

Significance
Sirtuin enzymes regulate metabolism and stress responses through deacetylation of specific protein lysine residues. Sirtuins are considered attractive drug targets, but selective inhibitors are rare, and their mechanisms mostly unknown. We report the mechanism of Sirtuin inhibition by Ex-527, a potent Sirt1 inhibitor widely used in physiological studies. A set of Sirtuin/ligand crystal structures, together with activity and binding data, reveals that the compound inhibits by forming a trimeric Sirtuin complex with a NAD+-derived coproduct. Our results yield insights in the unique Sirtuin catalytic mechanism and how it is exploited by Ex-527, and they provide essential information for rational drug development.
Abstract
Sirtuins are protein deacetylases regulating metabolism and stress responses. The seven human Sirtuins (Sirt1–7) are attractive drug targets, but Sirtuin inhibition mechanisms are mostly unidentified. We report the molecular mechanism of Sirtuin inhibition by 6-chloro-2,3,4,9-tetrahydro-1H-carbazole-1-carboxamide (Ex-527). Inhibitor binding to potently inhibited Sirt1 and Thermotoga maritima Sir2 and to moderately inhibited Sirt3 requires NAD+, alone or together with acetylpeptide. Crystal structures of several Sirtuin inhibitor complexes show that Ex-527 occupies the nicotinamide site and a neighboring pocket and contacts the ribose of NAD+ or of the coproduct 2’-O-acetyl-ADP ribose. Complex structures with native alkylimidate and thio-analog support its catalytic relevance and show, together with biochemical assays, that only the coproduct complex is relevant for inhibition by Ex-527, which stabilizes the closed enzyme conformation preventing product release. Ex-527 inhibition thus exploits Sirtuin catalysis, and kinetic isoform differences explain its selectivity. Our results provide insights in Sirtuin catalysis and inhibition with important implications for drug development.
Sirtuins are protein deacetylases that regulate various cellular processes, such as metabolism and stress responses, and they have been implicated in aging processes and aging-related diseases (1, 2). Sirtuins uniquely hydrolyze one NAD+ cosubstrate per deacetylation reaction (3), coupling their activity to cellular energy levels. Mammals have seven Sirtuin isoforms, Sirt1–7 (1). The nuclear isoforms Sirt1, 6, and 7 regulate substrates involved in chromosome stability and transcription (1, 2). Mitochondrial Sirt3, 4, and 5 regulate metabolic enzymes and stress response mechanisms (4–6). The mainly cytosolic Sirt2 deacetylates, e.g., tubulin and the protein acetyltransferase p300 (7, 8).
Crystal structures of the ∼250 amino acids large, conserved Sirtuin catalytic domains revealed that the substrates, NAD+, and acetyl-Lys of the protein substrate enter the active site from opposite sides of a cleft between a large Rossmann fold domain and a small, structurally more variable Zn2+-binding domain (9, 10). Binding of the protein substrate induces cleft closure (11–13), and a “cofactor binding loop” gets ordered on NAD+ binding (9, 14) (Fig. 1A). The NAD+ nicotinamide position varies in different structures, but during formation of an alkylimidate intermediate between the ADP ribose 1′ position and the acetyl oxygen (15) (Fig. 1A), it appears to occupy a conserved pocket (16). Nicotinamide then dissociates from this so-called C-pocket, possibly assisted by further closure of the cofactor binding loop (16, 17). The intermediate rearranges to a 1′-2′-bicyclic form, which is finally hydrolyzed to the products 2′-O-acetyl-ADP ribose and deacetylated polypeptide. If nicotinamide rebinds to the C-site before alkylimidate conversion, however, it can initiate substrate reformation, leading to an apparent inhibition of the deacetylation reaction (15).
Fig. 1.

Potency and kinetics of Sirtuin inhibition by Ex-527. (A) Mechanism proposed for Sirtuin-dependent deacetylation, illustrated with crystal structures: apo-enzyme (Sirt3 3GLS) (13), substrate complex (Sir2Tm 2H4F) (16), alkylimidate intermediate (Sirt3 8GLT) (13), bicyclic intermediate (Sirt5 4F56) (57), and product complex (Hst2 1Q1A) (14). (B) Chemical structure of Ex-527, with ring systems labeled A to C (*stereocenter). (C) Ex-527 dose–response experiments for Sirt1, Sirt3, and Sir2Tm (fitted with a tight-binding equation). (D) Sirt5 deacetylation and desuccinylation activity in the presence and absence of Ex-527. (E and F) Peptide substrate-dependent activity of Sir2Tm (E) and Sirt3 (F) in the absence and presence of varying Ex-527 concentrations revealing noncompetitive inhibition. (G and H) NAD+-dependent activity of Sir2Tm (G) and Sirt3 (H) at varying Ex-527 concentrations revealing uncompetitive inhibition. (D, E, and G) Error bars represent SEs of linear fits to time series experiments. (C, F, and H) Regression coefficients of linear fits to time series experiments were >0.95.
Sirtuins are considered attractive drug targets for (among others) the treatment of metabolic syndrome (e.g., Sirt1 activators), cancer (e.g., Sirt1 and Sirt3 inhibitors), and neurological disorders (e.g., Sirt2 inhibitors) (18–20). Physiological studies, target confirmation, and drug development have been hampered, however, by shortcomings of available Sirtuin inhibitors, which mostly show limited potency and/or isoform specificity and exploit unknown binding sites and mechanisms (21, 22). The widely used inhibitor sirtinol, for example, has an IC50 of 38 μM against Sirt2 and no effect on Sirt5, inhibits Sirt1 only approximately threefold weaker, and its effect on other isoforms and its mechanism are unknown (23–25). Ex-527 (6-chloro-2,3,4,9-tetrahydro-1H-carbazole-1-carboxamide; Fig. 1B), a Sirt1 inhibitor used in many physiological studies, is one of few compounds for which initial mechanistic data are available and that combine high potency with significant isoform selectivity (21, 26). Ex-527 inhibits Sirt1 ∼100-fold more potently than Sirt2 and Sirt3 and has no effect on Sirt5′s deacetylation activity (27, 28). Kinetic data suggest that Ex-527 allows alkylimidate formation and nicotinamide release before it blocks catalysis (27). A recent crystal structure of a Sirt1/NAD+/Ex-527 complex showed the inhibitor in the C-pocket (29), a binding site also speculated on for the Sirt2 inhibitor AGK2 (18), but the Sirt1 structure does not explain Ex-527’s isoform specificity and is incompatible with the kinetic results. The inhibition mechanisms of Ex-527, AGK2, and most other Sirtuin inhibitors thus remain enigmatic, hindering the development of inhibitors with improved potency and specificity for functional studies and therapeutic applications.
Here, we describe the molecular basis of Sirtuin inhibition by Ex-527. Binding studies, activity data, and crystal structures of complexes of a potently inhibited bacterial Sirt1 homolog and of the less sensitive human Sirt3 identify inhibitor binding site and coligand requirements, revealing a Sirtuin-specific inhibition mechanism and a kinetic basis for the compound’s isoforms selectivity. Our results yield insights into Sirtuin catalysis, including the geometry of the catalytic alkylimidate intermediate, and have major implications for structural analysis and further development of Sirtuin modulators.
Results
Ex-527 Is a Selective Sirtuin Inhibitor and Requires NAD+ for Inhibition.
