

Abstract
Cells respond to stress and starvation by adjusting their growth rate and enacting stress defense programs. In eukaryotes this involves inactivation of TORC1, which in turn triggers downregulation of ribosome and protein synthesis genes and upregulation of stress response genes. Here we report that the highly conserved inositol pyrophosphate second messengers (including 1-PP-IP5, 5-PP-IP4, and 5-PP-IP5) are also critical regulators of cell growth and the general stress response, acting in parallel to the TORC1 pathway to control the activity of the class I HDAC Rpd3L. In fact, yeast cells that cannot synthesize any of the PP-IPs mount little to no transcriptional response in osmotic, heat, or oxidative stress. Furthermore, PP-IP dependent regulation of Rpd3L occurs independently of the role individual PP-IPs (such as 5-PP-IP5) play in activating specialized stress/starvation response pathways. Thus, the PP-IP second messengers simultaneously activate and tune the global response to stress and starvation signals.
INTRODUCTION
To thrive when conditions are favorable and survive when they are stressful, cells must set their growth rate based on the level and combination of numerous intracellular and extracellular stimuli (Schmelzle and Hall, 2000). How this is accomplished remains unclear.
What is known is that in eukaryotes cell growth depends to a large degree on a single kinase called TOR (Laplante and Sabatini, 2012; Loewith and Hall, 2011). When conditions are favorable, TOR drives mass accumulation by promoting all aspects of protein and ribosome synthesis. Conversely, in stress conditions or when hormone or nutrient levels fall outside of an ideal range TOR activity is repressed. This triggers inhibition of protein synthesis and activation of numerous stress and starvation response pathways.
Studies in the budding yeast S. cerevisiae have begun to reveal the precise mechanisms underlying TOR dependent regulation of growth. In particular it is now clear that TOR is part of a multisubunit complex called TORC1 (Loewith et al., 2002), and that this complex signals through two distinct channels to regulate a global gene expression program known as the Environmental Stress Response or ESR (Airoldi et al., 2009; Brauer et al., 2008; Gasch et al., 2000; Loewith et al., 2002).
In one channel, active TORC1 promotes the expression of 650 genes involved in ribosome and protein synthesis by regulating the activity of the S6 kinase Sch9 and numerous transcription factors, including Sfp1, Fhl1, Maf1, Dot6, and Tod6 (Huber et al., 2011; Lempiainen et al., 2009; Lippman and Broach, 2009; Marion et al., 2004; Martin et al., 2004). When TORC1 is inactivated, dephosphorylation of the TORC1 and Sch9 dependent transcription factors triggers recruitment of the Class I histone deacetylase (HDAC) Rpd3L to ribosome and protein synthesis genes, leading to their repression (Alejandro-Osorio et al., 2009; Huber et al., 2011).
In the other channel, active TORC1 blocks the expression of 600 stress and starvation response genes by binding and sequestering a key regulator of the PP2A phosphatases, Tap42 (Alejandro-Osorio et al., 2009; Huber et al., 2011). In stress and starvation conditions, Tap42 is released from TORC1 and activates PP2A (Yan et al., 2012; Yan et al., 2006). This in turn triggers the dephosphorylation and activation of transcription factors that promote amino acid synthesis, nitrogen metabolism, the TCA cycle, and the general stress response, including: Gln3, Gat1, Rtg1/3, and Msn2/4 (Beck and Hall, 1999; Huber et al., 2009). Together, the PP2A dependent transcription factors function by converting the HDAC Rpd3L from a repressor to an activator at the stress/starvation response genes (Alejandro-Osorio et al., 2009).
What remains to be determined, in both S. cerevisiae and other organisms, is how stress and starvation signals are transmitted to TORC1 and which pathways (if any) cooperate with TORC1 to regulate growth and metabolism. Answering these questions is a prerequisite to building a realistic model of the cellular growth control circuitry, and ultimately to understanding how cells decide how fast to grow in different environments, how they keep growth and metabolism balanced, and how malfunction of the growth control system leads to diseases such as cancer and diabetes (Laplante and Sabatini, 2012; Loewith and Hall, 2011).
Here, to gain insight into the structure and function of the eukaryotic growth control network, we use DNA microarray analysis to examine the influence that 17 signaling proteins, known to be (de)phosphorylated during stress in S. cerevisiae (Fig. S1, (Soufi et al., 2009), have on the ESR. Surprisingly, our data reveal that only one of these factors, the inositol kinase Vip1, plays a significant role in regulating the ESR and thus cell growth.
Previous studies have shown that Vip1 is part of the inositol pyrophosphate synthesis pathway, which is conserved throughout eukaryotes (Fig. 1). In the first step of this pathway, Phosphatidylinositol 2-Phosphate (PIP2) is cleaved by Phospholipase C (Plc1) to release the lipid diacylglycerol and the soluble inositol head group IP3 (Shears, 2009). IP3 itself is a signaling molecule with a well-established role in calcium signaling (Berridge et al., 2000; Streb et al., 1983). More recently, however, it has been shown that IP3 can be converted to IP4 and IP5 in the nucleus by the inositol polyphosphate multikinase Arg82, and then to IP6 by the inositol polyphosphate kinase Ipk1 (Burton et al., 2009; Chakraborty et al., 2011; Shears, 2009; Tsui and York, 2009). IP5 and IP6 can also be pyrophosphorylated by Kcs1 to create a diphosphate at the 5-position (5-PP-IP4/5), while IP6 can be pyrophosphorylated by Vip1 to create a diphosphate at the 1-position (1-PP-IP5; (Fridy et al., 2007; Mulugu et al., 2007; Shears, 2009). The diphosphorylated inositols synthesized by Vip1 and Kcs1 are known as the inositol pyrophosphates or PP-IPs.
