

Summary
Introduction
Hyperglycemia‐induced oxidative stress has been implicated in diabetic vascular complications in which NADPH oxidase is a major source of reactive oxygen species (ROS) generation. Resveratrol is a naturally occurring polyphenol, which has vasoprotective effects in diabetic animal models and inhibits high glucose (HG)–induced oxidative stress in endothelial cells.
Aims
We aimed to examine whether HG‐induced NADPH oxidase activation and ROS production contribute to glucotoxicity to endothelial cells and the effect of resveratrol on glucotoxicity.
Results
Using a murine brain microvascular endothelial cell line bEnd3, we found that NADPH oxidase inhibitor (apocynin) and resveratrol both inhibited HG‐induced endothelial cell apoptosis. HG‐induced elevation of NADPH oxidase activity and production of ROS were inhibited by apocynin, suggesting that HG induces endothelial cell apoptosis through NADPH oxidase–mediated ROS production. Mechanistic studies revealed that HG upregulated NADPH oxidase subunit Nox1 but not Nox2, Nox4, and p22phox expression through NF‐κB activation, which resulted in elevation of NADPH oxidase activity and consequent ROS production. Resveratrol prevented HG‐induced endothelial cell apoptosis through inhibiting HG‐induced NF‐κB activation, NADPH oxidase activity elevation, and ROS production.
Conclusions
HG induces endothelial cell apoptosis through NF‐κB/NADPH oxidase/ROS pathway, which was inhibited by resveratrol. Our findings provide new potential therapeutic targets against brain vascular complications of diabetes.
Keywords: Apoptosis, Hyperglycemia, NADPH oxidase, Resveratrol, Vascular endothelial cells
Introduction
A substantial body of evidence implicates oxidative stress as an important pathogenic factor in diabetic vascular complications. The drivers of the oxidative stress include hyperglycemia, hyperinsulinemia, and the elevated free fatty acids and lipids that are usually associated with diabetes. Reactive oxygen species (ROS) production emanates from several different sources, including NADPH oxidase, mitochondrial electron transport system, xanthine oxidase, cytochrome p450, uncoupled nitric oxide synthase, and myeloperoxidase 1. NADPH oxidase has been implicated as the major source of ROS generation in the vasculature in response to high glucose (HG) 2, 3. Therefore, inhibition of ROS production through the inhibitors against NADPH oxidase is proposed as an alternative approach to conventional antioxidant therapies against diabetes and vascular complications 4.
Resveratrol (3, 4′, 5‐trihydroxystilbene) is a natural polyphenol found in grapes, red wine, berries, knotweed, peanuts, and other plants. Collected evidence suggests that resveratrol has cardioprotective, anticancer, antiinflammatory, and antioxidative properties 5. Resveratrol exerts significant vasoprotective effects in animal models of type 2 diabetes 6. In cultured coronary arterial endothelial cells, resveratrol attenuates HG–induced oxidative stress through induction of mitochondrial content 7, upregulation of manganese superoxide dismutase expression, and increase in cellular GSH content through activation of SIRT1 8, as well as activation of nuclear factor‐E2‐related factor‐2 (Nrf2), a transcription factor that regulates the expression of numerous ROS detoxifying and antioxidant genes 9. However, the effect of resveratrol on HG–induced NADPH oxidase activation and ROS production in vascular endothelial cells is not clear. In this study, we investigated the effect of resveratrol on NADPH oxidase–mediated oxidative stress and apoptosis of brain microvascular endothelial cells in response to HG and explored the underlying mechanisms.
Materials and Methods
Cell Culture and Treatment
Murine brain microvascular endothelial cell line bEnd3 was cultured in Dulbecco's Modified Eagle Medium (DMEM) containing 1000 mg/L (5.6 mM) glucose, 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin in a humidified atmosphere at 37°C with 5% CO2. High‐glucose treatment was performed by incubating cells in DMEM containing 4500 mg/L (25 mM) glucose for the indicated times. For ROS measurements and Western blot assays, cells were cultured overnight in DMEM containing 5.6 mM glucose and 0.5% bovine serum albumin before experiments.
Cell Viability Assay
Cell viability was measured using 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyl tetrazolium bromide (MTT; Sigma, St. Louis, MO, USA) according to the manufacturer's protocol. Briefly, endothelial cells were cultured in 96‐well plates with medium containing 5.6 mM glucose or 25 mM glucose with/without different concentrations of resveratrol or apocynin for 24 h. MTT (0.5 μg/μL per well) was added and incubated at 37°C for 4 h. The intracellular purple formazan product was dissolved with hydrogen chloride/isopropanol (0.04 mM). The absorbance at 570 nm (reference at 630 nm) was determined by a microplate reader Multiskan JX (Thermo LabSystems, Franklin, MA, USA). Each experiment was repeated three times in triplicate samples. The cell viability was expressed as a percentage of the cells cultured in medium containing 5.6 mM glucose.
