

Significance
Mutations in the RNA binding protein TDP-43 cause amyotrophic lateral sclerosis and frontotemporal dementia. Through expressing disease-causing mutants in mice and genome-wide RNA splicing analyses, mutant TDP-43 is shown to retain normal or enhanced activity for facilitating splicing of some RNA targets, but “loss-of-function” for others. These splicing changes, as well as age-dependent, mutant-dependent lower motor neuron disease, occur without loss of nuclear TDP-43 or accumulation of insoluble aggregates of TDP-43.
Keywords: neurodegeneration, RNA binding proteins, frontotemporal dementia
Abstract
Transactivating response region DNA binding protein (TDP-43) is the major protein component of ubiquitinated inclusions found in amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration (FTLD) with ubiquitinated inclusions. Two ALS-causing mutants (TDP-43Q331K and TDP-43M337V), but not wild-type human TDP-43, are shown here to provoke age-dependent, mutant-dependent, progressive motor axon degeneration and motor neuron death when expressed in mice at levels and in a cell type-selective pattern similar to endogenous TDP-43. Mutant TDP-43-dependent degeneration of lower motor neurons occurs without: (i) loss of TDP-43 from the corresponding nuclei, (ii) accumulation of TDP-43 aggregates, and (iii) accumulation of insoluble TDP-43. Computational analysis using splicing-sensitive microarrays demonstrates alterations of endogenous TDP-43–dependent alternative splicing events conferred by both human wild-type and mutant TDP-43Q331K, but with high levels of mutant TDP-43 preferentially enhancing exon exclusion of some target pre-mRNAs affecting genes involved in neurological transmission and function. Comparison with splicing alterations following TDP-43 depletion demonstrates that TDP-43Q331K enhances normal TDP-43 splicing function for some RNA targets but loss-of-function for others. Thus, adult-onset motor neuron disease does not require aggregation or loss of nuclear TDP-43, with ALS-linked mutants producing loss and gain of splicing function of selected RNA targets at an early disease stage.
Amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration with ubiquitinated inclusions (FTLD-U) are progressive, adult-onset neurodegenerative diseases with overlapping clinical and pathological features (1–3). ALS is characterized by the selective loss of upper and lower motor neurons, leading to progressive fatal paralysis and muscle atrophy. A large majority (∼90%) of ALS and FTLD-U cases are without a known genetic cause. Importantly, in these sporadic cases, the appearance of ubiquitinated inclusions within the affected neurons of the nervous system characterizes both ALS and FTLD-U patients, suggesting an overlapping mechanism underlying both diseases. Biochemical characterization of brains and spinal cords from ALS and FTLD-U patients identified transactivating response region (TAR) DNA binding protein (TDP-43) as the major protein component of these ubiquitinated inclusions (4, 5). The discovery of ALS-linked mutations in the glycine-rich C-terminal domain of TDP-43 (6-8) demonstrated a pathological role of TDP-43 in both diseases. The subsequent identification of mutations in a structurally and functionally related nucleic acid binding protein, FUS/TLS (fused in sarcoma/translocated in liposarcoma) (9, 10), further implicated defects in RNA processing in ALS pathogenesis.
TDP-43 is a multifunctional nucleic acid binding protein. Within the nervous system, TDP-43 binds to >6,000 pre-mRNAs and affects the levels of ∼600 mRNAs and the splicing patterns of another 950 (11). Structurally, the 414-aa protein consists of two RNA recognition motifs (RRM1 and RRM2) (12, 13), nuclear import and export signal (14), and a glycine-rich region implicated in protein–protein interactions (15, 16) that include components of the RNA splicing machinery (17, 18).
Disruption in mice of the highly conserved Tardbp gene is embryonically lethal (19–22). Similarly, postnatal inactivation of Tardbp (by Cre recombinase-mediated gene excision encoded by a ubiquitously-expressed CAG-Cre transgene) results in rapid postnatal death accompanied by defects in fat metabolism (22). TDP-43 autoregulates its own RNA level (11, 23) at least in part by stimulating excision of an intron in its 3′ untranslated region, thereby making the spliced RNA a substrate for nonsense-mediated RNA degradation (11). Furthermore, transgenic rodent models have been used to demonstrate that overriding the autoregulatory mechanism by overexpression of unregulated wild-type (24–28) or disease-linked mutant (26, 28–35) TDP-43 transgenes can produce neurodegeneration in mice.
ALS and FTLD-U patient brain and spinal cord samples are characterized by the accumulation of cytoplasmic TDP-43 aggregates accompanied by a distinct clearing of nuclear TDP-43 within affected neurons and glia (36, 37), implicating possible loss of nuclear TDP-43 function in disease pathogenesis. In human disease, TDP-43 has been reported to be abnormally phosphorylated, ubiquitinated, and cleaved to produce C-terminal fragments (4, 5, 38, 39). Ectopic expression of these C-terminal fragments in cell-culture models (40–42) has shown that they are aggregation-prone and confer an intrinsic toxicity. However, the extent of the contribution of these C-terminal fragments to disease pathogenesis is undetermined. Indeed, double-immunofluorescent labeling of ALS patient spinal cords using N-terminal–specific and C-terminal–specific antibodies suggests that inclusions in spinal cord motor neurons are comprised primarily of full-length TDP-43 (37). Importantly, retention of ability to bind RNA by full-length TDP-43 has been demonstrated to be required for toxicity in yeast, fly, and Caenorhabditis elegans models (43–46).
Nevertheless, it remains unresolved whether toxicity to motor neurons from mutations in TDP-43 is mediated through a gain of toxic property, loss-of-function, or a combination of both. By generation of transgenic mice encoding levels of wild-type or mutant human TDP-43 comparable to endogenous TDP-43, we demonstrate mutant-dependent, age-dependent motor neuron disease from ALS-linked TDP-43 mutants in the absence of overexpression, cytoplasmic accumulation of a 35 kDa TDP-43 fragment, or insoluble TDP-43 aggregates. Accompanying autoregulation-mediated reduction of endogenous wild-type TDP-43 are splicing alterations previously identified to be TDP-43–dependent (11). Additional splicing alterations are identified by systematic genome-wide analyses of alternative splicing that are indicative of both enhancement and loss-of-function by the TDP-43 mutants for individual RNA substrates, from which we conclude that ALS-linked mutations confer both loss- and gain-of-function properties to TDP-43, and that these act intranuclearly to induce splicing alterations that may underlie age-dependent motor neuron disease.
Results
Establishment of TDP-43 Transgenic Mice Expressing Wild-Type and Mutant TDP-43 Broadly in the Central Nervous System.
Transgenic mice were produced that express either wild-type or ALS-linked mutant TDP-43 broadly throughout the central nervous system, using the murine prion-promoter (47) previously reported to drive transgene expression most abundantly in the central nervous system, both in neurons and astrocytes (48). cDNAs encoding wild-type or either of two ALS-linked mutants of TDP-43 (6) [Q331K (glutamine to lysine substitution at amino acid position 331) and M337V (methionine to valine substitution at amino acid position 337)] were fused to an N-terminal myc-tag under control of the murine prion promoter (Fig. 1A). From >20 founders for each gene, three lines were selected and established for TDP-43Wild-Type, TDP-43Q331K, and TDP-43M337V, respectively.
Fig. 1.

Generation of multiple lines of transgenic mice expressing wild-type, Q331K, or M337V-mutant human TDP-43. (A) Schematic of the PrP-TDP-43 transgene. (B) Immunoblotting using an antibody that recognizes both mouse and human TDP-43 with equal affinity in whole-organ lysates from transgenic animals. Br, brain; Cb, cerebellum; SC, spinal cord; Hrt, heart; Mu, skeletal muscle; Liv, liver; Kid, kidney. Equal protein amounts were loaded per lane. (C) Immunoblotting of whole spinal cord lysates from transgenic mice with the mouse/human TDP-43 antibody. (D) Accumulated transgene-encoded mRNA levels of in spinal cords of PrP-TDP-43 mice. (E) Representative single-plane confocal images of the ventral horn of lumbar-level spinal cord from 2-mo-old animals using a rabbit polyclonal antibody (Sigma C3956) to the myc tag on the transgene encoded protein and costained for the neuronal marker NeuN.
