

Abstract
Estrogen receptor alpha (ERα, ESR1) is a pivotal transcriptional regulator of breast cancer physiology and is targeted by endocrine therapies. Loss of ERα activity or expression is an indication of endocrine resistance and is associated with increased risk of tumor recurrence and worse prognosis. In this study we sought to investigate whether elements of the tumor microenvironment, namely macrophages, would impact on ERα and we found that macrophage-derived factors caused loss of ERα expression in breast cancer cells. Conditioned media from macrophages caused activation of several intracellular pathways in breast cancer cells of which c-Src, PKC and MAPK were essential for loss of ERα expression. Moreover, a prolonged hyper-activation of MAPK was observed. The activation of this kinase cascade resulted in recruitment of Extracellular signal Regulated Kinase 2 (ERK2) directly to chromatin at the ESR1 gene locus in a process that was dependent upon activation and recruitment of the c-Jun transcription factor. Thus, we identify a novel mechanism for loss of ERα expression in breast cancer cells via macrophage activation of kinase cascades in the cancer cells causing transcriptional repression of the ESR1 gene by a direct chromatin action of a c-Jun/ERK2 complex. The findings in this study support an alternative mechanism, not intrinsic to the tumor cell but derived from the cross-talk with the tumor microenvironment, that could lead to endocrine resistance and might be targeted therapeutically to prevent loss of ERα expression in breast tumors.
Keywords: Estrogen Receptor, Macrophages, MAPK, Breast Cancer, Jun
INTRODUCTION
Breast cancer is the second leading cause of death among US women and is a major cause of morbidity and death world-wide (Jemal et al., 2009). One of the most important prognostic factors and therapeutic targets in breast cancer is the estrogen receptor alpha (ERα), a member of the nuclear receptor super-family of ligand-dependent transcription factors. ERα positive (ER+) breast tumors, which comprise ca. 70% of primary breast cancers, have better prognosis and are targeted by endocrine therapies like Selective Estrogen Receptor Modulators (SERMs, e.g. tamoxifen, raloxifene), aromatase inhibitors (e.g. letrozole) and Selective Estrogen Receptor Down-regulators (SERDs, e.g. fulvestrant) which have been designed to oppose estrogen receptor action (Jordan & O’Malley, 2007; Musgrove & Sutherland, 2009).
However, the most aggressive breast tumors, for which effective treatments are lacking, tend to be resistant to endocrine therapies and are generally ERα negative (ER-). This group of malignancies represents ca. 30% of the total number of breast cancer cases. Moreover, up to half of ER+ primary tumors lose ERα expression in case of recurrence and ca. 30% of metastatic tumors that initially respond to tamoxifen therapy develop resistance by losing ERα expression (Cheung et al., 1997; Johnston, 1997; Johnston et al., 1995). Apart from ER-related mechanisms, several additional factors have been shown to cause endocrine resistance including deregulation of transcription factors and tyrosine kinase receptors, and alterations of intracellular signaling pathways (Brinkman & El-Ashry, 2009; Musgrove & Sutherland, 2009). For these reasons, understanding the mechanisms that cause loss of ERα expression is of paramount importance, while the development of novel strategies to prevent receptor loss or restore its level and activity are of therapeutic potential.
Macrophages are functionally dynamic elements of the immune system that are present in the tumor microenvironment and have been correlated with breast cancer progression, ER status and prognosis (Beck et al., 2009; Leek et al., 1999; Leek et al., 1996; Pupa et al., 1996; Shabo et al., 2008; Sharma et al., 2009). Their functional profile is highly susceptible to dynamic changes in response to environmental cues (Benoit et al., 2008; Lewis & Pollard, 2006; Martinez et al., 2008; Sica et al., 2008b; Watkins et al., 2007) and it ranges from the M1 (pro-inflammatory) to the M2 (anti-inflammatory) states that can be elicited by lipopolysaccharides (LPS) and/or interferon gamma (IFNG) or IL-4 and IL-13, respectively (Allavena et al., 2008a; Allavena et al., 2008b; Mantovani et al., 2004; Mantovani et al., 2002; Martinez et al., 2006; Qian & Pollard, 2010; Sica et al., 2008a; Sica et al., 2008b; Sica et al., 2006; Yuan et al., 2008). The macrophage population that is present at the tumor site is usually referred to as tumor-associated macrophages (TAMs) and mostly has M2-like properties (Allavena et al., 2008a; Movahedi et al., 2010; Sica et al., 2008b; Sica et al., 2006). TAMs have been shown to promote tumor growth, invasion and metastasis in some types of cancer, while showing anti-tumor activities in others (Chen et al., 2005; Lewis & Pollard, 2006; Ma et al., ; Ohri et al., 2009; Redente et al.).
In this study, we investigated whether macrophages and their polarization (i.e. M1 vs. M2) would impact estrogen receptor activity in breast cancer cells. For this purpose, we generated and characterized an in vitro model of macrophage polarization using the monocytic THP-1 cell line and have found that conditioned media (CM) from differentiated and polarized THP-1 cells caused loss of ERα expression in MCF-7 breast cancer cells. We then analyzed in detail the molecular mechanisms underlying this phenomenon and found that the macrophage-elicited ERα down-regulation was dependent on activation of the c-Src, MAPK and PKC pathways which lead to recruitment of ERK2 and c-Jun to the ESR1 genomic locus. Thus, we demonstrate a previously unknown cross-talk between macrophages and breast cancer cells involving macrophage activation of kinase cascades in the breast cancer cells that leads to loss of ERα via a direct transcriptional repression mechanism involving the recruitment to chromatin of ERK2 and c-Jun. These findings support an alternative mechanism, not intrinsic to the tumor cell but derived from the cross-talk with the tumor microenvironment, that could lead to endocrine resistance and might be targeted therapeutically to prevent loss of ERα expression in breast tumors.