A first kinetic analysis of Sirt1 inhibition by Ex-527 (27, 28) was done with the Fluor-de-Lys (FdL) substrate, a peptide carrying a fluorophore that potentially causes artifacts (12, 30). To investigate the molecular inhibition mechanism, we first tested selectivity and kinetics using a continuous assay (31) and nonmodified peptides derived from physiological substrates for Sirt1 (p53), Sirt3 [acetyl-CoA synthetase 2 (ACS2)], and Sirt5 [carbamoyl phosphate synthethase 1 (CPS1)]. Because inhibition was proposed to be uncompetitive with NAD+ (27), we adjusted NAD+ concentrations according to the respective KM values to allow comparisons. The IC50 values are 0.09 ± 0.03 µM for Sirt1 and 22.4 ± 2.7 µM for Sirt3 (Fig. 1C), in agreement with the FdL values (0.1 and 49 µM, respectively) (27). Because Sirt1 crystals became available only recently, we included the bacterial homolog Sir2 from Thermotoga maritima (Sir2Tm) in our investigation. Sir2Tm was efficiently inhibited by Ex-527 (IC50 0.9 ± 0.3; Fig. 1C), and we thus used it as a representative of the potently inhibited Sirtuins for structural studies. Furthermore, Ex-527 had no pronounced effect on Sirt5-dependent deacetylation, consistent with FdL tests (28), and showed no inhibition of Sirt5-dependent desuccinylation (Fig. 1D), the dominant Sirt5 activity identified recently (32).
To identify the enzyme state recognized by Ex-527, and thus suitable ligands for cocrystallization, we analyzed inhibition kinetics. Activity assays in presence of varying Ex-527 concentrations showed that inhibition of Sir2Tm and Sirt3 by Ex-527 is noncompetitive with substrate peptide (Fig. 1 E and F; Fig. S1A; Sir2Tm Ki = 1.8 ± 0.4 µM, Sirt3 Ki = 33.4 ± 4.4 µM). Analogous assays for the cosubstrate NAD+ showed no competition (Fig. 1 G and H; Fig. S1A), but for both Sirtuins inhibition was uncompetitive (Fig. S1 B–G; Sir2Tm: Ki = 3.3 ± 0.4 µM, Sirt3 Ki = 31.3 ± 2.1 µM), indicating that NAD+ assists in inhibition. These results are consistent with FdL data on Sirt1 (27) and show that Ex-527 inhibits the potent targets Sirt1/Sir2Tm, as well as the less sensitive Sirt3, with the same, NAD+-dependent and apparently peptide-independent mechanism.
The S Enantiomer of Ex-527 Occupies the C-Pocket and Interacts with ADP Ribose.
The presence of peptide substrate appears optional for Ex-527 binding, but NAD+ acts as an essential coligand. Because no Sir2Tm crystals could thus far be obtained in the absence of peptide, we used the following strategy to obtain the high-affinity Sir2Tm/inhibitor complex: we soaked crystals of a Sir2Tm/acetylated peptide complex, obtained in the presence of 1.5 mM Ex-527, with 1 mM NAD+ to enable cosubstrate binding and subsequent inhibitor association. Stopping the soak by freezing crystals after 2 min yielded good diffraction data (1.9 Å resolution; Table 1) for a Sir2Tm complex that contained Ex-527 and NAD+ derivatives (see below). Human Sirt3, in contrast, readily crystallizes in different states (12, 13), and we thus added Ex-527 to the running reaction of Sirt3, NAD+, and acetylated substrate before crystallization. Well-diffracting crystals were obtained (2.0 Å resolution; Table 1) that revealed Ex-527 and a coligand in the NAD+ pocket (see below).
Table 1.
Data collection and refinement statistics
| Sirt3 NAD+ Ex-243 | Sirt3 ADPr Ex-243 | Sirt3 thio-intermediate 1 | Sirt3 thio-intermediate 2 | Sirt3 native intermediate | Sirt3 2OacADPr Ex-243 | Sir2Tm soaking | Sir2Tm products Ex-243 | |
| Data processing | ||||||||
| Space group | P212121 | P212121 | C2221 | C2221 | C2221 | P212121 | P212121 | P43 |
| Unit cell (a/b/c) (Å) | 60.9/63.4/66.4 | 61.5/64.2/67.8 | 81.0/127.0/76.5 | 81.0/127.6/76.3 | 78.0/131.3/76.5 | 63.7/66.5/201.2 | 46.8/59.9/109.0 | 46.0/46.0/241.9 |
| Resolution (outermost shell) | 45.8–2.0 (2.05–2.0) | 46.6–2.0 (2.05–2.0) | 34.1–2.05 (2.1–2.05) | 33.7–2.7 (2.77–2.70) | 38.18–2.5 (2.6–2.5) | 34.4–1.9 (1.95–1.9) | 46.8–1.9 (2.0–1.90) | 60.47–3.3 (3.4–3.3) |
| Unique reflections (o. s.) | 17,814 (1,196) | 18,341 (1,203) | 25,087 (1,840) | 11,091 (797) | 12,915 (1,436) | 68,289 (5,051) | 24,821 (3,478) | 7,476 (617) |
| Observations (o. s.) | 106,296 (6,371) | 73,129 (4,227) | 103,459 (7,678) | 55,148 (3,870) | 72,942 (8,079) | 323,175 (24,139) | 109,585 (15,445) | 31,261 (2,688) |
| Completeness (o. s.) | 99.3 (95.4) | 98.1 (93.1) | 99.8 (99.9) | 99.8 (99.9) | 92.4 (93.9) | 99.8 (100) | 99.8 (100) | 99.8 (100) |
| Mutliplicity (o. s.) | 6.0 (5.3) | 4.0 (3.5) | 4.1 (4.2) | 5.0 (4.9) | 5.6 (5.6) | 4.7 (4.8) | 4.4 (4.4) | 4.2 (4.4) |
| Rmeas (o. s.)* | 11.0 (64.7) | 7.3 (37.2) | 11.1 (56.7) | 12.9 (55.6) | 7.2 (52.3) | 7.7 (49.0) | 6 (46.9) | 11.0 (105.3) |
| I/sigma (o. s.) | 15.6 (2.9) | 18.6 (3.9) | 13.2 (2.9) | 11.8 (3.0) | 20.8 (3.8) | 16.3 (3.5) | 20.7 (3.5) | 11.5 (1.5) |
| Refinement | ||||||||
| Amino acids (aa range) | 270 (122-391) | 273 (122-394) | 273 (121-394) | 273 (122-394) | 273 (122-394) | 273 (121-393) | 237 (1–246) | 245 (2-246) |
| Protein atoms | 2,183 | 2,207 | 2,214 | 2,152 | 2,144 | 6,470 | 1,954 | 3,651 |
| Peptide/intermediate atoms | 0 | 0 | 92 | 83 | 101 | 0 | 69 | 88 |
| Ligand atoms | 97 | 53 | 0 | 0 | 0 | 165 | 101 | 112 |
| Waters | 123 | 151 | 222 | 82 | 57 | 490 | 200 | 0 |
| Solvent atoms | 18 | 0 | 17 | 4 | 63 | 29 | 4 | 0 |
| Metals | 1 | 1 | 1 | 1 | 1 | 3 | 1 | 1 |
| Resolution | 45.8–2.0 | 46.6–2.0 | 34.1–2.05 | 33.7–2.7 | 38.18–2.50 | 34.4–1.9 | 40.3–1.9 | 60.47–3.30 |
| Rcryst†/Rfree‡ | 16.3/20.9 | 17.0/23.2 | 16.2/20.8 | 16.8/23.0 | 18.2/24.8 | 17.2/22.6 | 16.0/19.4 | 24.5/31.8 |
| B factor protein (Å2) | 20.0 | 19.2 | 19.5 | 33.6 | 41.2 | 26.7 | 19.7 | 114.8 |
| B factor peptide/intermediate (Å2) | — | — | 24.8 | 42.5 | 53.5 | — | 48.9 | 101.3 |
| B factor ligands incl. Zn (Å2) | 16.7 | 13.5 | 13.7 | 21.8 | 28.3 | 20.6 | 25.5 | 116.8 |
| B factor water (Å2) | 25.0 | 26.5 | 27.6 | 29.8 | 39.6 | 32.9 | 38.0 | — |
| B factor solvent atoms (Å2) | 37.7 | — | 49.0 | 50.2 | 63.7 | 44.2 | 52.9 | — |
| RMSD bond lengths/target (Å) | 0.018/0.019 | 0.020/0.020 | 0.020/0.019 | 0.013/0.019 | 0.015/0.020 | 0.020/0.019 | 0.019/0.020 | 0.010/0.020 |
| RMSD angles/target (°) | 2.009/2.039 | 2.139/2.019 | 2.108/2.017 | 1.765/2.010 | 1.936/2.025 | 2.077/2.019 | 2.011/2.047 | 2.133/2.025 |
| RMSD planes/target (Å) | 0.009/0.021 | 0.012/0.022 | 0.011/0.022 | 0.007/0.022 | 0.009/0.022 | 0.012/0.022 | 0.009/0.021 | 0.008/0.021 |
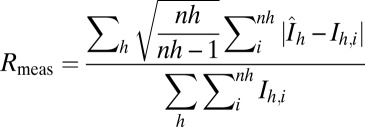 , with
, with 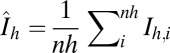 .