Apoptosis Determination
The morphological change in the nuclei in apoptotic cells was examined under fluorescence microscope as described previously with modification 10. Briefly, cells cultured on glass slides were washed with PBS and fixed with 4% (wt/vol) of neutral buffered formalin for 30 min, then washed with PBS, and stained with 0.6 mg/mL Hoechst 33258 (Sigma). The images were taken at 400× magnification under a fluorescence microscope (Olympus BX61, Tokyo, Japan). The percentage of apoptotic bodies (fragmented and condensed nuclei) were examined with 500 nuclei from control and experimental cells, respectively.
RNA Extraction and RT‐PCR
Total RNA was extracted from bEnd3 cells using the TRIZOL reagent (Invitrogen, Carlsbad, CA, USA) and depleted contaminating DNA with RNase‐free Dnase. cDNA was synthesized from 2 μg RNA with M‐MuLV reverse transcriptase and random hexamer according to the manufacturer's instructions (Fermentas, Burlington, ON, Canada). The cDNA fragment was amplified by PCR using the following specific primers: Nox1: 5′‐ATCCTGATTCCTGTGTGTCG‐3′ (sense), 5′‐GGCTTCTTCTGTAGCGTTCG‐3′ (antisense); Nox2: 5′‐CTCAGGGGTTCCAGTGCGTG‐3′ (sense), 5′‐CCATTTCCAAGTCATAGGAGG‐3′(antisense); p22phox: 5′‐GGGGAAAGAGGAAAAAGGG‐3′ (sense), 5′‐CAACAGGAAGTGGAGGGCA ‐3′ (antisense); β‐actin: 5′‐TGTGATGGTGGGAATGGGTCAG‐3′ (sense), 5′‐TTTGATGTCACGCACGATTTCC‐3′ (antisense). PCR products were visualized by ethidium bromide staining in 1.5% agarose gel and quantified using the Dolphin‐Doc image analyzer (WEALTEC Corp., Sparks, NV, USA). Amplification of the target cDNA was normalized to β‐actin expression.
Western Blot
The cells were lysed with cold lysis buffer and, the proteins were electrophoresed on 10% SDS‐PAGE gel and transferred onto polyvinylidene difluoride membrane 11. The membranes were blocked with 5% nonfat milk and then were incubated with primary antibodies overnight at 4°C. After incubation with a horseradish peroxidase–conjugated secondary antibody, the protein bands were detected with a Supersignal West Pico chemiluminescent substrate (Pierce, Rockford, IL, USA) and X‐Omat BT film (Eastman Kodak Co., Rochester, NY, USA). Polyclonal antibody against Nox1 was from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibody against phosphorylated IκBα was from Cell Signaling Technology (New England Biolabs, Beverly, MA, USA). Monoclonal antibody against β‐actin was bought from Abcam (Abcam, Cambridge, UK).
NADPH Oxidase Assay
NADPH oxidase activity was measured by the lucigenin chemiluminescence method 12. The cells were washed three times with ice‐cold PBS and scraped from the plate followed by centrifugation at 1000 × g at 4°C for 10 min. The cell pellets were resuspended in lysis buffer containing 20 mM KH2PO4, pH 7.0, 1 mM ethylene glycol tetraacetic acid (EGTA), 1 mM phenylmethylsulfonyl fluoride, 10 μg/mL aprotinin, and 0.5 μg/mL leupeptin. Cell suspensions were homogenized with 100 strokes in a Dounce homogenizer on ice. Hundred microliters of homogenate was added into 900 μL of phosphate buffer (50 mM, pH 7.0), containing 1 mM EGTA, 150 mM sucrose, 5 μM lucigenin (TCI, Tokyo, Japan), and 100 μM NADPH. Photon emission was measured every 15 second for 5 min in a luminometer (Berthold, Bad Wildbad, Germany). A buffer blank was subtracted from each reading before calculation of the data. NADPH oxidase activity was defined as relative chemiluminescence (light) units per second per milligram of protein.
Measurement of Intracellular ROS Production
The membrane permeable indicator 2′,7′‐dichlorodihydrofluorescein diacetate (H2DCF‐DA) (Invitrogen) was used to detect intracellular ROS production by bEnd3 cells. The cells were cultured in medium containing 5.6 mM or 25 mM glucose with or without different concentrations of apocynin or resveratrol for the indicated length of time, then were loaded with 10 μM H2DCF‐DA in serum‐free DMEM containing 5.6 mM or 25 mM glucose at 37°C for 30 min, and washed twice with PBS. Intracellular ROS production was detected by the FlexStation II384 fluorometric imaging plate reader (Molecular Devices, Sunnyvale, CA, USA) at an excitation wavelength of 488 nm and an emission wavelength of 525 nm.
