

Abstract
Upregulation of ubiquitin ligase atrogin1/MAFbx and muscle wasting are hallmarks of cancer cachexia; however, the underlying mechanism is undefined. Here, we describe a novel signalling pathway through which Lewis lung carcinoma (LLC) induces atrogin1/MAFbx upregulation and muscle wasting. C2C12 myotubes treated with LLC-conditioned medium (LCM) rapidly activates p38 MAPK and AKT while inactivating FoxO1/3, resulting in atrogin1/MAFbx upregulation, myosin heavy chain loss, and myotube atrophy. The p38α/β MAPK inhibitor SB202190 blocks the catabolic effects. Upon activation, p38 associates with C/EBPβ resulting in its phosphorylation and binding to a C/EBPβ-responsive cis-element in the atrogin1/MAFbx gene promoter. The promoter activity is stimulated by LCM via p38β-mediated activation of the C/EBPβ-responsive cis-element, independent of the adjacent FoxO1/3-responsive cis-elements in the promoter. In addition, p38 activation is observed in the muscle of LLC tumour-bearing mice, and SB202190 administration blocks atrogin1/MAFbx upregulation and muscle protein loss. Furthermore, C/EBPβ−/− mice are resistant to LLC tumour-induced atrogin1/MAFbx upregulation and muscle wasting. Therefore, activation of the p38β MAPK–C/EBPβ signalling pathway appears a key component of the pathogenesis of LLC tumour-induced cachexia.
Keywords: cachexia, Lewis lung carcinoma, proteolysis, p38 MAPK
Introduction
Cachexia, a wasting disease characterized by loss of muscle mass with or without loss of fat mass, is a complex metabolic syndrome seen in about 50% of cancer patients. The prominent clinical feature of cachexia is weight loss with anorexia, inflammation, insulin resistance, and increased muscle protein breakdown, which cannot be corrected by nutritional measures (Evans et al, 2008). Patients with gastric, pancreatic, colorectal, or lung cancer often experience significant weight loss at the time of diagnosis, due largely to loss of skeletal muscle mass with or without anorexia (Dewys et al, 1980; O’Gorman et al, 1999). Cancer patients with cachexia respond poorly to chemotherapy with more frequent and severe toxicity (Dewys et al, 1980; Andreyev et al, 1998). It is estimated that cachexia is the immediate cause of death in 20–40% of cancer patients (Bruera, 1997; Tisdale, 2009). Thus, the development of cachexia predicts poor prognosis for cancer patients. However, there is no approved medication for cancer cachexia due to the limited understanding of the underlying pathogenesis of this disorder.
Excessive protein degradation, particularly the loss of myofibrillar proteins, has been considered the major cause of the muscle weakness, fatigue, and muscle mass loss seen in cachexia. Recent research effort on cancer cachexia has been focused on the regulation of the ubiquitin/proteasome pathway (UPP; Acharyya and Guttridge, 2007; Tisdale, 2009), a common pathway for accelerated muscle protein degradation induced by various catabolic conditions including cachexia as well as physiological muscle atrophy due to fasting, denervation, or disuse (Lecker et al, 2006). The expression of muscle-specific ubiquitin ligase (E3 protein) atrogin1/MAFbx and MuRF1 that are rate-limiting for UPP-mediated muscle mass loss (Bodine et al, 2001; Gomes et al, 2001) is upregulated in diverse animal models of muscle atrophy including cancer models in mice (Lecker et al, 2004; Baltgalvis et al, 2009; Penna et al, 2009; Zhou et al, 2010). MuRF1 directly targets myofibrillar proteins for degradation (Clarke et al, 2007; Cohen et al, 2009). On the other hand, atrogin1/MAFbx induce myosin heavy chain (MHC, a myofibrillar protein) loss via an indirect mechanism through degrading MyoD (Lagirand-Cantaloube et al, 2009). Interestingly, the signalling mechanisms through which the ubiquitin ligases are upregulated in cachexia associated with certain types of cancer appear different from muscle atrophy induced by physiological stimuli (fasting, denervation, and disuse). The upregulation of atrogin1/MAFbx and MuRF1 in fasting, denervation, and disuse is mediated by a reduction in the activity of the PI3K/AKT signalling pathway, which activates FoxO1/3 transcription factors (Sandri et al, 2004; Stitt et al, 2004; Sacheck et al, 2007). On the other hand, MuRF1 upregulation in tumour-bearing mice is mediated by the transcription factor NF-κB that is activated by TNF-α and other pro-inflammatory cytokines (Cai et al, 2004). The existing data on cancer-induced atrogin1/MAFbx upregulation are confounding. Activation of AKT and inactivation of FoxO1/3 were observed in the muscle of mice bearing C26 colon carcinoma, which was supposed to downregulate the ubiquitin ligases; but the ubiquitin ligases were actually upregulated (Penna et al, 2009). Furthermore, the pro-inflammatory cytokine TNF-α has been shown to activate AKT and upregulate atrogin1/MAFbx via a mechanism depending on FoxO4 expression but not AKT/FoxO1/3 signalling (Moylan et al, 2008). It is possible that additional signalling mechanisms independent of AKT/FoxO1/3 signalling may mediate atrogin1/MAFbx upregulation by some types of cancer.
Previously, p38 MAPK was shown to mediate atrogin1/MAFbx upregulation by TNF-α (Li et al, 2005) or lipopolysaccharide (Doyle et al, 2011), both of which are reportedly elevated in the circulation of certain cancer patients (Nakashima et al, 1995; Han et al, 2007). The present study is designed to determine whether and how p38 MAPK mediates atrogin1/MAFbx upregulation and muscle wasting in a mouse model of cancer. We show that Lewis lung carcinoma (LLC) induces atrogin1/MAFbx upregulation and muscle protein loss through a p38β MAPK-regulated C/EBPβ-responsive cis-element in the atrogin1/MAFbx gene promoter. In addition, experimental therapy of cancer cachexia has been carried out successfully by targeting this signalling pathway.
Results
LLC-conditioned medium induces atrogin1/MAFbx upregulation and myofibrillar protein loss via the activation of p38 MAPK in myotubes
Because patients with certain types of cancer including lung, pancreatic, colorectal, or gastric cancer are more prone to cachexia (Dewys et al, 1980; O’Gorman et al, 1999), we postulate that certain types of cancer cells release factors that are capable of stimulating muscle proteolysis through p38 MAPK-regulated cis-element(s) in the atrogin1/MAFbx gene promoter, and that by targeting this signalling pathway cancer cachexia can be intervened. To test our hypothesis, C2C12 myotubes were treated with culture medium of LLC cells (LLC-conditioned medium, LCM) that are highly cachectic (Cai et al, 2004; Acharyya et al, 2005). LCM induced activation of p38 MAPK and AKT within 30 min, which was followed by inactivation of FoxO1/3 (hyperphosphorylation) within 1 h (Figure 1A). The atrogin1/MAFbx mRNA and protein were upregulated within 2 h, peaked around 4 and 8 h, respectively, and returned to basal levels around 24 h (Figure 1B). On the other hand, MuRF1 expression was not altered up to 24 h. As an additional control, we observed that conditioned medium from the non-tumourigenic human lung epithelial cell line NL20 did not alter the expression of atrogin1/MAFbx and MuRF1 (data not shown). Atrogin1/MAFbx upregulation by LCM was significantly attenuated by a specific inhibitor of the α and β isoforms of p38 MAPK, SB202190 (Figure 1C). Although atrogin1/MAFbx has not been shown to directly target muscle contractile proteins as MuRF1 does, elevated atrogin1/MAFbx can induce MHC loss via downregulating MHC expression through the degradation of MyoD (Lagirand-Cantaloube et al, 2009). Indeed, we observed a loss of total MHC proteins in myotubes treated with LCM for 72 h in a p38α/β MAPK-dependent manner (Figure 1D). This effect is much delayed considering that LCM induces atrogin1/MAFbx upregulation in just a few hours, suggesting an indirect effect of atrogin1/MAFbx on MHC content. Because the C2C12 cell line was derived from murine fast-twitch skeletal muscle, we measured the mRNA levels of the fast isoform of MHC, MHC2B, and found that the mRNA level of MHC2B was downregulated progressively by 80% in 72 h (Supplementary Figure S1), which suggests that atrogin1/MAFbx mediates LCM-induced MHC loss through downregulating its expression. Consistent to MHC loss, myoblasts treated with LCM became thinner, which was largely prevented by SB202190 without affecting the differentiation state of myotubes (Figure 1E). These observations support a key role for p38α/β MAPK in mediating LLC-induced atrogin1/MAFbx upregulation and muscle catabolism, and suggest that LLC cells release factor(s) that are capable of upregulating atrogin1/MAFbx expression and inducing muscle mass loss via rapid activation of p38α/β MAPK, independent of host-released factors.
Figure 1.

