

Summary
Upon detection of viral RNA, retinoic acid inducible gene I (RIG-I) undergoes TRIM25-mediated Lys-63 linked ubiquitination, leading to type-I interferon (IFN) production. In this study, we demonstrate that the linear ubiquitin assembly complex (LUBAC), comprised of two RING-IBR-RING (RBR)-containing E3 ligases HOIL-1L and HOIP, independently targets TRIM25 and RIG-I to effectively suppress virus-induced IFN production. RBR E3 ligase domains of HOIL-1L and HOIP bind and induce proteosomal degradation of TRIM25, whereas the NZF domain of HOIL-1L competes with TRIM25 for RIG-I binding. Consequently, both actions by the HOIL-1L/HOIP LUBAC potently inhibit RIG-I ubiquitination and anti-viral activity, but in a mechanistically separate manner. Conversely, the genetic deletion or depletion of HOIL-1L and HOIP robustly enhances virus-induced type-I IFN production. Taken together, the HOIL-1L/HOIP LUBAC specifically suppresses RIG-I ubiquitination and activation by inducing TRIM25 degradation and inhibiting TRIM25 interaction with RIG-I, resulting in the comprehensive suppression of the IFN-mediated anti-viral signaling pathway.
Introduction
The host has evolved at least two classes of pattern recognition receptors (PRRs) that differ fundamentally with respect to their cellular localization to detect viruses: the transmembrane-localized Toll-like receptors (TLRs) and the cytosolic retinoic acid-inducible gene-I (RIG-I) and melanoma differentiation-associated gene 5 (MDA5) receptors (Kawai and Akira, 2008). While TLRs detect incoming virions in the endosomes or phagosomes of specialized immune cells, RIG-I and MDA5 sense actively replicating viruses in the cytoplasm of various cell types (Kato et al., 2006; Yoneyama et al., 2004). RIG-I and MDA5 are members of the DExD/H box RNA helicase family, which serve as the primary intracellular sensors for viral RNA and subsequently initiate signaling cascades leading to type-I interferon (IFN) production, thereby establishing an anti-viral state (Kato et al., 2006; Nakhaei et al., 2009a; Yoneyama et al., 2004). We have recently demonstrated that tripartite motif protein 25 (TRIM25) interacts with the N-terminal caspase recruitment domains (CARDs) of RIG-I, resulting in the effective delivery of the Lys63 (K63)-linked ubiquitin moiety to Lys117 of RIG-I (Gack et al., 2007). This gives rise to an efficient interaction with MAVS/VISA/IPS-1/Cardif, a crucial downstream adaptor protein (Kawai et al., 2005; Meylan et al., 2005; Seth et al., 2005; Xu et al., 2005), leading to the recruitment of signaling molecules such as the TBK1 complex to MAVS. Recruited signaling molecules then activate IRF3 and NF-κB transcription factors to induce IFN production. A recent study also demonstrates that TRIM25 can activate RIG-I in an in vitro reconstituted cell free system (Zeng et al., 2010).
As seen with TRIM25, the ubiquitination system plays an important role in the regulation of IFN signal transduction (Bhoj and Chen, 2009). In polyubiquitin chains, ubiquitin monomers are usually linked via isopeptide bonds between an internal Lys of one monomer and the C-terminal Gly of the other monomer (Pickart and Fushman, 2004). Recently, a protein complex consisting of two RING finger proteins, HOIL-1L and HOIP, has been shown to exhibit a unique ubiquitin polymerization activity, forming ubiquitin polymers not by Lys linkages, but by linkages between the C- and N-termini of ubiquitin molecules to assemble a head-to-tail linear polyubiquitin chain. Thus, this complex is designated as LUBAC (linear ubiquitin assembly complex) (Kirisako et al., 2006). Recent studies have demonstrated that LUBAC activates the canonical NF-κB pathway by binding to NEMO (NF-κ essential modulator, also called IKKγ) to conjugate linear polyubiquitin chains in an Ubc13-independent manner (Rahighi et al., 2009; Tokunaga et al., 2009).