Analysis of whole-tissue lysates by immunoblotting with a polyclonal anti–TDP-43 antibody recognizing recombinant mouse and human TDP-43 protein with equal affinity confirmed that transgene expression was confined primarily to the brain and spinal cord, with very low to no expression in some other tissues (Fig. 1B). Immunoblotting of protein extracts or real-time quantitative RT-PCR (RT-qPCR) of RNAs, respectively, from whole brain identified levels of transgene expression for each of the nine lines (Fig. S1). Analysis of cortex and spinal cord identified lines expressing comparable levels of human TDP-43 mRNA, ranging from 1× to 1.5× the level of endogenous TDP-43 in nontransgenic mice, which was recapitulated in the accumulated protein levels from whole brain and spinal cord lysates (Fig. 1 C and D, and Fig. S1A). Immunofluorescent staining with myc antibody to detect human TDP-43 showed that it accumulated in neurons and glia of the spinal cord (Fig. 1E) in a pattern that mimicked the endogenous expression pattern of TDP-43.
Mice Expressing Mutant Human TDP-43 Develop Adult-Onset Motor Deficits and Hindlimb Weakness.
Four lines of mice were selected in which wild-type TDP-43, TDP-43M337V, or TDP-43Q331K were accumulated to levels comparable to endogenous TDP-43 in nontransgenic mice, as well as one line with a lower level (referred TDP-43Q331K-low). At birth, mice expressing wild-type human TDP-43 or either TDP-43 mutant were indistinguishable from their nontransgenic littermates. However, in contrast to their nontransgenic littermates or TDP-43Wild-Type mice, TDP-43Q331K transgenic mice developed a tremor at as early as 3 mo of age (Fig. S2A). This tremor worsened with aging (Movie S1) and was accompanied by the development of hindlimb clasping indicative of spastic motor impairment (27, 49) (Fig. S2B).
Motor performance of TDP-43Wild-Type, TDP-43Q331K-Low, TDP-43Q331K, and TDP-43M337V mice was followed during aging. Motor performance (on an accelerating rotarod) was normal at the earliest time points analyzed in each of the mutant TDP-43–expressing lines (Fig. 2 A and B). In contrast with nontransgenic and TDP-43Wild-Type mice, all three mutant TDP-43 lines developed significant age-dependent motor deficits by 10 mo of age, with the most severe deficit and earliest onset (3 mo) in the higher expressing TDP-43Q331K line (Fig. 2B). A significant decrease in hindlimb grip strength by 10 mo of age accompanied age-dependent motor impairment in TDP-43Q331K animals (Fig. 2C). Analysis of animals at later time points (e.g., after 17 mo of age) showed no further exacerbation of motor impairment (Fig. S3A), indicating a window of adult onset, active degeneration up to 10 mo of age, after which there is little further worsening of motor phenotype despite continued transgene accumulation in the remaining motor neurons and surrounding glia (see, for example, below and Fig. 5).
Fig. 2.

TDP-43Q331K and TDP-43M337V mice develop age-dependent, progressive motor deficits. (A and B) Motor deficits measured by rotarod. n ≥ 11 for each genotype and each time point. **P < 0.01 and ***P < 0.001 using one-way ANOVA at each time point with Bonferroni’s post hoc test. (C) The 10- to 12-mo-old TDP-43Q331K high-expressing animals developed hindlimb weakness as measured by a hindlimb grip-strength assay (P = 0.0002 by Student’s t test). Data shown are the average ± SEM. n = 12 per genotype.
Fig. 5.
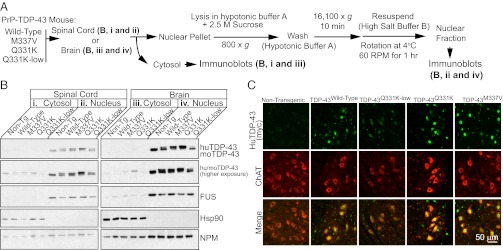
Neither wild-type nor mutant TDP-43 show aberrant cytosolic localization in the brain and spinal cords of TDP-43 transgenic mice. (A) Experimental scheme for the nuclear and cytosolic fractionation of spinal cords (B, i and ii) or brains (B, iii and iv) of nontransgenic and TDP-43 transgenic mice. (B) Immunoblotting of enriched cytosolic or nuclear extracts from the brain or spinal cord of 10- to 12-mo-old nontransgenic, wild-type, and mutant TDP-43 transgenic animals using an antibody recognizing both mouse and human TDP-43 with equal affinity. Note enrichment of Hsp90 in the cytosolic extract and FUS in the nuclear extract, indicating the efficiency of separation. (C) Immunofluorescent localization of endogenous ChAT and transgene-encoded TDP-43 in 10-mo-old TDP-43 transgenic animals.
To determine whether the motor deficits and hindlimb weakness observed in TDP-43Q331K transgenic mice were accompanied by neuromuscular abnormalities similar to those clinically observed in human ALS, we performed electrophysiological analyses on TDP-43Wild-Type and TDP-43Q331K transgenic mice (Fig. 3A). Resting electromyographic (EMG) recordings were obtained from the gastrocnemius muscle in the absence of any stimulus (in isoflurane-anesthetized animals). As expected, in SOD1G37R symptomatic mice that will go on to develop fatal paralytic disease, high-frequency spontaneous firings of the motor units (fibrillations) were recorded (Fig. 3 B, v), consistent with the known widespread denervation. Recordings in TDP-43Q331K mice (Fig. 3 B, iii) revealed the presence of muscle fibrillations similar to those observed in presymptomatic SOD1G37R mice (Fig. 3 B, iv), indicating neuromuscular denervation and motor unit degeneration and regeneration. These spontaneous firings were found only in the presence of mutant TDP-43–dependent mice, as recordings in nontransgenic and expression matched TDP-43Wild-Type mice (Fig. 3 B, i and ii) lacked such spontaneous EMG activity.
Fig. 3.

Electrophysiological measures identify lower motor neuron deficits in aged mutant-expressing TDP-43 mice. (A) Schematic of measurement of MEPs elicited by electrical stimulation of motor cortex and extrapyramidal system, with responses recorded from the exposed T12 spinal segments (spinal cord surface MEPs) or from the gastrocnemius muscle (MMEPs). (B) Resting EMG recording from the gastrocnemius muscle in isoflurane anesthetized animals in the absence of any stimulus. (C) MMEP recordings from the gastrocnemius muscle. **P < 0.01, ***P < 0.001 by Student's t test. (D) Spinal cord surface MEP recordings in nontransgenic, TDP-43Wild-Type and TDP-43Q331K mice.
Because both upper and lower motor neuron deficits can develop in ALS patients, we tested if the spontaneous EMG activity associated with neuromuscular denervation observed in TDP-43Q331K mice was accompanied by upper motor neuron deficits. To this end, myogenic motor evoked potentials (MMEPs), which report a measure of connectivity of the entire neuromuscular unit, including the motor cortex, interneurons, α-motorneurons, and the neuromuscular junction, were recorded from the gastrocnemius muscle after electrical stimulation of the motor cortex (Fig. 3C). Use of this measure in SOD1G37R mice revealed that, as expected, MMEPs were decreased (from ∼2.4 mV in nontransgenic animals) to 1.5 mV at presymptomatic stages, and were almost completely lost in symptomatic SOD1G37R mice (0.03 mV). TDP-43Q331K transgenic mice also developed significant decreases (to 0.18 mV; n = 3) in MMEP amplitude (Fig. 3C) compared with nontransgenic (2.4 mV; n = 4) and TDP-43Wild-Type transgenic mice (1.8 mV; n = 3), indicating a disruption of the neuromuscular unit.
To determine whether the decrease in MMEPs reported above involved disruption of the connectivity between the upper and lower motor neurons, spinal cord motor-evoked potentials (MEPs) were then recorded from the dorsal surface of an exposed thoracic (T12) segment after electrical stimulation of the motor cortex. MEPs consist of multiple waves, with the two earliest peaks (N1, N2) corresponding to the activation of the extrapyramidal system. No significant differences were found in the amplitude of the N1 wave when comparing nontransgenic (0.20 mV; n = 4), TDP-43Wild-Type (0.21 mV; n = 5), and TDP-43Q331K transgenic mice (0.20 mV; n = 4) (Fig. 3D). Quantification of immunofluorescently labeled Ctip2+ upper motor neurons in cortex layer V in TDP-43Q331K transgenic animals showed no loss of upper motor neurons, consistent with the retention of upper motor neuron function indicated by the persistence of cortical MEPs (Fig. S4A). Therefore, spontaneous muscle firing recorded using EMGs, accompanied by decreased MMEP amplitudes, must reflect motor dysfunction associated primarily with lower motor neuron degeneration in TDP-43Q331K mice.