Fig. 1. The PP-IP synthesis pathway.

See text for details.
Identification of Vip1 as an important regulator of the ESR led us to investigate the role that inositol pyrophosphates play in regulating cell growth. To do this we measured the influence that each enzyme in the PP-IP synthesis pathway (Arg82, Ipk1, Vip1 and Kcs1) has on gene expression, in both log growth and stress conditions. Through this analysis we discovered that the three major PP-IPs cooperate to activate the ESR. In fact, a strain that is unable to synthesize any PP-IP (kcs1Δvip1Δ) mounts little to no transcriptional response in heat, osmotic, or oxidative stress.
To determine how the PP-IPs regulate the ESR and cell growth, we also measured signaling at each stage of the TORC1 pathway in kcs1Δvip1Δ and wild-type strains. These experiments revealed that the PP-IPs act in parallel with the canonical TORC1 signaling pathway to activate the HDAC Rpd3L. PP-IP activation of Rpd3L may be direct since we found that mutation of residues in an inositol binding site on the surface of Rpd3, identified when a crystal structure of human HDAC3-SMART complex was found to contain IP4, has the same general effect on cell signaling as blocking the production of the PP-IPs themselves.
Taken together, our data show that the inositol pyrophosphates are critical regulators of the ESR and cell growth, with an overall impact similar to that of TORC1 itself. Moreover, our discovery that the PP-IPs activate the HDAC Rpd3L reveals the core function of these conserved second messengers in cell signaling. These results have important implications for understanding cell growth control in eukaryotes as well as the regulation of HDAC activity in health and disease.
RESULTS
Vip1 Regulates the Environmental Stress Response
To identify proteins that regulate the ESR, and thus cell growth, we performed a directed screen. In the first step, we searched a recently published phosphoproteomics dataset (Soufi et al., 2009) to find proteins that are rapidly (<5 min) phosphorylated or dephosphorylated in osmotic stress (at p<0.05, Fig. S1). Through this analysis we generated a list of 24 signaling proteins likely to regulate the ESR, but that have not been studied in detail previously (see Supplement for details). We then created a series of strains, each missing one of the 17 nonessential genes on our list, and measured their response to osmotic stress using DNA microarrays. These data revealed that deletion of Vip1, an evolutionarily conserved inositol pyrophosphate synthase, inhibits the ESR (238 genes≥ 1.5-fold change, Fig. S2).
Inositol Pyrophosphates act redundantly to regulate the Environmental Stress Response
Our discovery that Vip1 regulates the ESR led us to study the role that inositol phosphates and pyrophosphates play in stress signaling.
First, we asked if Vip1 influences the ESR through 1-PP-IP5 by measuring gene expression in a strain carrying a catalytically dead Vip1 mutant (D487A; (Mulugu et al., 2007). We found that vip1D487A and vip1Δ strains have similar expression patterns, both with defects in their stress response (Fig. S3 and Table S1). Thus, 1-PP-IP5 and/or other PP-IPs synthesized by Vip1 are required for activation of the ESR.
Next, we asked if inositol phosphates besides 1-PP-IP5 influence the ESR. To do this we measured the impact that each enzyme in the PP-IP synthesis pathway (Fig. 1) has on gene expression, in both log growth and osmotic stress conditions. Remarkably, these data show that there are 1647 genes activated/repressed (≥2-fold) by Arg82, Ipk1, Kcs1 and/or Vip1. Most of these genes (1272/1647) are part of the ESR (Fig. 2a and Fig S4), in line with our results for the vip1Δ and vip1D487A strains. The remaining genes are primarily known targets of the PP-IPs, including the phosphate-signaling pathway, a target of 1-PP-IP5 via Pho80/85 (Lee et al., 2008; Lee et al., 2007b), and the glycolysis pathway, a target of 5-PP-IP4/5 via Gcr1 (Szijgyarto et al., 2011), as described in detail in the Supplement (Fig. S4).
Fig. 2. The role of the Inositol Phosphates and Pyrophosphates in Gene Regulation.