Tight regulation of the immune signaling pathways is essential for a successful immune response against viral infections. Whereas positive regulatory mechanisms lead to the rapid activation of IFN signaling upon viral infection, negative regulatory mechanisms are required to prevent unwanted or excessive production of IFNs or pro-inflammatory cytokines. In this report, we unveil the potential feedback inhibitory role of the HOIL-1L/HOIP LUBAC complex through the down-regulation of TRIM25 protein level as well as its competition with TRIM25 for RIG-I binding, which ultimately leads to the suppression of the K63-linked ubiquitination and signaling activity of RIG-I.
Results
HOIL-1L/HOIP LUBAC independently targets TRIM25 and RIG-I
A yeast two-hybrid screen using a TRIM25 mutant lacking the N-terminal RING domain (TRIM25 ΔRING) as bait revealed that HOIL-1L, a member of the RING-IBR-RING (RBR) E3 ligase family, is a binding partner of TRIM25. Co-immunoprecipitation (co-IP) showed that TRIM25 strongly interacts with HOIL-1L (Figure 1A). Due to its significant similarity to HOIL-1L, HOIP also showed a strong interaction with TRIM25 (Figure 1B). To determine whether HOIL-1L and HOIP also interact with RIG-I, HEK293T cells were transfected with a mammalian glutathionine-S-transferase-RIG-I-2CARD (GST-RIG-I-2CARD) fusion construct together with HOIL-1L and/or HOIP, followed by GST-pulldown or co-IP assay. This showed that RIG-I efficiently binds to HOIL-1L but not HOIP when they are individually expressed, while co-expression of HOIL-1L and HOIP enables RIG-I to interact with HOIP (Figure 1C and D), suggesting that the HOIP and RIG-I-2CARD interaction is mediated by HOIL-1L.
Figure 1. HOIL-1L/HOIP interacts with TRIM25 and RIG-I.
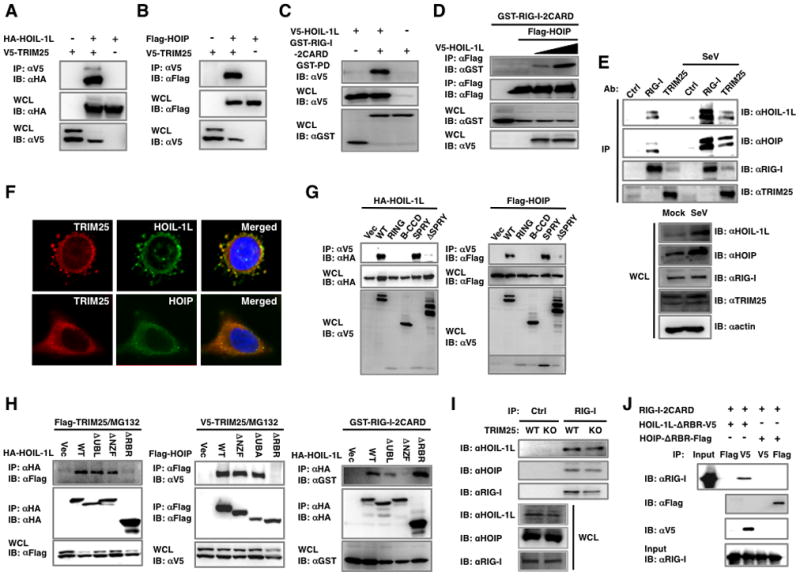
(A–B) Interactions between TRIM25 and HOIL-1L or HOIP. 293T were transfected with V5-TRIM25 together with (A) HA-HOIL-1L or (B) Flag-HOIP, followed by co-IP and IB. (C) HOIL-1L interacts with RIG-I. 293T cells transfected with GST-RIG-2CARD and V5-HOIL-1L or vector were used for GST-pull down (PD) assay and IB. (D) HOIP requires HOIL-1L to interact with RIG-I. GST-RIG-2CARD and Flag-HOIP were transfected with increasing amounts of HOIL-1L, followed by IP with anti-Flag. (E) Endogenous HOIL-1L/HOIP interacts with RIG-I and TRIM25. WT MEFs were mock-infected or infected with SeV for 6 h and treated with MG132 for 4 h before harvest, followed by IP with control IgG, anti-RIG-I or anti-TRIM25. (F) Co-localization of HOIL-1L/HOIP with TRIM25. HeLa cells were mock-treated or infected with SeV for 10 h, followed by fixation and staining with anti-TRIM25, anti-HOIL-1L or anti-HOIP antibody. Co-localization between TRIM25 and HOIL-1L, Pearson’s correlation=0.871 and Mander’s overlap=0.885; co-localization between TRIM25 and HOIP, Pearson’s correlation=0.702 and Mander’s overlap=0.955 (G) TRIM25 SPRY