LCM induces atrogin1/MAFbx upregulation and myofibrillar protein loss via p38 MAPK in C2C12 myotubes. (A) LCM activates p38 MAPK and AKT, and inactivates FoxO1/3a. C2C12 myotubes were treated with LCM for indicated time periods as described in Materials and methods. Myotubes were lysed and phosphorylation of p38 MAPK, AKT, and FoxO1/3a were evaluated by western blot analysis with total p38 MAPK, AKT, and GAPDH as controls. (B) LCM upregulates atrogin1/MAFbx expression. The mRNA levels of atrogin1/MAFbx and MuRF1 in C2C12 myotubes treated with LCM for indicated periods were determined by using real-time PCR. *Denotes a difference (P<0.05) from 0 h determined by ANOVA. The protein levels of the ubiquitin ligases were determined by western blot. (C) Upregulation of atrogin1/MAFbx by LCM is mediated by p38 MAPK. C2C12 myotubes were treated with LCM for 4 h with or without the presence of SB202190 (SB, 10 μM). The mRNA level of atrogin1/MAFbx and MuRF1 was determined by real-time PCR. (D) LCM induces MHC loss via p38 MAPK. C2C12 myotubes were treated with LCM for 72 h with or without the presence of SB202190 (10 μM). Western blot analysis was carried out using an antibody against total MHC (MF-20). (E) LCM induces myotube atrophy via p38 MAPK. C2C12 myotubes were treated with LCM as described in (D). Immunofluorescence was performed to stain myotubes with MF-20. Diameter of myotubes was then determined as described previously (Doyle et al, 2011). Bar represents 100 μm. *Denotes a difference (P<0.05) from PBS control (PBS/vehicle) and † denotes a difference from LCM control (LCM/vehicle) determined by ANOVA.
The atrogin1/MAFbx gene is upregulated by p38β MAPK through a C/EBPβ-responsive cis-element in its 5′ promoter region
To elucidate the molecular mechanism through which p38α/β MAPK regulates atrogin1/MAFbx expression, the 5′ promoter region of the mouse, rat, and human atrogin1/MAFbx gene was examined by using a database (http://www.cbil.upenn.edu/cgi-bin/tess), and a conserved putative C/EBPβ-binding motif TTGTGCAA in the 5′ promoter region within 200 bp from the transcription start site was identified (Figure 2A). C/EBPβ was shown to be a p38 substrate in vitro, and p38 stimulates the transactivation potential of C/EBPβ in 3T3-L1 cells (Engelman et al, 1998). C/EBPβ is activated by such inflammatory mediators as LPS, TNF-α, IL-6, and IL-1 (Ramji and Foka, 2002) that mediate muscle wasting (Tisdale, 2009), and its DNA-binding activity is increased in septic muscle (Penner et al, 2002). Therefore, we hypothesized that the putative C/EBPβ-binding motif is a C/EBPβ-responsive cis-element that mediates p38α/β MAPK upregulation of the atrogin1/MAFbx gene, and that the activation of this cis-element is critical to the development of cancer cachexia. To test our hypothesis, we first evaluated whether C/EBPβ is a p38α/β MAPK substrate in C2C12 myotubes by determining if p38 MAPK interacts with, phosphorylates, and activates C/EBPβ. To activate p38 MAPK, an adenovirus encoding a constitutively active mutant of MKK6 which activates all four isoforms of p38 MAPK, MKK6bE (Huang et al, 1997), was transduced into myotubes. Expression of MKK6bE and activation of p38 MAPK in myotubes were verified by western blot analysis of the cell lysates (Figure 2B), which did not alter the differentiation state of the myotubes judged by morphology (data not shown). To evaluate p38 MAPK-mediated phosphorylation of C/EBPβ an antibody specific for C/EBPβ that is phosphorylated at Thr-188, a known site of p38 and ERK1/2 MAPK-mediated phosphorylation (Piwien-Pilipuk et al, 2002; Kim et al, 2007), was utilized along with a pan C/EBPβ antibody in western blot analysis. A modest increase in both Thr-188 phosphorylation and total C/EBPβ level by overexpressed MKK6bE were observed, and the increases were blunted by SB202190 (Figure 2B). The lysates were then subjected to immunoprecipitation assay with antibodies against either pan p38 MAPK or pan C/EBPβ. Upon p38 MAPK activation by MKK6bE, the two proteins were co-precipitated with either antibody and the co-precipitation was abolished by SB202190 (Figure 2C). Considering that C/EBPβ contains multiple serine and threonine-phosphorylation sites (Piwien-Pilipuk et al, 2002; Kim et al, 2007), we assessed p38α/β MAPK-mediated phosphorylation of C/EBPβ by western blot analysis of immunoprecipitated C/EBPβ using antibodies against phospho-serine or phospho-threonine. Phosphorylation of serine and threonine residues in C/EBPβ was increased in MKK6bE-expressing myotubes, and SB202190 blocked the MKK6bE action (Figure 2D). Because C/EBPβ can be activated through a phosphorylation-dependent increase of its binding activity to its targeted gene promoters (Tang et al, 2005), we evaluated C/EBPβ binding to the putative binding motif present in the atrogin1/MAFbx promoter in C2C12 myotubes by using the chromatin immunoprecipitation (ChIP) assay. We observed that C/EBPβ binding to the atrogin1/MAFbx promoter containing the putative binding motif increased dramatically when p38 MAPK was activated by MKK6bE (Figure 2E). In addition, LCM induced C/EBPβ phosphorylation at Thr-188 in 2 h and an increase in C/EBPβ level (Figure 2F). These observations indicate that p38α/β MAPK mediates LCM activation of C/EBPβ in myotubes through activating its phosphorylation and binding to the atrogin1/MAFbx promoter.
Figure 2.

Activated p38 MAPK interacts with and phosphorylates C/EBPβ, activating the binding of C/EBPβ to the atrogin1/MAFbx promoter. (A) The 5′ promoter sequence of the mouse, rat, and human atrogin1/MAFbx gene contains a putative C/EBPβ-binding motif (TTGTGCAA) within 200 bp from the transcription start site. *Indicates conserved sequences. (B) Activation of p38 MAPK by adenovirus-mediated overexpression of MKK6bE. Adenovirus encoding MKK6bE or GFP was used to transduce C2C12 cells as described in Materials and methods. In 48 h, myotubes were lysed. Expression of HA-tagged MKK6bE and activation of p38 MAPK and C/EBPβ level were verified by western blot. C/EBPβ phosphorylation was evaluated by using an antibody specific for Thr-188 phosphorylated C/EBPβ (p-C/EBPβ). (C) Activated p38 MAPK interacts with C/EBPβ. C2C12 myotubes were transduced with adenovirus encoding MKK6bE or GFP. SB202190 (SB, 10 μM) was included in the culture medium as indicated. In 48 h, immunoprecipitation was carried out with antibody against either p38 MAPK or C/EBPβ. Precipitation of the two proteins was analysed by western blot. The input control of the assay is shown in (B). (D) Activation of p38 MAPK results in C/EBPβ phosphorylation. C/EBPβ was immunoprecipitated from cell lysate of C2C12 myotubes expressing MKK6bE or GFP and treated with SB202190 as described above. Phosphorylation of the serine and threonine residues in immunoprecipitated C/EBPβ was analysed by western blot using antibody specific for phosphorylated serine or threonine. (E) Activated p38 MAPK stimulates C/EBPβ binding to the putative C/EBPβ-binding motif identified in the atrogin1/MAFbx promoter. C2C12 myotubes were transduced with adenovirus encoding MKK6bE or GFP as described above. ChIP assay was carried out to evaluate the effect of MKK6bE activation of p38 MAPK on C/EBPβ binding to the putative C/EBPβ-binding motif present in the atrogin1/MAFbx promoter as described in Materials and methods. The inverted image is shown. (F) LCM stimulates the phosphorylation of C/EBPβ. C2C12 myotubes were treated with LCM for indicated time periods. Thr-188 phosphorylation of C/EBPβ (p-C/EBPβ) and total C/EBPβ were determined by western blot analysis of the cell lysates.
Next, to evaluate whether the C/EBPβ-binding motif identified above is a functional C/EBPβ-responsive cis-element regulated by p38α/β MAPK, luciferase reporter gene constructs under the control of the 5′-flanking promoter sequence of the mouse atrogin1/MAFbx gene with (pA) or without (pB) the C/EBPβ-binding motif were generated. As shown in Figure 3A, a construct with the C/EBPβ-binding motif mutated from TTGTGCAA to AGGGCCCA was also created (pA-C/EBPβ-M). It is noteworthy that two previously identified FoxO1/3-responsive cis-elements (Sandri et al, 2004) are present in the promoter region adjacent to the C/EBPβ-binding motif. Thus, a construct with mutations in these cis-elements was generated (pA-FoxO-M). As shown in Figure 3B, 24 h after transfection of the reporter gene constructs into C2C12 myoblasts, reporter gene activity generated by the constructs was detected (control). Because C/EBPβ is not expressed before the differentiation of C2C12 myoblasts (Mancini et al, 2007), this promoter activity is considered the basal activity that is C/EBPβ independent. Co-transfection of plasmids encoding LAP, the active form of C/EBPβ (Ramji and Foka, 2002), and p38 MAPK activator MKK6bE stimulated the reporter gene activity by three-fold, while co-transfection of either LAP or MKK6bE had little effect. Expression of the recombinant proteins was verified by western blot analysis (Supplementary Figure S2). Inclusion of SB202190 in the cell culture medium abolished LAP/MKK6bE-mediated upregulation of reporter gene expression. Mutation of the C/EBPβ-binding motif (pA-C/EBPβ-M) or deletion of the promoter sequence containing the C/EBPβ-binding motif (pB) abolished the upregulation of reporter gene activity by LAP and MKK6bE. On the other hand, mutation of the two FoxO1/3-responsive cis-elements (pA-FoxO-M) did not alter MKK6bE effect on promoter activity. Therefore, the C/EBPβ-binding motif identified in the atrogin1/MAFbx promoter is a bona fide C/EBPβ-responsive cis-element that mediates the upregulation of the atrogin1/MAFbx gene by p38 MAPK, and the adjacent FoxO 1/3-responsive cis-elements do not respond to p38 MAPK.
Figure 3.

