

Abstract
A human polyomavirus was recently discovered in Merkel cell carcinoma (MCC) specimens. The Merkel cell polyomavirus (MCPyV) genome undergoes clonal integration into the host cell chromosomes of MCC tumors and expresses small T antigen and truncated large T antigen. Previous studies have consistently reported that MCPyV can be detected in approximately 80% of all MCC tumors. We sought to increase the sensitivity of detection of MCPyV in MCC by developing antibodies capable of detecting large T antigen by immunohistochemistry. In addition, we expanded the repertoire of quantitative PCR primers specific for MCPyV to improve the detection of viral DNA in MCC. Here we report that a novel monoclonal antibody detected MCPyV large T antigen expression in 56 of 58 (97%) unique MCC tumors. PCR analysis specifically detected viral DNA in all 60 unique MCC tumors tested. We also detected inactivating point substitution mutations of TP53 in the two MCC specimens that lacked large T antigen expression and in only 1 of 56 tumors positive for large T antigen. These results indicate that MCPyV is present in MCC tumors more frequently than previously reported and that mutations in TP53 tend to occur in MCC tumors that fail to express MCPyV large T antigen.
Introduction
Merkel cell carcinoma (MCC) is a rare skin cancer with high risk for metastasis and death (1). MCC has features of a neuroendocrine tumor, with expression of synaptophysin (SYP) and chromogranin A (CHGA) (2). In addition, most MCCs express cytokeratin 20 (KRT20; CK20), which distinguishes it from other neuroendocrine tumors (3, 4). Risk factors for developing MCC include advanced age, prolonged life-long UV exposure, and an immunocompromised state from solid organ transplantation or HIV-1 infection (5–7). In addition, there is an increased incidence of MCC in patients with chronic lymphocytic leukemia (CLL) and other hematologic proliferative disorders (8, 9).
Recognition of the increased risk for developing MCC in immunocompromised patients led to a search for a pathogenic cause and the discovery of the Merkel cell polyomavirus (MCPyV). Sequencing of RNA from 4 MCC specimens revealed a novel transcript distantly related to large T antigen of the polyomavirus family (10). Sequencing of the complete MCPyV genome revealed that it had features typical of a polyomavirus, including a circular double-stranded DNA genome of 5,387 bp; an early region encoding genes for large and small T antigens; a late region with genes for the viral coat proteins VP1, VP2, and VP3; and a regulatory region that contained the viral origin of replication and a bidirectional promoter for the early and late genes.
MCC cells do not typically harbor episomal MCPyV viral DNA. Instead, the MCPyV viral genome is clonally integrated in the tumor chromosomal DNA, as first revealed by Southern blotting (10). Sequencing of the integrated MCPyV viral genomes from MCC has revealed mutations and deletions of the early region that result in truncation of the large T antigen to encode only the N-terminal 260–455 residues instead of the full-length 817 residues, while the small T antigen coding sequence remains intact (10–12). It is not known whether Merkel cells are permissive for MCPyV viral replication. However, expression of full-length MCPyV T antigen in a MCC cell line has been reported to promote replication of an integrated viral origin of replication (11, 13).
Since the original report, several studies have confirmed the presence of the virus in approximately 80% of MCC (14–17). In most studies, MCPyV DNA has been detected by PCR amplification of DNA extracted from formalin-fixed, paraffin-embedded (FFPE) tissue samples. This approach has led to the recognition that the number of copies of viral DNA integrated in a tumor can vary from less than one to several hundred copies per cell (18–20). In addition to detection of viral DNA in MCC, immunohistochemistry with antibodies specific for MCPyV large T antigen and small T antigen has been reported (20, 21). Similarly, immunohistochemistry studies of FFPE sections of MCC report that approximately 80% of MCCs express MCPyV large T antigen (17, 22).
Based on homology to other polyomaviruses, the MCPyV large and small T antigens are predicted to be oncogenic and contribute directly to the carcinogenesis of MCC. Given that approximately 80% of MCCs contain MCPyV viral DNA and express MCPyV large T antigen, the question arises whether the remaining MCC can be distinguished by any feature other than the absence of MCPyV. If an MCC tumor does not contain MCPyV DNA or express T antigens, then it would be predicted that these tumors contain mutations in oncogenes or tumor suppressor genes that contribute to oncogenesis. To date, few mutations in known oncogenes or tumor suppressors have been reported in MCC. Inactivating mutations in the p53 tumor suppressor gene (TP53) have been reported to occur rarely in MCC (23, 24). In addition, activating mutations in PIK3CA have also been reported in a few MCC tumors (25, 26).
This study was performed to determine whether the sensitivity of detection of MCPyV in MCC could be improved, with the ultimate goal of enabling high-confidence discrimination between virus-positive and -negative tumors. To accomplish this goal, we analyzed 75 archival FFPE MCC tumor specimens obtained from 60 patients. We performed immunohistochemistry staining with a newly developed mouse monoclonal antibody specific for MCPyV large T antigen. We also developed several quantitative PCR (qPCR) primer and probe sets to determine the viral copy number per tumor cell. Furthermore, we performed mass spectrometry based genotyping of 112 oncogenes and tumor suppressor genes from DNA extracted from these same tumor samples.
Results
Cases.
FFPE specimens corresponding to 60 patients presenting with MCC to a referral specialty clinic were included in this study (Supplemental Table 1; supplemental material available online with this article; doi: 10.1172/JCI64116DS1). The patients included 25 females and 35 males who ranged in age from 45 to 90 years (median, 74). Fourteen patients had a prior history, comorbidity, or medication consistent with an immunocompromised state from malignancy, autoimmune disease, or solid organ transplantation.
From this cohort, a total of 75 archival MCC samples were available for study that included 44 primary cutaneous, 12 metastatic lymph node, and 6 metastatic visceral tumors obtained at initial presentation and 13 recurrent tumors obtained after initial therapy (Supplemental Table 1). Review of clinical pathology reports indicated that 59 of 60 MCC patient specimens were positive for CK20 immunostaining. Only one specimen, from unique patient identifier (UPI) 31, had no reported CK20 staining. DNA was extracted from all samples, and FFPE sections from 58 patients corresponding to 72 samples were available for immunohistochemistry staining.
Western blotting of MCPyV large T antigen.