domain is responsible for HOIL-1L/HOIP interaction. HA-HOIL-1L or Flag-HOIP was transfected with TRIM25 deletion mutants, followed by co-IP. (H) Binding ability of HOIL-1L or HOIP mutants. Deletion mutants of HOIL-1L or HOIP were transfected with TRIM25 or GST-RIG-I 2CARD as indicated. Cells were treated with MG132 for 6 h, followed by Co-IP and IB. (I) RIG-I interaction with HOIL-1L/HOIP in TRIM25−/− MEFs. WT and TRIM25−/− MEFs were infected with SeV for 10 h, followed by co-IP using control IgG or anti-RIG-I. (J) In vitro interaction between RIG-I-2CARD and HOIL-1L. Bacterially purified GST fusion proteins of RIG-I-2CARD, HOIL-1L ΔRBR-V5 and HOIP ΔRBR-Flag were used for in vitro binding, followed by co-IP and IB as indicated. See also Figure S1.
We further examined the interactions between endogenous TRIM25 and RIG-I with HOIL-1L and HOIP in mouse embryonic fibroblast cells (MEFs). Since Sendai virus (SeV) infection readily induces RIG-I-mediated type-I IFN signaling, we performed co-IP assays with or without SeV infection. RIG-I interactions with HOIL-1L or HOIP were apparent in normal conditions, and these interactions were significantly enhanced by SeV infection (Figure 1E). In contrast, TRIM25 interactions with HOIL-1L or HOIP were detected only after SeV infection (Figure 1E). Confocal microscopy analysis showed that upon viral infection, endogenous HOIL-1L and HOIP have diffuse and punctate patterns in the cytoplasm where they apparently co-localize with endogenous TRIM25 (Figure 1F). These results collectively show that TRIM25 and RIG-I independently interact with HOIL-1L and HOIP in different manners.
HOIL-1L and HOIP share an Npl4-type zinc-finger domain (NZF) and a C-terminal RBR domain, whereas HOIL-1L and HOIP carry the ubiquitin like (UBL) domain and the ubiquitin associated (UBA) domain, respectively, which is responsible for their interaction (Kirisako et al., 2006) (Figures S1A). Mutational analysis showed that the C-terminal SPRY domain of TRIM25 is responsible for its interaction with either HOIL-1L or HOIP, whereas the N-terminal two CARDs of RIG-I sufficiently interacted with HOIL-1L (Figure 1G and C). To map the regions of HOIL-1L and HOIP responsible for their interactions with TRIM25 and RIG-I, various deletion mutants of HOIL-1L and HOIP were constructed and tested for their ability to bind to TRIM25 and RIG-I. Both HOIL-1L ΔRBR and HOIP ΔRBR, which lack their respective RBR domains, lost their TRIM25 binding abilities (Figure 1H). Interestingly, the HOIP ΔUBA mutant that no longer forms a complex with HOIL-1L was still able to interact with TRIM25 (Fig. 1H). Furthermore, not only was TRIM25 and HOIP interaction detected in HOIL-1L−/− MEFs (Figure S1B), but a bacterially purified TRIM25 also bound in vitro translated HOIL-1L or HOIP (Figure S1C). These indicate that HOIP interacts with TRIM25 in a HOIL-1L independent manner.