TDP-43Q331K Transgenic Mice Develop Age-Dependent Motor Neuron Loss and Motor Axon Degeneration.
Having observed the development of motor deficits and electrophysiological abnormalities, we examined the central nervous system for signs of neurodegeneration. Immunofluorescent staining for the astrocytic marker GFAP and the microglial marker Iba-1 revealed immunoreactive astrocytes and infiltrating microglia (Fig. S4B) in the ventral horn of the spinal cord in 10- to 12-mo-old TDP-43Q331K transgenic mice. Consistent with the initial identification of the Q331K mutation in an ALS patient with classic lower motor-neuron involvement (6), quantification of choline acetyl-transferase (ChAT)-positive lower motor neurons in lumbar spinal cords from TDP-43Q331K mice revealed a significant, age-dependent decrease (P = 0.04, n = 3) in motor neuron number. Loss of motor neurons initiated before 2 mo of age and continued until about 12 mo of age, yielding a reduction of ∼35% compared with age-matched nontransgenic or TDP-43Wild-Type transgenic animals (Fig. 4A). Additionally, the milder motor deficits seen by 10–12 mo of age in both TDP-43Q331K-Low and TDP-43M337V mice were accompanied by a trend in age-dependent reduction in ChAT-positive motor neurons compared with nontransgenic or TDP-43Wild-Type transgenic animals in the ventral lumbar horns at 10–12 mo of age (Fig. 4A).
Fig. 4.
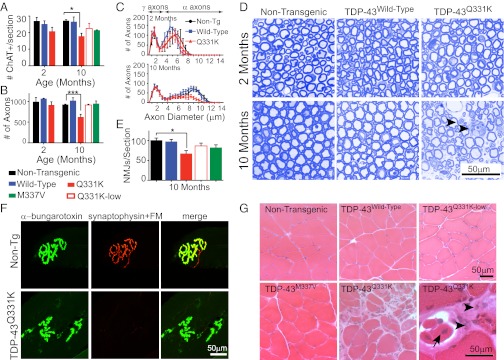
TDP-43Q331K transgenic mice develop age-dependent lower motor neuron degeneration. (A) Quantification of ChAT-positive α-motor neurons in lumbar spinal cords from TDP-43 transgenic mice (average ± SD, n = 3 per genotype per time point; *P = 0.04). (B) Quantification of motor axons shown in D (at 10–12 mo of age, n ≥ 3 per genotype per time point; ***P < 0.001). (C) Distributions of diameters of L5 motor axons determined from images in D. (D) Representative images of the L5 motor axon roots from TDP-43Wild-Type or TDP-43Q331K transgenic animals stained with Toluidine blue. (arrowheads) Degenerating axons in TDP-43Q331K mice. (E) Quantification of average neuromuscular junctions per section in 10- to 12-mo-old TDP-43 transgenic animals (± SEM; *P = 0.02, n ≥ 3 per genotype). (F) Representative neuromuscular junctions from nontransgenic and TDP-43Q331K–expressing animals. FM, fluoromyelin. (G) Hematoxylin and eosin staining of grastrocnemius muscles. (arrowheads) Atrophied fibers of varying sizes; (arrows) regenerating fibers with centralized nuclei. All statistics performed using one-way ANOVA with Bonferroni’s post hoc test.
Quantification of axons remaining in the fifth lumbar (L5) roots of TDP-43Q331K transgenic mice revealed a significant reduction (623 vs. 923 in nontransgenic, ***P < 0.001) of total motor axons by 10–12 mo of age (Fig. 4B), with the most significant reduction in large caliber α-motor axons (>3.5 μm) that innervate muscle (50) (Fig. 4C). Morphological examination of the remaining motor axons revealed the presence of degenerating axons, characterized by the appearance of vacuolization and myelin defects (Fig. 4D). Similar to what was demonstrated behaviorally, there was no further loss of L5 motor axons at 20 mo of age (Fig. S3 B–D), indicating a period of active degeneration up to 10 mo of age.
We therefore focused our remaining analyses within this disease period. Quantification of postsynaptic neuromuscular junctions using α-bungarotoxin staining of the gastrocnemius muscle revealed a significant reduction (30%; P = 0.02) in the number of neuromuscular junction endplates in of TDP-43Q331K mice (Fig. 4E). In contrast, TDP-43Q331K-Lowand TDP-43M337V transgenic mice displayed numbers of α-bungarotoxin–positive neuromuscular junction endplates that were not significantly different from nontransgenic or TDP-43Wild-Type animals, consistent with functional deficits without overt structural changes in lower motor neuron synapses. Morphological examination of hematoxylin and eosin (H&E)-stained sections of gastrocnemius muscle from 10-mo-old TDP-43Q331K animals revealed regions of damaged muscle fibers and regions of regeneration characterized by the appearance of centralized nuclei (Fig. 4G). TDP-43Wild-Type, TDP-43Q331K-low and TDP-43M337V animals lacked similar morphological abnormalities in the muscle fibers. Additionally, neuromuscular junctions in TDP-43Q331K mice appeared malformed with an abnormal bleb-like appearance (Fig. 4F). Examination of the descending corticospinal tracts in the dorsal and lateral columns of the spinal cords revealed minor degeneration in all mutant-expressing lines, including TDP-43Q331K-Low and TDP-43M337V, suggesting that degeneration of other regions of the motor circuit is responsible for the phenotypes observed in the low mutant-expressing lines.
Cytoplasmic Mislocalization of TDP-43 Is Not Required for the Development of Motor Neuron Disease.
A primary pathological feature in ALS patients is the accumulation of cytoplasmic, ubiquitinated and insoluble TDP-43–containing aggregates within the neurons of the nervous system (4). To test whether the development of motor neuron disease was accompanied by a similar alteration in TDP-43 localization, we first performed nuclear and cytoplasmic fractionation of whole spinal cord and brain from 10- to 12-mo-old TDP-43 transgenic mice (Fig. 5A). Immunoblotting for mouse and human TDP-43 in enriched fractions from either cytoplasm (Fig. 5 B, i and iii) or nuclei (Fig. 5 B, ii and iv) derived from cortex or spinal cord revealed that the majority of human TDP-43 remained in the nuclear fraction, just as it did in nontransgenic animals. Examination of longer exposures revealed a similarly small proportion of both endogenous mouse and human TDP-43 fractionated within the cytosol.
Immunofluorescent staining in lumbar spinal cord sections of TDP-43Q331K mice from 2 to 10 mo of age revealed that the majority of mutant TDP-43 remained nuclear (Fig. 5C). Additionally, analysis of sequential biochemical extraction using high-salt and urea buffers revealed that the majority of both mouse and human TDP-43 was extracted in the high-salt soluble fraction (Fig. S5 B and C). Analysis for nontransgenic and all transgenic lines of a final extraction of the nuclear pellets with SDS demonstrated that only a small fraction (Fig. S5D) of human and endogenous mouse TDP-43 was in an initially insoluble form that could be solubilized with SDS.
Widespread Splicing Changes from Altered TDP-43 Levels Within the Central Nervous System.
To evaluate if normal patterns of alternative splicing were disrupted upon reduction of mouse TDP-43 and its replacement with wild-type or mutant human TDP-43 at or above the level of endogenous TDP-43 in normal mice, RNA extracted from cortices of 2-mo-old mice were examined using Affymetrix splicing-sensitive microarrays (Fig. 6A). Analysis of splicing changes in cortices of TDP-43Wild-Type, TDP-43Q331K, and TDP-43Q331K-Low revealed that 824, 1,195, and 208 exons, respectively, were differentially spliced relative to mRNA from nontransgenic mice, using a statistical threshold that captured as many significant changes as possible (absolute separation score of <0.3) (51, 52) (Fig. 6A).
Fig. 6.