The C/EBPβ-binding motif in the atrogin1/MAFbx promoter is a functional cis-element regulated by p38β MAPK. (A) Generation of reporter gene constructs controlled by the atrogin1/MAFbx promoter. Reporter constructs were generated by inserting fragments of the atrogin1/MAFbx promoter into a luciferase reporter vector as described in Materials and methods. The location of the C/EBPβ-binding motif and two previously identified FoxO-responsive cis-elements are as indicated. (B) The C/EBPβ-binding motif mediates MKK6bE stimulation of atrogin1/MAFbx promoter activity. Reporter gene constructs were co-transfected with plasmids encoding the active form of C/EBPβ (LAP) and/or MKK6bE into C2C12 myoblasts as indicated. In 24 h, cells were lysed and luciferase activity was determined. (C) p38β MAPK, but not other isoforms of p38 MAPK, stimulates atrogin1/MAFbx promoter activity in the presence of C/EBPβ. Plasmids encoding an active mutant of p38 MAPK isoform and/or LAP were co-transfected with the pA reporter gene construct into C2C12 myoblasts as indicated. Luciferase activity in myotubes was determined in 24 h. (D) The C/EBPβ-binding motif mediates p38β stimulation of atrogin1/MAFbx promoter activity. Reporter gene constructs were co-transfected with plasmids encoding LAP and/or active p38β MAPK into C2C12 myoblasts as indicated. In 24 h, cells were lysed and luciferase activity was determined. (E) The C/EBPβ-binding motif mediates LCM stimulation of atrogin1/MAFbx promoter activity. Reporter gene constructs were co-transfected with a plasmid encoding LAP into C2C12 myoblasts as indicated. In 24 h, cells were treated with LCM for 3 h. Luciferase activity in lysed cells was then determined. *Indicates difference (P<0.05) from control based on ANOVA.
Because the p38 MAPK family of proteins has four known isoforms, α, β, γ and δ, and muscle cells express at least three of the isoforms (α, β and γ) (Enslen et al, 1998), it is of interest to identify the p38 MAPK isoform(s) that is responsible for activating the C/EBPβ-responsive cis-element. To individually activate each of the p38 isoforms, plasmids encoding the active mutant of each of the p38 MAPK isoforms (Askari et al, 2009) were individually transfected into C2C12 myoblasts. Expression of the active isoforms and activation of the p38 substrate ATF-2 by each of the expressed isoforms were verified by western blot analysis (Supplementary Figure S3). By co-transfecting a plasmid encoding the active mutant of one of the p38 MAPK isoforms with the plasmid encoding LAP, we observed that only active p38β MAPK, not the other three isoforms of p38 MAPK, was capable of upregulating C/EBPβ-mediated reporter gene activity of pA (Figure 3C). Similarly to MKK6bE, p38β MAPK upregulation of the reporter gene activity was blocked by SB202190 and mutation or deletion of the C/EBPβ-binding motif, but not by mutation of FoxO1/3-responsive cis-elements (Figure 3D). In addition, we observed that LCM stimulated atrogin1/MAFbx promoter activity in a similar manner to MKK6bE and p38β, and mutation of the FoxO1/3-responsive cis-elements did not alter its effect (Figure 3E). Therefore, LCM stimulates atrogin1/MAFbx promoter activity via the p38β MAPK-regulated C/EBPβ-responsive cis-element, independent of the adjacent FoxO1/3-responsive cis-elements.
We further verified whether p38β MAPK and LCM induce upregulation of endogenous atrogin1/MAFbx gene via C/EBPβ utilizing siRNA-mediated C/EBPβ knock down in C2C12 myotubes (Figure 4A). As shown in Figure 4B, ectopically expressed active p38β MAPK or MKK6bE, which did not alter the differentiation state of C2C12 cells (data not shown), upregulated atrogin1/MAFbx mRNA level in myotubes that were transfected with control siRNA, but not in myotubes in which C/EBPβ was knocked down by C/EBPβ-specific siRNA. In addition, LCM failed to induce atrogin1/MAFbx upregulation and MHC loss in myotubes in which C/EBPβ was knocked down (Figure 4C). To verify the specificity of C/EBPβ siRNA-mediated knockdown, we observed that C/EBPα level was not affected by the siRNA and ectopically expressed LAP increased atrogin1/MAFbx levels and restored LCM upregulation of atrogin1/MAFbx in myotubes where C/EBPβ had been knocked down by the siRNA (Supplementary Figure S4). In addition, C/EBPβ siRNA did not affect the differentiation of myoblasts (data not shown). Thus, C/EBPβ is a crucial mediator of p38β MAPK- and LCM-induced atrogin1/MAFbx upregulation.
Figure 4.

C/EBPβ mediates upregulation of endogenous atrogin1/MAFbx by p38β MAPK and LCM in C2C12 myotubes. (A) C/EBPβ knockdown by C/EBPβ-specific siRNA. C2C12 myoblasts were transfected with either control or C/EBPβ-specific siRNA. After differentiation for 72 h, C/EBPβ protein level in myotubes was determined by western blot analysis. (B) C/EBPβ mediates p38β MAPK upregulation of the atrogin1/MAFbx gene. In C/EBPβ knockdown myotubes that were co-transfected with a plasmid encoding a constitutively active p38β or transduced with an adenovirus encoding MKK6bE, atrogin1/MAFbx mRNA level was determined by real-time PCR. *Denotes a difference (P<0.05) from control/control siRNA and † denotes a difference from p38β/control siRNA or MKK6bE/control siRNA, correspondently, based on ANOVA. (C) C/EBPβ mediates LLC-induced atrogin1/MAFbx upregulation and MHC loss. Myotubes in which C/EBPβ was knocked down as shown in (A) were treated with control medium or LCM. Atrogin1/MAFbx and MHC levels were determined by western blot in 8 and 72 h, respectively. *Denotes a difference (P<0.05) from control/control siRNA and † denotes a difference from LCM/control siRNA or MKK6bE/control siRNA, correspondently, based on ANOVA.
The p38β MAPK–C/EBPβ signalling pathway mediates muscle wasting in LLC tumour-bearing mice
We evaluated whether p38 MAPK is activated in the muscle of LLC tumour-bearing mice and whether blocking p38β MAPK is an effective therapeutic strategy for LLC tumour-induced muscle wasting. In 14 days of LLC implant, when the LLC tumour-bearing C57BL/6 mice had developed cachexia, activation of p38 MAPK was detected in tibialis anterior (TA, Figure 5A). To evaluate the effect of LLC tumour on C/EBPβ phosphorylation in muscle, we utilized the existing antibody specific for C/EBPβ with phosphorylated Thr-188 in western blot analysis. TA from LLC tumour-bearing mice displayed a higher level of phosphorylated C/EBPβ along with a modest increase in total C/EBPβ. These increases were inhibited by the administration of p38α/β MAPK inhibitor SB202190, in the lack of a p38β MAPK-specific inhibitor (Figure 5B). SB202190 did not affect tumour growth (Figure 5C). However, SB202190 blunted LLC tumour-induced atrogin1/MAFbx upregulation (Figure 5D), loss of net body weight gain (Figure 5E), muscle mass (TA, Figure 5F; extensor digitorum longus (EDL), Figure 5G), and tyrosine release from EDL (Figure 5H). Consequently, SB202190 blocked the shrinkage of TA fibre cross-sectional area caused by LLC tumour (Figure 5I). Consistent to data from myotubes, MuRF1 expression was not altered in LLC tumour-bearing mice (Figure 5D). These in vivo data, consistent to above in vitro data, support p38β MAPK as a key mediator of LLC tumour-induced atrogin1/MAFbx upregulation and muscle mass loss, and prove in principle that p38β MAPK inhibition could be an effective therapeutic intervention for cancer cachexia.
Figure 5.

Inhibition of p38α/β MAPK blocks LLC tumour-induced muscle catabolism. LLC cells or PBS (control) was injected subcutaneously into the right flank of C57BL/6 male mice (8 weeks of age) as described in Materials and methods. SB202190 was i.p. injected daily (5 mg/kg) from day 5 of LLC implant with equal volume of vehicle as control. In 14 days, mice were weighed and euthanized. Tumour and muscle samples were immediately collected and analysed. (A) p38 MAPK is activated in the muscle of LLC tumour-bearing mice. Phosphorylation state of p38 MAPK in TA was analysed by western blot. *Indicates difference (P<0.05) from control based on Student's t-test. (B) C/EBPβ undergoes a p38α/β MAPK-dependent phosphorylation in LLC tumour-bearing mice. C/EBPβ phosphorylation at Thr-188 and total C/EBPβ in the TA of LLC tumour-bearing mice was evaluated by western blot analysis. (C) LLC tumour growth (tumour volume) is not affected by SB202190. Net body weight gain is the difference of body weight between the time of LLC implant and the time of euthanization minus tumour weight. (D) LLC-induced atrogin1/MAFbx upregulation is blocked by SB202190. Atrogin1/MAFbx mRNA isolated from TA was determined by real-time PCR and atrogin1/MAFbx protein levels were determined by western blot analysis. (E) LLC-induced loss of net body weight gain is blocked by SB202190. (F) LLC-induced loss of TA mass is attenuated by SB202190. (G) SB202190 attenuates LLC-induced loss of EDL mass. (H) LLC-induced tyrosine release from EDL is blocked by SB202190. Tyrosine release was determined as described previously (Doyle et al, 2011). *Denotes a difference (P<0.05) from PBS control (PBS/vehicle) and † denotes a difference from LLC control (LLC/vehicle) determined by ANOVA. (I) SB202190 blocks LLC-induced shrinkage in TA fibre cross-sectional area (CSA). CSA of H&E stained TA cross-sections was measured as described in Materials and methods. Bar represents 50 μm.