While the C-terminal RBR domain of HOIL-1L was required for TRIM25 interaction, the central NZF domain of HOIL-1L was necessary for RIG-I interaction (Figure 1H). Consistent with this, HOIL-1L and RIG-I binding was detected in TRIM25−/− MEFs and a yeast two-hybrid screen of HOIL-1L ΔRBR also revealed RIG-I as a binding partner (Figure 1I and data not shown). RIG-I-2CARD readily bound bacterially purified HOIL-1L ΔRBR but showed little or no interaction with HOIP ΔRBR in an in vitro binding assay (Figure 1J). Furthermore, the apparent interaction between HOIL-1L and the RIG-I-2CARD K172R mutant that no longer undergoes K63-linked ubiquitination (Gack et al., 2007) suggests that RIG-I ubiquitination is not required for this interaction (Figure S1D). Finally, unlike TRIM25 binding for which the first CARD of RIG-I is sufficient (Gack et al., 2008), HOIL-1L binding requires both the first and second CARD of RIG-I (Figure S1E). These results collectively indicate that HOIL-1L and HOIP interact with TRIM25 and RIG-I in distinct manners.
HOIL-1L/HOIP complex negatively regulates RIG-I-mediated type-I IFN induction
Either HOIL-1L or HOIP markedly inhibited the IFN-β and NF-κB promoter activity induced by RIG-I-2CARD, a constitutively active form of RIG-I, in a dose-dependent manner (Figure 2A and 2B). Accordingly, HOIL-1L or HOIP expression also suppressed the IFN-β promoter activation induced by a full length RIG-I upon SeV infection (Figure 2C). To test the effect of HOIL-1L and HOIP on TRIM25-mediated activation of RIG-I, IFN-β, NF-κB or ISRE promoter activities were measured from cells transfected with RIG-I, TRIM25, HOIL-1L and/or HOIP. This showed that HOIL-1L or HOIP effectively nullified the TRIM25 ability to induce RIG-I-mediated IFN-β, NF-κB or ISRE promoter activation (Figure 2D, E, and F).
Figure 2. HOIL-1L/HOIP complex negatively regulates RIG-I mediated IFN-β signaling.
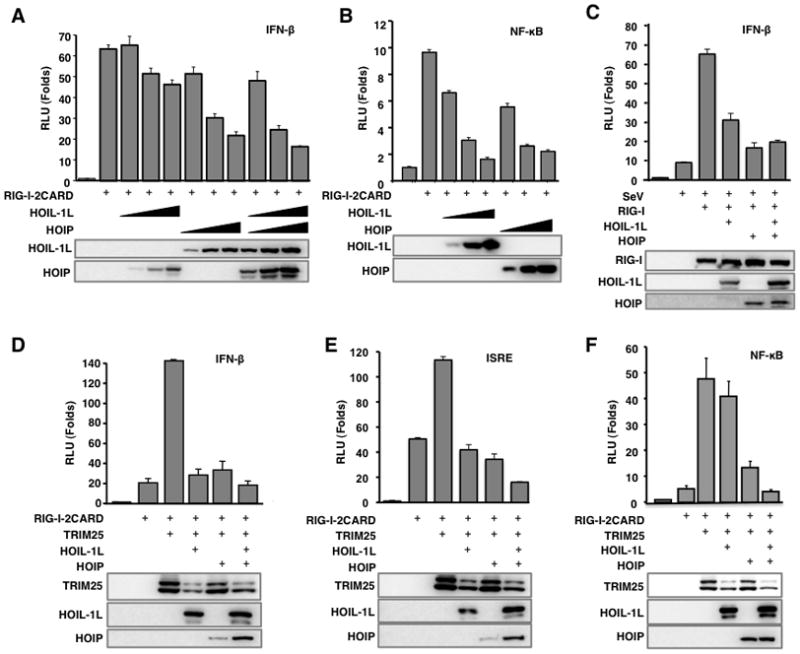
(A–B) Inhibition of RIG-I-2CARD induced IFN-β and NF-κB promoter activities by HOIL-1L/HOIP. 293T cells were transfected with RIG-I-2CARD with increasing amounts of HOIL-1L, HOIP, or both together with IFN-β (A) or NF-κB promoter (B). (C) Inhibition of SeV induced IFN-β promoter activity by HOIL-1L/HOIP. IFN-β promoter activities were measured from 293T cells transfected with full-length RIG-I and HOIL-1L and/or HOIP. At 24 h after transfection, cells were mock-infected or infected with SeV (40 HAU/ml) for 10 h before luciferase assay. (D-F) RIG-I-2CARD, TRIM25, HOIL-1L and HOIP plasmids were transfected together with IFN-β (D), ISRE (E), or NF-κB (F) reporter plasmids into 293T. All luciferase assays were performed at least three times and graphs show the mean ± SD. Values are normalized by pRL-TK Renilla.