Cell Transfection
bEnd3 cells were transfected with IκBα‐2N, a dominant‐negative IκBα expressing plasmid, or control vector flag‐zeo 13 (a kind gift from Dr. R. Lin (McGill University, Montreal, QC, Canada) using SuperFect Transfection Reagent (Qiagen, Valencia, CA, USA). Thirty‐six hours after transfection, the cells were stimulated with 25 mM glucose for 36 h, and Nox1 protein expression was detected by Western blot.
Inhibition of Nox1 Expression by RNA Interference
siRNA against mouse Nox1 and the control siRNA were synthesized by GenePharma (Shanghai, China). The sequences of Nox1 siRNA are 5′‐CCUUACUGGAGUGAUUGCCACUGUA‐3′ (sense) and 5′‐UACAGUGGCAAUCACUCCAGUAAGG‐3′ (antisense) 14. The sequences for control siRNA are 5′‐UUCUCCGAACGUGUCACGUTT‐3′ (sense) and 5′‐ACGUGACACGUUCGGAGAATT‐3′ (antisense). bEnd3 cells were transfected with siRNA at final concentration of 100 nM using SuperFect Transfection Reagent (Qiagen, Hilden, Germany). After transfection for 36 h, the cells were cultured in medium containing 5.6 mM or 25 mM glucose for additional 24 h and then Nox1 protein expression and NADPH oxidase activity were examined.
Statistical Analysis
Data are presented as means ± SD. Statistical differences between groups were analyzed by unpaired Student's t‐test.
Results
Resveratrol Inhibits High Glucose–Induced Endothelial Cell Apoptosis
We cultured bEnd3 cells in medium containing 5.6 mM or 25 mM glucose and found that 25 mM glucose significantly reduced endothelial cell viability detected by MTT assay (Figure 1A) and increased cell apoptosis measured by nuclear staining with Hoechst 33258 (Figure 1B). These results are consistent with previous reports 15, 16. Apocynin, an NADPH oxidase inhibitor, and resveratrol both protected bEnd3 cells from the toxicity of 25 mM glucose, while these two compounds had no effect on viability of bEnd3 cells cultured in medium containing 5.6 mM glucose (Figure 1A,B). These results indicate that NADPH oxidase activation–driven oxidative stress mediates HG–induced endothelial apoptosis. As resveratrol has been reported to inhibit the basal expression of NADPH oxidase subunit Nox4 in human umbilical vein endothelial cells 17, the protective effect of resveratrol from HG–induced apoptotic cell death may be mediated thorough inhibition of NADPH oxidase activation by HG.
Figure 1.
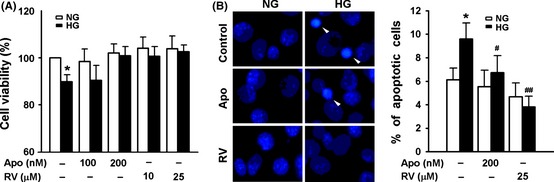
Resveratrol inhibits high glucose (HG)–induced vascular endothelial apoptosis. bEnd3 cells were cultured in medium containing normal glucose (NG, 5.6 mM) or HG (HG, 25 mM) in the presence or absence of different concentrations of apocynin (Apo) or resveratrol (RV) for 24 h, cell viability were examined with MTT assay (A), and apoptotic cells were observed under fluorescence microscope after staining with Hoechst 33258 (B). Results represent the mean ± SD of three independent experiments. *P < 0.05 compared with cells cultured in medium containing NG alone; # P < 0.05, ## P < 0.01 compared with cells cultured in medium containing HG alone. The representative images of Hoechst 33258 staining are shown in the left part of B.
Resveratrol Inhibits High Glucose–Induced NADPH Oxidase Activation and ROS Production in Endothelial Cells
We then examined the effect of resveratrol on NADPH oxidase activation and ROS production by HG. High‐glucose (25 mM) treatment resulted in significant increase in NADPH oxidase activity in bEnd3 cells (Figure 2A), which could be markedly inhibited by 25 μM resveratrol or 200 nM apocynin (Figure 2B). Further studies showed that HG triggered ROS production in a time‐dependent manner (Figure 3A), which was also reversed by apocynin (Figure 3B). Similarly, resveratrol dose dependently inhibited ROS production induced by HG (Figure 3C). These results indicate that resveratrol inhibits HG–induced ROS production through inhibition of NADPH oxidase activation by HG.
Figure 2.