.
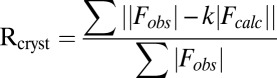 ;
; 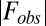 is the observed and
is the observed and 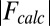 the calculated structure factor amplitude.
the calculated structure factor amplitude.
Rfree was calculated from 5% of measured reflections omitted from refinement.
The overall structures of the Ex-527 complexes of Sir2Tm and Sirt3 (Fig. 2A) differ only in the cofactor binding loop conformations (see below) from previous structures of these proteins (11–13, 16). In the Sir2Tm/Ex-527 complex, cocrystallized peptide and the ADP ribose part of the soaked-in NAD+ are visible. The electron density reveals a mixture of ligands due to the dynamic processes started by NAD+ soaking. Density analysis and rerefinement with combinations of substrates, products, intermediate, and inhibitor (Fig. S2 A–H) revealed the following complexes: an Ex-527 molecule is clearly indicated by electron density, with a C-ring and carbamide group in the C-pocket (Fig. 2 B and C). Part of the pocket, however, is occupied by nicotinamide from NAD+ that has not reacted and thus has acetylated peptide as coligand (40% occupancy). Coligands for Ex-527 are the products deacetylated peptide and 2′-O-acetyl-ADP ribose (60% occupancy for all three ligands), which provides part of the binding site (Fig. 2C). To confirm that products and inhibitor can form a complex with Sir2Tm, we then crystallized Sir2Tm mixed with deacetylated peptide, 2′-O-acetyl-ADP ribose, and Ex-527. The obtained structure (3.3 Å resolution; Table 1) revealed Ex-527 and 2'-O-acetyl-ADP ribose (Fig. 2D) arranged as observed in the soaked complex and partial occupation (∼50%) for the deacetylated peptide. In addition to its interaction with the coproduct, Ex-527 binds with its cyclohexenylcarbamide in the Sir2Tm C-pocket (Fig. 2 B–E), mimicking nicotinamide binding (33). The density reveals exclusively the S enantiomer of the Ex-527 racemic mixture as ligand, consistent with the finding that only one stereoisomer, called Ex-243, acts as inhibitor and that the S-form is the active isomer of a related compound (27–29). Only the kinked C-ring of the S isomer allows simultaneous positioning of the carbamide in the C-site and the aromatic rings in an almost perpendicularly oriented pocket formed by Gln98, Ile159, Phe48, and His116. We propose the term extended C-site (ECS) inhibitors for ligands exploiting both pockets. The environment of the Ex-243 chlorine is mainly nonpolar (Phe48 and Ile159), but the Val160 backbone carboxyl acts as a polar interaction partner for the chlorine, possibly forming a halogen bond (distance 3.6 Å, 117° angle to the carbonyl).
Fig. 2.

Crystal structures of Ex-243 complexes with Sir2Tm and Sirt3. (A) Overall structures of Sir2Tm/Ex-243 complex (brown), Sirt3/NAD+/Ex-243 (green), and apo Sirt3 (PDB ID 3GLS, blue) (13). (B) Active site of the Sir2Tm structure obtained through NAD+/Ex-527 soaking and enzymatic turnover resulting in Sir2Tm/substrates and Sir2Tm/products/Ex-243 complexes. (C) Sigma-A–weighted Fo-Fc density for products and Ex-243 contoured at 2.5 σ. (D) Structure of the Sir2Tm/products/Ex-243 complex obtained by cocrystallization. (E) Schematic view of the interactions between Ex-243 and Sir2Tm. (F) Structure of Sirt3 in complex with NAD+ in elongated and nonproductive conformation and Ex-243 bound to the C-pocket. (G) Structure of the Sirt3/ADP ribose/Ex-243 complex. (A–G) Proteins are shown in cartoon representation; ligands as sticks colored by atom type. (D, F, and G) The sigma-A–weighted 2Fo-Fc maps are contoured at 1.0 σ.
In contrast to the Sir2Tm/Ex-243 complex, the Sirt3 complex comprises a complete NAD+ molecule in addition to the well-defined inhibitor, and no density for a peptide (Fig. 2F; Fig. S2 I and J). The density again identifies the Ex-527 S enantiomer, Ex-243, in the C-site and the neighboring hydrophobic pocket (Fig. 2F; Fig. S2K). In addition to the contact to NAD+-derived ribose, the Ex-243 A-ring interacts with the nicotinamide of NAD+, which is in a nonproductive, extended conformation placing the nicotinamide in the acetyl-Lys binding site (Fig. S2L), as recently described for a Sirt1/NAD+/Ex-527 complex (29). Because Ex-243 binds to a binary Sirt3/NAD+ complex, we cocrystallized these partners in the absence of peptide (2.0 Å resolution; Table 1). Under these crystallization conditions, Ex-243 bound to a complex of Sirt3 and ADP ribose (Fig. 2G), likely formed from NAD+ through nonenzymatic hydrolysis, showing that even the ribose moiety of ADP ribose is sufficient for generating an Ex-243 binding site. We conclude that effective Ex-243 binding requires the cosubstrate binding pocket to be occupied, but our structures indicate two possibilities for the Ex-243 coligand under physiological conditions: NAD+ and 2′-O-acetyl-ADP ribose.
Ex-243 Binds Preferentially After Intermediate Formation and Allows Product Formation.
To identify the major Ex-243 coligand in solution, we performed binding experiments with Sirt3 and Sir2Tm using microscale thermophoresis (Fig. 3 A and B). Ex-527 showed negligible affinities (Sir2Tm: Kd > 180 μM; Sirt3 Kd > 330 µM) to apo-enzymes and to the Sirtuins saturated with 1 mM peptide substrate (Sir2Tm: Kd > 170 μM; Sirt3: Kd > 180 µM). Adding saturating NAD+ concentrations (5 mM), however, improved Ex-527 binding, with Kd values of 6.0 ± 0.4 µM for Sir2Tm and 16.5 ± 2.9 µM for Sirt3. Strikingly, addition of both substrates resulted in even higher affinities, with Kd values of 4.9 ± 0.5 µM (Sir2Tm) and 10.0 ± 1.4 µM (Sirt3). Thus, peptide substrate alone has no influence on Ex-243 binding, but it promotes tighter binding in presence of the cosubstrate, which enables alkylimidate formation. These results and the inhibition kinetics (see above) show that the arrangement in the Sirt3/NAD+/Ex-243 complex is not relevant for inhibition, because the inhibitor forces nicotinamide in the acetyl-Lys pocket, which would lead to competition between inhibitor and both substrates (Fig. S2L). Instead, these data suggest that Ex-243 binding after intermediate formation dominates inhibition, which is supported by the observation that nicotinamide release foregoes Sirt1 inhibition by Ex-243 (27).
Fig. 3.