Finally, whether C/EBPβ is a crucial mediator of LLC-induced muscle wasting was evaluated utilizing C/EBPβ−/− mice (Sterneck et al, 1997). The body weight of C/EBPβ−/− mice (in C57BL/6 background) at the time of LLC implant (8 weeks of age) was significantly less than WT littermates (18.6±0.6 g versus 23.4±0.9 g), consistent with a previous report (Staiger et al, 2009). LLC tumour growth measured by tumour weight at 14 days of implant was found to be comparable in WT and C/EBPβ−/− mice (Figure 6A). Atrogin1/MAFbx was upregulated in the TA of LLC tumour-bearing WT mice, however, not in LLC tumour-bearing C/EBPβ−/− mice (Figure 6B). LLC tumour-bearing WT mice developed cachexia as indicated by a dramatic loss in net body weight gain as compared with mice received phosphate-buffered saline (PBS) injection, whereas C/EBPβ−/− mice were resistant to cachexia as indicated by essentially normal net body weight gain (Figure 6C). In addition, muscle wasting was apparent in WT mice as indicated by shrinkage in muscle mass (TA, Figure 6D; EDL, Figure 6E), increased tyrosine release in EDL (Figure 6F), and smaller fibre cross-sectional area in TA (Figure 6G). In contrast, C/EBPβ−/− mice did not develop muscle wasting as indicated by preserved muscle mass, essentially unaltered tyrosine release and fibre cross-sectional area. These data indicate that C/EBPβ mediates LLC-induced atrogin1/MAFbx upregulation and muscle catabolism.
Figure 6.

C/EBPβ is crucial to the development of muscle wasting in LLC tumour-bearing mice. LLC cells or PBS (control) was injected subcutaneously into the right flank of male C/EBPβ−/− and WT mice. In 14 days, mice were weighed and euthanized. Tumour and muscle samples were immediately collected and analysed. (A) LLC tumour growth (tumour volume) in C/EBPβ−/− and WT mice is comparable. (B) LLC tumour-induced atrogin1/MAFbx upregulation is blocked in C/EBPβ−/− mice. Atrogin1/MAFbx levels in TA were determined by real-time PCR and western blot. (C) LLC-induced loss of net body weight gain is attenuated in C/EBPβ−/− mice. Net body weight gain is the difference of body weight between the time of LLC implant and the time of euthanization minus tumour weight. (D) LLC-induced loss of TA mass is abolished in C/EBPβ−/− mice. (E) LLC does not induce loss of EDL mass in C/EBPβ−/− mice. (F) LLC-induced tyrosine release from EDL is blocked in C/EBPβ−/− mice. Tyrosine release was determined as described previously (Doyle et al, 2011). *Denotes a difference (P<0.05) from PBS control (PBS/vehicle) and † denotes a difference from LLC control (LLC/vehicle) determined by ANOVA. (G) LLC-induced shrinkage in TA fibre cross-sectional area (CSA) is blocked in C/EBPβ−/− mice. CSA of H&E stained TA cross-sections was measured as described in Materials and methods. Bar represents 50 μm.
Discussion
Although atrogin1/MAFbx-mediated proteolysis by the UPP has been known for several years as a key contributor to muscle mass loss induced by cancer, the molecular mechanism through which cancer induces atrogin1/MAFbx upregulation was undefined. Combining in vitro and in vivo approaches, the current study demonstrates for the first time that LLC cells induce atrogin1/MAFbx upregulation and muscle mass loss by activating C/EBPβ binding to a cis-element in the atrogin1/MAFbx gene promoter via the activation of p38β MAPK. Our in vitro results suggest that the initiation of this chain of events does not require the input of immune cells. In addition, the rapid activation of p38 MAPK and upregulation of atrogin1/MAFbx in myotubes by LCM indicate that no synthesis of host factors by muscle cells is needed for this action either. Our in vivo studies demonstrated that the p38 MAPK–C/EBPβ signalling is essential for atrogin1/MAFbx upregulation and the development of muscle wasting in LLC tumour-bearing mice. Based on our data, we propose a signalling mechanism that mediates LLC tumour-induced muscle wasting as depicted in Figure 7. We recognize that the in vivo effect of LLC tumour on muscle metabolism is more complicated than the in vitro effect of LCM and is likely to involve some host response. For example, a recent study showed that adipose triglyceride lipase plays a role in the cachexia induced by LLC tumour (Das et al, 2011). Whether there is a connection between adipose triglyceride lipase-mediated adipolysis and p38β MAPK-mediated muscle catabolism is an interesting question for future studies.
Figure 7.
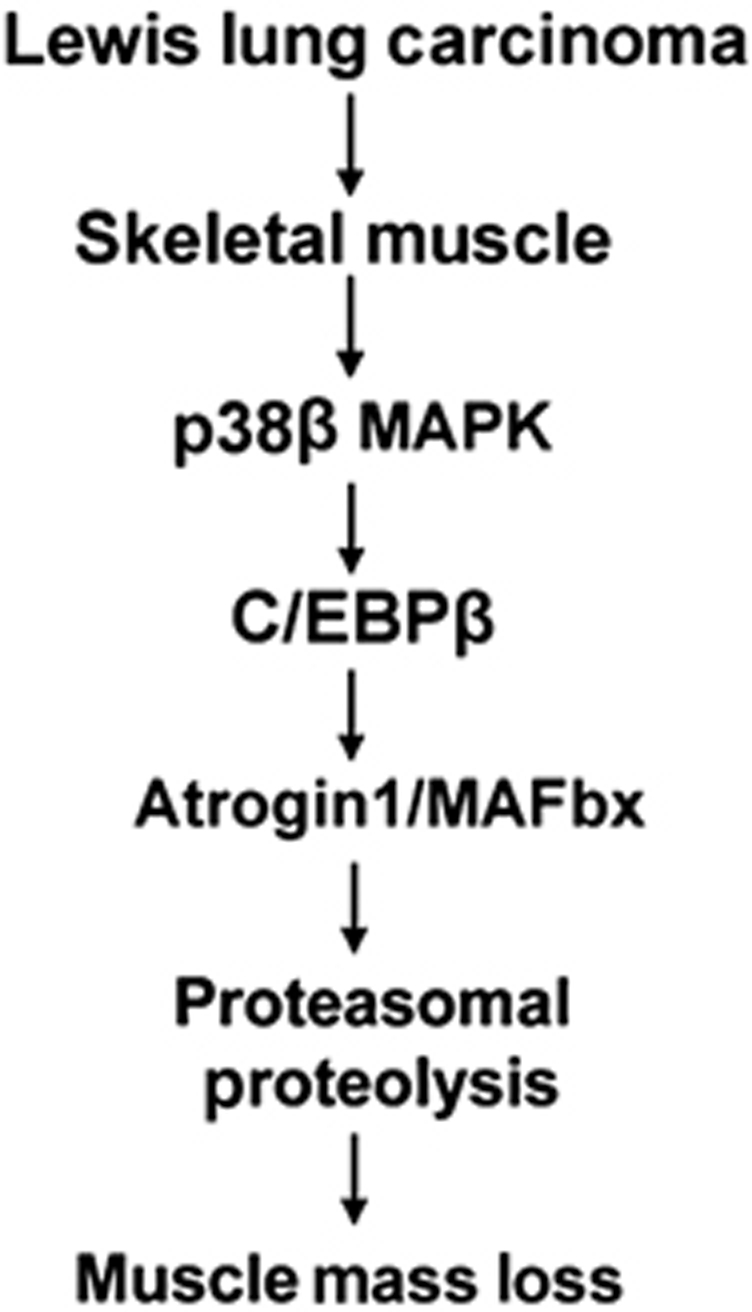
A working model of the signalling mechanism through which LLC induces muscle mass loss.
Although both atrogin1/MAFbx and MuRF1 are upregulated in the muscle of cachectic animals bearing Yoshida hepatoma (Lecker et al, 2004) or C26 colon carcinoma tumour (Zhou et al, 2010), our data indicate that only one of the ubiquitin ligases, atrogin1/MAFbx, is upregulated by LLC in vitro and in vivo. Similarly, Baltgalvis et al (2009) showed previously that atrogin1/MAFbx but not MuRF1 is upregulated in the muscle of Apcmin/+ mice that develop cachexia chronically due to spontaneous development of intestinal adenomas. The lack of MuRF1 upregulation in response to LLC is not surprising. Previous studies revealed that in humans suffering loss of muscle mass and MHC due to certain diseases only atrogin1/MAFbx, but not MuRF1, was upregulated (Leger et al, 2006; Ottenheijm et al, 2006). Although MuRF1, but not atrogin1/MAFbx, is known to target MHC for degradation in response to denervation (Cohen et al, 2009) or dexamethasone treatment (Clarke et al, 2007), our data confirmed that atrogin1/MAFbx upregulation induces MHC loss via suppressing the expression of MHC, particularly, the fast MHC. Thus, atrogin1/MAFbx upregulation alone can result in MHC loss. On the other hand, we cannot rule out the possibility that an unidentified ubiquitin ligase directly targets MHC for degradation in our system.