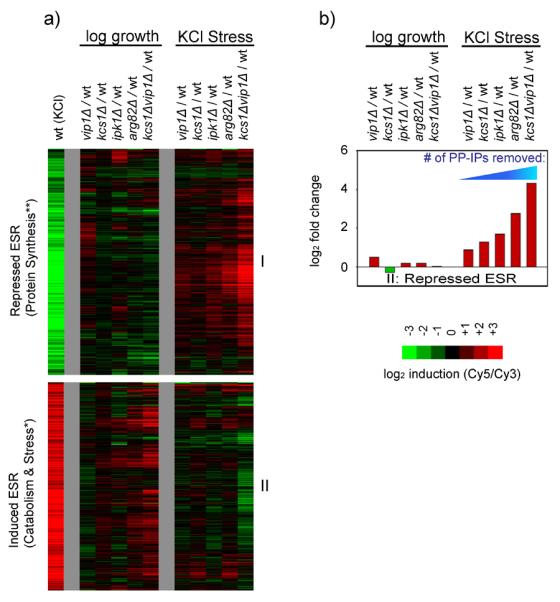
(a) DNA microarray data showing gene expression in log growth (YEPD, OD600= 0.6) or in response to osmotic stress (0.4 M KCl, 20 minutes) in strains missing one or more of the enzymes inositol polyphosphate/pyrophosphate (IP) synthesis pathway. Each experiment compares the mRNA levels in a mutant strain to those in the wild type strain, in identical conditions. In all of these experiments cDNA from mutant strains are labeled with Cy5 while the wild type strain is labeled with Cy3. Thus, genes that are upregulated in the mutant strains appear red while genes that are downregulated are green. The wild type stress response (osmotic stress sample labeled red, log growth labeled green) is shown at far left for comparison. The heat map shows data for all of the genes in the ESR based on the average of two to three replicate experiments per microarray (Table S1). Other genes regulated by the inositol phosphate pathway are described in the Supplement. Within the ESR, the correlation between the expression changes found in the kcs1Δvip1Δ strain and the arg82Δ strain are very strong, with a Pearson's r of 0.70 in YEPD, 0.77 for genes repressed in stress, and 0.63 for genes activated in stress. The correlation between the expression changes found in the kcs1Δvip1Δ and ipk1Δ strains are also strong for genes repressed in stress (r=0.69), but moderate in YEPD (r=0.45) and for genes activated in stress (r=0.49). Last, the expression changes found in the kcs1Δ and vip1Δ strains also correlate well with those found in the arg82Δ strain (r values ranging from 0.76 to 0.52, for the three gene groups listed above) except that there is little to no correlation between the influence of Vip1 and Arg82 in YEPD (r=0.12), suggesting that Vip1 is less important than Kcs1 for PP-IP synthesis in YEPD growth conditions. Overall, the strong correlation between the expression changes caused by deleting Kcs1 and Vip1, and removing their key substrates (IP5 and IP6) via deletion of Arg82 or Ipk1, indicates that it is the PP-IPs, and not other (unknown) molecules synthesized by Kcs1/Vip1, that regulate the ESR. (b) Bar graph showing the defect in Ribi gene repression for each mutant strain and condition in (a). Note that arg82Δ cells still produce some PP-IPs since Kcs1 phosphorylates and pyrophosphorylates IP3 to create PP-IP3, PP-IP4, and other PP-IP species, when IP5 and IP6 are not available as substrates (Seeds et al., 2005). Accordingly, the expected concentration of PP-IPs in kcs1Δvip1Δ < arg82Δ < kcs1Δ and ipk1Δ < vip1Δ (Fig. 1; Seeds et al., 2005).
The expression data for genes within the ESR were especially illuminating. First, the data show that deletion of the two inositol-pyrophosphate synthases in yeast, Kcs1 and Vip1, has a much bigger effect on the transcriptional response to stress than deletion of Vip1 alone (Fig. 2a). Specifically, the kcs1Δvip1Δ strain has a dramatic defect in: (1) the down-regulation protein synthesis genes in stress, (2) the repression of stress genes in log growth conditions, and (3) the activation of stress genes in stress conditions. Second, the data show that the enzymes upstream of Kcs1 and Vip1 in the PP-IP synthesis pathway (Arg82 and Ipk1) regulate nearly the same expression program as Kcs1 and Vip1 (Fig. 2a, see legend for Pearson's r values). However, the influence that each enzyme in the PP-IP synthesis pathway has on the level of gene expression is roughly proportional to the number of PP-IPs it helps to synthesize (Fig. 2b, S5). Taken together, these data indicate that the PP-IPs, including 1-PP-IP5 and 5-PP-IP4/5, act partially redundantly to regulate the ESR (see Supplement for further discussion).
Finally, we asked if the PP-IPs activate the ESR in conditions other than osmotic stress. We found that kcs1Δvip1Δ cells have similar, dramatic, defects in their response to osmotic, oxidative, and heat stress (Fig. 3). In fact, kcs1Δvip1Δ cells fail to mount any significant response to H2O2 stress (Fig. 3). Thus, Kcs1, Vip1, and the PP-IPs are among the most potent regulators of the Environmental Stress Response (ESR) identified to date, with an influence similar to that of TORC1 itself (Table S1).
Fig. 3. Inositol pyrophosphates (PP-IPs) Regulate the Environmental Stress Response.

DNA microarray data showing the gene expression programs activated in various stresses in wild type and kcs1Δvip1Δ strains. (a) Each column compares the mRNA levels in log growth conditions (YEPD, OD=0.6) to the mRNA levels in the same strain after 20 minutes of the indicated stress. In all of these experiments cDNA from cells collected prior to stress treatment were labeled with Cy3 (green) while cDNA from cells treated with stress stimuli were labeled with Cy5 (red). Thus, genes activated by 0.4M KCl, 42° C heat shock, or 0.4 mM H2O2 are red on the heat map while genes repressed in these stimuli are green. Only genes that are up or downregulated by >3-fold, in one or more experiment, are shown on the heat map. Clustering these data revealed five gene groups, each of which is labeled by its major gene ontology groups and the probability that a GO group was found by chance. Microarrays are the average of at least two replicates. (b) Each column shows the difference between the kcs1Δvip1Δ and wild-type response to the stress indicated to highlight the Kcs1 and Vip1 regulated genes.