Coligand for the inhibitory Ex-243 complex. (A and B) Thermophoresis binding experiments to determine Ex-243 affinities to different states of Sir2Tm (A) and Sirt3 (B). Error bars represent SEs of two independent measurements. (C–E) Product formation quantified by MS at various Sirt3 (C), Sirt1 (D), and Sir2Tm (E) concentrations in the presence or absence of Ex-243 and Ro-31-8220. The dashed line indicates the formation of one product molecule per Sirtuin molecule. Error bars represent SEs of two independent measurements. The control in E is saturated. (F) Structure of Sirt3 in complex with native alkylimidate intermediate. The sigma-A–weighted 2Fo-Fc map is contoured at 1.0 σ. (G) Overlay of Sirt3 bound to native alkylimidate intermediate or its thio-analog. Ex-243 was modeled into the C-site by overlaying the structures with the Sirt3/NAD+/Ex-243 complex revealing clashes between intermediate and Ex-243 (dashed line). (F and G) Proteins are shown in cartoon representation; ligands as sticks colored by atom type. rel. thermophoresis, relative thermophoresis; deac. peptide, decaetylated peptide.
Our Sir2Tm structures suggest that Ex-243 inhibits by stabilizing a Sirtuin complex with coproduct. To confirm the relevance of this coproduct complex for inhibition in solution, we analyzed product formation at saturating Ex-243 concentrations. Varying enzyme amounts were tested, and deacetylated peptide was quantified by MS. We find that for Sirt1, Sir2Tm, and Sirt3, product is indeed formed, but only up to one product molecule per Sirtuin molecule (Fig. 3 C–E; Fig. S3A). A control experiment with Sirt3 and the adenosine analog and potent Sirtuin inhibitor Ro-31-8220 [IC50 Sirt1 3.5 ± 0.4 µM (34); IC50 Sirt3 3.7 ± 0.7 µM, this study] yielded no product. We thus conclude that Ex-243 inhibition, in contrast to more common inhibition mechanisms, involves binding after intermediate formation and allows generation of the coproduct 2′-O-acetyl-ADP ribose.
Crystal Structure of a Native Intermediate Complex and Incompatibility with Ex-243 Binding.
Ex-243 might either target Sirtuin complexes with alkylimidates, with subsequent product formation, or it could directly bind to the coproduct complex. As a first test for an intermediate/Ex-243 interaction, we generated Sirt3 complexes with a more stable thio-analog of the alkylimidate (13, 35). Conformation and position of the thio-alkylimidate in the absence (2.05 Å resolution; Table 1; Fig. S3B) and presence of Ex-243 (2.7 Å resolution; Table 1; Fig. S3C) were indistinguishable (Fig. S3D). Also in presence of Ex-243, the inhibitor was not bound, and the C-pocket instead occupied by water molecules. To exclude the possibility that the artificial thio-modification causes the incompatibility of intermediate and Ex-243, we next generated a Sirt3 complex with native alkylimidate through soaking of Sirt3/acetyl-peptide crystals with a mix of NAD+ and Ex-243. The obtained Sirt3 complex with native alkylimidate (2.5 Å resolution; Table 1; Fig. 3F; Fig. S3E) is strong experimental evidence for this intermediate in Sirtuin-dependent deacetylation. It shows cis-configuration between Lys-ε-C-N and the glycosidic bond, but in contrast to NAD+ in productive conformation and the thio-intermediates with longer C-S bonds described above and in previous publications (13, 35), the ribose is flipped around (Fig. 3G). This feature of the native alkylimidate suggests that ribose rotation (and associated rearrangements in the diphosphate) enables moving the ribose C1′ from the C site to the acetyl-O for bond formation. However, this alkylimidate complex also did not contain bound Ex-243. Modeling Ex-243 into the C-pocket of the Sirt3/native intermediate complex by superposition with the Sirt3/NAD+/Ex-243 structure reveals a clash between inhibitor and the alkylimidate methyl group as molecular basis for the incompatibility (1.1 Å; Fig. 3G).
The second cyclic intermediate could also serve as a Ex-243 receptor, followed by product formation and trapping. If Ex-243 instead binds directly to the product complex, then product and inhibitor should increase each other’s affinity. Testing binding of 2′-O-acetyl-ADP ribose required >50 mM coproduct for signals, however, which is a concentration range where nonspecific effects prevent reliable quantification. The fact that we were able to assemble and crystallize a Sir2Tm/Ex-243/product complex at moderate 2′-O-acetyl-ADP ribose concentrations (5 mM) suggests, however, that the product complex is a major receptor. We conclude that Ex-243 inhibition involves binding after intermediate formation and nicotinamide release, likely to the coproduct complex, whereas the alkylimidate complex can be excluded as the receptor.
Ex-243 Induces the Closed Sirtuin Conformation.
Our Sir2Tm structures and biochemical data on Sirt1, Sir2Tm, and Sirt3 suggest Ex-243 binding to, and stabilization of, a Sirtuin/2′-O-acetyl-ADP ribose complex as its inhibition mechanism. To confirm that the inhibitor is compatible with this product in Sirt3, we solved a Sirt3 complex with 2′-O-acetyl-ADP ribose and Ex-243 (1.9 Å resolution; Table 1). The Sirt3 active site is fully occupied with 2′-O-acetyl-ADP ribose and Ex-243, with binding modes equivalent to the Sir2Tm complexes (Fig. 4A). The coproduct acetyl group appears to display partial rotational flexibility, because it is defined only in two of the three monomers of the asymmetric unit. The cofactor binding loop is trapped in a closed conformation instead of being open for product release. Comparison of our Ex-243 complexes with apo Sirt3 (PDB ID 3GLS) (13) and Sir2Tm/Sirt3 structures with occupied NAD+ pockets [Sir2Tm/NAD+/p53 peptide, 2H4F (16); Sirt3/intermediate, this study] indeed shows that Ex-243 stabilizes the loop’s closed conformation also adopted on NAD+ binding (Fig. 4B; Fig. S4). Sir2Tm-Phe33/Sirt3-Phe157, which was proposed to orient the NAD+ nicotinamide riboside and to shield the intermediate against nicotinamide and solvent (15, 16), is packed against the acetyl-ribose and interacts with the B- and C-ring of the inhibitor. The interaction and the associated Phe side chain rotation seem to contribute to this stabilization. Ex-243 thus appears to inhibit Sirtuins by trapping the protein in its closed conformation, preventing product release.
Fig. 4.

Structural mechanism of Ex-243 inhibition and isoform selectivity. (A) Structure of Sirt3 bound to 2′-O-acetyl ADP ribose and Ex-243. The sigma-A–weighted 2Fo-Fc map is contoured at 1.0 σ. (B) Comparison of the cofactor binding loop in the Sirt3/2′-O-acetyl ADP ribose/Ex-243 complex (green) with apo Sirt3 (PDB ID 3GLS, blue) (13) and Sirt3 in complex with thio-intermediate (this study, orange). Ex-243 and 2′-O-acetyl-ADP ribose are from the respective Sirt3 complex structure; ligands from the other structures are omitted for clarity. The loop’s movement and the rotation of the Phe157 side chain are indicated by arrows. (A and B) Proteins are shown in cartoon representation; ligands and residues involved in Ex-243 binding as sticks colored by atom type. (C) kcat/KM ratios obtained from NAD+ titration experiments with Sirt3 and Sir2Tm in the presence and absence of Ex-243. (D) Scheme describing the mechanism of Sirtuin inhibition by Ex-243.
Sirtuin Isoform Selectivity of Ex-243.
To identify Sirtuin features responsible for Ex-243 selectivity, we analyzed sequence alignments and structure comparisons. The insensitivity of Sirt5 against Ex-243 inhibition is likely due to the Sirt5-specific Arg-105 next to the C-pocket, which contributes to the Sirt5-specific response to nicotinamide (36). The only Sir2Tm-specific residue contacting Ex-243 in our complex structures is Met71. It is part of an additional helix turn in Sir2Tm, whereas mammalian Sirt1, 2, and 3 have identical chain lengths and residues in this region and no side chain directly corresponding to Met71. This residue thus does not explain the increased potency for Sirt1/Sir2Tm, and the Ex-243 binding sites are otherwise highly similar for isoforms that are inhibited potently (Sirt1, Sir2Tm) or only weakly (Sirt2, Sirt3). Consistently, Ex-243 affinities are comparable for the sensitive Sir2Tm and the less sensitive Sirt3 (see above). Therefore, factors other than isoform-specific residues interacting with Ex-243 have to cause selectivity.