In order to further dissect the role of LUBAC in the negative regulation of the RIG-I signaling, induction of the IFN-β promoter activity was examined in WT, HOIL-1L−/−, lentiviral-shRNA-mediated HOIP knockdown (HOIP KD), and HOIL-1L−/−-HOIP KD MEFs upon SeV infection (Figure 3A). Upon SeV infection, HOIL-1L−/− and HOIL-1L−/−-HOIP KD MEFs showed markedly increased levels of IFN-β promoter activity compared to WT MEFs. HOIP KD MEF also showed two to three times higher IFN-β promoter activity than WT MEFs (Figure 3B). Consistently, IFN-β production in response to SeV infection was significantly elevated in HOIL-1L−/− and HOIL-1L−/−-HOIP KD MEFs compared to WT MEFs (Figure 3C). In addition, the increased IFN-β response in HOIL-1L−/− cells was abrogated by HOIL-1L complementation (Figure 3D). When infected with VSV-eGFP, HOIL-1L−/− and HOIL-1L−/−-HOIP KD MEFs showed substantially fewer VSV-eGFP-positive cells and lower VSV yields (over 103–104 fold) compared with WT MEFs (Figure 3E). Accordingly, the HOIL-1L complementation of HOIL-1L−/− MEFs led to the robust recovery of VSV-eGFP replication. Moreover, HOIL-1L overexpression also led to the drastic increase of VSV-eGFP replication (Figure 3F and G). Collectively, these results indicate that LUBAC serves as a critical negative regulator of the RIG-I mediated anti-viral IFN response.
Figure 3. HOIL-1L/HOIP depletion increases IFN-β production and anti-viral response.
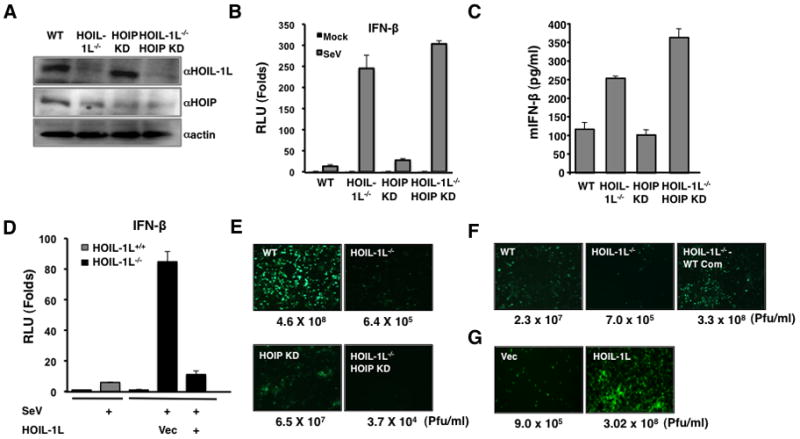
(A) WT, HOIL-1L−/−, HOIP knock-down MEF (HOIP KD), HOIL-1L−/−-HOIP KD MEFs were established as described in the Experimental Procedures. Depletion and reduction of HOIL-1L and HOIP, respectively, were confirmed by IB. (B) Enhanced IFN-β promoter activity in HOIL-1L/HOIP depleted MEFs. MEFs were infected with SeV (50 HAU/ml) for 10 h and then subjected to dual-luciferase assay. Experiments were performed in triplicate and graph shows the mean ± SD. (C) Increased IFN-β production in HOIL-1L/HOIP depleted MEFs. MEFs were infected with SeV (50 HAU/ml) for 12 h and supernatants were subjected to mouse IFN-β ELISA. Experiments were performed in triplicate and graph shows the mean ± SD. (D) Suppression of IFN-β promoter activity in HOIL-1L−/− MEFs by complementation of HOIL-1L. HOIL-1L was co-transfected with reporter plasmid as indicated. Virus infection and luciferase assay were performed similarly to (A). Experiments were performed in triplicate and graph shows the mean ± SD. (E–F) Viral replication in HOIL-1L/HOIP depleted MEFs. MEFs were infected with VSV-eGFP (M.O.I=0.02). Supernatants were taken at 48 h p.i. and subjected to plaque assay. (G) VSV-eGFP replication in 293T expressing vector alone or HOIL-1L.