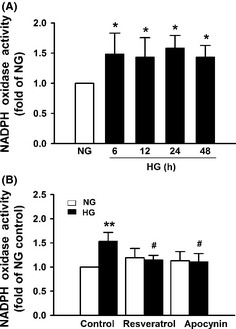
Resveratrol inhibits high glucose (HG)–induced NADPH oxidase activation in vascular endothelial cells. bEnd3 cells were cultured in medium containing 5.6 mM glucose (NG) or 25 mM glucose (HG) for different times (A) or for 12 h in the presence or absence of 200 nM apocynin or 25 μM resveratrol (B), and NADPH oxidase activity was measured by lucigenin chemiluminescence method. Results represent the mean ± SD of three independent experiments.*P < 0.05 compared with cells cultured in NG alone. # P < 0.05 compared with cells cultured in HG alone.
Figure 3.
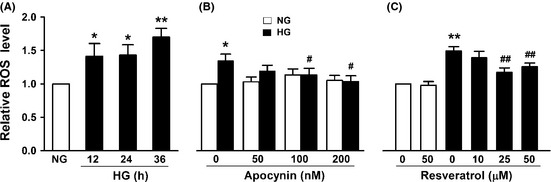
Resveratrol inhibits high glucose (HG)–induced reactive oxygen species (ROS) productions. bEnd3 cells were cultured in medium containing 5.6 mM glucose (NG) or 25 mM glucose (HG) for different times (A) or for 12 h in the presence or absence of different concentrations of apocynin (B) or resveratrol (C), and ROS production was examined with H2 DCF‐DA. Results represent the mean ± SD of three independent experiments. *P < 0.05, **P < 0.01 compared with cells exposed to NG alone. # P < 0.05, ## P < 0.01 compared with cells exposed to HG alone.
High Glucose Enhances NADPH Oxidase Activity in Endothelial Cells Through Upregulation of Nox1 Expression
We examined the effect of glucose on the expression of NADPH oxidase subunits Nox1, Nox2, NOx4, and p22phox and found that HG (25 mM) induced Nox1 expression at mRNA and protein levels in a time‐dependent manner but had no effect on Nox2, Nox4, and p22phox mRNA levels in bEnd3 cells (Figure 4A,B). Transfection of bEnd3 cells with Nox1 siRNA significantly inhibited HG–induced Nox1 protein expression (Figure 4C) and abolished HG–induced NADPH oxidase activation (Figure 4D). These results demonstrate that the augmentation of NADPH oxidase activity by HG is mediated by upregulation of Nox1.
Figure 4.
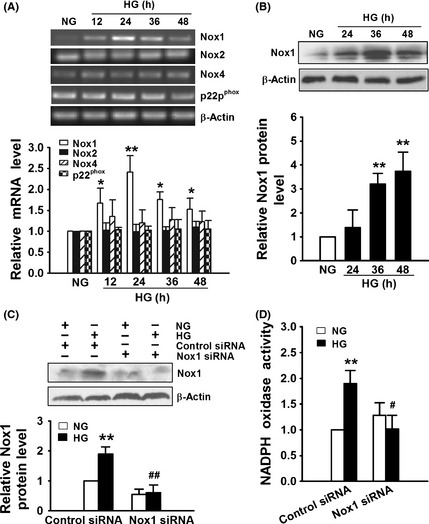
High glucose (HG) enhances NADPH oxidase activity in endothelial cells through upregulation of Nox1 expression. (A–B) bEnd3 cells were cultured in medium containing 5.6 mM glucose (NG) or 25 mM glucose (HG) for different periods of time, the mRNA levels of Nox1, Nox2, Nox4, and p22phox were examined by RT‐PCR (A), and protein levels of Nox1 were examined by Western blot (B). (C–D) bEnd3 cells transfected with control siRNA or Nox1 siRNA for 36 h were incubated with 5.6 mM glucose (NG) or 25 mM glucose (HG) for additional 36 h and then examined for Nox1 expression by Western blot (C) and NADPH oxidase activity (D). All results represent the mean ± SD of three independent experiments. The representative images of RT‐PCR and Western blot are shown in the upper part of A, B, and C, respectively. A–B: *P < 0.05, **P < 0.01 compared with cells cultured with NG. C–D: **P < 0.01 compared with control siRNA transfected cells cultured with NG. # P < 0.05, ## P < 0.01 compared with control siRNA transfected cells cultured with HG.
Resveratrol Attenuates Nox1 Upregulation in Endothelial Cells by High Glucose through Inhibition of NF‐κB Activation
We further investigated the effect of resveratrol on Nox1 upregulation by HG and explored the underlying mechanisms. Pretreatment of bEnd3 cells with 25 μM resveratrol significantly inhibited Nox1 mRNA and protein elevation by HG (25 mM) (Figure 5A,B). Sulfasalazine, an NF‐κB inhibitor, suppressed the increase in Nox1 protein by HG (Figure 5B). Transfection of bEnd3 cells with dominant‐negative IκBα plasmids also inhibited HG–induced Nox1 expression (Figure 5C). These results demonstrate that HG induces Nox1 expression through NF‐κB. We further found that sulfasalazine could inhibit HG–induced ROS production (Figure 5D) and reverse the inhibition of cell viability by HG (Figure 5E). These results combined with the previous results indicate that HG induces vascular endothelial cell death through NF‐κB‐mediated NADPH oxidase subunit Nox1 expression, NADPH oxidase activation, and subsequent ROS production.