RESULTS
THP-1 monocytic cells can be polarized into M1 vs. M2-like macrophage phenotypes
In order to study the cross-talk between macrophages and breast cancer cells, we generated and characterized a human macrophage-like model that is easy to manipulate and that recapitulates the macrophage phenotypes ranging from the M1 pro-inflammatory to the M2 anti-inflammatory. We chose for this study the monocytic (abbreviated “Mn” in Fig.1A) human THP-1 cell line which was shown, upon differentiation, to have characteristics similar to human macrophages (Auwerx, 1991; Daigneault et al., 2010; Tsuchiya et al., 1982). We then adapted a differentiation and polarization protocol (Fig.1A) that was shown to be effective for primary human macrophages (Martinez et al., 2006). THP-1 cells were differentiated with the phorbol ester PMA for 24 hours followed by a resting period of 6 days at the end of which fully differentiated macrophages were generated (denoted as “M0” in Fig.1). At this stage, differentiated THP-1 cells were treated with either interferon gamma (IFNG) or interleukin-4 (IL-4) for 24 hours to obtain M1- and M2-like phenotypes, respectively (denoted as “M1” and “M2”, Fig.1).
Fig.1. Polarization of the monocytic THP-1 cell line into M1- or M2-like macrophages.
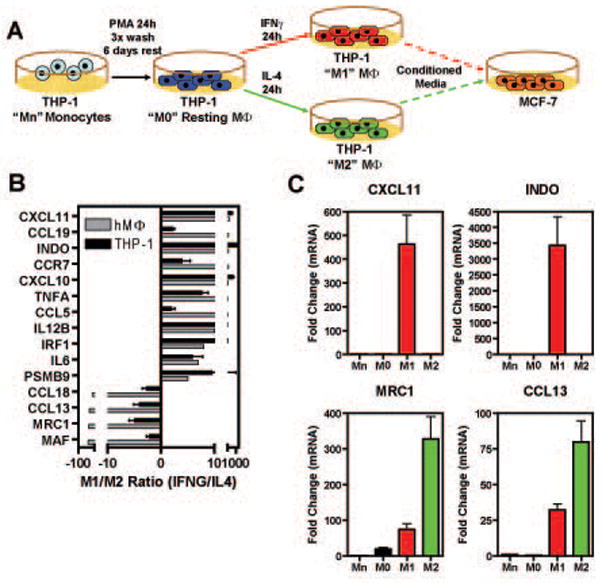
A) Schematic of experimental protocol (for details see Material and Methods) B) Comparison between q-PCR data obtained with the protocol used in panel A with data from expression cDNA microarrays in primary human macrophages (Martinez et al., 2006). Data are represented as M1/M2 ratio and are average +/− SEM of 6 independent experiments. C) q-PCR data for selected genes that are specific for M1- vs. M2- like macrophages in THP-1 cells.
To validate that this protocol was efficient in eliciting M1 and M2-like polarization in the THP-1 cells, we measured, by q-PCR, the expression level of selected genes that have been shown to discriminate between M1 and M2 in primary human macrophages (Martinez et al., 2006), and we found that the THP-1 cell system was comparable as shown by data represented as the M1/M2 ratio (Fig.1B) and by expression of representative stimulus-specific genes (Fig.1C). Thus, these data demonstrate that, based on gene expression profiles, we were able to generate M1 and M2 polarized macrophage-like populations that we could utilize in studying the cross-talk with breast cancer cells.
Conditioned medium from polarized THP-1 cells causes down-regulation of ERα
To analyze the cross-talk between estrogen receptor signaling in breast cancer cells and polarized macrophages, we first examined ERα mRNA and protein expression by q-PCR and Western blot, respectively, in MCF-7 breast cancer cells. Starting after 8 hours of treatment with conditioned media from polarized macrophages (e.g. M1-like, interferon gamma treated (CM1)), we observed down-regulation of ERα (Fig.2A–B). We also found that loss of ERα was occurring after treatment with conditioned media from both resting macrophages (CM0) and polarized ones (M1-like (CM1) or M2-like (CM2)), but that the M1-like (CM1) showed more profound and sustained effects over time, as observed in mRNA and protein time-course experiments (Fig.2A–B).
Fig.2. Conditioned Media (CMs) from polarized THP-1 cells, but not cytokines alone, cause loss of ERα mRNA and protein in a time-dependent manner in MCF-7 cells.
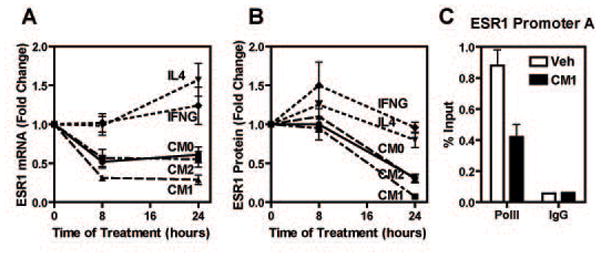
A-B) MCF-7 cells were treated with THP-1 CMs or cytokines for the times indicated and ERα mRNA and protein levels were assessed by q-PCR and Western blot. Panel B shows quantification of Western blot data for ERα protein from four independent experiments. C) ChIP assay was performed in MCF-7 cells after treatment with vehicle or CM1 for 45 min using RNA polymerase II or IgG control antibodies. Recovered DNA was used to assess the ESR1 promoter A. All experiments were performed a minimum of three times. Data are average +/− SEM. CM0: CM from vehicle treated THP-1 cells; CM1: CM from IFNG treated THP-1 cells; CM2: CM from IL4 treated THP-1 cells.