HOIL-1L/HOIP LUBAC reduces the TRIM25-induced K63-linked ubiquitination of RIG-I
Next, we tested whether HOIL-1L/HOIP LUBAC affects the TRIM25-mediated RIG-I ubiquitination and thereby suppresses its downstream signaling activity. GST-RIG-I-2CARD and TRIM25 were co-expressed together with increasing amounts of HOIL-1L and/or HOIP. While GST-RIG-I-2CARD underwent robust ubiquitination in the presence of TRIM25 (Gack et al., 2007), its ubiquitination levels were detectably decreased by either the individual expression of HOIL-1L or HOIP or further reduced by HOIL-1L and HOIP co-expression (Figures 4A and S2). In line with this, the ubiquitination of endogenous RIG-I was readily detected upon SeV infection, and this ubiquitination was profoundly reduced by HOIL-1L and HOIP overexpression (Figure 4B). Conversely, eliminating the HOIL-1L gene and/or silencing the HOIP gene enhanced the ubiquitination levels of endogenous RIG-I upon viral infection (Figure 4C). Furthermore, immunoblotting with a K63-linked polyubiquitin specific antibody (Wang et al., 2008) confirmed the increase of K63-linked polyubiquitinated RIG-I in HOIL-1L or HOIP depleted cells (Figure 4C). Consistent with the notion that K63-linked RIG-I ubiquitination plays a crucial role for its interaction with MAVS (Gack et al., 2007), HOIL-1L and HOIP expression efficiently inhibited the RIG-I-2CARD and MAVS-CARD interaction (Figure 4D). This indicates that LUBAC suppresses RIG-I ubiquitination, thereby blocking its interaction with MAVS.
Figure 4. HOIL-1L/HOIP LUBAC inhibits TRIM25-mediated RIG-I ubiquitination.
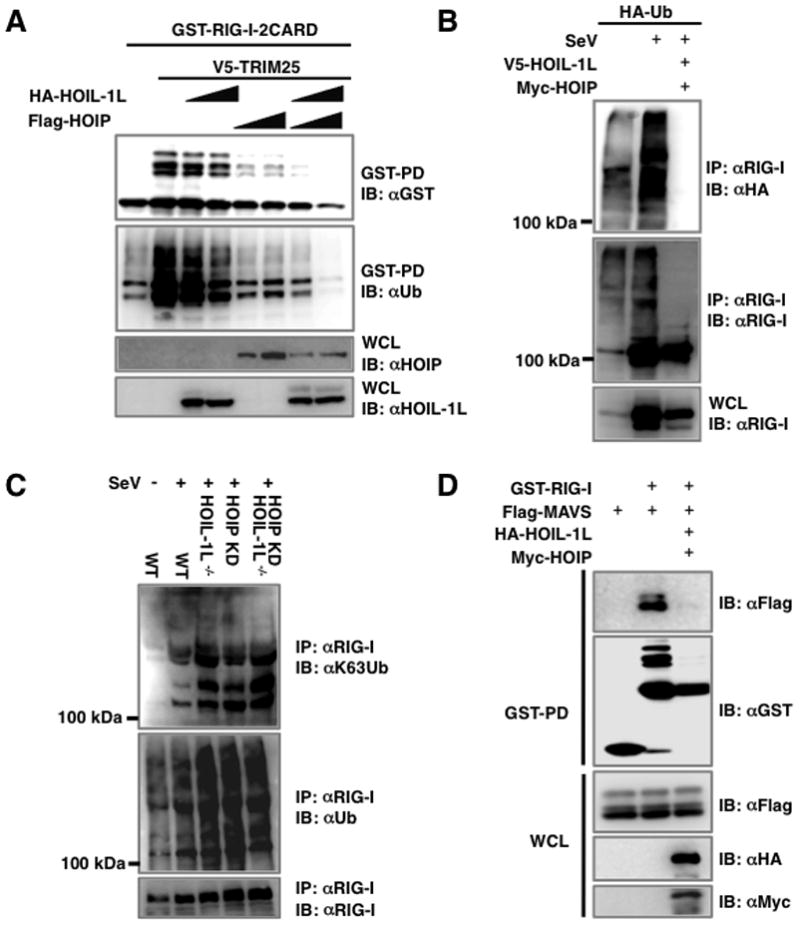
(A) Inhibition of RIG-I ubiquitination by HOIL-1L/HOIP. 293T cells transfected with GST-RIG-I-2CARD and TRIM25 together with HOIL-1L or HOIP were subjected to GST-PD and IB. (B) Inhibition of endogenous RIG-I ubiquitination by HOIL-1L/HOIP. 293T cells transfected with HA-ubiquitin with or without HOIL-1L/HOIP were mock-infected or infected with SeV for 10 h, followed by IP with anti-RIG-I. (C) RIG-I ubiquitination in HOIL-1L/HOIP depleted MEF. WT, HOIL-1L−/−, HOIP-KD and HOIL-1L−/−-HOIP-KD MEFs infected with SeV were used for IP and IB. Anti-ubiquitin (αUb) antibody and anti-K63-ubiquitin chain specific antibody (αK63) were used to detect RIG-I ubiquitination. (D) Decreased interaction between RIG-I and MAVS by HOIL-1L/HOIP expression. 