PP-IPs Regulate the ESR by activating the HDAC Rpd3L
How then do the PP-IP second messengers regulate the ESR? To answer this question we first sought to determine whether the PP-IPs act upstream or downstream of TORC1. We reasoned that if the PP-IPs act upstream of TORC1 then we should be able to rescue the stress response in a strain missing the PP-IPs (kcs1Δvip1Δ) by inhibiting TORC1 using the potent inhibitor rapamycin. Surprisingly, this was not the case. In fact, rapamycin has almost no effect on signaling in kcs1Δvip1Δ cells (Fig. 4a), indicating that the PP-IPs act at or below the level of TORC1.
Fig. 4. Inositol Pyrophosphates Regulate Rpd3L.
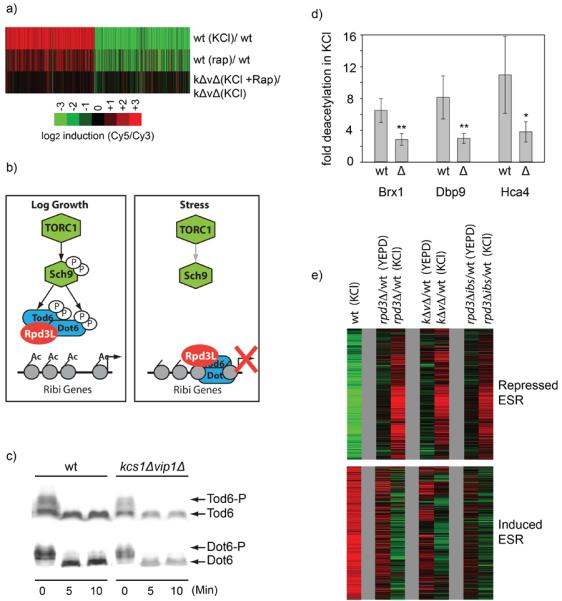
(a) Inositol pyrophosphates act at or below the level of TORC1. DNA microarray data showing genes up or downregulated >3-fold in response to 20 minutes of 0.4M KCl in wild type (top), or in kcs1Δvip1Δ strain (Cy3) compared to the same strain treated with 0.4M KCl and 300 nM rapamycin for 20 minutes (Cy5) (bottom). The gene expression changes induced by rapamycin in the wild-type strain are also shown for comparison (middle). If the PP-IPs acted upstream of TORC1 then treatment with rapmycin in stress would have rescued expression defect found in the kcs1Δvip1Δ strain, leading to dramatic changes in gene expression in rapamycin. Microarrays are the average of three replicates. (b) Known mechanism of stress induced repression of Ribosome Biogenesis (Ribi) genes. Inactivation of TORC1 leads to Tod6/Dot6 activation and recruitment of the histone deacetylase Rpd3L to Ribi promoters, as described in the text. (c) Stress signaling through the TORC1 pathway still occurs in the absence of the PP-IPs. The phosphorylation of Dot6-HA and Tod6-HA was monitored using band-shift analysis, both before and after treatment with 0.4M KCl. Here cells were treated with TCA, lysed and whole cell extracts run on SDS-PAGE. Dot6 and Tod6 were identified using a western blot with an anti-HA antibody (12-CA5). The band-shifts monitored here have previously been show to be due to phosphorylation by Sch9 (Huber et al., 2009). (d) PP-IPs are required for deacetylation at Ribi genes in 0.4M KCl stress. The acetylation level of three highly regulated Ribi genes was measured in the kcs1Δvip1Δ and wild-type strains, both before and after KCl stress, using an anti acetylated-H4 antibody and quantitative PCR. Plotted here is the ratio of the pre-stress to post-stress acetylation level. No significant changes in the pre-stress acetylation levels were found in the kcs1Δvip1Δ strain. However, stress triggers more deacetylation in the wild-type strain than it does in the kcs1Δvip1Δ strain; **(p<0.03) and *(p<0.1), t-test values for null hypothesis. (e) Rpd3, the PPIPs, and disrupting the inositol-phosphate binding site on Rpd3L have similar effects on the ESR. The DNA microarrays show the change of gene expression caused by deleting Rpd3, Kcs1 and Vip1, or mutating the inositol binding site in Rpd3 (rpd3Δibs), in both log growth and 0.4M KCl stress (after 20 min). In each case cDNA from the mutant strain is labeled with Cy5, while cDNA from the wild-type strain, grown in identical conditions, is labeled with Cy3.
To determine how the PP-IPs act downstream of TORC1, we next examined signaling through the Sch9 channel. Previous studies have shown that in log growth conditions TORC1 phosphorylates the S6 kinase Sch9, leading to its activation (Fig. 4b; (Urban et al., 2007)). Active Sch9, in turn, phosphorylates and inactivates the transcriptional repressors Dot6 and Tod6 (Lippman and Broach, 2009), both part of the HDAC complex Rpd3L (Huber et al., 2011; Shevchenko et al., 2008). When TORC1 is inactivated by stress, Sch9, Dot6, and Tod6 are all rapidly dephosphorylated and the protein synthesis genes are repressed by active Dot6/Tod6-Rpd3L. (Huber et al., 2009; Huber et al., 2011; Humphrey et al., 2004). Using bandshift analysis we monitored the stress dependent phosphorylation of Dot6 and Tod6 and found that the switch from active TORC1 in log growth conditions, to inactive TORC1 in stress conditions, occurs normally in the kcs1Δvip1Δ strain (Fig. 4c). Therefore TORC1 is inactivated by stress, leading to dephosphorylation of Dot6/Tod6, even in a strain missing all PP-IPs.