The CM2B4 mouse monoclonal antibody was generated against a peptide corresponding to residues 116–129 of MCPyV large T antigen contained within the second exon of large T antigen (20). For additional antibodies against MCPyV T antigens, we generated a mouse monoclonal antibody (Ab3) against a fragment of MCPyV large T antigen corresponding to the N-terminal 260 residues (i.e., 1–260) produced in bacteria (Figure 1A). Additional experiments confirmed that Ab3 recognized large T antigen and not small T antigen, indicating that residues between 79 and 260 were required for binding. Epitope mapping with an overlapping series of linear peptides corresponding to the entire large T antigen coding region indicated that Ab3 and CM2B4 could bind to the same peptides, suggesting that they recognize a similar epitope on MCPyV large T antigen (Yanis L. Tolstov, Yuan Chang, and Patrick S. Moore, University of Pittsburgh Cancer Institute, Pittsburgh, Pennsylvania, USA, personal communication).
Figure 1. Detection of MCPyV in MCC cell lines.

(A) Diagram of the MCPyV early region. Numbers correspond to nucleotide position in the MCPyV genome (HM355825). Rectangles show domain structure of large (LT) and small T (ST) antigen with approximate position of epitopes for CM2B4 and Ab3 antibodies and LXCXE (RB-binding) motif. Ab3 was generated against recombinant LT 1–260 corresponding to nucleotides 196–429 and 861–1,406. MCPyV isolated from MCC tumor contains truncations of LT that delete the helicase domain. PCR primer sets with nucleotide boundaries are indicated. (B) Western blot with Ab3 or CM2B4 of cell lysates prepared from MCC cell lines showing a predominant signal for truncated large T antigen between 36 and 50 kDa. U-2OS-LT contains a full-length large T antigen cDNA. Actin blot indicates protein loading. (C) MCPyV genome copy number in cell lines with indicated qPCR primer sets. All values were normalized to MKL-2. Error bars represent mean ± SEM.
We compared the Western blot signal obtained with Ab3 and CM2B4 using lysates prepared from 4 MCC cell lines, including MKL-1, MKL-2, MS-1, and WaGa. Two cell lines, UISO and U-2OS, without MCPyV as well as a U-2OS derivative containing a cDNA for MCPyV large T antigen served as controls (Figure 1B). Both Ab3 and CM2B4 detected a signal corresponding to MCPyV large T antigen in the 4 MCC cell lines but not in UISO or U-2OS cells. Notably, the size of the large T antigen varied in the 4 MCC cell lines, reflecting differences in the truncating mutations (27). The U-2OS cell line with large T antigen expressed full-length as well as a smaller version likely corresponding to the 57-kDa alternatively spliced form (11).
Immunohistochemistry staining of MCC specimens.
Sections from MCC tumor specimens were stained with Ab3 and CM2B4 (20). Both antibodies were optimized for signal strength and background staining using MCPyV-positive sections of MCC. Sections of MCC corresponding to 58 unique patients were available for immunostaining with Ab3 and 57 with CM2B4. Scoring was based on the intensity of signal, with 3+ indicating strong staining; 2+, moderate; 1+, weak; and 0, absent.
A total of 46 of 57 (81%) unique patient specimens stained positive with CM2B4, and 11 were negative (Figure 2A and Supplemental Table 1). When used at the lower concentration, Ab3 (0.6 μg/ml) detected a specific signal in 52 of 57 (91%) specimens. Specimens from four additional cases (UPI 38, 45, 46, and 57) stained positive when Ab3 was used at the higher concentration (2.4 μg/ml), raising the total Ab3 positive to 56 of 58 cases (97%) tested (Figure 2A and Supplemental Table 1).
Figure 2. Detection of MCPyV in MCC.

(A) Number of cases with immunohistochemical staining intensity for Ab3 and CM2B4. 3+, strong; 2+, moderate; 1+, weak, 0, negative. Ab3: n = 58; CM2B4: n = 57. (B) Histogram showing number of cases with highest MCPyV copy number value for any qPCR primer set. Copy number of viral genomes per tumor cell indicates less than or equal to the value indicated. n = 60.
In most cases, both antibodies detected a nuclear signal surrounded by a small rim of unstained cytoplasm (Figure 3A, UPI 26). Nuclear and cytoplasmic signals were detected in a few samples that strongly stained with Ab3 and CM2B4 (Figure 3A, UPI 18). In several cases, staining with higher concentrations of Ab3 increased the strength of the signal. For example, specimens from UPI 37 stained weakly with Ab3 at 0.6 μg/ml and moderately well with Ab3 at 2.4 μg/ml but negative when stained with CM2B4 (Figure 3B).
Figure 3. Comparison of Ab3 and CM2B4 immunohistochemistry staining of MCC.

(A) UPI 26 and UPI 18: 3+ staining with CM2B4 and Ab3; UPI 30: primary and metastatic (Met) lymph node–negative staining with CM2B4 and 2+ with Ab3; UPI 27: 1+ staining with CM2B4 and Ab3. (B) UPI 37: no staining with CM2B4, 1+ with Ab3 at 0.6 μg/ml, and 2+ with Ab3 at 2.4 μg/ml. (C) UPI 15 and UPI 42: negative staining with CM2B4 and Ab3. Original magnification, ×400; scale bar: 50 μm.
Samples from a total of 38 cases stained strongly (3+) or moderately (2+) with CM2B4, and each of these also stained strongly or moderately with Ab3 (Figure 2A and Supplemental Table 1). In contrast, Ab3 stained a total of 50 cases strongly or moderately. Samples from a total of 6 cases stained weakly with Ab3, and 2 of these also stained weakly with CM2B4, including UPI 27 (Figure 3A) and UPI 14. Ab3 detected a specific signal in 9 samples that was not detected with CM2B4 (for example, UPI 30 and UPI 37; Figure 3, A and B). Conversely, there were no specimens that stained positive with CM2B4 that were negative with Ab3. Overall, the staining intensity for large T antigen was higher with Ab3 compared with CM2B4 in 27 of 57 (47%) samples. Importantly, when different specimens from the same patient were stained, we observed consistent staining intensity between them. For example, the primary tumor and a metastatic lymph node from UPI 30 (Figure 3A) both stained positive with Ab3 but negative with CM2B4. Samples from only two cases, UPI 15 and UPI 42, were not positive when stained with either Ab3 or CM2B4 (Figure 3C). Overall, Ab3 staining intensity was significantly correlated with CM2B4 staining (Table 1).
Table 1.