Figure 5.
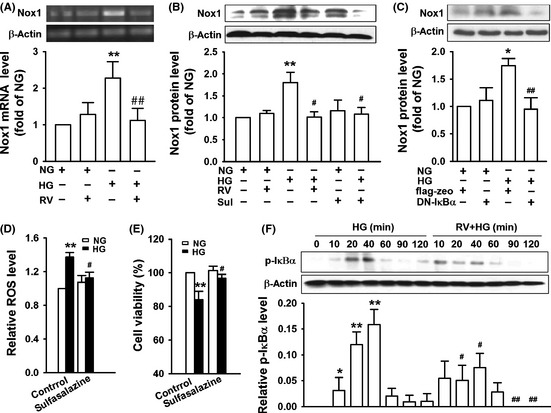
Resveratrol attenuates Nox1 upregulation in endothelial cells by high glucose (HG) through inhibition of NF‐κB activation. (A) bEnd3 cells pretreated with or without 25 μM resveratrol (RV) for 2 h were cultured in medium containing 5.6 mM glucose (NG) or 25 mM glucose (HG) for 36 h, and then, Nox1 mRNA level was examined by RT‐PCR. (B) bEnd3 cells pretreated with 25 μM resveratrol (RV) or 50 μM sulfasalazine (Sul) for 1 h were incubated with medium containing 5.6 mM glucose (NG) or 25 mM glucose (HG) for 36 h and then examined for Nox1 expression by Western blot. (C) bEnd3 cells transfected with dominant‐negative IκB (DN‐IκB) or control vector flag‐zeo for 36 h were treated with 5.6 mM glucose (NG) or 25 mM glucose (HG) for another 36 h and then examined for Nox1 expression by Western blot. (D–E) bEnd3 cells were exposed to 5.6 mM glucose (NG) or 25 mM glucose (HG) in the presence or absence of 50 μM sulfasalazine, reactive oxygen species production was examined with H2DCF‐DA after 12 h (D), and cell viability was examined with MTT assay after 24 h (E). (F) bEnd3 cells pretreated with or without 25 μM resveratrol for 1 h were incubated with medium containing 5.6 mM glucose (NG) or 25 mM glucose (HG) for the indicated times and then examined for IκBα phosphorylation by Western blot. All results represent the mean ± SD of three independent experiments. The representative images of RT‐PCR and Western blot are shown in the upper part of A–C and F. A, B, D, E: **P < 0.01 compared with cells cultured with NG alone; # P < 0.05, ## P < 0.01 compared with cells cultured with HG alone. C: *P < 0.05 compared with control plasmid flag‐zeo‐transfected cells cultured with NG; ## P < 0.01 compared with flag‐zeo‐transfected cells cultured with HG. F: *P < 0.05, **P < 0.01 compared with cells cultured with NG. # P < 0.05, ## P < 0.01 compared with cells treated with HG alone for corresponding time.
To elucidate whether resveratrol directly inhibit NF‐κB activation by HG, we performed Western blot to examine IκBα phosphorylation. As shown in Figure 5F, HG induced IκBα phosphorylation in a time‐dependent manner, and resveratrol significantly inhibited HG–induced IκBα phosphorylation. These results together with our previous results demonstrate that resveratrol inhibits HG–induced Nox1 elevation in endothelial cells through suppression of NF‐κB activation, and resveratrol protects endothelial cells from glucotoxicity through inhibiting the activation of NF‐κB/Nox1/ROS pathway by HG.
Discussion
In this study, we demonstrated that resveratrol attenuated hyperglycemia‐induced endothelial apoptosis through inhibition of oxidative stress. The protective effect of resveratrol is mediated by inhibiting the activation of NF‐κB/Nox1/NADPH oxidase/ROS pathway by HG.