As inactivation of TORC1 and the subsequent dephosphorylation of transcription factors, including Dot6 and Tod6, ultimately leads to activation of the ESR via the HDAC Rdp3L, we next asked whether the PP-IPs regulate Rpd3L itself. To do this we measured the acetylation of three of the most PP-IP dependent protein synthesis genes (shown previously to be deacetylated by Rpd3L during the ESR, (Alejandro-Osorio et al., 2009; Huber et al., 2011), both before and after osmotic stress, using a chromatin immunoprecipitation (ChIP) assay. These experiments revealed a significant defect in stress dependent histone deacetylation in the kcs1Δvip1Δ strain at all three genes (Fig, 4d), indicating that Rpd3L does not function appropriately in stress without the PP-IPs.
To test whether the PP-IPs influence the entire Rpd3-mediated ESR, we next measured the ESR in a strain missing the catalytic subunit in the Rpd3L complex (rpd3Δ), and compared it with the data for kcs1Δvip1Δ strain. We found that the expression profiles for the rpd3Δ and kcs1Δvip1Δ strains are remarkably similar; both in pre-stress conditions, where Rpd3 acts to represses the expression of stress genes, and in stress conditions, where Rpd3 switches to an activator of stress genes and a repressor of ribosome and protein synthesis genes (Fig. 4e). We therefore conclude that the PP-IPs are required for Rpd3L activity in both log growth and stress conditions.
Do PP-IPs activate Rpd3L directly?
After discovering that the inositol pyrophosphates activate Rpd3L in vivo, we wanted to determine if the PP-IPs function by binding directly to the Rpd3L complex. While this work was in progress, Watson et. al. published a crystal structure of human HDAC3 (Rpd3 in yeast) bound to the co-repressor SMRT. The structure revealed that the interaction between HDAC3 and SMRT depends on an inositol 1,4,5,6-phosphate (IP4) molecule located at the interface between the two proteins (Watson et al., 2012). Watson et. al. further showed that IP4 is required for activation of HDAC3 by SMRT, in vitro. Although it is unclear if there is a corepressor that acts like SMRT in yeast (see supplement for discussion), these findings led us to ask if the PP-IPs activate Rpd3L by binding to the “IP4” site described in the HDAC3-SMRT crystal structure. This seemed reasonable given that: (1) sequence alignment shows that the residues mediating the HDAC3-IP4 interaction are conserved across the class I HDACs, including in Rpd3 (Watson et al., 2012), and (2) Examination of the HDAC3-SMRT structure (pdb 4A69) shows that approximately half of the IP4 ring is exposed to solvent, suggesting that the inositol binding site on Rpd3 could also accommodate 1-PP-IP5 or 5-PP-IP4/5.
To test whether the PP-IPs activate the Rpd3L complex by binding to the inositol phosphate binding site on Rpd3, we created a strain missing the side chains of three solvent exposed residues that form salt bridges with IP4 in the HDAC3 structure (Rpd3K41A,R280A,R316A, rpd3Δibs for short) and studied its response to stress. We found that this rpd3Δibs strain has a similar expression profile to those of the kcs1Δvip1Δ and rpd3Δ strains, in both log growth and stress conditions (Fig. 4e). Thus, activation of Rpd3L to wild-type levels requires both the PP-IPs and an intact inositol-phosphate binding site on Rpd3. It therefore seems likely that the PP-IPs activate Rpd3L, at least in part, by binding to same pocket that is occupied by IP4 in the IP4-HDAC3 complex. However, further work is needed to confirm that the PP-IPs bind and activate Rpd3L directly since the rpd3Δibs mutation could inhibit Rpd3L by disrupting a PP-IP independent function of Rpd3.
DISCUSSION
Over the last five years, the inositol pyrophosphates have emerged as important signaling molecules in eukaryotic cells. First, it was discovered that yeast synthesize 1-PP-IP5 in phosphate starvation conditions and that this form of IP7 binds to Pho80/85, triggering activation of phosphate-scavenging genes (Lee et al., 2007a). Later, 5-PP-IP5 was shown to play a role in human insulin signaling, where it blocks activation of AKT by the lipid PIP3 (Chakraborty et al., 2010). Finally, a recent study in yeast revealed that 5-PP-IP5 inhibits transcription of glycolysis genes by regulating the transcription factor Gcr1 (Szijgyarto et al., 2011). Here we show that these specific roles of 1-PP-IP5 and 5-PP-IP5 are just one aspect of inositol pyrophosphate function. Significantly, the PP-IPs also act together (partially redundantly) to regulate a Class I HDAC and thus the global gene expression program. In yeast this means that 1-PP-IP5 activates both the phosphate starvation pathway (15 genes) and the ESR (>1200 genes), while 5-PP-IP4/5 downregulates glycolysis (50 genes) while activating the ESR.
Beyond uncovering a core function of the inositol pyrophosphates, our study provides important insight into the mechanisms underlying regulation of the ESR and cell growth in yeast. A key conclusion from the work here is that the PP-IPs act in parallel with the known master regulator of growth in eukaryotes, TORC1, to control Rpd3L. Furthermore, the influence that the PP-IPs have on gene expression is similar in both scale and impact to that of TORC1 itself (Figs. 4a and 4e).