Comparison of immunohistochemistry and qPCR values
To rule out the possibility of nonspecific staining with Ab3, we performed staining on sections of discarded tonsillar tissue (5 unique specimens) and reactive lymph nodes (2) from unrelated cases. Staining with Ab3 revealed no detectable signal from these 7 different normal tissue specimens. Representative stains with Ab3 of normal lymph node and tonsil are shown in Figure 4A. We tested 10 unique GI neuroendocrine tumors and detected no signal by immunohistochemistry staining with Ab3 (Supplemental Table 2). Two representative cases are shown in Figure 4B. In addition, immunostaining of 8 unique small cell lung carcinomas with CM2B4 and Ab3 were negative (Supplemental Table 2). Two representative cases are shown in Figure 4, C and D. Sections from an MCC case (UPI 36) served as a positive control for Ab3 and C2B4 staining performed at the same time as staining of the small cell lung carcinoma specimens (Figure 4E).
Figure 4. Negative staining with Ab3 of normal lymph node and tonsil and GI neuroendocrine and small cell lung carcinomas.
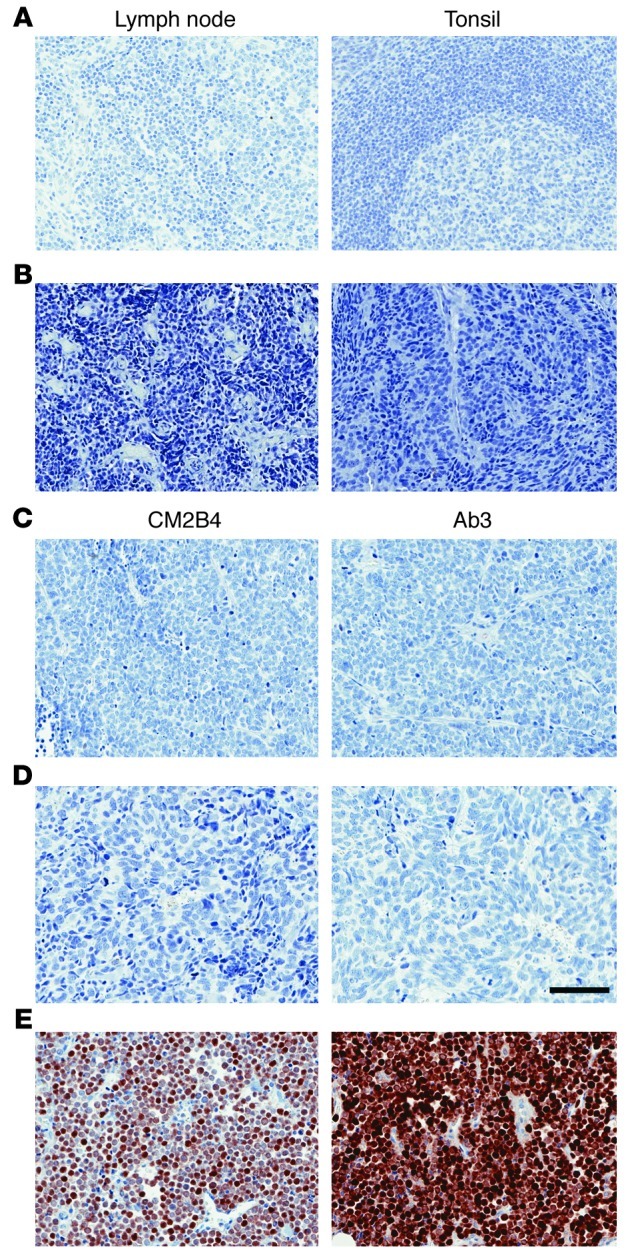
(A) Lymph node and tonsil stained with Ab3. (B) GI neuroendocrine tumors stained with Ab3. Results are shown for cases C (left) and E (right). (C) SCLC specimen Q and (D) N stained with CM2B4 or Ab3. (E) MCC (UPI 36) stained with CM2B4 and Ab3. Original magnification, ×200; scale bar: 100 μm.
qPCR detection of MCPyV DNA.
We developed a qPCR assay to determine the copy number of viral genomes per tumor cell. Two different qPCR primer sets were adapted from a previous report, and 3 additional quantitative PCR primer and probe sets were generated (15). All primer and probe sets were designed to amplify sequences corresponding to nucleotides 196–1,407 of MCPyV encoding small T antigen and the N-terminal 260 residues of LT (Table 2). This region of MCPyV genome was selected because it was present within all sequenced MCPyV variants obtained from MCC tumors. We designed 3 primer sets corresponding to the second exon (LT2, Set6, and Set7) of large T antigen and 2 primer sets (LT3 and Set9) corresponding to the unique region of small T antigen (Figure 1A). These primer sequences were identical to most MCPyV sequences and were not cross-reactive to any other DNA sequence in the NCBI database. Primers specific for the human TPO gene were used to normalize for diploid gene copy number, since this gene was reported to have infrequent copy number alterations in a study of MCC using array comparative genomic hybridization (28).
Table 2.
qPCR primer and probes

To validate the PCR primers, we tested them against DNA extracted from the 4 MCPyV-positive MCC cell lines and the U-2OS cell line containing the cDNA for MCPyV large T antigen (Figure 1C). As expected, no amplification with any MCPyV primer set was observed in DNA extracted from the parental U-2OS cell line. In contrast, the U-2OS cell line containing the large T antigen cDNA was amplified with the 3 primer sets specific for large T antigen (LT2, Set6, and Set7) but not with primer sets LT3 and Set9 specific for small T antigen (Figure 1C).
All 5 primer sets amplified DNA from the 4 MCC cell lines tested. The MKL-2 cell line contained the lowest copy number, with each of the MCPyV primer sets yielding Ct values approximately equal to the values corresponding to the TPO gene. Based on this observation, we normalized the values obtained from the MKL-2 cell line to be equivalent to a single copy of the viral genome per cell to compare with all other samples (Supplemental Table 1). The MS-1 cell line contained 3–5 copies of the viral genome per cell depending on the results from the 5 individual primer sets, while the MKL-1 cell line contained an average of 21 copies per cell. The WaGa cell line showed some variation in copy number, ranging from 12 to 52 copies per cell depending on the primer used. Of note, the relative MCPyV copy number in the 4 MCC cell lines corresponded roughly to the relative strength of the large T antigen signal by Western blotting, with the highest levels detected in WaGa and MKL-1 and the lowest in MKL-2 cells (Figure 1B).
Using these validated primer and probe sets, we performed qPCR with DNA extracted from the same FFPE tumor blocks used for immunohistochemistry. To conserve tumor DNA samples, we initially tested 3 MCPyV primer/probe sets: LT2, LT3, and Set7, together with TPO in triplicate for all samples. The copy number for each primer set was compared with the TPO value and normalized to the values obtained from the MKL-2 cell line (Supplemental Table 1). When the results indicated that the viral copy number per cell was less than 1.0 for any of the LT2, LT3, and Set7 primer sets, then Set6 and Set9 were also tested. Overall, the MCPyV genome copy number obtained from 75 specimens corresponding to 60 unique patients using the 5 primer/probe sets varied from less than 0.001 to 3,203 copies per cell.