Because the coproduct contributes to the Ex-243 binding site, isoform differences in binding geometry and affinity could contribute to selectivity. The conserved Sirtuin catalytic core and mechanism involve similar interactions to NAD+, indicated by several crystal structures and comparable KM and Kd values for different Sirtuins (this study and refs. 9 and 36). However, coproduct formation and cofactor loop closure are involved in Ex-243 inhibition, and factors influencing loop conformation and dynamics and catalytic rate thus can influence inhibition. Because Ex-243 inhibits through formation of a complex with Sirtuin and coproduct, analyzing changes in kcat/KM on inhibition shows whether product release is rate limiting, as previously shown for yeast Hst2 (37), or whether a step preceding inhibitor binding is rate limiting (38). Indeed, for the less Ex-243–sensitive Sirt3, kcat/KM remains unchanged (Fig. 4C), indicating inhibition before the rate-limiting step. In contrast, kcat/KM increases for the highly Ex-243–sensitive Sir2Tm (Fig. 4C), indicating inhibitor binding after the rate-limiting catalytic step, which leads to more effective inhibition. Different kinetics of catalysis by Sir2Tm/Sirt1 and Sirt3 thus appear to cause their different Ex-243 sensitivities, and it seems challenging or impossible to identify individual isoform features responsible for the respective inhibitor sensitivity from the many subtle contributions to active site dynamics. This result suggests that rather than optimizing the observed interactions, novel contacts to more remote, isoform-specific residues need to be incorporated in Ex-243 to obtain highly isoform selective Sirtuin inhibitors, and our structures provide a molecular basis for designing such derivatives.
Discussion
Sirtuins are attractive drug targets (2), and specific Sirtuin inhibitors would be excellent tools for in vivo studies, but few potent and selective compounds are available, and they are not mechanistically well understood (21, 26). Our results indeed show that understanding the mechanisms of Sirtuin catalysis and regulation is essential for the efficient development of improved drugs. The Ex-243 binding pocket includes the C-site and cosubstrate-derived ADP ribose, and Ex-243 inhibits uncompetitive to NAD+ and noncompetitive to the substrate. Thus, Ex-243 inhibition requires an NAD+-derived coligand (Fig. 4D) and nicotinamide release under conditions allowing activity, i.e., when both substrates are present (otherwise it would compete with NAD+ and peptide), so that Sirtuin complexes with intermediate or product, respectively, could be the receptors. Our Sirt3/native alkylimidate complex provides improved evidence for—and information on—this Sirtuin reaction intermediate compared with previous studies that used artificial thio-peptides (13, 15). Kinetic data suggest that the Sirtuin intermediate can exist for seconds (39), which would allow nicotinamide dissociation and Ex-243 binding, but the steric incompatibility between inhibitor and intermediate suggest the coproduct complex as direct Ex-243 receptor. In any case, the dependence on the unique catalytic mechanism makes this compound Sirtuin specific, consistent with the lack of Ex-243 effects on other enzyme families (27), and should render it an excellent starting point for drug development. We show, however, that there are no molecular differences between most Sirtuins in the site exploited by Ex-243 and that the compound’s moderate isoform selectivity appears to be based on kinetic differences, implying that isoform selectivity can even change under physiological conditions depending on substrate availability. Because the Ex-243 site lacks isoform-specific features, larger substitutions that can reach such isoform-specific regions will be required for improving the Sirt1 selectivity of Ex-243 or to develop compounds selective for other Sirtuins based on this binding site, and our complexes provide structural information for generating such specific interactions. The Ex-243 binding site likely also contributes to binding of other compounds, such as the Sirt2 inhibitor AGK2 (18) and the Sirt5 desuccinylation inhibitor GW5074 (25), indicating its potential to contribute to specific inhibition also of isoforms other than Sirt1. The identified mechanism and technical approaches will allow further identification of ECS inhibitors and solving of their Sirtuin complex structures to obtain further information on how to optimize binding to this site and how to add specific interactions. Obvious candidates, besides AGK2 and GW5074, are nicotinamide-related, Sirtuin-inhibiting 2-anilinobenzamides and dihydropyridin amides (26, 40, 41). Nonaromatic carbamides might in fact be favorable ECS ligands for two reasons. First, they allow carbamide rotation for optimal C-site binding. Second, they enable kinked conformations, as observed in Ex-243, simplifying simultaneous binding to C-site and the neighboring ECS pocket. Consistently, Ex-243 potency against Sirt1 is sensitive to the carboxamide position (27). Our results rationalize such data and suggest improvement strategies for further development of Ex-243 and other Sirtuin inhibitors.
In the physiological C-site ligand nicotinamide, the observed deviations from coplanarity of ring and carbamide are energetically demanding, which was proposed to contribute to NAD+ destabilization during catalysis (15). Interestingly, the carbamide is twisted in our Sirt3/NAD+/Ex-243 complex, although the inhibitor blocks the C-pocket and the nicotinamide thus occupies a larger pocket. Nevertheless, the NAD+ conformation appears more relaxed than with nicotinamide in the C-site due to a favorable bond angle at C1′, and it likely resembles the initially binding conformer in absence of peptide. The substrate acetyl-Lys site overlaps with the NAD+ nicotinamide, however (Fig. S2L), and would push it into the C-pocket during catalysis, explaining the missing peptide density in our Sirt3/NAD+/Ex-243 complex and the reported nicotinamide competition with NAD+ and peptide in Sirt3 inhibition (42). Moving nicotinamide in the C-site will bend the glycosidic bond, facilitating separation of nicotinamide and ribose, and a peptide contribution to this transition would minimize NAD+ glycohydrolase activity, an activity we indeed did not observe for Sirt3. Such a “pushing mechanism” was previously proposed based on Sir2Af structures (43), and our results suggest a general relevance of this mechanism and of an alkylimidate intermediate for Sirtuin-catalyzed deacetylation (15). The hydrophobic pocket that contributes to Ex-243 accommodation is conserved in Sirtuins and likely provides the route for nicotinamide from the initial docking position to the C-pocket. Our results thus reveal mechanistic details of Sirtuin catalysis and how to exploit them for inhibition, which provides essential information for Sirtuin-targeted drug development.
Methods
Chemicals and Peptides.
Chemicals were from Sigma if not stated otherwise. O-acetyl-ADP ribose (mixture of 2′ and 3′ form) (44) was a gift from Sirtris, a GSK company. Peptides were synthesized and HPLC purified by GL Biochem: ACS2-K642-peptide [TRSG-Lys(Ac)-VMRRL], p53-K382-peptide [RHK-Lys(Ac)-LMFK], deacetylated p53-K382 peptide (STSRHK-Lys-LMFKTE), H3-K116-peptide [IHA-Lys(Ac)-RVT], and CPS1-K527-peptide [FKRGVL-Lys(Ac)-EYGVKV]. Thio-acetyl-ACS2-K642-peptide [TRSG-Lys(thioAc)-VMRRL] was synthesized by standard Fmoc-based solid phase peptide synthesis and purified using RP-8 HPLC. Fluorenylmethoxycarbonyl-Lys(thioAc)-OH was used for introducing the thioacetylated lysine residue.
Protein Expression and Purification.
Sir2Tm was expressed and purified as previously described using a full-length construct in pET11a (Addgene plasmid 25815) (31, 33).