This raises the question, why would the cell use two parallel signaling systems to control growth and the ESR, with TORC1 targeting Rpd3L to the appropriate promoters and the PP-IPs regulating Rpd3L activity? We favor two nonexclusive possibilities. First, this AND gate may filter noise in the TORC1 and PP-IP synthesis pathways, preventing unintentional and transient reprogramming of one-fifth of the genome. Second, PP-IP signaling may tune or control the dynamics of the response to TORC1 inhibition. This latter point may be especially important, as total Rpd3L activity increases dramatically in stress conditions (Fig. 4e).
Distinguishing between these and other models of PP-IP and TORC1 cooperation will require a more detailed view of the way PP-IP synthesis is regulated. Currently, no upstream regulators of Kcs1 or Vip1 have been identified in yeast, and it is only possible to measure the bulk level of inositol phosphates and pyrophosphates in the cell (Shears, 2009). These bulk measurements are unlikely to provide a realistic view of PP-IP production as synthesis may occur at specific locations within the cell and the PP-IPs are known to turn over rapidly (Menniti et al., 1993). It is clear, however, that PP-IP levels increase in some stress and starvation conditions (Lee et al., 2007a; Nagata et al., 2005; Pesesse et al., 2004), in line with a model where PP-IP levels increase in stress to upregulate Rpd3L activity.
The results presented here also have important implications for cancer research. Studies in human cells have shown that IP6K2, a human homologue of Kcs1, is required for efficient induction of apoptosis in stress conditions, and is missing in some squamous cell carcinomas (Morrison et al., 2001; Morrison et al., 2009; Nagata et al., 2005). Cells missing the PP-IPs fail to activate apoptosis in stress because they erroneously upregulate cell cycle arrest genes when they should only activate pro-apoptotic genes (Koldobskiy et al., 2010). In other words, the cells arrest before they can apoptose. Our discovery that the PP-IPs are required for HDAC activation sheds light on why this happens since it is known that HDAC1 (another homologue of Rpd3L) must cooperate with p53 to downregulate the cell cycle arrest genes in stress (Lagger et al., 2003; Ocker and Schneider-Stock, 2007). This suggests that small molecules that mimic PP-IP4/5 and activate HDAC1 may help push cancer cells away from arrest and towards apoptosis in stresses such as chemotherapy.
EXPERIMENTAL PROCEDURES
Yeast were grown from OD600 = 0.10 to 0.60 in YEPD at 30 °C and then harvested for further analysis (log growth samples) or treated with stress (0.4M KCl, 42 °C final temperature or 0.4 mM H2O2) and harvested after 5/10 min to examine signaling/histone acetylation or after 20 min to examine mRNA levels. For microarrays, mRNA was extracted from the cells using hot phenol, purified using a poly A sepharose column, and converted to aa-UTP labeled cDNA using StrataScript reverse transcriptase. The cDNA was then labeled with Cy3 or Cy5, and transcript levels measured using Agilent G4813A DNA microarrays and an Axon 4000B scanner. For band-shift experiments TCA treated cells were lysed by bead-beating in urea buffer, the cell extracts run on a SDS-PAGE gel, and Dot6 and Tod6 mobility measured using Western Blotting and the Li-Cor infrared imaging system. ChIP samples were purified using standard procedures and the enrichment levels measured using real-time PCR. A more detailed description of the Methods, including all buffers and reagents used, is included in the Expanded Experimental Procedures.
Supplementary Material
Highlights
Inositol pyrophosphates regulate the 1600 gene environmental stress response in yeast
Inositol pyrophosphates function by activating the Class I HDAC Rpd3L
Mutations in a conserved inositol-phosphate binding site on Rpd3 inhibit its activity
ACKNOWLEDGEMENTS
We thank Jim Hughes Hallett and Tushar Chawala for help with the initial microarray based screen. We are also grateful to Roy Parker, Ted Weinert and Rod Capaldi for critical reading of the manuscript, and to the Parker lab for use of equipment and reagents. This work was supported by grants 5T32GM008659 and 1R01GM097329 from the NIGMS.
Footnotes
Publisher's Disclaimer: This is a PDF le of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its nal citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
ACCESSION NUMBERS The GEO accession number for the microarray data reported in this paper is GSE45370.