Most samples showed good agreement in copy number when different specimens from the same patient were tested (Supplemental Table 1). Furthermore, there was good agreement for a given specimen when different primer sets were tested (Table 1). However, there were several specimens that showed agreement between 2 or more primer sets but not with other sets. For example, UPI 59 showed copy number values between 5 and 15 with LT2, LT3, Set6, and Set9 but less than 0.1 for Set7. Remarkably, UPI 11 revealed values more than 2,000 with LT3, Set7, and Set9 but less than 0.1 with LT2 and Set6.
For 48 of 60 cases (80%), at least one primer set detected greater than 2 copies per cell, consistent with the clonal integration of MCPyV DNA in every tumor cell (Supplemental Table 1 and Figure 2B). Of the remaining 12 cases with less than 2 copies of MCPyV genome detected, 7 cases had viral copy number greater than 0.1 detected with at least one primer set. For 2 of these cases, UPI 15 and 42, immunohistochemistry staining for large T antigen was negative with both antibodies (Figure 3C). An additional 5 cases (UPI 27, 47, 49, 57, and 60) yielded viral copy number values greater than 0.03 but less than 0.1. Despite the low copy number values, samples from all 5 of these cases were stained positive with Ab3, including 3 that stained moderately (2+). In addition, samples from 2 of these cases also stained positive with CM2B4.
To rule out nonspecific amplification of DNA using MCPyV-specific primers from tissues, we extracted DNA from FFPE tissue blocks of 3 tonsillar and 3 reactive lymph nodes from unrelated cases. We used DNA extracted from the MKL-2 cell line as a positive control. From all control tissues, we were able to amplify DNA using the primer and probe set for the TPO gene with Ct values similar to MKL-2 DNA. In contrast, we found no signal for MCPyV with any of the 5 primers and probe sets from the tonsillar or reactive lymph node specimens. These results indicate that viral copy numbers as low as 0.03 (MCC UPI 60) were specific for detection of MCPyV DNA. Furthermore, the absence of staining with Ab3 in normal tonsil and lymph node sections as well as in GI neuroendocrine and small cell lung carcinoma specimens support the conclusion that positive staining in MCC was specific for the presence of MCPyV large T antigen.
We prepared a nonredundant list of patient values using the highest reported value for each assay including immunohistochemistry and qPCR and then performed a pairwise Spearman rank correlation. As shown in Table 1, we observed a significant correlation between every combination of antibody staining and qPCR primer sets. The highest degree of correlation was between Ab3 and CM2B4 staining intensity, as well between qPCR values for LT3 and Set7, LT3 and Set9, and LT2 and Set7. No differences were observed in overall survival or time to disease progression in patients with high versus low MCPyV copy number or large T antigen expression (data not shown).
OncoMap.
We employed a mass spectrometry–based genotyping method to detect specific point substitution mutations in 112 commonly mutated oncogenes and tumor suppressor genes (29, 30). This approach was designed to detect more than 1,000 polymorphic and mutated alleles corresponding to these genes. DNA isolated from the 60 unique patients encompassing a total of 75 samples plus the 4 MCC cell lines were evaluated. After initial screening, all potential mutations were validated for the entire tumor set by homogenous mass extension (hME) (29, 30).
From 75 tumor specimens, only 4 tumor specimens contained a validated point mutation in any of the 112 oncogenes and tumor suppressor genes evaluated (Supplemental Table 1). Two samples with negative staining for MCPyV large T antigen by immunohistochemistry with Ab3 and CM2B4 and PCR copy number less than 0.7 for all 5 primer sets contained an inactivating point mutation, R248Q, in TP53 (UPI 42 and 15). In addition, a tumor specimen (UPI 18) with strong staining for large T antigen with both Ab3 and CM2B4 and copy number between 2 and 4 contained a different inactivating point substitution mutation, R273C, in TP53. The presence of inactivating point mutations in p53 in both tumors without large T antigen expression and in only 1 of 58 specimens that expressed large T antigen is highly significant (Fisher’s exact P = 0.0017). A fourth tumor specimen (UPI 38) contained a point substitution in ATM P604S (31). Notably, none of the other tumor specimens nor the 4 MCC cell lines showed the presence of any additional point mutations or polymorphisms tested in this panel.
Discussion
The discovery of MCPyV and the detection of viral DNA and T antigens in MCC provided an important new insight into the oncogenesis of this highly aggressive cancer (10, 20). Since this initial discovery, many centers from around the world have confirmed the presence of MCPyV in approximately 80% of MCCs. A variety of methods have been used to detect the presence of the virus, including Southern blot analysis (10), PCR amplification of viral DNA, DIPS (detection of integrated polyomavirus sequences), in situ hybridization (16), DNA sequencing by hybrid capture (12), RT-PCR, and immunohistochemistry of the viral T antigens (20).
One goal of this study was to determine whether detection of the MCPyV and viral gene products could be improved. A new monoclonal antibody, Ab3, was developed that has markedly increased sensitivity in detecting MCPyV large T antigen (97%) in MCC compared with CM2B4 (80%) (20). In addition to its improved sensitivity in detecting MCPyV large T antigen expression in MCC, Ab3 does not stain lymphocytes, even when used at higher concentrations or when staining tonsillar and reactive lymph node tissue specimens. The lack of nonspecific staining may be especially useful in clinical specimens, since MCC tumors frequently contain infiltrating lymphocytes (32). Furthermore, Ab3 did not stain tumor specimens from GI neuroendocrine tumors or small cell lung cancer, supporting its usefulness as a clinical biomarker for MCC. It should be noted that CK20 staining was positive in 59 of the 60 cases tested, and the CK20 status of the remaining case was not tested. Therefore, it is not known whether Ab3 can detect MCPyV large T antigen in the rare cases of CK20-negative MCC.
Our study is consistent with an earlier report that the CM2B4 antibody could not detect MCPyV large T antigen in every case of MCC (33). It was noted that the CM2B4 antibody failed to detect large T antigen in several cases of MCC but that many of these negative specimens stained positive with the monoclonal antibody CM5E1 specific for MCPyV small T antigen (33). The combined use of CM2B4 and CM5E1 led to detection of MCPyV T antigens in 47 of 51 (92%) cases of MCC, whereas CM2B4 alone detected large T antigen in 75% of cases (33). Our study is compatible with this small T antigen expression study, where use of CM2B4 alone underestimated the fraction of MCC cases that express the MCPyV T antigens.