We also checked if ERα down-regulation was due to direct action of the cytokines used to polarize THP-1 cells (e.g. IL-4 or IFNG) and we found this not to be the case (Fig.2A). Moreover, we examined if the effect of CM1 was due to transcriptional repression by monitoring RNA polymerase II levels at the ERα gene (ESR1) promoter A which has been previously shown to be the most active of the characterized ESR1 promoters in MCF-7 cells (Kos et al., 2001). As shown in Fig.2C, treatment of MCF-7 cells for 45 min with CM1 caused RNA polymerase II dismissal from the ESR1 promoter A, indicating that the reduction of ERα mRNA is primarily due to transcriptional repression.
Activation of MAPK, c-Src and PKC is required for macrophage-mediated down-regulation of ERa in breast cancer cells
We next investigated which intracellular pathways might be activated by THP-1 CM1 and which ones would be necessary for down-regulating ERα expression. To do this, we utilized a combined approach with small molecule pathway inhibitors and phospho-specific antibodies to probe selected intracellular pathways (e.g. MAPK, PKC). After 15 min of treatment, conditioned media from M1-like THP-1 cells (CM1) caused phosphorylation of several kinases, including c-Src, MAPK, p38MAPK, p90RSK, SAPK/JNK, PKC, Akt and GSK3β (Fig.3A). To elucidate which of these pathways was essential for ERα mRNA down-regulation, we pre-treated MCF-7 cells with small molecule inhibitors followed by treatment with CM1. In examining the effect of the small molecule pathway inhibitors on ERα mRNA, we found that the c-Src (PP2), MEK1 (U0126) and PKC (Gö6976) inhibitors were all able to prevent the CM1-mediated reduction of ERα mRNA (Fig.3B). This was observed also at the protein level as shown by Western blot after 24 h treatment with the MAPK (U0126), c-Src (PP2) and PKC (Gö6976) inhibitors (Fig.3C). These experiments thus highlighted the role of c-Src, MAPK and PKC as essential pathways involved in CM-mediated ERα down-regulation. To understand the hierarchical activation of intracellular pathways by THP-1 conditioned media, we combined small molecule inhibitors with activation of the pathways by using phospho-specific antibodies. After 15 min of treatment, the c-Src inhibitor PP2 was able to prevent activation of all the pathways involved in ERα down-regulation, demonstrating c-Src to be the most upstream kinase which then regulates the activation of two parallel downstream pathways (PKC and MAPK) (Fig.3D–E).
Fig.3. MAPK, c-Src and PKC pathways are required for loss of ERα elicited by polarized THP-1 cell conditioned media.
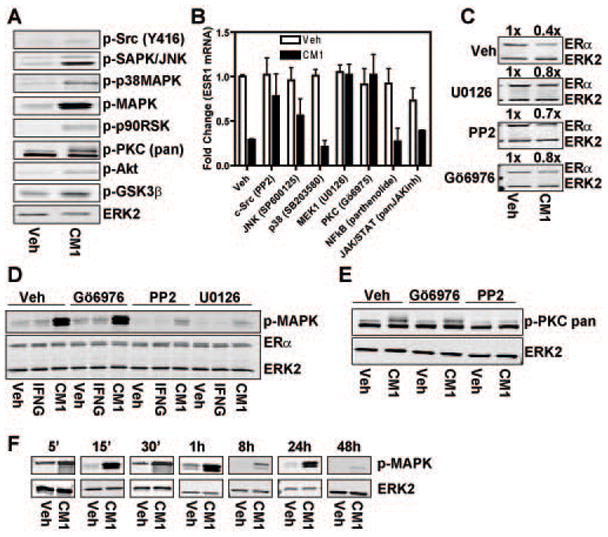
A) MCF-7 cells were treated with conditioned media from IFNG-treated THP-1 cells (CM1) for 15 min and activation of various pathways was tested by Western blot using phospho-specific antibodies. B) MCF-7 cells were pre-treated for 1 h with the indicated pathway inhibitors and then treated with CM1 for 8 h. ERα mRNA was quantified by q-PCR. Data are average +/− SEM of three independent experiments. C) MCF-7 cells were pre-treated for 1 h with the indicated pathway inhibitors and then treated with CM1 for 24 h. ERα protein was then quantified by Western blot. Values for CM1 treated samples are expressed relative to control vehicle treated samples which are set at 1x. D–E) MCF-7 cells were pretreated for 1 h with PKC (Gö6976), c-Src (PP-2) or MEK1 (U0126) inhibitors prior to 15 min of CM1 or IFNG 20ng/ml treatment. Activated pathways were probed with phospho-specific antibodies via Western blot. F) Time-course of MAPK activation in MCF-7 cells after treatment with CM1.
Because one of the most downstream kinases activated by the conditioned media was the MAPK and its constitutive activation has been shown to be involved in generation of ERα negative tumors (Bayliss et al., 2007; Creighton et al., 2006; Oh et al., 2001), we wanted to verify if its activation would be transient or sustained. As shown in Fig.3F, time-course analysis evidenced a strong and sustained phosphorylation of MAPK up to 48 h of treatment with CM1. These observations regarding the activation of c-Src and MAPK by macrophage-derived factors may be very relevant to breast cancer biology because hyper-activation of both pathways has been linked to loss of ERα in primary breast tumors and breast cancer cell lines (Bayliss et al., 2007; Chu et al., 2007; Creighton et al., 2006).