We demonstrate in the present study that while both p38 MAPK and AKT are activated in myotubes by LCM, it is p38 MAPK activity that determines the level of atrogin1/MAFbx expression and muscle catabolism. A diverse array of stimuli can activate p38 MAPK, and unsurprisingly p38 mediates a wide range of cellular events (New and Han, 1998). Paradoxically, p38 MAPK responds to both catabolic and anabolic stimuli in skeletal muscle cells. On one hand, LPS, catabolic cytokines, and oxidative stress can activate p38 MAPK, upregulate atrogin1/MAFbx, and synergistically stimulate muscle protein breakdown (Li et al, 2003, 2005; Baltgalvis et al, 2009; McClung et al, 2010; Doyle et al, 2011). On the other hand, p38 MAPK is also activated by exercise and muscle contraction (Long et al, 2004), and p38 MAPK activation is crucial to satellite cell-mediated myogenesis (Lluis et al, 2006), which are of anabolic nature. The presence of isoforms of p38 MAPK family contributes to its ability to transduce a variety of extracellular signals into distinct nuclear responses (Enslen et al, 1998; Loesch and Chen, 2008; Remy et al, 2010). The α isoform of p38 MAPK has been shown to mediate myogenic differentiation (Perdiguero et al, 2007; Palacios et al, 2010), and the γ isoform of p38 MAPK has been shown to regulate the expansion of myogenic precursor cells (Gillespie et al, 2009), endurance exercise-induced mitochondrial biogenesis and angiogenesis (Pogozelski et al, 2009), and glucose uptake (Ho et al, 2004). However, the function of the β isoform of p38 MAPK was previously unknown. Our data presented here not only identify for the first time that p38β MAPK is the only member of the p38 MAPK family that mediates atrogin1/MAFbx upregulation and muscle catabolism, but also resolves the issue why the p38 MAPK family of kinases is capable of exerting the paradoxical effects on muscle protein homeostasis. In addition, this finding provides a relatively specific therapeutic target for the intervention of cancer cachexia, as previous studies indicated that p38α activity but not p38β activity mediates acute or chronic inflammatory responses (O’Keefe et al, 2007) and ischaemic myocardial injury (Sicard et al, 2010).
Because p38 MAPK is activated by inflammatory mediators and reactive oxygen species in muscle (Li et al, 2005; Jin and Li, 2007; McClung et al, 2010; Doyle et al, 2011), and increased p38 MAPK activity in muscle has been reported in several pro-catabolic states in humans and laboratory animals, including type 2 diabetes (Koistinen et al, 2003), acute quadriplegic myopathy (Di Giovanni et al, 2004), neurogenic atrophy (Di Giovanni et al, 2004), and sarcopenia (Williamson et al, 2003), p38β MAPK activation may be responsible for the muscle catabolism seen in multiple pro-catabolic states. Therefore, safe inhibitors of p38 MAPK, preferably, p38β MAPK-specific inhibitors may be developed to combat muscle wasting associated with cancer and other catabolic conditions.
C/EBPβ is phosphorylated at various sites by several protein kinases, which has a key role in the regulation of C/EBPβ activity (Ramji and Foka, 2002). The present study reveals that C/EBPβ phosphorylation by p38 MAPK in muscle cells enhances its transactivation of the atrogin1/MAFbx gene primarily through increasing its DNA-binding activity. Although we also observed that p38 MAPK and LLC increase the level of C/EBPβ, previous studies indicate that the DNA-binding activity of C/EBPβ is dependent on its phosphorylation state not on expression level (Tang et al, 2005). In deed, a most recent study by Gonnella et al (2011) demonstrated in L6 myotubes that silencing C/EBPβ blocks dexamethasone-induced increase in atrogin1/MAFbx expression; however, overexpressing C/EBPβ does not increase atrogin1/MAFbx expression. Therefore, we conclude that p38β MAPK-mediated activation of C/EBPβ accounts for the rapid increase of atrogin1/MAFbx expression within 2 h shown in Figure 1B. It is noteworthy that although we used the existing antibody against C/EBPβ with phosphorylated Thr-188 to assess the phosphorylation state of C/EBPβ, this site is likely only one of the potentially multiple p38 MAPK-mediated phosphorylation sites in C/EBPβ as suggested by Figure 2D. Our promoter assays indicate that LCM upregulation of atrogin1/MAFbx gene expression is mediated by the C/EBPβ-responsive cis-element but not the FoxO1/3-responsive elements in the promoter. These findings explain the seemingly paradoxical observation that LCM upregulates atrogin1/MAFbx while activating AKT (Figure 1A).
Utilizing C/EBPβ−/− mice we confirmed that C/EBPβ is a key mediator of atrogin1/MAFbx upregulation and muscle protein degradation induced by LLC tumour. Although it was reported previously that tumourigenicity by MC-38 colon adenocarcinoma cells was reduced in C/EBPβ−/− mice (Staiger et al, 2009), we observed unaltered LLC tumour growth in C/EBPβ−/− mice (Figure 6A), such difference is possibly due to the difference in tumour type. C/EBPβ has been shown to contribute to endocrine expression of IGF-1 and insulin, and lower serum levels of IGF-1 and insulin are measured in C/EBPβ−/− mice (Staiger et al, 2009). Lower serum IGF-1 and insulin levels would reduce muscle mass due to decreased protein synthesis and increased protein degradation. The latter may result from FoxO1/3 activation in response to lower PI3K/AKT activity. Consistently, we observed lower net weight gain, smaller muscle mass, and higher muscle proteolysis in C/EBPβ−/− mice (Figure 6). Notwithstanding, C/EBPβ−/− mice are resistant to LLC tumour-induced muscle proteolysis, which further supports the notion that p38β-mediated C/EBPβ activation has a predominant role in LLC tumour-induced upregulation of atrogin1/MAFbx and muscle catabolism. C/EBPβ is activated by such inflammatory mediators as LPS, TNF-α, IL-6, and IL-1 (Ramji and Foka, 2002) that mediate muscle wasting; thus, it is likely to mediate atrogin1/MAFbx upregulation by these inflammatory mediators. Interestingly, insulin that induces anabolic responses has been reported to inhibit transactivation by C/EBPβ via the PI3K pathway (Guo et al, 2001). Thus, C/EBPβ appears to be a key regulatory molecule upon which catabolic and anabolic signals converge.
Cancer is a highly diverse group of diseases and the mechanism by which cancer provokes the loss of host's muscle mass is highly complex and likely cancer-type dependent. Therefore, the signalling mechanism described here may not mediate cachexia in all types of cachectic cancer. For example, ERK, but not p38, was reportedly to mediate muscle wasting in C26 tumour-bearing mice (Penna et al, 2010). Activin appears to mediate cancer cachexia via the upregulation of atrogin1/MAFbx and MuRF1 through the Smad–AKT–FoxO1/3 pathway (Zhou et al, 2010). Previously, an MAC16 tumour cell-released factor known as proteolysis-inducing factor (PIF) was shown to induce muscle protein degradation via the activation of the UPP (Lorite et al, 1998). PIF activation of transcription factor NF-κB is crucial to its catabolic action; and although PIF upregulates the mRNA levels for several components of the UPP, PIF does not upregulate atrogin1/MAFbx (Tisdale, 2008). Thus, LLC-induced atrogin1/MAFbx upregulation and muscle wasting observed here are unlikely to involve PIF. Cancer cells may release such cytokines as TNF-α, IL-1, and IL-6 that are capable of activating p38 MAPK and inducing muscle catabolism (Malik, 1992; Tisdale, 2009). Nevertheless, the lack of MuRF1 upregulation in LLC-bearing mice argues against a significant role of cytokines, because they would have induced NF-κB-mediated MuRF1 expression. The identification of LLC cell-released factor(s) responsible for the activation of p38β MAPK and the ensuing atrogin1/MAFbx upregulation and muscle wasting will be pursued in our future projects.
Materials and methods
Myogenic cell culture
Murine C2C12 myoblasts (American Type Culture Collection) were cultured in growth medium (DMEM supplemented with 10% fetal bovine serum) at 37°C under 5% CO2. At 85–90% confluence, myoblast differentiation was induced by incubation for 96 h in differentiation medium (DMEM supplemented with 4% heat-inactivated horse serum) to form myotubes. Preconditioned medium from cultures of LLC cells (obtained from National Institute of Cancer) or non-tumourigenic human lung epithelial cell line NL20 (obtained from ATCC) that were cultured for 48 h were centrifuged and the supernatant was added to C2C12 cultures (25% final volume in fresh medium) when indicated. The conditioned medium was replaced with fresh one every 24 h. Normal culture medium was used as the control. Pretreatment of SB202190 (10 μM, dissolved in DMSO of 0.1% volume of culture medium which did not alter the parameters we measured) was carried out at 30 min prior to the experiment.
Animal use
Experimental protocols were approved in advance by the institutional Animal Welfare Committee at the University of Texas Health Science Center at Houston. For LLC-induced cancer cachexia model, 100 μl LLC cells (5 × 106) or an equal volume of PBS (control) was injected subcutaneously into the right flanks of 8-week-old male mice (C57BL/6). C/EBPβ−/− mice in C57BL/6 background were bred from C/EBPβ−/+ mice generated by Peter Johnson's group of NCI (Sterneck et al, 1997). When indicated, SB202190 (5 mg/kg) or an equal volume of vehicle (PBS containing 50% DMSO) was intraperitoneally (i.p.) injected daily from day 5 after LLC implant when the tumour became palpable. Mice were weighed daily and those with tumour size between 1 and 1.5 cm were sacrificed on day 14 of LLC implant. TA and EDL muscles were then collected immediately for analyses.
Adenovirus transduction
Ad5 cytomegalovirus encoding MKK6bE (Huang et al, 1997), a constitutively active form of mitogen-activated protein kinase kinase (MKK6), or green fluorescence protein (GFP) (prepared by The Vector Development Core of Baylor College of Medicine) was used at 800 MOI to transduce C2C12 cells that had been differentiated for 48 h. Cells were further incubated in differentiation medium for 48 h to form myotubes before experimenting.