To date, every report has detected MCPyV DNA in a significant proportion of MCC tumor specimens in their cohorts. Using PCR and related methods, most centers have reported that approximately 80% of MCCs contain detectable amounts of MCPyV viral DNA. In our study, more than 0.1 viral genome per cell was detected in 55 of 60 cases (92%), consistent with clonal integration of MCPyV DNA. The slightly higher than expected frequency of MCPyV DNA could be related to the use of 5 unique primer sets, which thereby increased the chances of detection. In the 5 remaining cases, MCPyV DNA was detected in a range from 0.03 to 0.08 copies per cell. These values exceeded those obtained from qPCR amplification of normal lymph node and tonsil tissue. This is consistent with a prior study that observed 60-fold-higher levels of MCPyV DNA in MCC specimens compared with the levels found in normal tissues (19). Therefore, in this series of MCC, there was evidence for specific detection of MCPyV DNA in every case. Further evidence supporting this conclusion is that in all 5 cases of MCC with less than 0.1 copy viral genome per cell, expression of large T antigen was detected with Ab3, and 2 also stained with CM2B4.
It should be noted that studies with extracted DNA from fresh frozen MCC samples have detected a higher frequency of MCPyV than studies with extracted DNA from FFPE tumor blocks. For example, one study detected MCPyV in all 12 cases of MCC when DNA was isolated from fresh frozen tumor specimens and in only 9 of 20 cases when DNA was extracted from FFPE specimens (34). It is likely that the formalin fixation process, long-term storage, and harsh extraction methods of archival MCC specimens reduce the likelihood of detection of MCPyV DNA by qPCR. Furthermore, the frequent mutations and polymorphisms observed in MCPyV DNA obtained from MCC reduce the efficiency of detection by qPCR (10).
The results of qPCR amplification revealed a large variation in MCPyV genome copy number per cell in MCC. Peak values ranged from 0.03 to several thousand copies per cell. Notably, we estimated copy number based on the assumption that the MKL-2 cell line had a single copy of MCPyV DNA per cell, since the Ct values for MCPyV were similar to those obtained for TPO. This assumption may have underestimated the copy number of MCPyV in the MKL-2 cell line, thereby systematically reducing the estimate of viral copy number in all other specimens. For example, if the MKL-2 tumor cell line contains 2 copies of MCPyV DNA per cell, then each tumor specimen would contain twice as many viral copies than we have reported here. In addition, it should be noted that normalization of each tumor specimen to its corresponding TPO DNA content may not be accurate in all cases, since some tumors may have undergone copy number alterations of this gene (28).
We observed a significant correlation in the intensity of immunohistochemistry staining with Ab3 and CM2B4 relative to the copy number of MCPyV per cell. Given that MCPyV integration occurs randomly in the host cell DNA, expression of the MCPyV T antigens is likely dependent on the local chromatin accessibility to transcription factors and RNA polymerases. Expression of MCPyV T antigens is likely to be higher when the viral DNA is integrated into open chromatin regions than into heterochromatin regions even when the viral copy number may be very high.
Determination of point substitution mutations in 112 oncogenes and tumor suppressor genes revealed the surprising result that most tumor samples (56 of 60; 93%) did not contain any point substitution mutations in the genes surveyed. In addition, we did not detect any mutations in 4 MCC cell lines that contain MCPyV. Consistent with previous studies, we did not find activating mutations in PDGFRA or KIT (23, 35–38). We did identify 3 tumors with p53 point substitutions, most notably in 2 specimens with absent expression of MCPyV large T antigen. Notably, the MKL-1 cell line had been reported to express wild-type p53 transcript (39). Other studies have reported infrequent p53 mutations (24, 40, 41). A previous study found p53 mutations in 11 of 23 MCC tumors that had no detectable large T antigen expression by immunohistochemistry using CM2B4 antibody and absence of p53 mutations in 16 large T antigen–positive MCC tumors (42). The OncoMap genotyping approach does not detect large deletions, and therefore it is possible that some MCC tumors in our study contained deletions of the TP53 gene. It should be noted that cervical cancers with evidence for human papilloma virus (HPV) typically do not contain p53 mutations, while HPV-negative cervical cancers often contain p53 mutations (43). Thus, while all MCC may contain MCPyV viral DNA, if the expression of the viral T antigens is absent, tumors may be selected for the presence of inactivating p53 mutations.
It should be noted that integration of MCPyV DNA into MCC typically results in truncation of the large T antigen viral oncogene. The mutations preserve the retinoblastoma protein binding or LXCXE motif but disrupt the helicase domain of large T antigen (11). The helicase domain of the related polyomavirus SV40 large T antigen binds specifically to p53 (44). It has not been reported whether wild-type, full-length MCPyV large T antigen can bind to p53 in a manner similar to other well-known polyomaviruses including SV40, BKPyV, and JCPyV (45). In any case, integration and mutation of MCPyV would be expected to delete this potential p53-binding domain of large T antigen. Therefore, if MCPyV-positive MCC tumors disrupt p53 signaling, then this is accomplished by another mechanism, one that does not involve the C terminus of large T antigen. It is possible that MCPyV small T antigen contributes to functional inactivation of p53 in a manner similar to mouse polyoma virus (46). We did not address the possibility that episomal viral DNA was present in these tumor specimens. However, mutation of the large T antigen helicase domain is expected to disable its ability to support episomal viral replication.
The significance of the point substitution P604S in ATM is unclear, since it is not within the catalytic domain. This may represent a normal polymorphism, although normal tissue was not available from this patient to exclude this possibility (31). Two recent reports have identified point substitution mutations in the PIK3CA gene in MCC specimens (25, 26). In a study of 60 MCC archival FFPE specimens, Nardi and colleagues identified TP53 mutations in 3 cases and PIK3CA mutations in 6 cases. Notably, 2 of the 3 cases with TP53 mutations and only 1 of the 6 cases with PIK3CA mutations contained MCPyV DNA according to PCR amplification. In the study by Becker and colleagues, 2 of 38 MCC specimens contained PIK3CA mutations (15). Both tumors with the PIK3CA mutation tested positive for MCPyV by PCR amplification of viral DNA. Notably, this group did not find mutations in the PIK3CA gene in the MKL-1 and WaGa cell lines.