Recombinant human Sirt1, Sirt3, and Sirt5 were purified as described previously (12, 45). Briefly, His-tagged full-length Sirt1 (in pET15b), His-Trx-tagged Sirt3 residues 118–399 (in pVFT3S), and His-tagged Sirt5 residues 34–302 (in pET151/D-TOPO) were expressed in Escherichia coli BL21DE3 Rosetta2 (Novagen) and purified by affinity chromatography with Talon resin (Clontech). For Sirt3, the tag was removed by incubation with Tobacco Etch Virus protease, and the protein was purified through a second affinity chromatography. Finally, proteins were subjected to gel filtration on Superose12 column (GE Healthcare), concentrated, and stored at −80 °C. Sirtuin concentrations were determined through UV absorption at 280 nm, and Sirt1 concentrations were corrected for contaminants by SDS/PAGE and densitometry. His-tagged Sirt3 for activity assays (residues 114–380) was produced as previously described (46).
Crystallization and Structure Solution.
Sirtuin complexes were crystallized by the vapor diffusion method. Protein solution (Sir2Tm, 10 mg/mL; Sirt3, 11 mg/mL) was mixed with an equivalent volume of reservoir and equilibrated against reservoir at 20 °C. If not stated differently, crystals were transferred to cryoprotectant solution [reservoir supplemented with 25% (vol/vol) glycerol and the respective ligands] and flash frozen in liquid nitrogen.
Sir2Tm was crystallized in complex with acetyl-p53-peptide (1 mM) and Ex-243 (1.5 mM) in the presence of 5% (vol/vol) DMSO and with 25% (wt/vol) PEG 4000, 50 mM Li2SO4, and 100 mM Tris, pH 8.5, as reservoir. Crystals were then soaked with NAD+ (1 mM) for 2 min in reservoir supplemented with acetyl-p53-peptide (1 mM), Ex-527 (1.5 mM), and 25% (vol/vol) ethylene glycol, and crystals were flash frozen in liquid nitrogen. Sir2Tm in complex with products and Ex-243 was cocrystallized by mixing protein with deacetylated p53-peptide (5 mM), O-acetyl-ADP ribose (5 mM), Ex-527 (1 mM), and 10% (vol/vol) DMSO and using 20% (wt/vol) PEG 6000 and 0.1 M Bis-Tris, pH 5.9 as reservoir.
For the Sirt3/NAD+/Ex-243 complex, protein was mixed with acetyl-ACS2-peptide (1 mM), NAD+ (10 mM), Ex-527 (1 mM), and 10% (vol/vol) DMSO and the reservoir was 20% (wt/vol) PEG 3350 and 0.2 M ammonium nitrate. The structure of Sirt3 in complex with ADP ribose and Ex-243 was obtained by crystallizing Sirt3 in presence of NAD+ (5 mM), Ex-527 (1 mM), and 10% (vol/vol) DMSO in reservoir [0.08 M KH2PO4, 18% (wt/vol) PEG 8000, and 20% (vol/vol) glycerol] providing cryoprotection. The Sirt3/thio-intermediate complexes were obtained by soaking Sirt3/thio-acetyl-ACS2-peptide complex crystals (1 mM thio-acetyl-ACS2 peptide; reservoir: 25% (wt/vol) PEG 3350, 0.2 M LiSO4, 0.1 M Na citrate, pH 5.6) with NAD+ (1.25 mM), and optionally Ex-527 (1 mM) and 10% (vol/vol) DMSO in cryoprotectant solution [reservoir plus 25% (vol/vol) glycerol] for several hours. The native intermediate complex with Sirt3 was produced similar to its thio-analog, using 5 mM acetyl-ACS2-peptide and 0.2 M (NH4)2SO4, 0.1 M Bis-Tris, pH 5.5, and 21% (wt/vol) PEG 3350 as reservoir. The Sirt3/acetyl-ACS2-peptide crystals were soaked with NAD+ (5 mM), Ex-527 (1 mM), and 10% (vol/vol) DMSO in reservoir supplemented with 25% (vol/vol) glycerol for 80 min. Sirt3/2′-O-acetyl-ADP ribose/Ex-243 complex was crystallized by mixing Sirt3 with O-acetyl-ADP ribose (10 mM), Ex-527 (1 mM), and 10% (vol/vol) DMSO and as reservoir 20% (wt/vol) PEG 6000 and 0.1 M Hepes, pH 7.
X-ray diffraction data were collected at 100 K at beamline MX14.1 (operated by Helmholtz-Zentrum) at the BESSY II electron storage ring (47) using an MX-225 CCD detector (Rayonix). Diffraction data were integrated, scaled, and merged using X-ray Detector Software (XDS) (48) (Table 1). Structures were solved through Patterson searches with Phaser (49) or Molrep (50) using the protein parts of a Sir2Tm/peptide/nicotinamide complex (PDB ID 1YC5) or Sir2Tm/NAD+/p53-peptide complex (PDB ID 2H4F) as a search model for Sir2Tm and of a human Sirt3/thio-acetyl-ACS2-peptide alkylimidate intermediate complex (PDB ID 3GLT) as a search model for Sirt3. Structures were refined using Refmac (51), and models were manually rebuilt in Coot (52), with 5% of the reflections excluded from refinement for Rfree calculation (53). Individual isotropic Debye–Waller factors were refined, and solvent molecules were added in a later stage of model refinement. For the low-resolution Sir2Tm/products/Ex-243 structure, non-crystallographic symmetry and translation libration screw-motion restraints and a high-resolution reference structure (Sir2Tm/p53-peptide complex, PDB ID 3JR3) were included in refinement. Parameter files for building and refining the ligands Ex-243, reaction intermediate, and 2′-O-acetyl-ADP ribose were generated using MarvinSketch (www.chemaxon.com) and ProDrg (54). Model quality was assessed using MolProbity (55) and validation tools in Coot.
Continuous Deacetylation Assay.
The coupled deacetylation assay was performed as described (31). Briefly, reaction mixtures containing substrate peptide, NAD+, inhibitor, and 5% (vol/vol) DMSO were supplemented with 0.75 µM Sirt1, 2.5 µM Sir2Tm, or 2.5 µM Sirt3, respectively, and the decrease in absorbance at 340 nm was monitored using a Bio-Tek plate reader. For IC50 value determination, substrate and NAD+ concentrations were adjusted to the respective Sirtuin’s KM values (∼10-fold KM: Sirt1, 500 µM acetyl-p53-peptide, 1 mM NAD+; Sir2Tm, 1 mM acetyl-p53-peptide, 1 mM NAD+; Sirt3 1 mM acetyl-ACS2-peptide, 3.5 mM NAD+), and Ex-527 concentrations were varied. Peptide and NAD+ titration experiments with Sirt3 were performed using 1 mM NAD+ and 500 µM acetyl-ACS2-peptide, respectively, at varying Ex-527 concentrations. Product formation rates were determined by linear regression and fitted with GraFit7 (Erithacus Software) using a tight binding equation for IC50 determination and noncompetitive (peptide titration) or uncompetitive (NAD+ titration) inhibition equations for kinetics.
MS-Based Deacetylation Assay.
Peptide and NAD+ titration experiments with Sir2Tm were performed using an MS deacetylation assay as described (36), with 2.5 µM Sir2Tm and 1 mM NAD+ or 500 µM acetyl-p53-peptide, respectively, varying Ex-527 concentrations, and 5% (vol/vol) DMSO. Briefly, aliquots of a reaction were stopped after different time points, and substrate and product were separated and quantified by nano-LC-electrospray ionization mass spectrometry on an LTQ XL (Thermo Fisher scientific), followed by linear fitting to obtain product formation rates. Turnover rates were fitted with GraFit7 using equations for noncompetitive (peptide titration) or uncompetitive (NAD+ titration) inhibition.