ERK2 is recruited to the ESR1 genomic locus and is required for loss of ERa expression
Our next step was to ask which transcription factors and coregulators might be involved in the transcriptional repression of ERα. Because of the strong and persistent activation of MAPK we observed upon exposure to THP-1 conditioned media and based on our identification of genome-wide ERK2 binding sites via ChIP-chip technology (Madak-Erdogan et al., 2011), we were able to identify five putative ERK2 interacting regions in the ESR1 genomic locus (Fig.4A). We first verified by conventional ChIP assay if these regions would recruit ERK2 upon treatment of MCF-7 cells for 45 min with CM1. We detected an increase in ERK2 recruitment at three of the five putative ERK2 binding sites (sites 245, 246 and 248, Fig.4B). To confirm the role of ERK2 in ERα down-regulation, we performed RNAi experiments to reduce endogenous ERK2 levels. In Fig.4C, we transfected MCF-7 cells for 72 h with ERK2 or GL3 control siRNAs and then treated the cells with conditioned media from THP-1 cells for 8 h (mRNA) or 24 h (protein) and observed an almost complete recovery of ERα mRNA and protein, confirming the central role of ERK2 in the loss of ERα (Fig.4C). The knockdown efficiency of ERK2 siRNA was ca. 80% as measured by both mRNA and protein (Fig.4C).
Fig.4. ERK2 is recruited to the ERα promoter and its knock-down prevents loss of ERα after CM1 treatment.
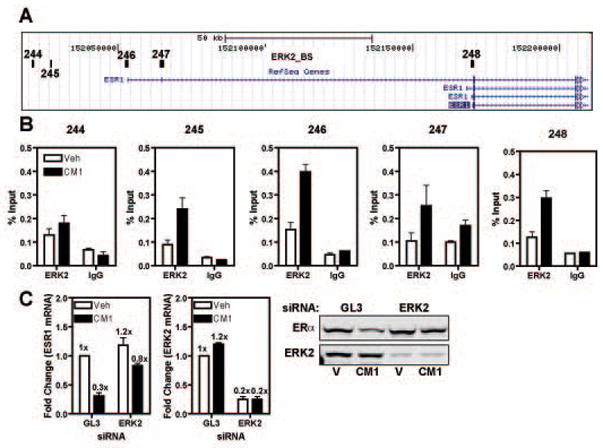
A) UCSC Genome Browser schematics of the ESR1 locus; the location of ERK2 binding sites (black boxes, 244–248) is based on (Madak-Erdogan et al., 2011). B) ChIP assay was performed in MCF-7 cells treated with vehicle or CM1 for 45 min; the isolated chromatin was probed with antibodies for ERK2 or IgG as control. C) MCF-7 cells were transfected with GL3 siRNA or ERK2 siRNA for 48 h prior to 8 h of CM1 treatment. ERα and ERK2 levels were analyzed via q-PCR and Western blot. Fold change values are expressed relative to that of vehicle control GL3 siRNA treated samples which are set at 1x. Data are average +/− SEM of three independent experiments.
The transcription factor c-Jun is activated and recruited to the ESR1 genomic locus where it plays a key role in the loss of ERα expression
To find candidate transcription factors that the kinase ERK2 might be using to tether to chromatin, we performed bioinformatics analysis of the ERK2 binding sites at the ESR1 locus using available programs (e.g. JASPAR, Match, PhastCons). Among the transcription factors whose motif was present in the ERK2 binding sites, we focused on c-Jun, a central part of the AP-1 transcription factor, because its over-expression in MCF-7 cells has been shown to lead to loss of ER expression and its activity is elevated in tamoxifen resistant tumors (Dumont et al., 1996; Johnston et al., 1999; Smith et al., 1999). To examine the role of c-Jun in the loss of ERα expression, we first monitored if c-Jun was being activated by treatment with THP-1 conditioned media. As shown in Fig.5A, c-Jun was activated by CM1, detected as an increase in phosphorylation of serine 73, in a biphasic and prolonged manner with a first peak at 15 min and then a second more prolonged peak after 1 h of treatment. Interestingly, we were also able to prevent c-Jun phosphorylation at serine 73 by pre-treating MCF-7 cells with the MAPK, c-Src and PKC inhibitors that also blocked ERα down-regulation (Fig.5B).
Fig.5. c-Jun is activated in a PKC-, c-Src- and MAPK-dependent manner, and is recruited to the ESR1 promoter and required for ERα loss upon CM1 treatment.
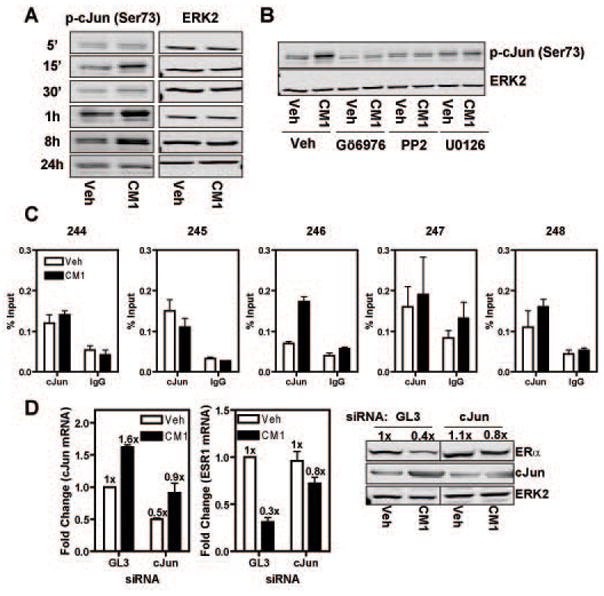
A) Activation of c-Jun was monitored by Western blot after 15 min of CM1 treatment in MCF-7 cells using phospho-specific antibody. B) MCF-7 cells were treated as in (A) except that they were pretreated for 1 h with the indicated pathway inhibitors. C) MCF-7 cells were treated with CM1 for 45 min and then ChIP was performed using c-Jun specific antibody. D) MCF-7 cells were transfected with GL3 or c-Jun siRNA for 48 h prior to CM1 treatment for 8 h (for mRNA) or 24 h (for protein). ERK2 was used as a loading control. Fold change values are expressed relative to that of vehicle control GL3 siRNA treated samples which are set at 1x. Data are average +/− SEM of three independent experiments.