In summary, this study revealed the presence of MCPyV DNA in all MCC tumors analyzed. Furthermore, the development of a new monoclonal antibody enabled the detection of MCPyV large T antigen expression in 97% of MCC cases tested. The two cases without detectable levels of large T antigen contained an inactivating mutation in TP53. We conclude that the presence of MCPyV in MCC is more common than previously reported and that improved detection methods may reveal that all MCC specimens contain viral DNA. The results of this study support the model that the MCPyV T antigens contribute to the pathogenesis of MCC and when their expression is absent, somatic mutations in TP53 and potentially other genes may be selected during oncogenesis.
Methods
Immunohistochemistry.
Immunohistochemistry was performed using 4-μm-thick FFPE tissue sections. Briefly, slides were soaked in xylene, passed through graded alcohols, and put in distilled water. Slides were then pretreated with 1.0 mM EDTA, pH 8.0 (Zymed), in a steam pressure cooker (Decloaking Chamber, Biocare Medical) as per the manufacturer’s instructions, followed by washing in distilled water. All further steps were performed at room temperature in a hydrated chamber. Slides were pretreated with Peroxidase Block (Dako) for 5 minutes to quench endogenous peroxidase activity. Mouse monoclonal antibodies Ab3 and CM2B4 (Santa Cruz Biotechnology Inc.) were applied at the indicated final concentration (0.6 or 2.4 μg/ml for Ab3; 2 μg/ml for CM2B4) in Dako diluent for 1 hour. Slides were washed in 50 mM Tris-HCl, pH 7.4, and detected with anti-mouse Envision+ kit (DAKO) as per the manufacturer’s instructions. After further washing, immunoperoxidase staining was developed using a DAB chromogen (Dako) and counterstained with hematoxylin. Images were acquired using an Aperio ScanScope Digital Slide Scanner and ImageScope Viewer.
DNA extraction and quantification.
FFPE tumor blocks were sectioned and stained with hematoxylin and eosin, and tumors were identified. DNA was extracted from 7- to 10-μm sections of macro-dissected tumors using QIAGEN QIAamp DNA FFPE following the manufacturer’s instructions, with the following exceptions: CitriSolv (Fisher) was used for deparaffinization and DNA samples were treated with RNAse A after Proteinase K digestion was performed. DNA concentration was determined with PicoGreen dsDNA Quantitation Reagent (Life Technologies).
Purification of total DNA from cell lines was performed by using a DNeasy Blood & Tissue kit (QIAGEN) following the manufacturer’s instructions. DNA concentration was determined with a NanoDrop spectrophotometer (NanoDrop Technologies).
qPCR.
Primer selection was made by comparing sequences from several MCPyV strains, including MKL-1, MCC339, R17a, MCC350, MCV322, 344, Mpt-LS2, Mpt-2, and Mpt-NLS4. Sequence numbering corresponds to MCPyV isolate MCC350 (GenBank EU375803.1). The primers were selected to amplify amplicons in the 50- to 150-bp range. Primer melting temperature (Tm) was 58–60°C, with the probe Tm 8–10°C higher. All primers were compared to the NCBI nucleotide collection database by BLAST.
The qPCR assays were performed with 96-well optical grade plates and caps (Agilent Technologies) with a Stratagene Mx3005P real-time PCR machine programmed for 50°C × 2 minutes with an initial denaturation at 95°C × 15 minutes, followed by 45 cycles at 95°C × 15 seconds and 60°C × 60 seconds. Reactions were performed in triplicate with 15 ng input DNA. MCPyV copy number was calculated with the assumption that the MKL-2 cell line contained 1 copy of viral DNA per cell, and results were analyzed using the 2–ΔΔCt method, where ΔΔCt = (Ct MCPyV primer set – Ct TPO) – (Ct MCPyV primer set – Ct TPO) MKL-2 (47).
OncoMap.
OncoMap v3.0 (Dana-Farber Cancer Institute) genotyping was performed on 112 genes corresponding to 1,047 mutations tested, as previously described (30). Briefly, DNA extracted from FFPE tumor specimens and MCC cell lines was subjected to whole genome amplification, followed by multiplex PCR to amplify regions of interest. After denaturation, PCR products were incubated with specific oligonucleotides, and a primer extension reaction was performed in the presence of chain-terminating dideoxynucleotides. Primer extension products were analyzed by MALDI-TOF mass spectrometry to determine the mutation status. The validation chemistry (hME) was performed on unamplified DNA to preclude any artifact introduced by the whole genome amplification process.
Statistics.
A non-redundant list of patient values using the highest reported value for each immunohistochemistry and copy number assay was prepared, and then a pairwise Spearman rank correlation was performed (Table 1). P values less than 0.05 were considered significant.
Study approval.
This study was conducted according to the principles expressed in the Declaration of Helsinki. Work on human tissue was conducted on excess archival human material after obtaining approval from the Dana-Farber Cancer Institute Institutional Review Board and informed consent from each subject. The patients’ medical records were reviewed to collect demographic data, clinical history, presentation, and staging.
Supplementary Material
Acknowledgments
We appreciate the gift of the MCC cell lines MKL-1, MKL-2, and MS-1 from Masahiro Shuda, Yuan Chang, and Patrick Moore (University of Pittsburgh Cancer Institute, Pittsburgh, Pennsylvania, USA) and WaGa from Jürgen Becker (Medical University of Graz, Graz, Austria). OncoMap profiling was performed under the direction of Laura MacConaill and Emanuele Palescandolo at the Center for Cancer Genome Discovery, Dana-Farber Cancer Institute. Tumor specimens were processed by Helen Kuo, Translational Research Resource Core, Department of Dermatology, Brigham and Women’s Hospital. DNA extraction from tumor specimens was performed by Candace Guiducci and Shannon Powers, Biological Samples Platform, Broad Institute, Cambridge, Massachusetts, USA. We thank the Monoclonal Antibody, Specialized Histopathology, and Tissue Microarray and Imaging Core facilities of the Dana-Farber/Harvard Cancer Center (DF/HCC). We acknowledge the support of the Claudia Adams Barr Program in Innovative Basic Cancer Research at the Dana-Farber Cancer Institute to J.A. DeCaprio, DF/HCC Development Award to T.S. Kupper, L.C. Wang, and J.A. DeCaprio, and support by the Skin Cancer SPORE (P50 CA93683) to T.S. Kupper. A. Martinez-Fernandez is supported by a fellowship from Fundación Caja Madrid, Spain. We gratefully acknowledge the support of the Eilian Family Research Fund for Merkel Cell Carcinoma at the Dana-Farber Cancer Institute.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2012;122(12):4645–4653. doi:10.1172/JCI64116.