For testing product formation under saturating Ex-527 or Ro-31-8220 concentrations, respectively, varying Sirtuin amounts (Sirt1, 3.4/6.8/13.6 µM; Sir2Tm, 4/8/16 µM; Sirt3, 5/10/20 µM) were mixed with substrate peptide (Sirt1 and Sir2Tm, 200 µM acetyl-p53-peptide; Sirt3 50/200 µM acetyl-ACS2-peptide), 500 µM NAD+, inhibitor (Sirt1 and Sir2Tm, 150 µM Ex-527; Sirt3, 500 µM Ex-527/Ro-31-8220), and 5% (vol/vol) DMSO and incubated at 37 °C, and the reaction was stopped after 30 min by addition of 0.25% (vol/vol) trifluoroacetic acid. Samples were analyzed, and product was quantified as described previously (36). Calibration lines (Fig. S3 F and G; regression coefficient >0.95) were generated with different ratios of the respective peptide pairs (deacetylated/acetylated) for absolute quantification.
Binding Analysis by Microscale Thermophoresis.
Affinities were measured by microscale thermophoresis (56). Sirtuins were labeled by incubation with twofold molar excess of FITC (Thermo Fischer) in 20 mM Hepes, pH 7.5, and 150 mM NaCl at 4 °C overnight. Free dye was removed with a Nap25 column (GE Healthcare). Sirtuin was mixed with varying concentrations of Ex-527, and thermophoresis was measured (excitation wavelength 470 nm, emission wavelength 520 nm, LED-power 10–20%, laser-power 10%) using a Monolith NT 115 (NanoTemper Technologies) in the absence and presence of 5 mM NAD+ and 1 mM acetylated peptide (Sir2Tm, acetyl-H3-peptide; Sirt3, acetyl-ACS2-peptide). Dissociation constants were determined with GraFit7 (Erithacus Software) by nonlinear fitting (one-site and two-site fitting equations). Each experiment was repeated at least twice.
Supplementary Material
Acknowledgments
We thank Felix Husslik, Norbert Grillenbeck, Martina Czapko, Katharina Ruhnke, and the beamline staff of BESSY MX14.1 (Helmholtz-Zentrum Berlin) for technical assistance and Dr. John Denu for helpful discussions. We thank Sirtris, a GSK company, and in particular Dr. Bruce Szczepankiewicz (Sirtris), for providing us with O-acetyl-ADP-ribose. Part of this work was supported through Deutsche Forschungsgemeinschaft Grant STE1701/5 (to C.S.), Bundesministerium für Bildung und Forschung Grant ProNet-T3 (to M.S.), and Elite Network Bavaria (G.T.T.N., M.L., C.S.).
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org [PDB ID codes 4BV3 (Sirt3/NAD+/Ex-243), 4BVB (Sirt3/ADPr/Ex-243), 4BVE (Sirt3 thio-intermediate 1), 4BVF (Sirt3 thio-intermediate 2), 4BVG (Sirt3 native intermediate), 4BVH (Sirt3/2OacADPr/Ex-243), 4BUZ (Sir2Tm soaking), and 4BV2 (Sir2Tm products Ex-243)].
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1303628110/-/DCSupplemental.
References
- 1.Michan S, Sinclair D. Sirtuins in mammals: Insights into their biological function. Biochem J. 2007;404(1):1–13. doi: 10.1042/BJ20070140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Haigis MC, Sinclair DA. Mammalian sirtuins: Biological insights and disease relevance. Annu Rev Pathol. 2010;5:253–295. doi: 10.1146/annurev.pathol.4.110807.092250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sauve AA, Wolberger C, Schramm VL, Boeke JD. The biochemistry of sirtuins. Annu Rev Biochem. 2006;75:435–465. doi: 10.1146/annurev.biochem.74.082803.133500. [DOI] [PubMed] [Google Scholar]
- 4.Gertz M, Steegborn C. Function and regulation of the mitochondrial sirtuin isoform Sirt5 in Mammalia. Biochim Biophys Acta. 2010;1804(8):1658–1665. doi: 10.1016/j.bbapap.2009.09.011. [DOI] [PubMed] [Google Scholar]
- 5.Bell EL, Guarente L. The SirT3 divining rod points to oxidative stress. Mol Cell. 2011;42(5):561–568. doi: 10.1016/j.molcel.2011.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lakshminarasimhan M, Steegborn C. Emerging mitochondrial signaling mechanisms in physiology, aging processes, and as drug targets. Exp Gerontol. 2011;46(2-3):174–177. doi: 10.1016/j.exger.2010.08.024. [DOI] [PubMed] [Google Scholar]
- 7.North BJ, Marshall BL, Borra MT, Denu JM, Verdin E. The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol Cell. 2003;11(2):437–444. doi: 10.1016/s1097-2765(03)00038-8. [DOI] [PubMed] [Google Scholar]
- 8.Black JC, Mosley A, Kitada T, Washburn M, Carey M. The SIRT2 deacetylase regulates autoacetylation of p300. Mol Cell. 2008;32(3):449–455. doi: 10.1016/j.molcel.2008.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sanders BD, Jackson B, Marmorstein R. Structural basis for sirtuin function: What we know and what we don’t. Biochim Biophys Acta. 2010;1804(8):1604–1616. doi: 10.1016/j.bbapap.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moniot S, Weyand M, Steegborn C. Structures, substrates, and regulators of Mammalian sirtuins - opportunities and challenges for drug development. Front Pharmacol. 2012;3:16. doi: 10.3389/fphar.2012.00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cosgrove MS, et al. The structural basis of sirtuin substrate affinity. Biochemistry. 2006;45(24):7511–7521. doi: 10.1021/bi0526332. [DOI] [PubMed] [Google Scholar]
- 12.Gertz M, et al. A molecular mechanism for direct sirtuin activation by resveratrol. PLoS ONE. 2012;7(11):e49761. doi: 10.1371/journal.pone.0049761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jin L, et al. Crystal structures of human SIRT3 displaying substrate-induced conformational changes. J Biol Chem. 2009;284(36):24394–24405. doi: 10.1074/jbc.M109.014928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhao K, Chai X, Marmorstein R. Structure of the yeast Hst2 protein deacetylase in ternary complex with 2′-O-acetyl ADP ribose and histone peptide. Structure. 2003;11(11):1403–1411. doi: 10.1016/j.str.2003.09.016. [DOI] [PubMed] [Google Scholar]
- 15.Sauve AA. Sirtuin chemical mechanisms. Biochim Biophys Acta. 2010;1804(8):1591–1603. doi: 10.1016/j.bbapap.2010.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hoff KG, Avalos JL, Sens K, Wolberger C. Insights into the sirtuin mechanism from ternary complexes containing NAD+ and acetylated peptide. Structure. 2006;14(8):1231–1240. doi: 10.1016/j.str.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 17.Chang JH, et al. Structural basis for the NAD-dependent deacetylase mechanism of Sir2. J Biol Chem. 2002;277(37):34489–34498. doi: 10.1074/jbc.M205460200. [DOI] [PubMed] [Google Scholar]
- 18.Outeiro TF, et al. Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson’s disease. Science. 2007;317(5837):516–519. doi: 10.1126/science.1143780. [DOI] [PubMed] [Google Scholar]
- 19.Sanchez-Fidalgo S, Villegas I, Sanchez-Hidalgo M, de la Lastra CA. Sirtuin modulators: Mechanisms and potential clinical implications. Curr Med Chem. 2012;19(15):2414–2441. doi: 10.2174/092986712800269272. [DOI] [PubMed] [Google Scholar]
- 20.Lavu S, Boss O, Elliott PJ, Lambert PD. Sirtuins—novel therapeutic targets to treat age-associated diseases. Nat Rev Drug Discov. 2008;7(10):841–853. doi: 10.1038/nrd2665. [DOI] [PubMed] [Google Scholar]
- 21.Cen Y. Sirtuins inhibitors: The approach to affinity and selectivity. Biochim Biophys Acta. 2010;1804(8):1635–1644. doi: 10.1016/j.bbapap.2009.11.010. [DOI] [PubMed] [Google Scholar]
- 22.Neugebauer RC, Sippl W, Jung M. Inhibitors of NAD+ dependent histone deacetylases (sirtuins) Curr Pharm Des. 2008;14(6):562–573. doi: 10.2174/138161208783885380. [DOI] [PubMed] [Google Scholar]