Fluorescence microscopy and histology study
C2C12 myotubes were stained with anti-MHC antibody (MF-20, Development Studies Hybridoma Bank at the University of Iowa, Iowa City, IA) and FITC-conjugated secondary antibody, and examined using a Zeiss Axioskop 40 microscope and a Zeiss Axiocam MRM camera system controlled by Axiovision Release 4.6 imaging software. Acquired images were edited using the Photoshop software. Myotube diameter was measured in MHC-stained myotubes as previously described (Doyle et al, 2011). Cross-sectional area of H&E stained muscle sections was quantified by using the ImageJ software (NIH). Five view-fields with ∼100 myofibres per field in each section were measured. Data are expressed as frequency histogram.
Immunoprecipitation
Cells were washed three times with ice-cold PBS and lysed in RIPA buffer (50 mM Tris–HCl (pH 7.5), 150 mM NaCl, 2 mM EDTA, 1% NP-40, 0.1% SDS, 2 mM phenylmethylsulphonylfluoride (PMSF), 0.5% sodium deoxycholate, 1 mM NaF, 1/100 protease inhibitor cocktail, and 1/100 phosphatase inhibitor cocktail (Sigma-Aldrich)). The lysate was centrifuged for 10 min at 4°C at 16 000 g and protein concentration was determined using the Bio-Rad protein assay with bovine serum albumin (BSA) as standard. Lysate (2 mg protein) was precleared with protein A/G agarose beads (Thermo Scientific) and then incubated with an antibody against C/EBPβ (H-7, Santa Cruz Biotechnology, Santa Cruz, CA) or p38 MAPK (Cell Signaling Technology, Beverly, MA) overnight at 4°C, followed by incubation with 20 μl protein A/G agarose beads for 2 h at 4°C. The beads were centrifuged down, washed five times with 1% NP-40–PBS, and boiled in SDS sample buffer. After brief centrifugation, the supernatant was analysed by western blot.
Western blot analysis
Western blot analysis was carried out as described previously (Li et al, 2005). Antibodies to total and/or phosphorylated p38 MAPK (T181/Y182), AKT (S473), FoxO1 (T24)/FoxO3a (T32), atrogin1/MAFbx, and ATF2 were from Cell Signaling Technology. Antibodies to C/EBPβ (H-7), C/EBPβ with phosphorylated Thr-188, C/EBPα, and MuRF1 were from Santa Cruz Biotechnology. Antibodies specific for phospho-serine and phospho-threonine were from Invitrogen. Data were normalized to GAPDH.
Promoter assays
Luciferase reporter gene constructs under the control of the 5′-flanking promoter sequence of the mouse atrogin1/MAFbx gene with (pA) or without (pB) the conserved putative C/EBPβ-binding motif TTGTGCAA were constructed by inserting DNA fragment generated by PCR using mouse genomic DNA as the template into the pGL4.10 vector (Promega, Madison, WI). A construct with mutations within the C/EBPβ-binding motif from 5′-TTGTGCAA-3′ to 5′-AGGGCCCA-3′ was created (pA-C/EBPβ-M) by using a site-directed mutagenesis kit (Stratagene). Similarly, a construct with mutations within the two FoxO-binding motives (pA-FoxO-M) was created according to Sandri et al (2004). Primers used for generating these constructs were as follows: pB sense: 5′-GGGGTACCGGCGAGCCTATAAACAAAGCC-3′, antisense: 5′-GGAAGATCTTGGTACAGAGCGCGGACGCG-3′; pA sense: 5′-gtcggtacccgagggtcagcgggacatc-3′, antisense: 5′-GGAAGATCTTGGTACAGAGCGCGGACGCG-3′; pA-C/EBPβ-M sense: 5′-CTGGTCCTTCCTGGAAGGGCCCAACCTGTGACTCTTG-3′, antisense: 5′-CAAGAGTCACAGGTTGGGCCCTTCCAGGAAGGACCAG-3′; pA-FoxO-M sense-1: 5′-GGG CAG CGG CCC GGG TAC CGT ACA GTG CTC GGG CAG-3′, antisense-1: 5′-CTG CCC GAG CAC TGT ACG GTA CCC GGG CCG CTG CCC-3′, sense-2: 5′-GCC TCG GAA AAC AAG GCT AGC CTA TAA GCT CAG CCA CGT GGC CTC-3′, antisense-2: 5′-GAG GCC ACG TGG CTG AGC TTA TAG GCT AGC CTT GTT TTC CGA GGC-3′. Plasmids were transfected into C2C12 myoblasts at ∼50% confluence by using deacylated polyethylenimine (PEI) 2200, a gift from Dr Guangwei Du, University of Texas Health Science Center at Houston. Briefly, C2C12 myoblasts were cultured in 6-well plates for 24 h and were co-transfected with 0.5 μg of a luciferase reporter construct, plasmid encoding the LAP form of C/EBPβ (provided by Peter Johnson of NCI via Addgene), MKK6bE (a gift from J Han of the Scripps Research Institute), or isoforms of p38 MAPK (gifts from David Engelberg of Hebrew University), along with 0.2 μg of a plasmid encoding Renilla luciferase (phRL-TK-luc, Promega). After culturing in growth medium for 24 h, luciferase activity in cell lysate was measured using the Dual-Luciferase Reporter Assay system (Promega) in Synergy 2 Multi-Mode Microplate Reader (Biotek Instruments).
ChIP assay
C2C12 myotubes were crosslinked with 1% formaldehyde for 15 min at room temperature and washed three times with ice-cold PBS containing 1 mM PMSF and 1/100 protease inhibitor cocktail. The cells were lysed in lysis buffer (50 mM HEPES (pH 8.0), 140 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 1 mM PMSF, and 1/100 protease inhibitor cocktail). The lysate was vortexed and sonicated 10 times for 10 pulses at power setting 3 (VibraCell Sonicator) with a 30-s interval on ice. The average length of DNA fragments yielded ranged between 300 and 800 bp. The lysate was then clarified by centrifugation and diluted five-fold in ChIP buffer (15 mM Tris (pH 8.0), 1% Triton X-100, 0.01% SDS, 1 mM EDTA, 150 mM NaCl, 1 mM PMSF, and 1/100 protease inhibitor cocktail). The samples were precleared using 1 μg/ml sonicated salmon sperm DNA and protein A/G Agarose beads for 1 h at 4°C. One hundred microlitre of each sample was used for input control. The samples were then immunoprecipitated with anti-C/EBPβ antibody or control Rabbit IgG (Santa Cruz Biotechnology), and the immune complexes were washed with low-salt buffer (20 mM Tris (pH 8.0), 150 mM NaCl, 1 mM EDTA, 0.1% SDS, 1% Triton X-100, 0.5 mM PMSF, and 1/100 protease inhibitor cocktail), high-salt buffer (50 mM HEPES (pH 8.0), 500 mM NaCl, 1 mM EDTA, 0.1% SDS, 1% Triton X-100, 0.1% Na-deoxycholate, 0.5 mM PMSF, and 1/100 protease inhibitor cocktail), LiCl buffer (20 mM Tris (pH 8.0), 1 mM EDTA, 250 mM LiCl, 0.5% NP-40, 0.5% Na-deoxycholate, 1.0 mM PMSF, and 1/100 protease inhibitor cocktail), and TE buffer (10 mM Tris–HCl (pH 8) and 1 mM EDTA). DNA was then eluted from the immune complex with elution buffer (50 mM Tris–HCl (pH 8), 1.0 mM EDTA, 1% SDS, and 50 mM NaHCO3). Immunoprecipitated DNA was reverse crosslinked at 65°C for 4 h in the presence of 0.2 M NaCl and purified using phenol/chloroform/isoamylalcohol. A total of 5 μl of the purified DNA was subjected to PCR amplification of a 190-bp fragment using the specific primers derived from the promoter region of the atrogin1/MAFbx gene containing the C/EBP-binding motif (sense primer: 5′-TCTTTGTTGCCGGAAGAGC-3′; antisense primer: 5′-CGAGGGTCAGCGGGACATC-3′). PCR products were analysed by agarose gel electrophoresis. Ethidium bromide-stained gels were photographed under ultraviolet illumination using a Kodak Gel Logic 200 imaging system.
Real-time PCR
Real-time PCR was performed as described previously (Doyle et al, 2011). Sequences of specific primers are atrogin1/MAFbx (sense: 5′-CACATTCTCT-CCTGGAAGGGC-3′, antisense: 5′-TTGATAAAGTCTTGAGGGGAAAGTG-3′); MuRF1 (sense: 5′-CACGAAGACGAGAAGATCAACATC-3′, antisense: 5′-AGCCCCAAACACCTTGCA-3′); MHC2B (sense: 5′-AGTCCCAGGTCAACAAGCTG-3′, antisense: 5′-TTTCTCCTGTCACCTCTCAACA-3′); and GAPDH (sense: 5′-CATGGCCTTCCGTGTTCCTA-3′, antisense: 5′-GCGGCACGTCAGATCCA-3′). Data were normalized to GAPDH.
Transfection of siRNA
The on-target smart pool siRNA specific for C/EBPβ and control siRNA was purchased from Dharmacon (Denver, CO, USA) and Ambion (Austin, TX, USA), respectively, and were introduced into C2C12 myoblasts by electroporation (5 μg) with the Nucleofector system (Lonza, Walkersville, MD, USA), according to the manufacturer's protocol. In 24 h, myoblasts were differentiated and experiment was started in 72 h when myotubes were formed.
Tyrosine release assay
Tyrosine release was measured using a protocol modified from (Fulks et al, 1975) as described previously (Doyle et al, 2011).
Statistical analysis
Data were analysed with one-way ANOVA or Student's t-test using the SigmaStat software as indicated. When applicable, control samples from independent experiments were normalized to a value of 1 without showing variations (actual variations were within a normal range). A P-value <0.05 was considered to be statistically significant. Data are presented as the mean±s.e.