Linda C. Wang’s present address is: Institute for Cancer Care at Mercy Medical Center, Baltimore, Maryland, USA.
References
- 1.Goessling W, McKee PH, Mayer RJ. Merkel cell carcinoma. J Clin Oncol. 2002;20(2):588–598. doi: 10.1200/JCO.20.2.588. [DOI] [PubMed] [Google Scholar]
- 2.Chan JK, Suster S, Wenig BM, Tsang WY, Chan JB, Lau AL. Cytokeratin 20 immunoreactivity distinguishes Merkel cell (primary cutaneous neuroendocrine) carcinomas and salivary gland small cell carcinomas from small cell carcinomas of various sites. Am J Surg Pathol. 1997;21(2):226–234. doi: 10.1097/00000478-199702000-00014. [DOI] [PubMed] [Google Scholar]
- 3.Moll R, Franke WW. Cytoskeletal differences between human neuroendocrine tumors: a cytoskeletal protein of molecular weight 46,000 distinguishes cutaneous from pulmonary neuroendocrine neoplasms. Differentiation. 1985;30(2):165–175. doi: 10.1111/j.1432-0436.1985.tb00528.x. [DOI] [PubMed] [Google Scholar]
- 4.Moll R, Lowe A, Laufer J, Franke WW. Cytokeratin 20 in human carcinomas. A new histodiagnostic marker detected by monoclonal antibodies. Am J Pathol. 1992;140(2):427–447. [PMC free article] [PubMed] [Google Scholar]
- 5.Engels EA, Frisch M, Goedert JJ, Biggar RJ, Miller RW. Merkel cell carcinoma and HIV infection. Lancet. 2002;359(9305):497–498. doi: 10.1016/S0140-6736(02)07668-7. [DOI] [PubMed] [Google Scholar]
- 6.Gooptu C, et al. Merkel cell carcinoma arising after therapeutic immunosuppression. Br J Dermatol. 1997;137(4):637–641. doi: 10.1111/j.1365-2133.1997.tb03802.x. [DOI] [PubMed] [Google Scholar]
- 7.Penn I, First MR. Merkel’s cell carcinoma in organ recipients: report of 41 cases. Transplantation. 1999;68(11):1717–1721. doi: 10.1097/00007890-199912150-00015. [DOI] [PubMed] [Google Scholar]
- 8.Quaglino D, et al. Association between chronic lymphocytic leukaemia and secondary tumours: unusual occurrence of a neuroendocrine (Merkell cell) carcinoma. Eur Rev Med Pharmacol Sci. 1997;1(1–3):11–16. [PubMed] [Google Scholar]
- 9.Koljonen V, et al. Chronic lymphocytic leukaemia patients have a high risk of Merkel-cell polyomavirus DNA-positive Merkel-cell carcinoma. Br J Cancer. 2009;101(8):1444–1447. doi: 10.1038/sj.bjc.6605306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Feng H, Shuda M, Chang Y, Moore PS. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science. 2008;319(5866):1096–1100. doi: 10.1126/science.1152586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shuda M, et al. T antigen mutations are a human tumor-specific signature for Merkel cell polyomavirus. Proc Natl Acad Sci U S A. 2008;105(42):16272–16277. doi: 10.1073/pnas.0806526105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Duncavage EJ, et al. Hybrid capture and next-generation sequencing identify viral integration sites from formalin-fixed, paraffin-embedded tissue. J Mol Diagn. 2011;13(3):325–333. doi: 10.1016/j.jmoldx.2011.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Feng H, et al. Cellular and viral factors regulating merkel cell polyomavirus replication. PLoS One. 2011;6(7):e22468. doi: 10.1371/journal.pone.0022468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kassem A, et al. Frequent detection of Merkel cell polyomavirus in human Merkel cell carcinomas and identification of a unique deletion in the VP1 gene. Cancer Res. 2008;68(13):5009–5013. doi: 10.1158/0008-5472.CAN-08-0949. [DOI] [PubMed] [Google Scholar]
- 15.Becker JC, Houben R, Ugurel S, Trefzer U, Pfohler C, Schrama D. MC polyomavirus is frequently present in Merkel cell carcinoma of European patients. J Invest Dermatol. 2009;129(1):248–250. doi: 10.1038/jid.2008.198. [DOI] [PubMed] [Google Scholar]
- 16.Sastre-Garau X, et al. Merkel cell carcinoma of the skin: pathological and molecular evidence for a causative role of MCV in oncogenesis. J Pathol. 2009;218(1):48–56. doi: 10.1002/path.2532. [DOI] [PubMed] [Google Scholar]
- 17.Paik JY, et al. Immunohistochemistry for Merkel cell polyomavirus is highly specific but not sensitive for the diagnosis of Merkel cell carcinoma in the Australian population. Hum Pathol. 2011;42(10):1385–1390. doi: 10.1016/j.humpath.2010.12.013. [DOI] [PubMed] [Google Scholar]
- 18.Garneski KM, Warcola AH, Feng Q, Kiviat NB, Leonard JH, Nghiem P. Merkel cell polyomavirus is more frequently present in North American than Australian Merkel cell carcinoma tumors. J Invest Dermatol. 2009;129(1):246–248. doi: 10.1038/jid.2008.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Loyo M, et al. Quantitative detection of Merkel cell virus in human tissues and possible mode of transmission. Int J Cancer. 2010;126(12):2991–2996. doi: 10.1002/ijc.24737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shuda M, et al. Human Merkel cell polyomavirus infection I. MCV T antigen expression in Merkel cell carcinoma, lymphoid tissues and lymphoid tumors. Int J Cancer. 2009;125:1243–1249. doi: 10.1002/ijc.24510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Busam KJ, et al. Merkel cell polyomavirus expression in merkel cell carcinomas and its absence in combined tumors and pulmonary neuroendocrine carcinomas. Am J Surg Pathol. 2009;33(9):1378–1385. doi: 10.1097/PAS.0b013e3181aa30a5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schrama D, et al. Merkel cell polyomavirus status is not associated with clinical course of Merkel cell carcinoma. J Invest Dermatol. 2011;131(8):1631–1638. doi: 10.1038/jid.2011.115. [DOI] [PubMed] [Google Scholar]
- 23.Waltari M, et al. Association of Merkel cell polyomavirus infection with tumor p53, KIT, stem cell factor, PDGFR-alpha and survival in Merkel cell carcinoma. Int J Cancer. 2011;129(3):619–628. doi: 10.1002/ijc.25720. [DOI] [PubMed] [Google Scholar]