Oxidative stress mediated by hyperglycemia‐induced generation of ROS contributes significantly to the development and progression of diabetes and related vascular complications 18. NAD(P)H oxidase has been implicated as the major source of ROS generation in the vasculature in response to HG 19. Vascular NADPH oxidase is a multicomponent enzyme composed of membrane‐bound subunits (Nox protein and p22phox) and cytosolic subunits (p47phox, p67phox, and Rac). Nox protein acts as the catalytic subunit of NADPH oxidase. NADPH oxidases can be divided into different isoforms according to Nox proteins. Nox1, Nox2, and Nox4 are expressed significantly in the vascular system 20. NADPH oxidase subunits have been reported to be elevated in different vascular beds of rodent diabetic models, including Nox2 in retinal vessels; Nox2, Nox4, p22phox, p47phox, p67phox, and Rac1 in kidney vessels; Nox1, Nox2, Nox4, p22phox, and Rac1 in aorta vessels 19. Expression of p22phox is increased in mouse pancreatic islet endothelial cell line and rat coronary microvascular endothelial cells exposed to HG 21, 22. High glucose induces p22phox, p47phox, p67phox, and Rac‐1 expression in human umbilical endothelial cells and upregulates Nox4 level in human aortic endothelial cells 16, 23, 24. As mouse cerebral vascular system expresses Nox1, Nox2, and Nox4, and different NADPH oxidase isoforms has a common p22phox subunit but different cytosolic subunits 20, we examined the effect of HG on the expression of Nox1, Nox2, Nox4, and p22phox and activation of NADPH oxidase. We found that HG induced ROS production in mouse brain microvascular endothelial cells through upregulation of Nox1 expression and enhanced NADPH oxidase activity. The difference in NADPH oxidase subunits upregulation by HG may be due to the differences in species and cell origin.
Accumulating evidences support that NF‐kB activation is involved in the pathogenesis of diabetes and diabetic vascular complications. In patients with type 2 diabetes, NF‐κB activation is associated with increased transcription of the p65 subunit of NF‐κB 25. The activation of NF‐κB in human kidney biopsies has been shown to correlate with severity of proteinuria and degree of glycemic control in diabetic nephropathy 26. NF‐κB was activated in endothelial cells, retina, heart, and kidneys of diabetic rats 27, and administration of NF‐κB inhibitor protects retinal capillary cells from apoptotic death 28, 29. In vitro studies showed that through activation of NF‐κB in endothelial cell, HG induced the expression of adhesion molecules and chemokines and promoted apoptotic cell death 30, 31, 32. Our present studies demonstrated that through activation of NF‐κB in brain vascular endothelial cells, HG induced NADPH subunit Nox1 expression, which resulted in NADPH activation, ROS production, and apoptotic cell death. High glucose has been reported to activate NF‐κB in human glomerular endothelial cells through IκBα phosphorylation and p65 nuclear translation 33. Our results revealed that the activation of NF‐κB by HG in murine endothelial cells was also mediated by phosphorylation of IκBα.
Resveratrol has been reported to improve vascular responses in streptozotocin ‐induced diabetic rats 34. In a mouse model of diabetes, resveratrol restored endothelial function and vascular responses by inhibiting TNF‐α‐induced activation of NAD(P)H oxidase and preserving endothelial nitric oxide synthase phosphorylation 6. In vitro studies showed that resveratrol attenuated HG–induced oxidative stress in endothelial cells through multiple pathways as described in the introduction section 7, 8, 9. Our new finding is that resveratrol protects endothelial cells from HG–induced apoptosis through inhibition of NADPH oxidase–driven oxidative pathway.
Resveratrol has been reported to inhibit NF‐κB activation by various stimuli through inhibition of IκB phosphorylation and degradation and/or p65 phosphorylation and nuclear localization 5, 35. Recently, Kumar et al. 36 reported that resveratrol protects diabetic neuropathy through inhibition of the expression of p65, IκBα, and inflammatory mediators. Our study revealed that through inhibition of IκB phosphorylation, resveratrol protects brain vascular endothelial cells from HG–induced apoptosis through inhibiting Nox1 upregulation and subsequent NADPH oxidase activation and ROS production.
In summary, using a murine brain microvascular endothelial cell line, we demonstrated that resveratrol protected endothelial cells from HG–induced apoptosis through inhibition of NF‐κB/Nox1/NADPH oxidase/ROS pathway. Further studies with mouse primary brain microvascular endothelial cells and related animal models will be helpful to confirm our in vitro results and to identify signaling molecule(s) in this pathway as target to prevent brain vascular complications under hyperglycemic conditions.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgments
We thank Dr. Rongtuan Lin (McGill University, Montreal, Canada) for providing plasmid IκBα‐DN and control vector flag‐zeo. This work has been supported by research grants from National Basic Research Program of China (973 Program) (2011CB504002) and the Knowledge Innovation Program of the Chinese Academy of Sciences (KSCX2‐EW‐R‐10).
