positive patients. J Clin Pathol. 1998;51:667–71. doi: 10.1136/jcp.51.9.667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu X, Bai L, Chen W, Padilla MT, Liu Y, Kim KC, et al. MUC1 contributes to BPDE-induced human bronchial epithelial cell transformation through facilitating EGFR activation. PloS one. 2012;7:e33846. doi: 10.1371/journal.pone.0033846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koga T, Kuwahara I, Lillehoj EP, Lu W, Miyata T, Isohama Y, et al. TNF-alpha induces MUC1 gene transcription in lung epithelial cells: its signaling pathway and biological implication. Am J Physiol Lung Cell Mol Physiol. 2007;293:L693–701. doi: 10.1152/ajplung.00491.2006. [DOI] [PubMed] [Google Scholar]
- 20.Correa I, Plunkett T, Vlad A, Mungul A, Candelora-Kettel J, Burchell JM, et al. Form and pattern of MUC1 expression on T cells activated in vivo or in vitro suggests a function in T-cell migration. Immunology. 2003;108:32–41. doi: 10.1046/j.1365-2567.2003.01562.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Taki C, Kitajima S, Sueyoshi K, Yonezawa S, Tanaka S, Sakoda K, et al. MUC1 mucin expression in follicular dendritic cells and lymphoepithelial lesions of gastric mucosa-associated lymphoid tissue lymphoma. Pathology international. 2002;52:691–701. doi: 10.1046/j.1440-1827.2002.01411.x. [DOI] [PubMed] [Google Scholar]
- 22.Wang Q, Chen W, Xu X, Li B, He W, Padilla MT, et al. RIP1 potentiates BPDE-induced transformation in human bronchial epithelial cells through catalase-mediated suppression of excessive reactive oxygen species. Carcinogenesis. 2013 doi: 10.1093/carcin/bgt143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ramirez RD, Sheridan S, Girard L, Sato M, Kim Y, Pollack J, et al. Immortalization of human bronchial epithelial cells in the absence of viral oncoproteins. Cancer research. 2004;64:9027–34. doi: 10.1158/0008-5472.CAN-04-3703. [DOI] [PubMed] [Google Scholar]
- 24.Chen W, Xu X, Bai L, Padilla MT, Gott KM, Leng S, et al. Low-dose gamma-irradiation inhibits IL-6 secretion from human lung fibroblasts that promotes bronchial epithelial cell transformation by cigarette-smoke carcinogen. Carcinogenesis. 2012;33:1368–74. doi: 10.1093/carcin/bgs159. [DOI] [PubMed] [Google Scholar]
- 25.March TH, Wilder JA, Esparza DC, Cossey PY, Blair LF, Herrera LK, et al. Modulators of cigarette smoke-induced pulmonary emphysema in A/J mice. Toxicol Sci. 2006;92:545–59. doi: 10.1093/toxsci/kfl016. [DOI] [PubMed] [Google Scholar]
- 26.Park YS, Guang W, Blanchard TG, Chul Kim K, Lillehoj EP. Suppression of IL-8 production in gastric epithelial cells by MUC1 mucin and peroxisome proliferator-associated receptor-gamma. American journal of physiology. 2012;303:G765–74. doi: 10.1152/ajpgi.00023.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Park YS, Lillehoj EP, Kato K, Park CS, Kim KC. PPARgamma inhibits airway epithelial cell inflammatory response through a MUC1-dependent mechanism. Am J Physiol Lung Cell Mol Physiol. 2012;302:L679–87. doi: 10.1152/ajplung.00360.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuwahara I, Lillehoj EP, Koga T, Isohama Y, Miyata T, Kim KC. The signaling pathway involved in neutrophil elastase stimulated MUC1 transcription. American journal of respiratory cell and molecular biology. 2007;37:691–8. doi: 10.1165/rcmb.2007-0072OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shalom-Barak T, Nicholas JM, Wang Y, Zhang X, Ong ES, Young TH, et al. Peroxisome proliferator-activated receptor gamma controls Muc1 transcription in trophoblasts. Molecular and cellular biology. 2004;24:10661–9. doi: 10.1128/MCB.24.24.10661-10669.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mullberg J, Althoff K, Jostock T, Rose-John S. The importance of shedding of membrane proteins for cytokine biology. European cytokine network. 2000;11:27–38. [PubMed] [Google Scholar]
- 31.Nakamuta M, Enjoji M, Uchimura K, Ohta S, Sugimoto R, Kotoh K, et al. Bisphenol a diglycidyl ether (BADGE) suppresses tumor necrosis factor-alpha production as a PPARgamma agonist in the murine macrophage-like cell line, RAW 264.7. Cell biology international. 2002;26:235–41. doi: 10.1006/cbir.2001.0838. [DOI] [PubMed] [Google Scholar]
- 32.Amoruso A, Bardelli C, Gunella G, Fresu LG, Ferrero V, Brunelleschi S. Quantification of PPAR-gamma protein in monocyte/macrophages from healthy smokers and non-smokers: a possible direct effect of nicotine. Life sciences. 2007;81:906–15. doi: 10.1016/j.lfs.2007.07.017. [DOI] [PubMed] [Google Scholar]
- 33.Ueno K, Koga T, Kato K, Golenbock DT, Gendler SJ, Kai H, et al. MUC1 mucin is a negative regulator of toll-like receptor signaling. American journal of respiratory cell and molecular biology. 2008;38:263–8. doi: 10.1165/rcmb.2007-0336RC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shen Y, Chen L, Wang T, Wen F. PPARgamma as a Potential Target to Treat Airway Mucus Hypersecretion in Chronic Airway Inflammatory Diseases. PPAR research. 2012;2012:256874. doi: 10.1155/2012/256874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Caito S, Yang SR, Kode A, Edirisinghe I, Rajendrasozhan S, Phipps RP, et al. Rosiglitazone and 15-deoxy-Delta12,14-prostaglandin J2, PPARgamma agonists, differentially regulate cigarette smoke-mediated pro-inflammatory cytokine release in monocytes/macrophages. Antioxidants & redox signaling. 2008;10:253–60. doi: 10.1089/ars.2007.1889. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.