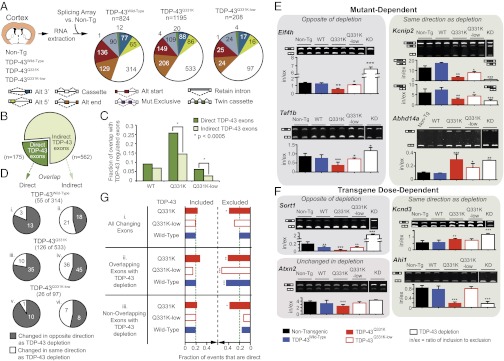
Both enhanced activity and loss-of-function for specific RNA splicing targets directly bound by TDP-43Q331K. (A) Experimental strategy to identify differentially regulated splicing events using Affymetrix splicing-sensitive microarrays to analyze RNAs extracted from cortices of 2-mo-old nontransgenic, TDP-43Wild-Type, TDP-43Q331K, or TDP-43Q331K-Low transgenic mice. (B) TDP-43–regulated cassette exons identified from TDP-43 depletion experiments in mouse (11). Cassette exons are divided into direct targets, bound by TDP-43 within 2 kb, and indirect targets, not bound by TDP-43. (C) Bar plot displaying the fraction of overlap of TDP-43–regulated exons, direct (dark green) and indirect (light green), with exons that changed upon expression of TDP-43Wild-Type, TDP-43Q331K, and TDP-43Q331K-low. The TDP-43Q331K and TDP-43Q331K-low overlapping exons show a significant enrichment (by χ2 analysis: P < 0.0005) in the fraction that are direct TDP-43–regulated exons compared with the fraction that are indirect TDP-43–regulated exons. No enrichment was found in TDP-43Wild-Type. (D) Overlap of significantly changed cassette exons observed in the cortices from TDP-43 transgenic mice with TDP-43–regulated cassette exons. Direct and indirect exons that are also regulated in TDP-43 transgenic mice are represented as exons that are changed in the opposite (colored gray) or same (included or excluded in both, colored white) direction of TDP-43 depletion. (E and F) RT-PCR validation for a subset of alternate cassette exons identified in cortex by splicing-sensitive microarrays and that are (E) mutant-dependent or (F) transgene dose-dependent and that change in the same or opposite direction following knockdown (KD) in TDP-43 following antisense oligonucleotide infusion within striatum. Bar plots show the ratio of inclusion to exclusion calculated from the mean intensities (n ≥ 3 biological replicates, ± SD). Representative gel analyses are images shown with duplicate biological replicates. *P < 0.05, **P < 0.01, and ***P < 0.001 with Student’s t test; in/ex, ratio of inclusion to exclusion. (G) Bar graph depicting the percentages of differentially included or excluded exons upon human TDP-43 expression (green dashed line). The fraction of unchanged exons in any cortex experiment (TDP-43Wild-Type, TDP-43Q331K, TDP-43Q331K-low) that have TDP-43 binding. (i) “All Changing Exons” includes all exons that were either included or excluded (25% of these were direct TDP-43 targets). (ii) “Overlapping Exons with TDP-43 KD” includes only exons that changed following TDP-43 depletion (i.e., exons regulated by endogenous TDP-43). A significant increase was found in the percentage of direct targets (∼43% for TDP-43Q331K and TDP-43Wild-Type, and 53% for TDP-43Q331K-low) for cassette exons that are excluded upon human TDP-43 expression, with only a modest ∼20% of included exons that are direct targets. (iii) “Non-overlapping exons with TDP-43 KD” includes only exons not previously shown to be regulated by endogenous TDP-43 depletion, in which 39% of excluded exons in the TDP-43Q331K animals were direct targets.
We focused on the largest represented class of alternative splicing events, namely cassette exons (i.e., exons that can be included or excluded in the final mRNA) (white slice of pie charts in Fig. 6A). For comparison, we reanalyzed our previous splicing array data (11) using a consistent threshold (absolute separation score of <0.3), identifying 737 cassette exons that were alternatively spliced upon depletion of endogenous mouse TDP-43 in the central nervous system (Fig. S6). These exons, the abundance of which is normally regulated by TDP-43 levels, were then compared with the 314, 533, and 97 cassette exons that were misregulated in human TDP-43Wild-Type, TDP-43Q331K, and TDP-43Q331K-Low transgenic animals.
To determine if misregulated exons were associated with endogenous TDP-43 binding [as measured by cross-linking and immunoprecipitation followed by deep sequencing (11)], we separated the 737 exons into “direct” (175, which contained TDP-43 binding sites within 2 kb of the exon) and “indirect” (562 without TDP-43 binding sites near the exon) targets (Fig. 6B). Notably, when comparing the percentages of direct and indirect targets that overlapped with TDP-43Q331K–dependent misregulated exons, we observed a statistically significant (P < 0.0005 by χ2 analysis) 1.8-fold enrichment of direct targets [26% (45/175)] compared with indirect ones [14% (81/562)]. A similar (2.4-fold) enrichment was seen for altered splicing of direct targets in TDP-43Q331K-Low [6% of direct targets (11/175) compared with 2.6% of indirect targets (15/562)]. No significant enrichment was seen in a similar comparison for TDP-43Wild-Type [9% (16/175) to 7% (39/562)] (Fig. 6C). Thus, the human mutant transgenes preferentially affect direct, endogenous TDP-43–dependent alternative splicing events.
Mutant TDP-43 Retention or Enhancement of TDP-43 Function in Splicing of Most pre-mRNAs.
To test whether the splicing changes for cassette exons reflected a loss or gain of normal TDP-43 function, we compared the directionality of exon inclusion when TDP-43 was depleted or in response to expression in mice of the transgene-encoded human TDP-43 (Fig. 6D). Of the cassette exon-splicing changes observed in the TDP-43 transgenic mice, we found that 55 of 314 in TDP-43Wild-Type (Fig. 6 D, i and ii), 126 of 533 in TDP-43Q331K (Fig. 6 D, iii and iv), and 26 of 97 TDP-43Q331K-Low (Fig. 6 D, v and vi) overlapped with cassette exon changes identified following TDP-43 depletion. For directly bound exon targets, the splicing of which was altered by expression of human TDP-43, the majority of the affected cassette exons overlapping with direct targets changed in a direction opposite to loss of TDP-43 [TDP-43Wild-Type (13/16), TDP-43Q331K (35/45), TDP-43Q331K-Low (10/11)] (in gray, Fig. 6 D, i, iii, and v).
Furthermore, at higher TDP-43Q331K levels (and the correspondingly stronger reduction in endogenous mouse TDP-43), splicing of 35 of 45 directly bound cassette exons (Fig. 6 D, iii) changed in a manner consistent with an elevated normal function of endogenous mouse TDP-43 (the opposite of TDP-43 depletion). In other words, these cassette exons, whose inclusion (or exclusion) was previously shown to be dependent on mouse TDP-43, were more included (or excluded) when human TDP-43Q331K was expressed, consistent with this mutant retaining normal (or enhanced) TDP-43 activity for most pre-mRNAs.
Mutant TDP-43–Dependent Loss-of-Function for Splicing of Specific pre-mRNAs.
Ten direct exon targets in TDP-43Q331K mice were altered in a manner indicative of a reduction in endogenous TDP-43 function in facilitating the splicing of these TDP-43 target exons (Fig. 6 D, iii). Exons whose splicing were reported altered by microarray analysis, as well as ones of biological interest but not represented on the array, were then analyzed by semiquantitative RT-PCR (Fig. 6 E and F, Fig. S7, and Table S1). To identify and validate splicing changes that are specifically affected by ALS-linked mutant TDP-43, we focused on changes occurring only in both TDP-43Q331K and TDP-43Q331K-Low-transgenic mice, but not in TDP-43Wild-Type mice (the accumulated transgene level of which is similar to that of TDP-43Q331K-Low transgenic mice). Within these changes, the presence of TDP-43 mutant produced splicing changes in Kcnip2, Abhd14a, Ctnnd1, and Atp2b1, which mimicked reduction of endogenous TDP-43 activity (Fig. 6E, Fig. S7A, and Table S1), demonstrating a loss-of-normal function of human TDP-43Q331K, which even at a high level of expression did not replace the endogenous mouse TDP-43 for the splicing of these exons.