- 24.Lill C, et al. P53 mutation is a rare event in Merkel cell carcinoma of the head and neck. Eur Arch Otorhinolaryngol. 2011;268(11):1639–1646. doi: 10.1007/s00405-011-1529-7. [DOI] [PubMed] [Google Scholar]
- 25.Hafner C, et al. Activation of the PI3K/AKT pathway in Merkel cell carcinoma. PLoS One. 2012;7(2):e31255. doi: 10.1371/journal.pone.0031255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nardi V, et al. Activation of PI3K signaling in Merkel cell carcinoma. Clin Cancer Res. 2012;18(5):1227–1236. doi: 10.1158/1078-0432.CCR-11-2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Houben R, et al. Merkel cell polyomavirus-infected Merkel cell carcinoma cells require expression of viral T antigens. J Virol. 2010;84(14):7064–7072. doi: 10.1128/JVI.02400-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Paulson KG, et al. Array-CGH reveals recurrent genomic changes in Merkel cell carcinoma including amplification of L-Myc. J Invest Dermatol. 2009;129(6):1547–1555. doi: 10.1038/jid.2008.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.MacConaill LE, et al. Profiling critical cancer gene mutations in clinical tumor samples. PLoS One. 2009;4(11):e7887. doi: 10.1371/journal.pone.0007887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matulonis UA, et al. High throughput interrogation of somatic mutations in high grade serous cancer of the ovary. PLoS One. 2011;6(9):e24433. doi: 10.1371/journal.pone.0024433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liberzon E, et al. Molecular variants of the ATM gene in Hodgkin’s disease in children. Br J Cancer. 2004;90(2):522–525. doi: 10.1038/sj.bjc.6601522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Paulson KG, et al. Transcriptome-wide studies of merkel cell carcinoma and validation of intratumoral CD8+ lymphocyte invasion as an independent predictor of survival. J Clin Oncol. 2011;29(12):1539–1546. doi: 10.1200/JCO.2010.30.6308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shuda M, Kwun HJ, Feng H, Chang Y, Moore PS. Human Merkel cell polyomavirus small T antigen is an oncoprotein targeting the 4E-BP1 translation regulator. J Clin Invest. 2011;121(9):3623–3634. doi: 10.1172/JCI46323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Touze A, et al. Merkel cell polyomavirus strains in patients with merkel cell carcinoma. Emerg Infect Dis. 2009;15(6):960–962. doi: 10.3201/eid1506.081463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Andea AA, et al. Merkel cell carcinoma: correlation of KIT expression with survival and evaluation of KIT gene mutational status. Hum Pathol. 2010;41(10):1405–1412. doi: 10.1016/j.humpath.2010.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Swick BL, Ravdel L, Fitzpatrick JE, Robinson WA. Merkel cell carcinoma: evaluation of KIT (CD117) expression and failure to demonstrate activating mutations in the C-KIT proto-oncogene - implications for treatment with imatinib mesylate. J Cutan Pathol. 2007;34(4):324–329. doi: 10.1111/j.1600-0560.2006.00613.x. [DOI] [PubMed] [Google Scholar]
- 37.Brunner M, et al. Expression of VEGF-A/C, VEGF-R2, PDGF-alpha/beta, c-kit, EGFR, Her-2/Neu, Mcl-1 and Bmi-1 in Merkel cell carcinoma. Mod Pathol. 2008;21(7):876–884. doi: 10.1038/modpathol.2008.63. [DOI] [PubMed] [Google Scholar]
- 38.Kartha RV, Sundram UN. Silent mutations in KIT and PDGFRA and coexpression of receptors with SCF and PDGFA in Merkel cell carcinoma: implications for tyrosine kinase-based tumorigenesis. Mod Pathol. 2008;21(2):96–104. doi: 10.1038/modpathol.3800980. [DOI] [PubMed] [Google Scholar]
- 39.Van Gele M, et al. Mutation analysis of P73 and TP53 in Merkel cell carcinoma. Br J Cancer. 2000;82(4):823–826. doi: 10.1054/bjoc.1999.1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schmid M, et al. p53 abnormalities are rare events in neuroendocrine (Merkel cell) carcinoma of the skin. An immunohistochemical and SSCP analysis. Virchows Arch. 1997;430(3):233–237. doi: 10.1007/BF01324807. [DOI] [PubMed] [Google Scholar]
- 41.Lassacher A, Heitzer E, Kerl H, Wolf P. p14ARF hypermethylation is common but INK4a-ARF locus or p53 mutations are rare in Merkel cell carcinoma. J Invest Dermatol. 2008;128(7):1788–1796. doi: 10.1038/sj.jid.5701256. [DOI] [PubMed] [Google Scholar]
- 42.Sihto H, Kukko H, Koljonen V, Sankila R, Bohling T, Joensuu H. Merkel cell polyomavirus infection, large T antigen, retinoblastoma protein and outcome in merkel cell carcinoma. Clin Cancer Res. 2011;17(14):4806–4813. doi: 10.1158/1078-0432.CCR-10-3363. [DOI] [PubMed] [Google Scholar]
- 43.Crook T, Wrede D, Tidy JA, Mason WP, Evans DJ, Vousden KH. Clonal p53 mutation in primary cervical cancer: association with human-papillomavirus-negative tumours. Lancet. 1992;339(8801):1070–1073. doi: 10.1016/0140-6736(92)90662-M. [DOI] [PubMed] [Google Scholar]
- 44.Lilyestrom W, Klein MG, Zhang R, Joachimiak A, Chen XS. Crystal structure of SV40 large T-antigen bound to p53: interplay between a viral oncoprotein and a cellular tumor suppressor. Genes Dev. 2006;20(17):2373–2382. doi: 10.1101/gad.1456306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Poulin DL, Kung AL, DeCaprio JA. p53 targets simian virus 40 large T antigen for acetylation by CBP. J Virol. 2004;78(15):8245–8253. doi: 10.1128/JVI.78.15.8245-8253.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.O’Shea CC, Fried M. Modulation of the ARF-p53 pathway by the small DNA tumor viruses. Cell Cycle. 2005;4(3):449–452. doi: 10.4161/cc.4.3.1555. [DOI] [PubMed] [Google Scholar]
- 47.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 48.Houben R, et al. Comparable expression and phosphorylation of the Retinoblastoma protein in Merkel cell polyoma virus positive and negative Merkel cell carcinoma. Int J Cancer. 2010;126(3):796–798. doi: 10.1002/ijc.24790. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.