- 23.Mai A, et al. Design, synthesis, and biological evaluation of sirtinol analogues as class III histone/protein deacetylase (Sirtuin) inhibitors. J Med Chem. 2005;48(24):7789–7795. doi: 10.1021/jm050100l. [DOI] [PubMed] [Google Scholar]
- 24.Grozinger CM, Chao ED, Blackwell HE, Moazed D, Schreiber SL. Identification of a class of small molecule inhibitors of the sirtuin family of NAD-dependent deacetylases by phenotypic screening. J Biol Chem. 2001;276(42):38837–38843. doi: 10.1074/jbc.M106779200. [DOI] [PubMed] [Google Scholar]
- 25.Suenkel B, Fischer F, Steegborn C. Inhibition of the human deacylase Sirtuin 5 by the indole GW5074. Bioorg Med Chem Lett. 2013;23(1):143–146. doi: 10.1016/j.bmcl.2012.10.136. [DOI] [PubMed] [Google Scholar]
- 26.Chen L. Medicinal chemistry of sirtuin inhibitors. Curr Med Chem. 2011;18(13):1936–1946. doi: 10.2174/092986711795590057. [DOI] [PubMed] [Google Scholar]
- 27.Napper AD, et al. Discovery of indoles as potent and selective inhibitors of the deacetylase SIRT1. J Med Chem. 2005;48(25):8045–8054. doi: 10.1021/jm050522v. [DOI] [PubMed] [Google Scholar]
- 28.Solomon JM, et al. Inhibition of SIRT1 catalytic activity increases p53 acetylation but does not alter cell survival following DNA damage. Mol Cell Biol. 2006;26(1):28–38. doi: 10.1128/MCB.26.1.28-38.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhao X, et al. The 2.5 Å crystal structure of the SIRT1 catalytic domain bound to nicotinamide adenine dinucleotide (NAD+) and an indole (EX527 analogue) reveals a novel mechanism of histone deacetylase inhibition. J Med Chem. 2013;56(3):963–969. doi: 10.1021/jm301431y. [DOI] [PubMed] [Google Scholar]
- 30.Kaeberlein M, et al. Substrate-specific activation of sirtuins by resveratrol. J Biol Chem. 2005;280(17):17038–17045. doi: 10.1074/jbc.M500655200. [DOI] [PubMed] [Google Scholar]
- 31.Smith BC, Hallows WC, Denu JM. A continuous microplate assay for sirtuins and nicotinamide-producing enzymes. Anal Biochem. 2009;394(1):101–109. doi: 10.1016/j.ab.2009.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Du J, et al. Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science. 2011;334(6057):806–809. doi: 10.1126/science.1207861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Avalos JL, Bever KM, Wolberger C. Mechanism of sirtuin inhibition by nicotinamide: Altering the NAD(+) cosubstrate specificity of a Sir2 enzyme. Mol Cell. 2005;17(6):855–868. doi: 10.1016/j.molcel.2005.02.022. [DOI] [PubMed] [Google Scholar]
- 34.Trapp J, et al. Adenosine mimetics as inhibitors of NAD+-dependent histone deacetylases, from kinase to sirtuin inhibition. J Med Chem. 2006;49(25):7307–7316. doi: 10.1021/jm060118b. [DOI] [PubMed] [Google Scholar]
- 35.Hawse WF, et al. Structural insights into intermediate steps in the Sir2 deacetylation reaction. Structure. 2008;16(9):1368–1377. doi: 10.1016/j.str.2008.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fischer F, et al. Sirt5 deacylation activities show differential sensitivities to nicotinamide inhibition. PLoS ONE. 2012;7(9):e45098. doi: 10.1371/journal.pone.0045098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Borra MT, Langer MR, Slama JT, Denu JM. Substrate specificity and kinetic mechanism of the Sir2 family of NAD+-dependent histone/protein deacetylases. Biochemistry. 2004;43(30):9877–9887. doi: 10.1021/bi049592e. [DOI] [PubMed] [Google Scholar]
- 38.Stein RL. Kinetics of Enzyme Action. New York: Wiley; 2011. [Google Scholar]
- 39.Blum CA, et al. SIRT1 modulation as a novel approach to the treatment of diseases of aging. J Med Chem. 2011;54(2):417–432. doi: 10.1021/jm100861p. [DOI] [PubMed] [Google Scholar]
- 40.Suzuki T, et al. Design, synthesis, enzyme inhibition, and tumor cell growth inhibition of 2-anilinobenzamide derivatives as SIRT1 inhibitors. Bioorg Med Chem. 2009;17(16):5900–5905. doi: 10.1016/j.bmc.2009.07.001. [DOI] [PubMed] [Google Scholar]
- 41.Mai A, et al. Study of 1,4-dihydropyridine structural scaffold: Discovery of novel sirtuin activators and inhibitors. J Med Chem. 2009;52(17):5496–5504. doi: 10.1021/jm9008289. [DOI] [PubMed] [Google Scholar]
- 42.Jin L, et al. Biochemical characterization, localization, and tissue distribution of the longer form of mouse SIRT3. Protein Sci. 2009;18(3):514–525. doi: 10.1002/pro.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Avalos JL, Boeke JD, Wolberger C. Structural basis for the mechanism and regulation of Sir2 enzymes. Mol Cell. 2004;13(5):639–648. doi: 10.1016/s1097-2765(04)00082-6. [DOI] [PubMed] [Google Scholar]
- 44.Szczepankiewicz BG, Koppetsch KJ, Perni RB. One-step, nonenzymatic synthesis of O-acetyl-ADP-ribose and analogues from NAD and carboxylates. J Org Chem. 2011;76(16):6465–6474. doi: 10.1021/jo2008466. [DOI] [PubMed] [Google Scholar]
- 45.Lakshminarasimhan M, Rauh D, Schutkowski M, Steegborn C. Sirt1 activation by resveratrol is substrate sequence-selective. Aging (Albany NY) 2013;5(3):151–154. doi: 10.18632/aging.100542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schlicker C, et al. Substrates and regulation mechanisms for the human mitochondrial sirtuins Sirt3 and Sirt5. J Mol Biol. 2008;382(3):790–801. doi: 10.1016/j.jmb.2008.07.048. [DOI] [PubMed] [Google Scholar]
- 47.Mueller U, et al. Facilities for macromolecular crystallography at the Helmholtz-Zentrum Berlin. J Synchrotron Radiat. 2012;19(Pt 3):442–449. doi: 10.1107/S0909049512006395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kabsch W. Xds. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 2):125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McCoy AJ, et al. Phaser crystallographic software. J Appl Cryst. 2007;40(Pt 4):658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vagin AA, Isupov MN. Spherically averaged phased translation function and its application to the search for molecules and fragments in electron-density maps. Acta Crystallogr D Biol Crystallogr. 2001;57(Pt 10):1451–1456. doi: 10.1107/s0907444901012409. [DOI] [PubMed] [Google Scholar]
- 51.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53(Pt 3):240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 52.Emsley P, Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 12 Pt 1):2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 53.Brünger AT. Free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature. 1992;355(6359):472–475. doi: 10.1038/355472a0. [DOI] [PubMed] [Google Scholar]
- 54.Schüttelkopf AW, van Aalten DM. PRODRG: A tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 8):1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- 55.Chen VB, et al. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 1):12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wienken CJ, Baaske P, Rothbauer U, Braun D, Duhr S. Protein-binding assays in biological liquids using microscale thermophoresis. Nat Commun. 2010;1:100. doi: 10.1038/ncomms1093. [DOI] [PubMed] [Google Scholar]
- 57.Zhou Y, et al. The bicyclic intermediate structure provides insights into the desuccinylation mechanism of human sirtuin 5 (SIRT5) J Biol Chem. 2012;287(34):28307–28314. doi: 10.1074/jbc.M112.384511. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.