We then wanted to check if c-Jun was present and/or recruited to the ERK2 binding sites at the ESR1 locus upon treatment with CM1. By ChIP analysis, we observed that c-Jun was recruited uniquely at the ERK2_246 site while being detected (but not affected by treatment) at three other ERK2 binding sites (Fig.5C). To further confirm the importance of c-Jun in regulating ERα expression, we performed c-Jun knock-down in MCF-7 cells. After 72 h of siRNA and 8 h (mRNA) or 24 h (protein) treatment with conditioned media, we were able to prevent much of the loss of ERα mRNA and protein despite only a ca. 50% knock-down efficiency, suggesting a central role for c-Jun in ERα down-regulation (Fig.5D).
The transcription factor c-Jun is required for ERK2 recruitment to the ESR1 locus
To determine if c-Jun was indeed the main factor affecting ERK2 recruitment at the ESR1 locus, we first performed ChIP-reChIP analysis which showed that ERK2 and c-Jun were present on chromatin in the same complex exclusively at the ERK2_246 binding site (Fig.6A). Secondly, we transfected MCF-7 cells with control GL3 or c-Jun siRNA and then performed ChIP assays using an ERK2 specific antibody. As shown in Fig.6B, reduction of c-Jun levels resulted in the inability of ERK2 to be recruited to chromatin, indicating that c-Jun is an important transcription factor involved in the recruitment of ERK2 to the ESR1 locus.
Fig.6. c-Jun is required for ERK2 recruitment to the ESR1 locus.
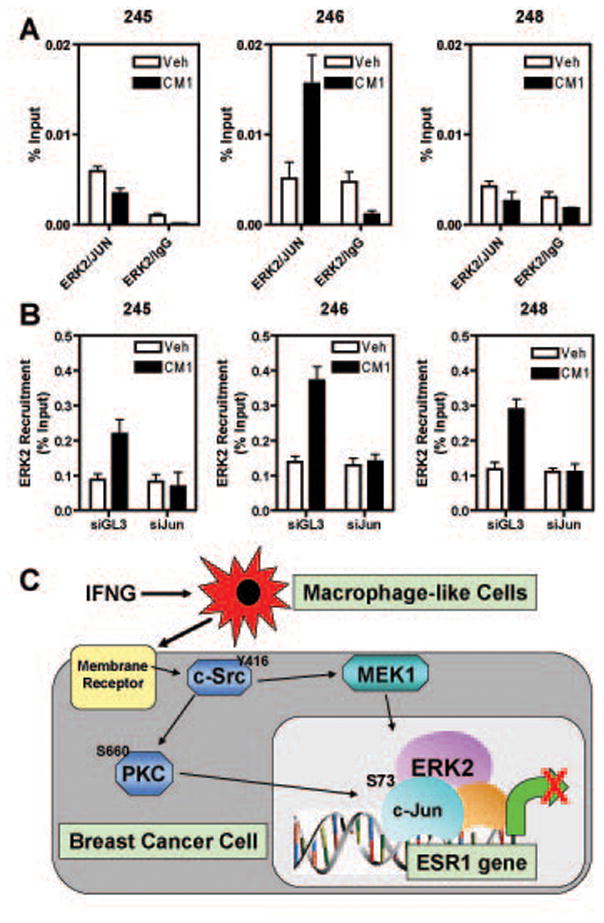
A) MCF-7 cells were treated with vehicle or CM1 for 45 min and then ChIP-reChIP was performed with the indicated antibodies. B) MCF-7 cells were transfected with GL3 control or cJun siRNA for 72 h prior to 45 min of vehicle or CM1 treatment. ERK2 ChIP was then performed. In both panels A and B, DNA was quantitated by q-PCR and the data are mean +/− SEM of 3 experiments. C) Proposed model for ERα down-regulation in breast cancer cells by macrophage-breast cancer cell interrelationships involving hyper-activation of MAPK and recruitment of c-Jun and ERK2 to the ESR1 genomic locus.
DISCUSSION
The estrogen receptor alpha (ERα) is a master regulator of breast cancer physiology, critically defining differences in the gene expression programs and the phenotypic properties of ER+ versus ER- breast cancers and, as such, ERα is also the main therapeutic target in ca. 70% of breast cancer patients. Unfortunately, the most aggressive and poor prognostic tumors either lack expression of ERα or have become resistant to endocrine therapies. Understanding the mechanisms underlying the loss of ERα expression and the development of endocrine resistance should enable promising avenues in the search for novel therapeutic approaches to treat these aggressive breast tumors for which efficient therapies are currently lacking.
The role of the tumor microenvironment as a causative effector of endocrine resistance has been largely understudied. In keeping with our findings here, studies in prostate cancer have shown important cross-talk between macrophages and tumor cells. In prostate cancer, this cross-talk was able to switch the action of SARMs (Selective Androgen Receptor Modulators) from antagonists to agonists of the androgen receptor, indicating that the tumor microenvironment can greatly impact on the nature of therapeutic drugs by altering the status of activation of intracellular pathways (Zhu et al., 2006).
Macrophages are central elements of the tumor microenvironment as they can form up to 50% of the tumor mass. Interest in tumor-associated macrophages (TAMs) stems from the fact that they are very dynamic cells that can have functions ranging from pro-inflammatory (M1-like population) to anti-inflammatory (M2-like population) depending largely on the nature of the microenvironmental cues (Allavena et al., 2008a; Mantovani et al., 2002; Martinez et al., 2008; Sica et al., 2008b; Sica et al., 2006). Moreover, they have been shown to be very attractive targets for immunotherapy and also drug delivery strategies.