Supplementary Material
Acknowledgments
This study was supported by an R01 grant from National Institute of Arthritis and Musculoskeletal and Skin Diseases to Y-P Li (AR052511). We thank Zekun Guo of Baylor College of Medicine for technical support. Thanks also go to Peter Johnson of National Cancer Institute for sharing the C/EBPβ−/− mice, Jiahuai Han of the Scripps Research Institute for sharing the adenovirus and plasmid constructs of MKK6bE, and David Engelberg of Hebrew University for sharing plasmids encoding the constitutively active mutants of p38 MAPK isoforms.
Author contributions: Y-PL designed experiments and wrote the manuscript. GZ conducted experiments, analysed data, and generated the figures. BJ conducted experiments.
Footnotes
The authors declare that they have no conflict of interest.
References
- Acharyya S, Butchbach ME, Sahenk Z, Wang H, Saji M, Carathers M, Ringel MD, Skipworth RJ, Fearon KC, Hollingsworth MA, Muscarella P, Burghes AH, Rafael-Fortney JA, Guttridge DC (2005) Dystrophin glycoprotein complex dysfunction: a regulatory link between muscular dystrophy and cancer cachexia. Cancer Cell 8: 421–432 [DOI] [PubMed] [Google Scholar]
- Acharyya S, Guttridge DC (2007) Cancer cachexia signaling pathways continue to emerge yet much still points to the proteasome. Clin Cancer Res 13: 1356–1361 [DOI] [PubMed] [Google Scholar]
- Andreyev HJ, Norman AR, Oates J, Cunningham D (1998) Why do patients with weight loss have a worse outcome when undergoing chemotherapy for gastrointestinal malignancies? Eur J Cancer 34: 503–509 [DOI] [PubMed] [Google Scholar]
- Askari N, Beenstock J, Livnah O, Engelberg D (2009) p38 is active in vitro and in vivo when monophosphorylated on Thr180. Biochemistry 48: 2497–2504 [DOI] [PubMed] [Google Scholar]
- Baltgalvis KA, Berger FG, Pena MM, Davis JM, White JP, Carson JA (2009) Muscle wasting and interleukin-6-induced atrogin-I expression in the cachectic Apc (Min/+) mouse. Pflugers Arch 457: 989–1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA, Poueymirou WT, Panaro FJ, Na E, Dharmarajan K, Pan ZQ, Valenzuela DM, DeChiara TM, Stitt TN, Yancopoulos GD, Glass DJ (2001) Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 294: 1704–1708 [DOI] [PubMed] [Google Scholar]
- Bruera E (1997) ABC of palliative care. Anorexia, cachexia, and nutrition. BMJ 315: 1219–1222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai D, Frantz JD, Tawa NE. Jr, Melendez PA, Oh BC, Lidov HG, Hasselgren PO, Frontera WR, Lee J, Glass DJ, Shoelson SE (2004) IKKbeta/NF-kappaB activation causes severe muscle wasting in mice. Cell 119: 285–298 [DOI] [PubMed] [Google Scholar]
- Clarke BA, Drujan D, Willis MS, Murphy LO, Corpina RA, Burova E, Rakhilin SV, Stitt TN, Patterson C, Latres E, Glass DJ (2007) The E3 Ligase MuRF1 degrades myosin heavy chain protein in dexamethasone-treated skeletal muscle. Cell Metab 6: 376–385 [DOI] [PubMed] [Google Scholar]
- Cohen S, Brault JJ, Gygi SP, Glass DJ, Valenzuela DM, Gartner C, Latres E, Goldberg AL (2009) During muscle atrophy, thick, but not thin, filament components are degraded by MuRF1-dependent ubiquitylation. J Cell Biol 185: 1083–1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das SK, Eder S, Schauer S, Diwoky C, Temmel H, Guertl B, Gorkiewicz G, Tamilarasan KP, Kumari P, Trauner M, Zimmermann R, Vesely P, Haemmerle G, Zechner R, Hoefler G (2011) Adipose triglyceride lipase contributes to cancer-associated cachexia. Science 333: 233–238 [DOI] [PubMed] [Google Scholar]
- Dewys WD, Begg C, Lavin PT, Band PR, Bennett JM, Bertino JR, Cohen MH, Douglass HO Jr, Engstrom PF, Ezdinli EZ, Horton J, Johnson GJ, Moertel CG, Oken MM, Perlia C, Rosenbaum C, Silverstein MN, Skeel RT, Sponzo RW, Tormey DC (1980) Prognostic effect of weight loss prior to chemotherapy in cancer patients. Eastern Cooperative Oncology Group. Am J Med 69: 491–497 [DOI] [PubMed] [Google Scholar]
- Di Giovanni S, Molon A, Broccolini A, Melcon G, Mirabella M, Hoffman EP, Servidei S (2004) Constitutive activation of MAPK cascade in acute quadriplegic myopathy. Ann Neurol 55: 195–206 [DOI] [PubMed] [Google Scholar]
- Doyle A, Zhang G, Abdel Fattah EA, Eissa NT, Li YP (2011) Toll-like receptor 4 mediates lipopolysaccharide-induced muscle catabolism via coordinate activation of ubiquitin-proteasome and autophagy-lysosome pathways. FASEB J 25: 99–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelman JA, Lisanti MP, Scherer PE (1998) Specific inhibitors of p38 mitogen-activated protein kinase block 3T3-L1 adipogenesis. J Biol Chem 273: 32111–32120 [DOI] [PubMed] [Google Scholar]
- Enslen H, Raingeaud J, Davis RJ (1998) Selective activation of p38 mitogen-activated protein (MAP) kinase isoforms by the MAP kinase kinases MKK3 and MKK6. J Biol Chem 273: 1741–1748 [DOI] [PubMed] [Google Scholar]
- Evans WJ, Morley JE, Argiles J, Bales C, Baracos V, Guttridge D, Jatoi A, Kalantar-Zadeh K, Lochs H, Mantovani G, Marks D, Mitch WE, Muscaritoli M, Najand A, Ponikowski P, Rossi Fanelli F, Schambelan M, Schols A, Schuster M, Thomas D et al. (2008) Cachexia: a new definition. Clin Nutr 27: 793–799 [DOI] [PubMed] [Google Scholar]
- Fulks RM, Li JB, Goldberg AL (1975) Effects of insulin, glucose, and amino acids on protein turnover in rat diaphragm. J Biol Chem 250: 290–298 [PubMed] [Google Scholar]
- Gillespie MA, Le Grand F, Scime A, Kuang S, von Maltzahn J, Seale V, Cuenda A, Ranish JA, Rudnicki MA (2009) p38-{gamma}-dependent gene silencing restricts entry into the myogenic differentiation program. J Cell Biol 187: 991–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes MD, Lecker SH, Jagoe RT, Navon A, Goldberg AL (2001) Atrogin-1, a muscle-specific F-box protein highly expressed during muscle atrophy. Proc Natl Acad Sci USA 98: 14440–14445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonnella P, Alamdari N, Tizio S, Aversa Z, Petkova V, Hasselgren PO (2011) C/EBPbeta regulates dexamethasone-induced muscle cell atrophy and expression of atrogin-1 and MuRF1. J Cell Biochem 112: 1737–1748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo S, Cichy SB, He X, Yang Q, Ragland M, Ghosh AK, Johnson PF, Unterman TG (2001) Insulin suppresses transactivation by CAAT/enhancer-binding proteins beta (C/EBPbeta). Signaling to p300/CREB-binding protein by protein kinase B disrupts interaction with the major activation domain of C/EBPbeta. J Biol Chem 276: 8516–8523 [DOI] [PubMed] [Google Scholar]
- Han DM, Zhang YQ, Bai QX, Chen XQ (2007) Assay of AVP, CRP, and LPS in leukemia. Int J Lab Hematol 29: 185–189 [DOI] [PubMed] [Google Scholar]
- Ho RC, Alcazar O, Fujii N, Hirshman MF, Goodyear LJ (2004) p38gamma MAPK regulation of glucose transporter expression and glucose uptake in L6 myotubes and mouse skeletal muscle. Am J Physiol Regul Integr Comp Physiol 286: R342–R349 [DOI] [PubMed] [Google Scholar]
- Huang S, Jiang Y, Li Z, Nishida E, Mathias P, Lin S, Ulevitch RJ, Nemerow GR, Han J (1997) Apoptosis signaling pathway in T cells is composed of ICE/Ced-3 family proteases and MAP kinase kinase 6b. Immunity 6: 739–749 [DOI] [PubMed] [Google Scholar]
- Jin B, Li YP (2007) Curcumin prevents lipopolysaccharide-induced atrogin-1/MAFbx upregulation and muscle mass loss. J Cell Biochem 100: 960–969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JW, Tang QQ, Li X, Lane MD (2007) Effect of phosphorylation and S-S bond-induced dimerization on DNA binding and transcriptional activation by C/EBPbeta. Proc Natl Acad Sci USA 104: 1800–1804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koistinen HA, Chibalin AV, Zierath JR (2003) Aberrant p38 mitogen-activated protein kinase signalling in skeletal muscle from Type 2 diabetic patients. Diabetologia 46: 1324–1328 [DOI] [PubMed] [Google Scholar]
- Lagirand-Cantaloube J, Cornille K, Csibi A, Batonnet-Pichon S, Leibovitch MP, Leibovitch SA (2009) Inhibition of atrogin-1/MAFbx mediated MyoD proteolysis prevents skeletal muscle atrophy in vivo. PLoS One 4: e4973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecker SH, Goldberg AL, Mitch WE (2006) Protein degradation by the ubiquitin-proteasome pathway in normal and disease states. J Am Soc Nephrol 17: 1807–1819 [DOI] [PubMed] [Google Scholar]
- Lecker SH, Jagoe RT, Gilbert A, Gomes M, Baracos V, Bailey J, Price SR, Mitch WE, Goldberg AL (2004) Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB J 18: 39–51 [DOI] [PubMed] [Google Scholar]