REFERENCES
- Airoldi EM, Huttenhower C, Gresham D, Lu C, Caudy AA, Dunham MJ, Broach JR, Botstein D, Troyanskaya OG. Predicting cellular growth from gene expression signatures. Plos Comput Biol. 2009;5:e1000257. doi: 10.1371/journal.pcbi.1000257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alejandro-Osorio AL, Huebert DJ, Porcaro DT, Sonntag ME, Nillasithanukroh S, Will JL, Gasch AP. The histone deacetylase Rpd3p is required for transient changes in genomic expression in response to stress. Genome Biol. 2009;10:R57. doi: 10.1186/gb-2009-10-5-r57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck T, Hall MN. The TOR signalling pathway controls nuclear localization of nutrient-regulated transcription factors. Nature. 1999;402:402–689. doi: 10.1038/45287. [DOI] [PubMed] [Google Scholar]
- Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol. 2000;1:1–11. doi: 10.1038/35036035. [DOI] [PubMed] [Google Scholar]
- Brauer MJ, Huttenhower C, Airoldi EM, Rosenstein R, Matese JC, Gresham D, Boer VM, Troyanskaya OG, Botstein D. Coordination of growth rate, cell cycle, stress response, and metabolic activity in yeast. Mol Biol Cell. 2008;19:19–352. doi: 10.1091/mbc.E07-08-0779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton A, Hu X, Saiardi A. Are inositol pyrophosphates signalling molecules? J Cell Physiol. 2009;220:220–8. doi: 10.1002/jcp.21763. [DOI] [PubMed] [Google Scholar]
- Chakraborty A, Kim S, Snyder SH. Inositol pyrophosphates as mammalian cell signals. Sci Signal. 2011;4:re1. doi: 10.1126/scisignal.2001958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty A, Koldobskiy MA, Bello NT, Maxwell M, Potter JJ, Juluri KR, Maag D, Kim S, Huang AS, Dailey MJ, et al. Inositol pyrophosphates inhibit akt signaling, thereby regulating insulin sensitivity and weight gain. Cell. 2010;143:143–897. doi: 10.1016/j.cell.2010.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridy PC, Otto JC, Dollins DE, York JD. Cloning and characterization of two human VIP1-like inositol hexakisphosphate and diphosphoinositol pentakisphosphate kinases. J Biol Chem. 2007;282:282–30754. doi: 10.1074/jbc.M704656200. [DOI] [PubMed] [Google Scholar]
- Gasch AP, Spellman PT, Kao CM, Carmel-Harel O, Eisen MB, Storz G, Botstein D, Brown PO. Genomic expression programs in the response of yeast cells to environmental changes. Mol Biol Cell. 2000;11:11–4241. doi: 10.1091/mbc.11.12.4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber A, Bodenmiller B, Uotila A, Stahl M, Wanka S, Gerrits B, Aebersold R, Loewith R. Characterization of the rapamycin-sensitive phosphoproteome reveals that Sch9 is a central coordinator of protein synthesis. Genes Dev. 2009;23:23–1929. doi: 10.1101/gad.532109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber A, French SL, Tekotte H, Yerlikaya S, Stahl M, Perepelkina MP, Tyers M, Rougemont J, Beyer AL, Loewith R. Sch9 regulates ribosome biogenesis via Stb3, Dot6 and Tod6 and the histone deacetylase complex RPD3L. Embo J. 2011;30:30–3052. doi: 10.1038/emboj.2011.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey EL, Shamji AF, Bernstein BE, Schreiber SL. Rpd3p relocation mediates a transcriptional response to rapamycin in yeast. Chem Biol. 2004;11:11–295. doi: 10.1016/j.chembiol.2004.03.001. [DOI] [PubMed] [Google Scholar]
- Koldobskiy MA, Chakraborty A, Werner JK, Jr., Snowman AM, Juluri KR, Vandiver MS, Kim S, Heletz S, Snyder SH. p53-mediated apoptosis requires inositol hexakisphosphate kinase-2. Proc Natl Acad Sci U S A. 2010;107:107–20947. doi: 10.1073/pnas.1015671107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagger G, Doetzlhofer A, Schuettengruber B, Haidweger E, Simboeck E, Tischler J, Chiocca S, Suske G, Rotheneder H, Wintersberger E, et al. The tumor suppressor p53 and histone deacetylase 1 are antagonistic regulators of the cyclin-dependent kinase inhibitor p21/WAF1/CIP1 gene. Mol Cell Biol. 2003;23:23–2669. doi: 10.1128/MCB.23.8.2669-2679.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:149–274. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YS, Huang K, Quiocho FA, O'Shea EK. Molecular basis of cyclin-CDK-CKI regulation by reversible binding of an inositol pyrophosphate. Nat Chem Biol. 2008;4:4–25. doi: 10.1038/nchembio.2007.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YS, Mulugu S, York JD, O'Shea EK. Regulation of a cyclin-CDK-CDK inhibitor complex by inositol pyrophosphates. Science. 2007a;316:316–109. doi: 10.1126/science.1139080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YS, Mulugu S, York JD, O'Shea EK. Regulation of a cyclin-CDK-CDK inhibitor complex by inositol pyrophosphates. Science. 2007b;316:316–109. doi: 10.1126/science.1139080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lempiainen H, Uotila A, Urban J, Dohnal I, Ammerer G, Loewith R, Shore D. Sfp1 interaction with TORC1 and Mrs6 reveals feedback regulation on TOR signaling. Mol Cell. 2009;33:33–704. doi: 10.1016/j.molcel.2009.01.034. [DOI] [PubMed] [Google Scholar]
- Lippman SI, Broach JR. Protein kinase A and TORC1 activate genes for ribosomal biogenesis by inactivating repressors encoded by Dot6 and its homolog Tod6. Proc Natl Acad Sci U S A. 2009;106:106–19928. doi: 10.1073/pnas.0907027106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewith R, Hall MN. Target of rapamycin (TOR) in nutrient signaling and growth control. Genetics. 2011;189:189–1177. doi: 10.1534/genetics.111.133363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewith R, Jacinto E, Wullschleger S, Lorberg A, Crespo JL, Bonenfant D, Oppliger W, Jenoe P, Hall MN. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol Cell. 2002;10:10–457. doi: 10.1016/s1097-2765(02)00636-6. [DOI] [PubMed] [Google Scholar]