In this study, we generated and characterized an easily manipulable in vitro system that would allow us to study the cross-talk between sub-populations of polarized macrophages and breast cancer cells. Using THP-1 cells (a human monocytic cell line) and a protocol we devised for their differentiation and polarization into M1- and M2-like populations (Fig.1A), and through the use of conditioned media from polarized macrophage-like populations, we found that soluble factors secreted from THP-1 cells elicited loss of ERα expression in breast cancer cells. The discovery and characterization of such factors and receptors through detailed proteomic analysis will be undertaken in future work. In this study, as depicted in the model in Fig.6C, we have uncovered a new mechanism for transcriptional repression of the ESR1 gene which occurs through hierarchical activation of multiple kinases (i.e. c-Src, PKC and MAPK) that results in recruitment to chromatin of an ERK2 and c-Jun containing complex that appears to be responsible for the loss of ERα expression. The mechanism described in this study fits very well with data from primary breast tumors where hyper-activation of MAPK and c-SRC, and high content of tumor associated macrophages (TAMs) are inversely correlated with ERα expression (Chu et al., 2007; Creighton et al., 2006; Leek et al., 1996; Oh et al., 2001). In keeping with the importance of ERK2 activation also in our system, we found that ERK2 was not recruited to the ESR1 genomic locus when the MEK1 inhibitor U0126, which blocks ERK2 activation, was used (data not shown).
Moreover, we have established for the first time a direct action of activated ERK2 at the ESR1 genomic locus where this kinase works at the chromatin level, tethered by the AP-1 transcription factor, to actively contribute to transcriptional repression of the ESR1 gene. Recently, we reported on an extensive genome-wide collaboration between ERα and ERK2 at the chromatin level that is important for estrogen regulation of gene expression and proliferation of breast cancer cells (Madak-Erdogan et al., 2011). These findings are in keeping with other cell systems where nuclear MAPK has been shown to contribute to both transcriptional stimulation and repression, depending on the environmental cues (Lawrence et al., 2009).
Thus, we demonstrate a previously unknown cross-talk between macrophages and breast cancer cells involving macrophage activation of kinase cascades in the breast cancer cells that leads to loss of ERα via a direct transcriptional repression mechanism involving the recruitment to chromatin of ERK2 and c-Jun. These findings support an alternative mechanism, not intrinsic to the tumor cell but derived from the cross-talk with the tumor microenvironment, that could lead to endocrine resistance and might be targeted therapeutically to prevent loss of ERα expression in breast tumors.
MATERIALS AND METHODS
Cell Culture, Treatments, RNA Extraction and Real-time Quantitative PCR
MCF-7 and THP-1 cells were obtained from ATCC (Manassas, VA) and cultured in DMEM (Sigma Chemical Co., St.Louis, MO) supplemented with 10% heat-inactivated calf serum (Hyclone, Logan, UT) and 1% antibiotics and RPMI supplemented with 10% heat-inactivated foetal bovine serum, 1% antibiotics, sodium bicarbonate and β-mercaptoethanol, respectively. Before experiments, the cells were cultured in phenol red-free DMEM containing 10% charcoal stripped calf serum. THP-1 monocytic cells were differentiated with phorbol-12-myristate-13-acetate (PMA, 100ng/ml, EMD-Calbiochem, La Jolla, CA) for 24 h, with media changed the next day and then every 2 days for 6 days prior to polarization. Polarization of these resting differentiated macrophages (“M0” cells) was performed as in (Martinez et al., 2006) using 20 ng/ml of interferon gamma (IFNG, R&D Systems, Minneapolis, MN) for M1 polarization or 30 ng/ml of IL-4 (R&D Systems, Minneapolis, MN) for M2 polarization. Conditioned media from differentially polarized THP-1 cells was harvested after 24 h and centrifuged for 10 min at maximum speed prior to addition to MCF-7 cells or was frozen at −80C (Figure 1A).
For pathway inhibitor experiments, MCF-7 cells were pre-treated with inhibitors for 1 h before addition of conditioned media. The inhibitors used were purchased from EMD-Calbiochem (MEK1 inhibitor (U0126, 20μM), c-Src inhibitor (PP2, 1μM), PKC inhibitor (Gö6976, 1μM), p38MAPK inhibitor (SB203580, 10μM), JAK/STAT inhibitor (panJAK inhibitor, 5μM); Sigma (NFkB inhibitor, (parthenolide, 10μM), and Cayman Chemical (JNK inhibitor (SP600125, 10μM).
Total RNA was isolated using Trizol® reagent (Invitrogen, Carlsbad, CA) following manufacturer’s instructions. Quantitative real-time PCR (q-PCR) was performed as previously described (Stossi et al., 2004). All PCR primer sequences are available upon request.
Chromatin Immunoprecipitation (ChIP) and reChIP Assays
ChIP assays were performed as described (Metivier et al., 2003). The antibodies used were from Santa Cruz Biotechnology (ERK2 (D-2), c-Jun (H-79), RNA polymerase II (N-20)). ChIP/reChIP experiments were performed as in (Stossi et al., 2009). After the first pull-down, immunoprecipitated material was recovered with 10mM DTT in IP buffer at 37C for 30 min, diluted and submitted to a second round of immunoprecipitation.
RNA Interference and Western Immunoblotting
MCF-7 cells were transfected with c-Jun or ERK2 SMARTpool or GL3 luciferase control siRNAs (Dharmacon) following the manufacturer’s instructions. After 72 h, cells were treated with THP-1 conditioned media for 8 or 24 h. Total RNA was harvested, prepared and analyzed as described in the previous section.