Dose-Dependent Enhanced Splicing of Some pre-mRNAs by Human TDP-43.
Within the exons that overlapped with those that changed upon TDP-43 depletion (Fig. 6 D, i, iii, and v), we found some that primarily displayed a dose-dependent TDP-43Q331K or TDP-43Wild-Type pattern of splicing in the opposite direction to what occurs after TDP-43 depletion (Fig. 6F, Sort1, and Fig. S7B, Ppm2c, AU040829, and Caly, the exons of which were more excluded, in contrast to more included in TDP-43 depletion). Added to this finding, for other pre-mRNA targets, enhancement of normal TDP-43 function in splicing was seen specifically for TDP-43Q331K, as demonstrated by dose-dependent splicing changes in a direction opposite what is seen with TDP-43 depletion (e.g., Eif4h and Taf1b, the exons of which were more excluded, in contrast to increased inclusion in TDP-43 depletion) (Fig. 6E and Table S1).
These findings for individual RNAs were supported globally by a genome-wide analysis of direct TDP-43 targets, the exons of which were included or excluded upon human TDP-43 expression (Fig. 6G). On average, ∼20% of unaffected exons represented on the array were direct targets (green line in Fig. 6G). However, when focusing on alternatively changing cassette exons, we found an overall increase in the percentages of direct TDP-43 exon targets (∼43%, TDP-43Q331K, and TDP-43Wild-Type; up to 53% in TDP-43Q331K-low) that were excluded in the presence of the human TDP-43 protein, compared with only ∼20% of the included exons (Fig. 6 G, ii). Surprisingly, 39% of excluded exons in the TDP-43Q331K animals not previously demonstrated as regulated by endogenous TDP-43 depletion were also direct targets (Fig. 6 G, iii). That is, some exons that were unchanged following TDP-43 depletion displayed a dose-dependent pattern of splicing in the direction of enhanced exclusion in the presence of the human TDP-43 (Fig. 6F, Atxn2, Fig. S7C, Zfp414, and Kcnj3, the exons of which were more excluded, in contrast to no change upon TDP-43 depletion, and Table S1). This finding supports a hypothesis that certain exons, although bound by TDP-43, are only misregulated upon elevation of wild-type or mutant TDP-43, but are not altered upon its loss. In other words, elevation or enhancement of TDP-43 activity through dose or mutation produces aberrant splicing events separate from those resulting from loss of TDP-43.
Mutant-Specific Splicing Alterations Accompany Motor Neuron Disease Development.
Having identified that the human transgenes cause both loss and enhancement of normal function, we focused on alternative splicing changes within the spinal cord, the tissue that develops the degenerative phenotype. RNA extracted from the spinal cords of 2-mo-old nontransgenic, TDP-43Wild-Type, and TDP-43Q331K animals before the onset of significant motor dysfunction were analyzed on the same splicing-sensitive microarrays (Fig. 7A). We identified 4,462 and 4,399 alternative splicing events in the TDP-43Wild-Type and TDP-43Q331K spinal cord conditions, respectively (Fig. 7A). Approximately 38% of changes in TDP-43Wild-Type cortex (118) and 42% of changes observed in TDP-43Q331K cortex (226) were also present in the spinal cord (Fig. 7B). Notably, with the exception of a cassette exon within the Kctd9 gene, altered splicing of all 1,060 cassette exons that overlapped between spinal cord in TDP-43Wild-Type and TDP-43Q331K mice occurred in the same direction, demonstrating that at least for this subset of RNA targets, the mutant remains functionally similar to wild-type TDP-43 (Fig. 7C).
Fig. 7.
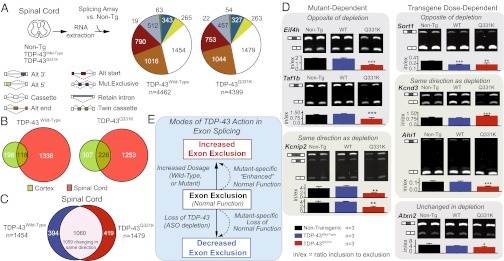
Unique splicing alterations in the spinal cord of TDP-43Q331K mice include changes in genes involved in neurological function and transmission. (A) Experimental strategy to identify differentially regulated splicing events in spinal cords of 2-mo-old nontransgenic, TDP-43Wild-Type, or TDP-43Q331K transgenic mice. Pie charts display total alternative splicing events significantly altered in the spinal cords of transgenic mice, relative to nontransgenic animals (colors representing the types of events on the array, defined at the right). (B) Diagram showing overlap between events changing in cortex and spinal cords of TDP-43Wild-Type and TDP-43Q331K animals. (C) Overlap of spinal cord TDP-43Wild-Type and TDP-43Q331K alternative cassette exons reveals a set of 1,060 common splicing events, 1,059 of which change in the same direction. (D) RT-PCR validation for a subset of spinal cord alternate cassette exons identified in A. Bar plots show mean intensities of n ≥ 3 biological replicates (± SD). Representative gel images shown, with duplicate biological replicates. (E) Modes of TDP-43 action in exon splicing. *P < 0.05, **P < 0.01, and ***P < 0.001 with Student's t test.
Use of semiquantitative RT-PCR validated a set of splicing changes that were consistent with those identified in the cortex (Fig. 7D and Table S1), again demonstrating that TDP-43Q331K mutant confers both loss and retention of normal TDP-43 function. Furthermore, among the 419 splicing events unique in TDP-43Q331K spinal cord were several RNAs, the encoded proteins of which are involved in neurological function and transmission (Table S2), including the synaptic cell-adhesion molecules neurexins 1 and 3 (Nrxn1 and Nrxn3), protein phosphatase 3 (also known as calcineurin, Ppp3ca), and glutamate receptor 2 (Gria2, encoding GluR2), which has been proposed to modify motor neuron vulnerability to excitotoxicity (53–55). In contrast, the 394 events unique to the TDP-43Wild-Type spinal cord condition did not include a similar representation from genes involved in neurological function.
Discussion
Although previous in vivo studies using transgenic rodent models (24–26, 28, 29, 31) have established that elevated levels of both wild-type and mutant TDP-43, or complete absence of wild-type TDP-43 (56), can be inherently toxic to neurons, it had not been determined (1) whether toxicity from high levels of TDP-43 (mutant or wild-type) reflects the toxicity arising in disease pathogenesis in ALS or FTLD, where TDP-43 is expressed at much lower levels (2), whether toxicity is mediated through a loss of TDP-43 function or a gain of aberrant toxic property, and (3) whether mutations in TDP-43 affect normal TDP-43 function in RNA splicing.
Our generation and analysis of mice expressing wild-type or ALS-linked mutants in TDP-43 at moderate levels (1–1.5× the normal level of endogenous mouse TDP-43) has revealed that mutant TDP-43 can provoke lower motor neuron disease in a mutant-dependent, age-dependent, dose-dependent manner. Furthermore, toxicity proceeds: (i) with loss of endogenous TDP-43 (the abundance of which is reduced by TDP-43 autoregulation), (ii) without loss of nuclear human TDP-43, (iii) without mutant TDP-43 redistribution or aggregation within the nucleus or cytoplasm, and (iv) without accumulation of a truncated portion of mutant TDP-43. Our findings are supported by evidence from a prior study (27), wherein neurodegeneration correlated not with cytoplasmic accumulation of human TDP-43 but rather with nuclear loss (from autoregulation) of endogenous mouse TDP-43 in the presence of either wild-type human TDP-43 or a TDP-43 variant not found in human disease but containing a mutation in a residue of its nuclear localization sequence.
Through expression of a moderate amount of human wild-type or mutant TDP-43 in the central nervous system in a pattern that mimics endogenous TDP-43 within neuronal and glial cells, we have demonstrated a mutant TDP-43–dependent contribution to adult-onset motor deficits. Both TDP-43Q331K-low and TDP-43M337V mice developed motor deficits by 10 mo of age, albeit without further exacerbation of the disease at later ages, whereas mice expressing comparable levels of human wild-type TDP-43 remained phenotypically normal. Furthermore, this motor phenotype was dose-dependent in relation to the amount of human TDP-43, as animals expressing a higher proportion of TDP-43Q331K (and correspondingly lower amount of mouse TDP-43) developed much more severe motor deficits, electrophysiological abnormalities characteristic of lower motor neuron disease, and loss of ∼35–40% of lower motor neurons and axons. Thus, motor neuron disease can initiate without robust overexpression of human TDP-43. Additionally, analysis of these mice has demonstrated that, despite the development of lower motor neuron disease, these mice lack significant alterations in either the biochemical solubility or nuclear localization of human and mouse TDP-43, contrary to what has been reported in human patients. Taken together, these data demonstrate that neither insoluble TDP-43 species nor the abnormal accumulation of cytosolic or nuclear TDP-43 is required for the development of lower motor neuron disease.