- Marion RM, Regev A, Segal E, Barash Y, Koller D, Friedman N, O'Shea EK. Sfp1 is a stress- and nutrient-sensitive regulator of ribosomal protein gene expression. Proc Natl Acad Sci U S A. 2004;101:101–14315. doi: 10.1073/pnas.0405353101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin DE, Soulard A, Hall MN. TOR regulates ribosomal protein gene expression via PKA and the forkhead transcription factor FHL1. Cell. 2004;119:119–969. doi: 10.1016/j.cell.2004.11.047. [DOI] [PubMed] [Google Scholar]
- Menniti FS, Miller RN, Putney JW, Jr., Shears SB. Turnover of inositol polyphosphate pyrophosphates in pancreatoma cells. J Biol Chem. 1993;268:268–3850. [PubMed] [Google Scholar]
- Morrison BH, Bauer JA, Kalvakolanu DV, Lindner DJ. Inositol hexakisphosphate kinase 2 mediates growth suppressive and apoptotic effects of interferon-beta in ovarian carcinoma cells. J Biol Chem. 2001;276:276–24965. doi: 10.1074/jbc.M101161200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison BH, Haney R, Lamarre E, Drazba J, Prestwich GD, Lindner DJ. Gene deletion of inositol hexakisphosphate kinase 2 predisposes to aerodigestive tract carcinoma. Oncogene. 2009;28:28–2383. doi: 10.1038/onc.2009.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulugu S, Bai W, Fridy PC, Bastidas RJ, Otto JC, Dollins DE, Haystead TA, Ribeiro AA, York JD. A conserved family of enzymes that phosphorylate inositol hexakisphosphate. Science. 2007;316:316–106. doi: 10.1126/science.1139099. [DOI] [PubMed] [Google Scholar]
- Nagata E, Luo HR, Saiardi A, Bae BI, Suzuki N, Snyder SH. Inositol hexakisphosphate kinase-2, a physiologic mediator of cell death. J Biol Chem. 2005;280:280–1634. doi: 10.1074/jbc.M409416200. [DOI] [PubMed] [Google Scholar]
- Ocker M, Schneider-Stock R. Histone deacetylase inhibitors: signalling towards p21cip1/waf1. Int J Biochem Cell Biol. 2007;39:39–1367. doi: 10.1016/j.biocel.2007.03.001. [DOI] [PubMed] [Google Scholar]
- Pesesse X, Choi K, Zhang T, Shears SB. Signaling by higher inositol polyphosphates. Synthesis of bisdiphosphoinositol tetrakisphosphate (“InsP8”) is selectively activated by hyperosmotic stress. J Biol Chem. 2004;279:279–43378. doi: 10.1074/jbc.C400286200. [DOI] [PubMed] [Google Scholar]
- Schmelzle T, Hall MN. TOR, a central controller of cell growth. Cell. 2000;103:103–253. doi: 10.1016/s0092-8674(00)00117-3. [DOI] [PubMed] [Google Scholar]
- Seeds AM, Bastidas RJ, York JD. Molecular definition of a novel inositol polyphosphate metabolic pathway initiated by inositol 1,4,5-trisphosphate 3-kinase activity in Saccharomyces cerevisiae. J Biol Chem. 2005;280:280–27654. doi: 10.1074/jbc.M505089200. [DOI] [PubMed] [Google Scholar]
- Shears SB. Diphosphoinositol polyphosphates: metabolic messengers? Mol Pharmacol. 2009;76:76–236. doi: 10.1124/mol.109.055897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevchenko A, Roguev A, Schaft D, Buchanan L, Habermann B, Sakalar C, Thomas H, Krogan NJ, Stewart AF. Chromatin Central: towards the comparative proteome by accurate mapping of the yeast proteomic environment. Genome Biol. 2008;9:R167. doi: 10.1186/gb-2008-9-11-r167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soufi B, Kelstrup CD, Stoehr G, Frohlich F, Walther TC, Olsen JV. Global analysis of the yeast osmotic stress response by quantitative proteomics. Mol Biosyst. 2009;5:5–1337. doi: 10.1039/b902256b. [DOI] [PubMed] [Google Scholar]
- Streb H, Irvine RF, Berridge MJ, Schulz I. Release of Ca2+ from a nonmitochondrial intracellular store in pancreatic acinar cells by inositol-1,4,5-trisphosphate. Nature. 1983;306:306–67. doi: 10.1038/306067a0. [DOI] [PubMed] [Google Scholar]
- Szijgyarto Z, Garedew A, Azevedo C, Saiardi A. Influence of inositol pyrophosphates on cellular energy dynamics. Science. 2011;334:334–802. doi: 10.1126/science.1211908. [DOI] [PubMed] [Google Scholar]
- Tsui MM, York JD. Roles of inositol phosphates and inositol pyrophosphates in development, cell signaling and nuclear processes. Adv Enzyme Regul. 2009;50:50–324. doi: 10.1016/j.advenzreg.2009.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban J, Soulard A, Huber A, Lippman S, Mukhopadhyay D, Deloche O, Wanke V, Anrather D, Ammerer G, Riezman H, et al. Sch9 is a major target of TORC1 in Saccharomyces cerevisiae. Mol Cell. 2007;26:26–663. doi: 10.1016/j.molcel.2007.04.020. [DOI] [PubMed] [Google Scholar]
- Watson PJ, Fairall L, Santos GM, Schwabe JW. Structure of HDAC3 bound to co-repressor and inositol tetraphosphate. Nature. 2012;481:481–335. doi: 10.1038/nature10728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan G, Lai Y, Jiang Y. The TOR complex 1 is a direct target of Rho1 GTPase. Mol Cell. 2012;45:45–743. doi: 10.1016/j.molcel.2012.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan G, Shen X, Jiang Y. Rapamycin activates Tap42-associated phosphatases by abrogating their association with Tor complex 1. Embo J. 2006;25:25–3546. doi: 10.1038/sj.emboj.7601239. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.