Western immunoblotting were performed on whole cell extracts following standard protocols. The antibodies used were purchased from Santa Cruz Biotechnology (ERα (F-10), ERK2 (D-2), c-Jun (H-79)), Cell Signaling Technology (PathScanR Multiplex Western Blot Cocktails I, II, III; Src antibody sampler kit, phospho-PKC sampler kit, phospho Akt pathway antibody sampler kit, phospho-c-Jun (ser 73). Imaging and quantitation of the Western blots used an Odyssey instrument (LI-COR Biosciences, Lincoln, NE) following manufacturer’s instructions.
Acknowledgments
This work was supported by a grant from the Breast Cancer Research Foundation and NIH P50 AT006268 from ODS, NCCAM, and NCI (to BSK).
Abbreviations
- ER
estrogen receptor
- E2
17β-estradiol
- IFNG
interferon gamma
- IL-4
interleukin 4
- PMA
phorbol-12-myristate-13-acetate
- CM
conditioned media
- TAMs
tumor-associated macrophages
Footnotes
Conflict of Interest:
F. S., Z. M. E, and B. S. K. have nothing to declare.
References
- Allavena P, Sica A, Garlanda C, Mantovani A. Immunol Rev. 2008a;222:155–61. doi: 10.1111/j.1600-065X.2008.00607.x. [DOI] [PubMed] [Google Scholar]
- Allavena P, Sica A, Solinas G, Porta C, Mantovani A. Crit Rev Oncol Hematol. 2008b;66:1–9. doi: 10.1016/j.critrevonc.2007.07.004. [DOI] [PubMed] [Google Scholar]
- Auwerx J. Experientia. 1991;47:22–31. doi: 10.1007/BF02041244. [DOI] [PubMed] [Google Scholar]
- Bayliss J, Hilger A, Vishnu P, Diehl K, El-Ashry D. Clin Cancer Res. 2007;13:7029–36. doi: 10.1158/1078-0432.CCR-07-0587. [DOI] [PubMed] [Google Scholar]
- Beck AH, Espinosa I, Edris B, Li R, Montgomery K, Zhu S, Varma S, Marinelli RJ, van de Rijn M, West RB. Clin Cancer Res. 2009;15:778–87. doi: 10.1158/1078-0432.CCR-08-1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benoit M, Desnues B, Mege JL. J Immunol. 2008;181:3733–9. doi: 10.4049/jimmunol.181.6.3733. [DOI] [PubMed] [Google Scholar]
- Brinkman JA, El-Ashry D. J Mammary Gland Biol Neoplasia. 2009;14:67–78. doi: 10.1007/s10911-009-9113-0. [DOI] [PubMed] [Google Scholar]
- Chen JJ, Lin YC, Yao PL, Yuan A, Chen HY, Shun CT, Tsai MF, Chen CH, Yang PC. J Clin Oncol. 2005;23:953–64. doi: 10.1200/JCO.2005.12.172. [DOI] [PubMed] [Google Scholar]
- Cheung KL, Willsher PC, Pinder SE, Ellis IO, Elston CW, Nicholson RI, Blamey RW, Robertson JF. Breast Cancer Res Treat. 1997;45:219–24. doi: 10.1023/a:1005828731462. [DOI] [PubMed] [Google Scholar]
- Chu I, Arnaout A, Loiseau S, Sun J, Seth A, McMahon C, Chun K, Hennessy B, Mills GB, Nawaz Z, Slingerland JM. J Clin Invest. 2007;117:2205–15. doi: 10.1172/JCI21739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creighton CJ, Hilger AM, Murthy S, Rae JM, Chinnaiyan AM, El-Ashry D. Cancer Res. 2006;66:3903–11. doi: 10.1158/0008-5472.CAN-05-4363. [DOI] [PubMed] [Google Scholar]
- Daigneault M, Preston JA, Marriott HM, Whyte MK, Dockrell DH. PLoS One. 2010;5:e8668. doi: 10.1371/journal.pone.0008668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumont JA, Bitonti AJ, Wallace CD, Baumann RJ, Cashman EA, Cross-Doersen DE. Cell Growth Differ. 1996;7:351–9. [PubMed] [Google Scholar]
- Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. CA Cancer J Clin. 2009;59:225–49. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- Johnston SR. Anticancer Drugs. 1997;8:911–30. doi: 10.1097/00001813-199711000-00002. [DOI] [PubMed] [Google Scholar]
- Johnston SR, Lu B, Scott GK, Kushner PJ, Smith IE, Dowsett M, Benz CC. Clin Cancer Res. 1999;5:251–6. [PubMed] [Google Scholar]
- Johnston SR, Saccani-Jotti G, Smith IE, Salter J, Newby J, Coppen M, Ebbs SR, Dowsett M. Cancer Res. 1995;55:3331–8. [PubMed] [Google Scholar]
- Jordan VC, O’Malley BW. J Clin Oncol. 2007;25:5815–24. doi: 10.1200/JCO.2007.11.3886. [DOI] [PubMed] [Google Scholar]
- Kos M, Reid G, Denger S, Gannon F. Mol Endocrinol. 2001;15:2057–63. doi: 10.1210/mend.15.12.0731. [DOI] [PubMed] [Google Scholar]
- Lawrence MC, Shao C, McGlynn K, Naziruddin B, Levy MF, Cobb MH. Proc Natl Acad Sci U S A. 2009;106:22181–6. doi: 10.1073/pnas.0912596106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leek RD, Landers RJ, Harris AL, Lewis CE. Br J Cancer. 1999;79:991–5. doi: 10.1038/sj.bjc.6690158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leek RD, Lewis CE, Whitehouse R, Greenall M, Clarke J, Harris AL. Cancer Res. 1996;56:4625–9. [PubMed] [Google Scholar]