Rather, systematic genome-wide splicing arrays and computational analyses have revealed that nearly complete replacement of endogenous TDP-43 with human TDP-43 is accompanied by widespread changes in alternative pre-mRNA splicing, with some changes reflecting an “enhancement of normal function” exacerbated by increased accumulation of TDP-43. Furthermore, we have found that a small subset of splicing changes is uniquely dependent on the TDP-43Q331K mutant protein. Within these mutant-dependent changes, splicing alterations include an enhancement of normal function, but others are characteristic of TDP-43 loss-of-function. Hence, the TDP-43Q331K mutant confers both gain- and loss-of-function properties, both of which are associated with mutant-dependent motor neuron disease. Additionally, the high overlap in splicing alterations in both TDP-43Wild-Type and TDP-43Q331K spinal cord further emphasizes that the TDP-43Q331K mutant remains functionally similar to its wild-type counterpart. Nevertheless, the TDP-43Q331K mutation also produces a subset of unique splicing alterations within the spinal cord, specifically in genes involved in neurological function.
The dependency of some splicing events solely on the level of human TDP-43, wild-type or mutant (e.g., Sort1), highlights a sensitivity in TDP-43–dependent splicing to levels of accumulated TDP-43 protein. Depletion of endogenous mouse TDP-43 has been demonstrated to have widespread effects on both splicing and levels of mRNAs in the central nervous system (11). Our work herein demonstrates that an increase in accumulated TDP-43 protein affects splicing just as broadly, results consistent with TDP-43’s previously observed role in exon repression in the nervous system (11). Taking these data together, we propose that maintenance of a homeostatic level of TDP-43 protein is critical for its splicing function in the central nervous system, and that disruption of this level produces widespread aberrant alternative splicing events.
Although it has been demonstrated that TDP-43 itself is intrinsically aggregation-prone (57) and contains prion-like properties within its C terminus (58–60), we have found that motor neuron disease develops in the TDP-43 mutant-expressing mice without detectable TDP-43 aggregation. Furthermore, the TDP-43 C terminus has been proposed by others to play a role in association with other proteins in splicing of target genes (16–18, 61), although the contribution of mutations in the C terminus to the disruption of TDP-43 splicing function was not well understood. Indeed, a study using a human cell-culture model expressing TDP-43Q331K and TDP-43M337V found that mutations in the TDP-43 C terminus do not alter the composition of complexes of known TDP-43–interacting hnRNPs, nor the splicing of a cystic fibrosis transmembrane conductance regulator-based splicing reporter (17). In contrast, using a true in vivo context in mice that develop progressive motor neuron disease, we have found widespread splicing alterations in the adult mammalian central nervous system in the presence of nuclear, mutant TDP-43. Furthermore, these splicing alterations occur at an early disease-stage (2–3 mo of age). Thus, nuclear loss or cytoplasmic TDP-43 accumulation is not a requirement for the initiation of neurodegeneration.
Finally, our systematic splicing analysis enables us to propose a molecular model for human TDP-43 function in which it drives: (i) dosage-enhanced increases in exon exclusion of normal endogenous mouse TDP-43 targets, (ii) mutant-enhanced increases in exon exclusion of normal TDP-43 targets, and (iii) mutant-specific decreases in exon exclusion, because of a loss-of-function on normal TDP-43 targets (Fig. 7E). Taken together, the unique splicing changes in TDP-43Q331K transgenic mice within genes in spinal cords could provide a molecular basis for selective vulnerability of motor neurons to such mutants.
Materials and Methods
Generation of Transgenic Mice Expressing Floxed Wild-Type and Mutant Human TDP-43.
Flanking SalI digestion sites were inserted by PCR into cDNAs containing N-terminal myc-tagged full-length wild-type or mutant (Q331K or M337V) TDP-43 and cloned into the XhoI site of MoPrP.XhoI (ATCC #JHU-2). The resultant construct was digested upstream of the minimal PrP promoter and downstream of the final PrP exon 3, subcloned into a shuttle vector containing loxP flanking sites (Fig. 1A), and linearized using XhoI. The final construct was injected into the pronuclei of fertilized C57Bl6/C3H hybrid eggs and implanted into pseudopregnant female mice. Founder mice were then backcrossed to C57Bl6 to establish lines (mice used in this report were backcrossed to C57Bl6 for a minimum of four generations).
RT-qPCR and Immunoblotting.
Total RNA from half of the mouse spinal cord was isolated using TRIzol (Invitrogen) extraction and mRNA levels were determined by RT-qPCR using the iQSYBR Green supermix (Bio-Rad). See details in SI Materials and Methods. The protein fraction from each TRIzol sample was immunoblotted (using nitrocellulose membranes) and bound proteins detected using antibodies to mouse and human TDP-43 (ProteinTech 12892, 1:1,000) or mouse GAPDH (AbCam clone 6C5, 1:20,000).
Immunohistochemistry.
Tissue preparation for immunohistochemistry was performed as described previously (50). Next, 30-μm free-floating sections were stained using standard protocols previously established in our laboratory with the indicated antibodies. See SI Materials and Methods for details. Confocal images were acquired on a Nikon Eclipse laser scanning confocal microscope using the Nikon EZ-C1 software.
Morphometric Analysis and Quantification of Motor Axons.
Roots from lumbar level 5 of the spinal cord (L5) were dissected from three to four mice per genotype at 2 mo and 10–12 mo of age, and thin sections (0.75 μm) were cut, stained with Toluidine blue, and quantified as previously described (50). L5 axons are reported as mean ± SD.
Quantification of Upper and Lower Motor Neurons.
ChAT-positive ventral horn motor neurons were counted from at least 30 sections per animal (minimum of three animals per genotype) at 2 and 12 mo of age and are reported as average ± SD. Quantification of Ctip2+ neurons in cortex layer five from 10- to 12-mo-old animals was performed on six consecutive sections of frontal cortex (∼2.34 mm Bregma), in the motor M1 region in an area of 0.08 mm2 and are reported as the average number of motor neurons per square millimeter ± SEM. Matching sections were chosen for each set of mice.
Quantification of Neuromuscular Junctions.
A total of ∼1,000 neuromuscular junctions were counted from at least 10 sections of gastrocnemius from 10- to 12-mo-old animals. To obtain the number of neuromuscular junctions per section, the average number of neuromuscular junctions was divided by the number of sections counted per animal, with three to four animals per genotype. The average number of neuromuscular junctions per section was reported as mean ± SEM. Statistical analysis was performed using one-way ANOVA with Bonferroni’s post hoc test.
Animal Behavior and Electrophysiology.
These studies were carried out under protocols approved by the Institutional Animal Care and Use Committee of the University of California at San Diego and were in compliance with Association for Assessment of Laboratory Animal Care guidelines for animal use. All studies were performed in such a manner as to minimize group size and animal suffering. See details in SI Materials and Methods.
Nuclear-Cytosolic Fractionation.
Nuclear-cytosolic fractionation of cortex and spinal cords from young and old mice was performed as described previously (26, 62). See SI Materials and Methods for details. Equivalent volumes were prepared for immunoblotting with the indicated antibodies.
Sequential Biochemical Fractionation.
Sequential biochemical fractionation on cortex and spinal cord was performed as described previously (63). For detailed protocols, see SI Materials and Methods. Equivalent volumes of samples were separated on 4–12% Bis-Tris gradient gels for immunoblotting with the indicated antibodies.
Microarray.
Microarray data analysis was performed as previously described (11). For each microarray condition, the log2 ratio of skipping intensities to inclusion intensities was estimated using least-squares analysis. Significantly changing splicing events between TDP-43Q331K, TDP-43Q331K-low, TDP-43Wild-Type, and control nontransgenic mice were identified using a q-value < 0.05 and an absolute separation score > 0.3.