References
- 1. Cave AC, Brewer AC, Narayanapanicker A, et al. NADPH oxidases in cardiovascular health and disease. Antioxid Redox Signal 2006;8:691–728. [DOI] [PubMed] [Google Scholar]
- 2. Christ M, Bauersachs J, Liebetrau C, Heck M, Günther A, Wehling M. Glucose increases endothelial‐dependent superoxide formation in coronary arteries by NAD(P)H oxidase activation: Attenuation by the 3‐hydroxy‐3‐methylglutaryl coenzyme A reductase inhibitor atorvastatin. Diabetes 2002;51:2648–2652. [DOI] [PubMed] [Google Scholar]
- 3. Inoguchi T, Li P, Umeda F, et al. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C–dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes 2000;49:1939–1945. [DOI] [PubMed] [Google Scholar]
- 4. Pérez‐Matute P, Zulet MA, Martínez JA. Reactive species and diabetes: Counteracting oxidative stress to improve health. Curr Opin Pharmacol 2009;9:771–779. [DOI] [PubMed] [Google Scholar]
- 5. Cucciolla V, Borriello A, Oliva A, Galletti P, Zappia V, Della Ragione F. Resveratrol: From basic science to the clinic. Cell Cycle 2007;6:2495–2510. [DOI] [PubMed] [Google Scholar]
- 6. Zhang H, Zhang J, Ungvari Z, Zhang C. Resveratrol improves endothelial function: Role of TNF{alpha} and vascular oxidative stress. Arterioscler Thromb Vasc Biol 2009;29:1164–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Csiszar A, Labinskyy N, Pinto JT, et al. Resveratrol induces mitochondrial biogenesis in endothelial cells. Am J Physiol Heart Circ Physiol 2009;297:H13–H20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ungvari Z, Labinskyy N, Mukhopadhyay P, et al. Resveratrol attenuates mitochondrial oxidative stress in coronary arterial endothelial cells. Am J Physiol Heart Circ Physiol 2009;297:H1876–H1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ungvari Z, Bagi Z, Feher A, et al. Resveratrol confers endothelial protection via activation of the antioxidant transcription factor Nrf2. Am J Physiol Heart Circ Physiol 2010;299:H18–H24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Calderoni AM, Biaggio V, Acosta M, Oliveros L, Mohamed F, Giménez MS. Cadmium exposure modifies lactotrophs activity associated to genomic and morphological changes in rat pituitary anterior lobe. Biometals 2010;23:135–143. [DOI] [PubMed] [Google Scholar]
- 11. Wang O, Cai K, Pang S, et al. Mechanisms of glucose‐induced expression of pancreatic‐derived factor in pancreatic beta‐cells. Endocrinology 2008;149:672–680. [DOI] [PubMed] [Google Scholar]
- 12. Meng D, Lv DD, Fang J. Insulin‐like growth factor‐I induces reactive oxygen species production and cell migration through Nox4 and Rac1 in vascular smooth muscle cells. Cardiovasc Res 2008;80:299–308. [DOI] [PubMed] [Google Scholar]
- 13. Zhao T, Yang L, Sun Q, et al. The NEMO adaptor bridges the nuclear factor‐kappaB and interferon regulatory factor signaling pathways. Nat Immunol 2007;8:592–600. [DOI] [PubMed] [Google Scholar]
- 14. Sasaki H, Yamamoto H, Tominaga K, et al. Receptor activator of nuclear factor‐kappaB ligand‐induced mouse osteoclast differentiation is associated with switching between NADPH oxidase homologues. Free Radic Biol Med 2009;47:189–199. [DOI] [PubMed] [Google Scholar]
- 15. Varma S, Lal BK, Zheng R, et al. Hyperglycemia alters PI3k and Akt signaling and leads to endothelial cell proliferative dysfunction. Am J Physiol Heart Circ Physiol 2005;289:H1744–H1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Quagliaro L, Piconi L, Assaloni R, Martinelli L, Motz E, Ceriello A. Intermittent high glucose enhances apoptosis related to oxidative stress in human umbilical vein endothelial cells: The role of protein kinase C and NAD(P)H‐oxidase activation. Diabetes 2003;52:2795–2804. [DOI] [PubMed] [Google Scholar]
- 17. Spanier G, Xu H, Xia N, et al. Resveratrol reduces endothelial oxidative stress by modulating the gene expression of superoxide dismutase 1 (SOD1), glutathione peroxidase 1 (GPx1) and NADPH oxidase subunit (Nox4). J Physiol Pharmacol 2009;60(Suppl 4):111–116. [PubMed] [Google Scholar]
- 18. Brownlee M. The pathobiology of diabetic complications: A unifying mechanism. Diabetes 2005;54:1615–1625. [DOI] [PubMed] [Google Scholar]