- Leger B, Vergani L, Soraru G, Hespel P, Derave W, Gobelet C, D’Ascenzio C, Angelini C, Russell AP (2006) Human skeletal muscle atrophy in amyotrophic lateral sclerosis reveals a reduction in Akt and an increase in atrogin-1. FASEB J 20: 583–585 [DOI] [PubMed] [Google Scholar]
- Li YP, Chen Y, John J, Moylan J, Jin B, Mann DL, Reid MB (2005) TNF-alpha acts via p38 MAPK to stimulate expression of the ubiquitin ligase atrogin1/MAFbx in skeletal muscle. FASEB J 19: 362–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YP, Chen Y, Li AS, Reid MB (2003) Hydrogen peroxide stimulates ubiquitin-conjugating activity and expression of genes for specific E2 and E3 proteins in skeletal muscle myotubes. Am J Physiol Cell Physiol 285: C806–C812 [DOI] [PubMed] [Google Scholar]
- Lluis F, Perdiguero E, Nebreda AR, Munoz-Canoves P (2006) Regulation of skeletal muscle gene expression by p38 MAP kinases. Trends Cell Biol 16: 36–44 [DOI] [PubMed] [Google Scholar]
- Loesch M, Chen G (2008) The p38 MAPK stress pathway as a tumor suppressor or more? Front Biosci 13: 3581–3593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long YC, Widegren U, Zierath JR (2004) Exercise-induced mitogen-activated protein kinase signalling in skeletal muscle. Proc Nutr Soc 63: 227–232 [DOI] [PubMed] [Google Scholar]
- Lorite MJ, Thompson MG, Drake JL, Carling G, Tisdale MJ (1998) Mechanism of muscle protein degradation induced by a cancer cachectic factor. Br J Cancer 78: 850–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik ST (1992) Tumour necrosis factor: roles in cancer pathophysiology. Semin Cancer Biol 3: 27–33 [PubMed] [Google Scholar]
- Mancini A, El Bounkari O, Norrenbrock AF, Scherr M, Schaefer D, Eder M, Banham AH, Pulford K, Lyne L, Whetton AD, Tamura T (2007) FMIP controls the adipocyte lineage commitment of C2C12 cells by downmodulation of C/EBP alpha. Oncogene 26: 1020–1027 [DOI] [PubMed] [Google Scholar]
- McClung JM, Judge AR, Powers SK, Yan Z (2010) p38 MAPK links oxidative stress to autophagy-related gene expression in cachectic muscle wasting. Am J Physiol Cell Physiol 298: C542–C549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moylan JS, Smith JD, Chambers MA, McLoughlin TJ, Reid MB (2008) TNF induction of atrogin-1/MAFbx mRNA depends on Foxo4 expression but not AKT-Foxo1/3 signaling. Am J Physiol Cell Physiol 295: C986–C993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakashima J, Tachibana M, Ueno M, Baba S, Tazaki H (1995) Tumor necrosis factor and coagulopathy in patients with prostate cancer. Cancer Res 55: 4881–4885 [PubMed] [Google Scholar]
- New L, Han J (1998) The p38 MAP kinase pathway and its biological function. Trends Cardiovasc Med 8: 220–228 [DOI] [PubMed] [Google Scholar]
- O’Gorman P, McMillan DC, McArdle CS (1999) Longitudinal study of weight, appetite, performance status, and inflammation in advanced gastrointestinal cancer. Nutr Cancer 35: 127–129 [DOI] [PubMed] [Google Scholar]
- O’Keefe SJ, Mudgett JS, Cupo S, Parsons JN, Chartrain NA, Fitzgerald C, Chen SL, Lowitz K, Rasa C, Visco D, Luell S, Carballo-Jane E, Owens K, Zaller DM (2007) Chemical genetics define the roles of p38alpha and p38beta in acute and chronic inflammation. J Biol Chem 282: 34663–34671 [DOI] [PubMed] [Google Scholar]
- Ottenheijm CA, Heunks LM, Li YP, Jin B, Minnaard R, van Hees HW, Dekhuijzen PN (2006) Activation of the ubiquitin-proteasome pathway in the diaphragm in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 174: 997–1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palacios D, Mozzetta C, Consalvi S, Caretti G, Saccone V, Proserpio V, Marquez VE, Valente S, Mai A, Forcales SV, Sartorelli V, Puri PL (2010) TNF/p38alpha/polycomb signaling to Pax7 locus in satellite cells links inflammation to the epigenetic control of muscle regeneration. Cell Stem Cell 7: 455–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penna F, Bonetto A, Muscaritoli M, Costamagna D, Minero VG, Bonelli G, Fanelli FR, Baccino FM, Costelli P (2009) Muscle atrophy in experimental cancer cachexia: Is the IGF-1 signaling pathway involved? Int J Cancer 127: 1706–1717 [DOI] [PubMed] [Google Scholar]
- Penna F, Costamagna D, Fanzani A, Bonelli G, Baccino FM, Costelli P (2010) Muscle wasting and impaired myogenesis in tumor bearing mice are prevented by ERK inhibition. PLoS One 5: e13604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penner G, Gang G, Sun X, Wray C, Hasselgren PO (2002) C/EBP DNA-binding activity is upregulated by a glucocorticoid-dependent mechanism in septic muscle. Am J Physiol Regul Integr Comp Physiol 282: R439–R444 [DOI] [PubMed] [Google Scholar]
- Perdiguero E, Ruiz-Bonilla V, Gresh L, Hui L, Ballestar E, Sousa-Victor P, Baeza-Raja B, Jardi M, Bosch-Comas A, Esteller M, Caelles C, Serrano AL, Wagner EF, Munoz-Canoves P (2007) Genetic analysis of p38 MAP kinases in myogenesis: fundamental role of p38alpha in abrogating myoblast proliferation. EMBO J 26: 1245–1256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piwien-Pilipuk G, MacDougald O, Schwartz J (2002) Dual regulation of phosphorylation and dephosphorylation of C/EBPbeta modulate its transcriptional activation and DNA binding in response to growth hormone. J Biol Chem 277: 44557–44565 [DOI] [PubMed] [Google Scholar]
- Pogozelski AR, Geng T, Li P, Yin X, Lira VA, Zhang M, Chi JT, Yan Z (2009) p38gamma mitogen-activated protein kinase is a key regulator in skeletal muscle metabolic adaptation in mice. PLoS One 4: e7934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramji DP, Foka P (2002) CCAAT/enhancer-binding proteins: structure, function and regulation. Biochem J 365: 561–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remy G, Risco AM, Inesta-Vaquera FA, Gonzalez-Teran B, Sabio G, Davis RJ, Cuenda A (2010) Differential activation of p38MAPK isoforms by MKK6 and MKK3. Cell Signal 22: 660–667 [DOI] [PubMed] [Google Scholar]
- Sacheck JM, Hyatt JP, Raffaello A, Jagoe RT, Roy RR, Edgerton VR, Lecker SH, Goldberg AL (2007) Rapid disuse and denervation atrophy involve transcriptional changes similar to those of muscle wasting during systemic diseases. FASEB J 21: 140–155 [DOI] [PubMed] [Google Scholar]
- Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A, Walsh K, Schiaffino S, Lecker SH, Goldberg AL (2004) Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 117: 399–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sicard P, Clark JE, Jacquet S, Mohammadi S, Arthur JS, O’Keefe SJ, Marber MS (2010) The activation of p38 alpha, and not p38 beta, mitogen-activated protein kinase is required for ischemic preconditioning. J Mol Cell Cardiol 48: 1324–1328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staiger J, Lueben MJ, Berrigan D, Malik R, Perkins SN, Hursting SD, Johnson PF (2009) C/EBPbeta regulates body composition, energy balance-related hormones and tumor growth. Carcinogenesis 30: 832–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterneck E, Tessarollo L, Johnson PF (1997) An essential role for C/EBPbeta in female reproduction. Genes Dev 11: 2153–2162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stitt TN, Drujan D, Clarke BA, Panaro F, Timofeyva Y, Kline WO, Gonzalez M, Yancopoulos GD, Glass DJ (2004) The IGF-1/PI3K/Akt pathway prevents expression of muscle atrophy-induced ubiquitin ligases by inhibiting FOXO transcription factors. Mol Cell 14: 395–403 [DOI] [PubMed] [Google Scholar]
- Tang QQ, Gronborg M, Huang H, Kim JW, Otto TC, Pandey A, Lane MD (2005) Sequential phosphorylation of CCAAT enhancer-binding protein beta by MAPK and glycogen synthase kinase 3beta is required for adipogenesis. Proc Natl Acad Sci USA 102: 9766–9771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tisdale MJ (2008) Catabolic mediators of cancer cachexia. Curr Opin Support Palliat Care 2: 256–261 [DOI] [PubMed] [Google Scholar]
- Tisdale MJ (2009) Mechanisms of cancer cachexia. Physiol Rev 89: 381–410 [DOI] [PubMed] [Google Scholar]
- Williamson D, Gallagher P, Harber M, Hollon C, Trappe S (2003) Mitogen-activated protein kinase (MAPK) pathway activation: effects of age and acute exercise on human skeletal muscle. J Physiol 547(Part 3): 977–987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Wang JL, Lu J, Song Y, Kwak KS, Jiao Q, Rosenfeld R, Chen Q, Boone T, Simonet WS, Lacey DL, Goldberg AL, Han HQ (2010) Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival. Cell 142: 531–543 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.