RT-PCR Validation of Splicing Targets Identified by Microarray.
RT-PCR was performed on cDNA generated from the cortex and spinal cords of 2-mo-old transgenic animals. See details in SI Materials and Methods. Gel imaging and quantification of the isoforms was performed using the Bio-Rad Chemidoc software. The intensity ratios between products including the cassette exon and skipping the cassette exon were averaged for a minimum of three biological replicates per genotype. Splicing alterations significantly different from the nontransgenic RNAs were determined with Student’s t test (*P < 0.05, **P < 0.01, and ***P < 0.001).
Supplementary Material
Acknowledgments
The authors thank Timothy Meerloo and Ying Jones (University of California at San Diego) and Janet Folmer (The Johns Hopkins University) for technical assistance in plastic thin sectioning and staining; Seiya Tokunaga, Sandra Lee, Anne Vetto, and Han Jin Park for technical assistance; and all members of the D.W.C. laboratory for helpful suggestions and discussion regarding the work. This work was supported by a Wellcome Trust grant (to D.W.C. and C.E.S.) and Grant NS069144 (to D.W.C.); salary support from the Ludwig Institute for Cancer Research (D.W.C.); University of California San Diego Genetics Training Grant National Institute of General Medical Sciences T32 GM008666 (to E.S.A.); National Institutes of Health Neuroplasticity of Aging Training Grant T32 AG 000216 (to S.-C.L.); a National Science Foundation Graduate Research Fellowship (to S.C.H.); a Milton Safenowitz Postdoctoral Fellowship from the Amyotrophic Lateral Sclerosis Association (to D.D.); a Muscular Dystrophy Association Career Development Award (to C.L.-T.); and National Institutes of Health Grants NS075449 and HG004659 (to G.W.Y.) and NS075216 (to M.P.). G.W.Y. is an Alfred P. Sloan Research Fellow.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1222809110/-/DCSupplemental.
References
- 1.Da Cruz S, Cleveland DW. Understanding the role of TDP-43 and FUS/TLS in ALS and beyond. Curr Opin Neurobiol. 2011;21(6):904–919. doi: 10.1016/j.conb.2011.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lomen-Hoerth C, Anderson T, Miller B. The overlap of amyotrophic lateral sclerosis and frontotemporal dementia. Neurology. 2002;59(7):1077–1079. doi: 10.1212/wnl.59.7.1077. [DOI] [PubMed] [Google Scholar]
- 3.Liscic RM, Grinberg LT, Zidar J, Gitcho MA, Cairns NJ. ALS and FTLD: Two faces of TDP-43 proteinopathy. Eur J Neurol. 2008;15(8):772–780. doi: 10.1111/j.1468-1331.2008.02195.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Neumann M, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314(5796):130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 5.Arai T, et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351(3):602–611. doi: 10.1016/j.bbrc.2006.10.093. [DOI] [PubMed] [Google Scholar]
- 6.Sreedharan J, et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319(5870):1668–1672. doi: 10.1126/science.1154584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gitcho MA, et al. TDP-43 A315T mutation in familial motor neuron disease. Ann Neurol. 2008;63(4):535–538. doi: 10.1002/ana.21344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kabashi E, et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet. 2008;40(5):572–574. doi: 10.1038/ng.132. [DOI] [PubMed] [Google Scholar]
- 9.Vance C, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323(5918):1208–1211. doi: 10.1126/science.1165942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kwiatkowski TJ, Jr, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323(5918):1205–1208. doi: 10.1126/science.1166066. [DOI] [PubMed] [Google Scholar]
- 11.Polymenidou M, et al. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat Neurosci. 2011;14(4):459–468. doi: 10.1038/nn.2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ou SH, Wu F, Harrich D, García-Martínez LF, Gaynor RB. Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. J Virol. 1995;69(6):3584–3596. doi: 10.1128/jvi.69.6.3584-3596.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Buratti E, et al. Nuclear factor TDP-43 and SR proteins promote in vitro and in vivo CFTR exon 9 skipping. EMBO J. 2001;20(7):1774–1784. doi: 10.1093/emboj/20.7.1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Winton MJ, et al. Disturbance of nuclear and cytoplasmic TAR DNA-binding protein (TDP-43) induces disease-like redistribution, sequestration, and aggregate formation. J Biol Chem. 2008;283(19):13302–13309. doi: 10.1074/jbc.M800342200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang HY, Wang IF, Bose J, Shen CK. Structural diversity and functional implications of the eukaryotic TDP gene family. Genomics. 2004;83(1):130–139. doi: 10.1016/s0888-7543(03)00214-3. [DOI] [PubMed] [Google Scholar]
- 16.Ayala YM, et al. Human, Drosophila, and C. elegans TDP43: Nucleic acid binding properties and splicing regulatory function. J Mol Biol. 2005;348(3):575–588. doi: 10.1016/j.jmb.2005.02.038. [DOI] [PubMed] [Google Scholar]
- 17.D’Ambrogio A, et al. Functional mapping of the interaction between TDP-43 and hnRNP A2 in vivo. Nucleic Acids Res. 2009;37(12):4116–4126. doi: 10.1093/nar/gkp342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Buratti E, et al. TDP-43 binds heterogeneous nuclear ribonucleoprotein A/B through its C-terminal tail: An important region for the inhibition of cystic fibrosis transmembrane conductance regulator exon 9 splicing. J Biol Chem. 2005;280(45):37572–37584. doi: 10.1074/jbc.M505557200. [DOI] [PubMed] [Google Scholar]
- 19.Sephton CF, et al. TDP-43 is a developmentally regulated protein essential for early embryonic development. J Biol Chem. 2010;285(9):6826–6834. doi: 10.1074/jbc.M109.061846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kraemer BC, et al. Loss of murine TDP-43 disrupts motor function and plays an essential role in embryogenesis. Acta Neuropathol. 2010;119(4):409–419. doi: 10.1007/s00401-010-0659-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu LS, et al. TDP-43, a neuro-pathosignature factor, is essential for early mouse embryogenesis. Genesis. 2010;48(1):56–62. doi: 10.1002/dvg.20584. [DOI] [PubMed] [Google Scholar]
- 22.Chiang PM, et al. Deletion of TDP-43 down-regulates Tbc1d1, a gene linked to obesity, and alters body fat metabolism. Proc Natl Acad Sci USA. 2010;107(37):16320–16324. doi: 10.1073/pnas.1002176107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ayala YM, et al. TDP-43 regulates its mRNA levels through a negative feedback loop. EMBO J. 2011;30(2):277–288. doi: 10.1038/emboj.2010.310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tsai KJ, et al. Elevated expression of TDP-43 in the forebrain of mice is sufficient to cause neurological and pathological phenotypes mimicking FTLD-U. J Exp Med. 2010;207(8):1661–1673. doi: 10.1084/jem.20092164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shan X, Chiang PM, Price DL, Wong PC. Altered distributions of Gemini of coiled bodies and mitochondria in motor neurons of TDP-43 transgenic mice. Proc Natl Acad Sci USA. 2010;107(37):16325–16330. doi: 10.1073/pnas.1003459107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wils H, et al. TDP-43 transgenic mice develop spastic paralysis and neuronal inclusions characteristic of ALS and frontotemporal lobar degeneration. Proc Natl Acad Sci USA. 2010;107(8):3858–3863. doi: 10.1073/pnas.0912417107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Igaz LM, et al. Dysregulation of the ALS-associated gene TDP-43 leads to neuronal death and degeneration in mice. J Clin Invest. 2011;121(2):726–738. doi: 10.1172/JCI44867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu YF, et al. Wild-type human TDP-43 expression causes TDP-43 phosphorylation, mitochondrial aggregation, motor deficits, and early mortality in transgenic mice. J Neurosci. 2010;30(32):10851–10859. doi: 10.1523/JNEUROSCI.1630-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wegorzewska I, Bell S, Cairns NJ, Miller TM, Baloh RH. TDP-43 mutant transgenic mice develop features of ALS and frontotemporal lobar degeneration. Proc Natl Acad Sci USA. 2009;106(44):18809–18814. doi: 10.1073/pnas.0908767106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stallings NR, Puttaparthi K, Luther CM, Burns DK, Elliott JL. Progressive motor weakness in transgenic mice expressing human TDP-43. Neurobiol Dis. 2010;40(2):404–414. doi: 10.1016/j.nbd.2010.06.017. [DOI] [PubMed] [Google Scholar]