- 19. Gao L, Mann GE. Vascular NAD(P)H oxidase activation in diabetes: A double‐edged sword in redox signalling. Cardiovasc Res 2009;82:9–20. [DOI] [PubMed] [Google Scholar]
- 20. Ago T, Kuroda J, Kamouchi M, Sadoshima J, Kitazono T. Pathophysiological roles of NADPH oxidase/nox family proteins in the vascular system. Review and perspective. Circ J 2011;75:1791–1800. [DOI] [PubMed] [Google Scholar]
- 21. Ding H, Aljofan M, Triggle CR. Oxidative stress and increased eNOS and NADPH oxidase expression in mouse microvessel endothelial cells. J Cell Physiol 2007;212:682–689. [DOI] [PubMed] [Google Scholar]
- 22. Weidig P, McMaster D, Bayraktutan U. High glucose mediates pro‐oxidant and antioxidant enzyme activities in coronary endothelial cells. Diabetes Obes Metab 2004;6:432–441. [DOI] [PubMed] [Google Scholar]
- 23. Sheu ML, Chiang CK, Tsai KS, et al. Inhibition of NADPH oxidase‐related oxidative stress‐triggered signaling by honokiol suppresses high glucose‐induced human endothelial cell apoptosis. Free Radic Biol Med 2008;44:2043–2050. [DOI] [PubMed] [Google Scholar]
- 24. Williams CR, Lu X, Sutliff RL, Hart CM. Rosiglitazone attenuates NF‐κB‐mediated Nox4 upregulation in hyperglycemia‐activated endothelial cells. Am J Physiol Cell Physiol 2012;303:C213–C223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bierhaus A, Schiekofer S, Schwaninger M, et al. Diabetes‐associated sustained activation of the transcription factor nuclear factor‐kappaB. Diabetes 2001;50:2792–2808. [DOI] [PubMed] [Google Scholar]
- 26. Sanchez AP, Sharma K. Transcription factors in the pathogenesis of diabetic nephropathy. Expert Rev Mol Med 2009;11:e13. [DOI] [PubMed] [Google Scholar]
- 27. Chen S, Khan ZA, Cukiernik M, Chakrabarti S. Differential activation of NF‐kappa B and AP‐1 in increased fibronectin synthesis in target organs of diabetic complications. Am J Physiol Endocrinol Metab 2003;284:E1089–E1097. [DOI] [PubMed] [Google Scholar]
- 28. Kowluru RA, Koppolu P, Chakrabarti S, Chen S. Diabetes‐induced activation of nuclear transcriptional factor in the retina, and its inhibition by antioxidants. Free Radic Res 2003;37:1169–1180. [DOI] [PubMed] [Google Scholar]
- 29. Zheng L, Howell SJ, Hatala DA, Huang K, Kern TS. Salicylate‐based anti‐inflammatory drugs inhibit the early lesion of diabetic retinopathy. Diabetes 2007;56:337–345. [DOI] [PubMed] [Google Scholar]
- 30. Piga R, Naito Y, Kokura S, Handa O, Yoshikawa T. Short‐term high glucose exposure induces monocyte‐endothelial cells adhesion and transmigration by increasing VCAM‐1 and MCP‐1 expression in human aortic endothelial cells. Atherosclerosis 2007;193:328–334. [DOI] [PubMed] [Google Scholar]
- 31. Ho FM, Lin WW, Chen BC, et al. High glucose‐induced apoptosis in human vascular endothelial cells is mediated through NF‐kappaB and c‐Jun NH2‐terminal kinase pathway and prevented by PI3K/Akt/eNOS pathway. Cell Signal 2006;18:391–399. [DOI] [PubMed] [Google Scholar]
- 32. Chen G, Shen X, Yao J, et al. Ablation of NF‐kappaB expression by small interference RNA prevents the dysfunction of human umbilical vein endothelial cells induced by high glucose. Endocrine 2009;35:63–74. [DOI] [PubMed] [Google Scholar]
- 33. Yang WS, Seo JW, Han NJ, et al. High glucose‐induced NF‐kappaB activation occurs via tyrosine phosphorylation of IkappaBalpha in human glomerular endothelial cells: Involvement of Syk tyrosine kinase. Am J Physiol Renal Physiol 2008;294:F1065–F1075. [DOI] [PubMed] [Google Scholar]
- 34. Silan C. The effects of chronic resveratrol treatment on vascular responsiveness of streptozotocin‐induced diabetic rats. Biol Pharm Bull 2008;31:897–902. [DOI] [PubMed] [Google Scholar]
- 35. Carluccio MA, Ancora MA, Massaro M, et al. Homocysteine induces VCAM‐1 gene expression through NF‐kappaB and NAD(P)H oxidase activation: Protective role of Mediterranean diet polyphenolic antioxidants. Am J Physiol Heart Circ Physiol 2007;293:H2344–H2354. [DOI] [PubMed] [Google Scholar]
- 36. Kumar A, Sharma SS. NF‐kappaB inhibitory action of resveratrol: A probable mechanism of neuroprotection in experimental diabetic neuropathy. Biochem Biophys Res Commun 2010;394:360–365. [DOI] [PubMed] [Google Scholar]