- Lewis CE, Pollard JW. Cancer Res. 2006;66:605–12. doi: 10.1158/0008-5472.CAN-05-4005. [DOI] [PubMed] [Google Scholar]
- Ma J, Liu L, Che G, Yu N, Dai F, You Z. BMC Cancer. 2010;10:112. doi: 10.1186/1471-2407-10-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madak-Erdogan Z, Lupien M, Stossi F, Brown M, Katzenellenbogen BS. Mol Cell Biol. 2011;31:226–36. doi: 10.1128/MCB.00821-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. Trends Immunol. 2004;25:677–86. doi: 10.1016/j.it.2004.09.015. [DOI] [PubMed] [Google Scholar]
- Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Trends Immunol. 2002;23:549–55. doi: 10.1016/s1471-4906(02)02302-5. [DOI] [PubMed] [Google Scholar]
- Martinez FO, Gordon S, Locati M, Mantovani A. J Immunol. 2006;177:7303–11. doi: 10.4049/jimmunol.177.10.7303. [DOI] [PubMed] [Google Scholar]
- Martinez FO, Sica A, Mantovani A, Locati M. Front Biosci. 2008;13:453–61. doi: 10.2741/2692. [DOI] [PubMed] [Google Scholar]
- Metivier R, Penot G, Hubner MR, Reid G, Brand H, Kos M, Gannon F. Cell. 2003;115:751–63. doi: 10.1016/s0092-8674(03)00934-6. [DOI] [PubMed] [Google Scholar]
- Movahedi K, Laoui D, Gysemans C, Baeten M, Stange G, Van den Bossche J, Mack M, Pipeleers D, In’t Veld P, De Baetselier P, Van Ginderachter JA. Cancer Res. 2010;70:5728–39. doi: 10.1158/0008-5472.CAN-09-4672. [DOI] [PubMed] [Google Scholar]
- Musgrove EA, Sutherland RL. Nat Rev Cancer. 2009;9:631–43. doi: 10.1038/nrc2713. [DOI] [PubMed] [Google Scholar]
- Oh AS, Lorant LA, Holloway JN, Miller DL, Kern FG, El-Ashry D. Mol Endocrinol. 2001;15:1344–59. doi: 10.1210/mend.15.8.0678. [DOI] [PubMed] [Google Scholar]
- Ohri CM, Shikotra A, Green RH, Waller DA, Bradding P. Eur Respir J. 2009;33:118–26. doi: 10.1183/09031936.00065708. [DOI] [PubMed] [Google Scholar]
- Pupa SM, Bufalino R, Invernizzi AM, Andreola S, Rilke F, Lombardi L, Colnaghi MI, Menard S. J Clin Oncol. 1996;14:85–94. doi: 10.1200/JCO.1996.14.1.85. [DOI] [PubMed] [Google Scholar]
- Qian BZ, Pollard JW. Cell. 2010;141:39–51. doi: 10.1016/j.cell.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redente EF, Dwyer-Nield LD, Merrick DT, Raina K, Agarwal R, Pao W, Rice PL, Shroyer KR, Malkinson AM. Am J Pathol. 2010;176:2972–85. doi: 10.2353/ajpath.2010.090879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shabo I, Stal O, Olsson H, Dore S, Svanvik J. Int J Cancer. 2008;123:780–6. doi: 10.1002/ijc.23527. [DOI] [PubMed] [Google Scholar]
- Sharma M, Beck AH, Webster JA, Espinosa I, Montgomery K, Varma S, van de Rijn M, Jensen KC, West RB. Breast Cancer Res Treat. 2009 doi: 10.1007/s10549-009-0654-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sica A, Allavena P, Mantovani A. Cancer Lett. 2008a;267:204–15. doi: 10.1016/j.canlet.2008.03.028. [DOI] [PubMed] [Google Scholar]
- Sica A, Larghi P, Mancino A, Rubino L, Porta C, Totaro MG, Rimoldi M, Biswas SK, Allavena P, Mantovani A. Semin Cancer Biol. 2008b;18:349–55. doi: 10.1016/j.semcancer.2008.03.004. [DOI] [PubMed] [Google Scholar]
- Sica A, Schioppa T, Mantovani A, Allavena P. Eur J Cancer. 2006;42:717–27. doi: 10.1016/j.ejca.2006.01.003. [DOI] [PubMed] [Google Scholar]
- Smith LM, Wise SC, Hendricks DT, Sabichi AL, Bos T, Reddy P, Brown PH, Birrer MJ. Oncogene. 1999;18:6063–70. doi: 10.1038/sj.onc.1202989. [DOI] [PubMed] [Google Scholar]
- Stossi F, Barnett DH, Frasor J, Komm B, Lyttle CR, Katzenellenbogen BS. Endocrinology. 2004;145:3473–86. doi: 10.1210/en.2003-1682. [DOI] [PubMed] [Google Scholar]
- Stossi F, Madak-Erdogan Z, Katzenellenbogen BS. Mol Cell Biol. 2009;29:1749–59. doi: 10.1128/MCB.01476-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchiya S, Kobayashi Y, Goto Y, Okumura H, Nakae S, Konno T, Tada K. Cancer Res. 1982;42:1530–6. [PubMed] [Google Scholar]
- Watkins SK, Egilmez NK, Suttles J, Stout RD. J Immunol. 2007;178:1357–62. doi: 10.4049/jimmunol.178.3.1357. [DOI] [PubMed] [Google Scholar]
- Yuan A, Chen JJ, Yang PC. Adv Clin Chem. 2008;45:199–223. [PubMed] [Google Scholar]
- Zhu P, Baek SH, Bourk EM, Ohgi KA, Garcia-Bassets I, Sanjo H, Akira S, Kotol PF, Glass CK, Rosenfeld MG, Rose DW. Cell. 2006;124:615–29. doi: 10.1016/j.cell.2005.12.032. [DOI] [PubMed] [Google Scholar]