- 31.Zhou H, et al. Transgenic rat model of neurodegeneration caused by mutation in the TDP gene. PLoS Genet. 2010;6(3):e1000887. doi: 10.1371/journal.pgen.1000887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Swarup V, et al. Pathological hallmarks of amyotrophic lateral sclerosis/frontotemporal lobar degeneration in transgenic mice produced with TDP-43 genomic fragments. Brain. 2011;134(Pt 9):2610–2626. doi: 10.1093/brain/awr159. [DOI] [PubMed] [Google Scholar]
- 33.Cannon A, et al. Neuronal sensitivity to TDP-43 overexpression is dependent on timing of induction. Acta Neuropathol. 2012;123(6):807–823. doi: 10.1007/s00401-012-0979-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu YF, et al. Expression of mutant TDP-43 induces neuronal dysfunction in transgenic mice. Mol Neurodegener. 2011;6:73. doi: 10.1186/1750-1326-6-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang C, Tong J, Bi F, Zhou H, Xia XG. Mutant TDP-43 in motor neurons promotes the onset and progression of ALS in rats. J Clin Invest. 2012;122(1):107–118. doi: 10.1172/JCI59130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Van Deerlin VM, et al. TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: A genetic and histopathological analysis. Lancet Neurol. 2008;7(5):409–416. doi: 10.1016/S1474-4422(08)70071-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Igaz LM, et al. Enrichment of C-terminal fragments in TAR DNA-binding protein-43 cytoplasmic inclusions in brain but not in spinal cord of frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Am J Pathol. 2008;173(1):182–194. doi: 10.2353/ajpath.2008.080003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Neumann M, et al. Phosphorylation of S409/410 of TDP-43 is a consistent feature in all sporadic and familial forms of TDP-43 proteinopathies. Acta Neuropathol. 2009;117(2):137–149. doi: 10.1007/s00401-008-0477-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Arai T, et al. Phosphorylated and cleaved TDP-43 in ALS, FTLD and other neurodegenerative disorders and in cellular models of TDP-43 proteinopathy. Neuropathology. 2010;30(2):170–181. doi: 10.1111/j.1440-1789.2009.01089.x. [DOI] [PubMed] [Google Scholar]
- 40.Zhang YJ, et al. Aberrant cleavage of TDP-43 enhances aggregation and cellular toxicity. Proc Natl Acad Sci USA. 2009;106(18):7607–7612. doi: 10.1073/pnas.0900688106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang YJ, et al. Progranulin mediates caspase-dependent cleavage of TAR DNA binding protein-43. J Neurosci. 2007;27(39):10530–10534. doi: 10.1523/JNEUROSCI.3421-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Igaz LM, et al. Expression Of TDP-43 C-terminal fragments in vitro recapitulates pathological features of TDP-43 proteinopathies. J Biol Chem. 2009;284(13):8516–8524. doi: 10.1074/jbc.M809462200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Elden AC, et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature. 2010;466(7310):1069–1075. doi: 10.1038/nature09320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Voigt A, et al. TDP-43-mediated neuron loss in vivo requires RNA-binding activity. PLoS ONE. 2010;5(8):e12247. doi: 10.1371/journal.pone.0012247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Johnson BS, McCaffery JM, Lindquist S, Gitler AD. A yeast TDP-43 proteinopathy model: Exploring the molecular determinants of TDP-43 aggregation and cellular toxicity. Proc Natl Acad Sci USA. 2008;105(17):6439–6444. doi: 10.1073/pnas.0802082105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ash PE, et al. Neurotoxic effects of TDP-43 overexpression in C. elegans. Hum Mol Genet. 2010;19(16):3206–3218. doi: 10.1093/hmg/ddq230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Borchelt DR, et al. A vector for expressing foreign genes in the brains and hearts of transgenic mice. Genet Anal. 1996;13(6):159–163. doi: 10.1016/s1050-3862(96)00167-2. [DOI] [PubMed] [Google Scholar]
- 48.Wang J, et al. Coincident thresholds of mutant protein for paralytic disease and protein aggregation caused by restrictively expressed superoxide dismutase cDNA. Neurobiol Dis. 2005;20(3):943–952. doi: 10.1016/j.nbd.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 49.Yazawa I, et al. Mouse model of multiple system atrophy alpha-synuclein expression in oligodendrocytes causes glial and neuronal degeneration. Neuron. 2005;45(6):847–859. doi: 10.1016/j.neuron.2005.01.032. [DOI] [PubMed] [Google Scholar]
- 50.Ilieva HS, et al. Mutant dynein (Loa) triggers proprioceptive axon loss that extends survival only in the SOD1 ALS model with highest motor neuron death. Proc Natl Acad Sci USA. 2008;105(34):12599–12604. doi: 10.1073/pnas.0805422105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Du H, et al. Aberrant alternative splicing and extracellular matrix gene expression in mouse models of myotonic dystrophy. Nat Struct Mol Biol. 2010;17(2):187–193. doi: 10.1038/nsmb.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sugnet CW, et al. Unusual intron conservation near tissue-regulated exons found by splicing microarrays. PLOS Comput Biol. 2006;2(1):e4. doi: 10.1371/journal.pcbi.0020004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Van Damme P, et al. Astrocytes regulate GluR2 expression in motor neurons and their vulnerability to excitotoxicity. Proc Natl Acad Sci USA. 2007;104(37):14825–14830. doi: 10.1073/pnas.0705046104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Van Damme P, Braeken D, Callewaert G, Robberecht W, Van Den Bosch L. GluR2 deficiency accelerates motor neuron degeneration in a mouse model of amyotrophic lateral sclerosis. J Neuropathol Exp Neurol. 2005;64(7):605–612. doi: 10.1097/01.jnen.0000171647.09589.07. [DOI] [PubMed] [Google Scholar]
- 55.Pizzi M, et al. Neuroprotection by metabotropic glutamate receptor agonists on kainate-induced degeneration of motor neurons in spinal cord slices from adult rat. Neuropharmacology. 2000;39(5):903–910. doi: 10.1016/s0028-3908(99)00257-9. [DOI] [PubMed] [Google Scholar]
- 56.Wu LS, Cheng WC, Shen CK. Targeted depletion of TDP-43 expression in the spinal cord motor neurons leads to the development of amyotrophic lateral sclerosis-like phenotypes in mice. J Biol Chem. 2012;287(33):27335–27344. doi: 10.1074/jbc.M112.359000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Johnson BS, et al. TDP-43 is intrinsically aggregation-prone, and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity. J Biol Chem. 2009;284(30):20329–20339. doi: 10.1074/jbc.M109.010264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cushman M, Johnson BS, King OD, Gitler AD, Shorter J. Prion-like disorders: Blurring the divide between transmissibility and infectivity. J Cell Sci. 2010;123(Pt 8):1191–1201. doi: 10.1242/jcs.051672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fuentealba RA, et al. Interaction with polyglutamine aggregates reveals a Q/N-rich domain in TDP-43. J Biol Chem. 2010;285(34):26304–26314. doi: 10.1074/jbc.M110.125039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang IF, et al. The self-interaction of native TDP-43 C terminus inhibits its degradation and contributes to early proteinopathies. Nat Commun. 2012;3:766. doi: 10.1038/ncomms1766. [DOI] [PubMed] [Google Scholar]
- 61.Zhang T, Hwang HY, Hao H, Talbot C, Jr, Wang J. Caenorhabditis elegans RNA-processing protein TDP-1 regulates protein homeostasis and life span. J Biol Chem. 2012;287(11):8371–8382. doi: 10.1074/jbc.M111.311977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ditsworth D, Zong WX, Thompson CB. Activation of poly(ADP)-ribose polymerase (PARP-1) induces release of the pro-inflammatory mediator HMGB1 from the nucleus. J Biol Chem. 2007;282(24):17845–17854. doi: 10.1074/jbc.M701465200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Giasson BI, et al. Neuronal alpha-synucleinopathy with severe movement disorder in mice expressing A53T human alpha-synuclein. Neuron. 2002;34(4):521–533. doi: 10.1016/s0896-6273(02)00682-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.