293T cells transfected with Flag-RIG-I-2CARD, GST-MAVS-CARD-proline-rich domain (PRD), V5-HOIL-1L and/or Myc-HOIP were used for co-IP and IB. See also Figure S2.
HOIL-1L/HOIP LUBAC induces the ubiquitination and degradation of TRIM25
To elaborate the effect of HOIL-1L/HOIP LUBAC on RIG-I and TRIM25 function, we tested whether HOIL-1L and HOIP inhibited RIG-I ubiquitination by lowering the TRIM25 level. We found that SeV-infected HOIP KD MEFs had a greater amount of endogenous TRIM25 compared to WT MEFs, with the highest levels found in HOIL-1L−/−-HOIP KD MEFs (Figure 5A). Conversely, HOIL-1L and/or HOIP expression led to a measurable reduction of endogenous TRIM25 (Figure 5B). Transient expression assay also showed that HOIL-1L and HOIP co-expression induced a detectable reduction of TRIM25 in a synergistic manner, but this reduction was efficiently reversed by MG132 proteasome inhibitor treatment (Figure 5C). In addition, HOIL-1L and HOIP expression increased the level of TRIM25 ubiquitination, which was more pronounced upon MG132 treatment (Figure 5D). While TRIM25 ubiquitination was potently induced in WT MEFs upon SeV infection, it was markedly reduced in HOIL-1L deleted and/or HOIP depleted MEFs (Figure 5E and F). Furthermore, TRIM25 ubiquitination was dependent on the HOIL-1L and HOIP E3 ligase activities, as mutating the conserved cysteines in the RING domains of HOIL-1L and HOIP into serine (RINGCS) rendered them unable to ubiquitinate TRIM25 (Figure 5G). Finally, the LUBAC-mediated degradation of TRIM25 was specific since LUBAC showed little or no effect on ectopically expressed RIG-I levels (Figure S3A). It should be noted that due to its negative role in IFN signal transduction, HOIL-1L/HOIP LUBAC expression affects the RIG-I mRNA and protein levels induced by SeV infection as seen with IFN-β (Figures 4B, C and S3B).
Figure 5. HOIL-1L/HOIP LUBAC induces TRIM25 ubiquitination and degradation.
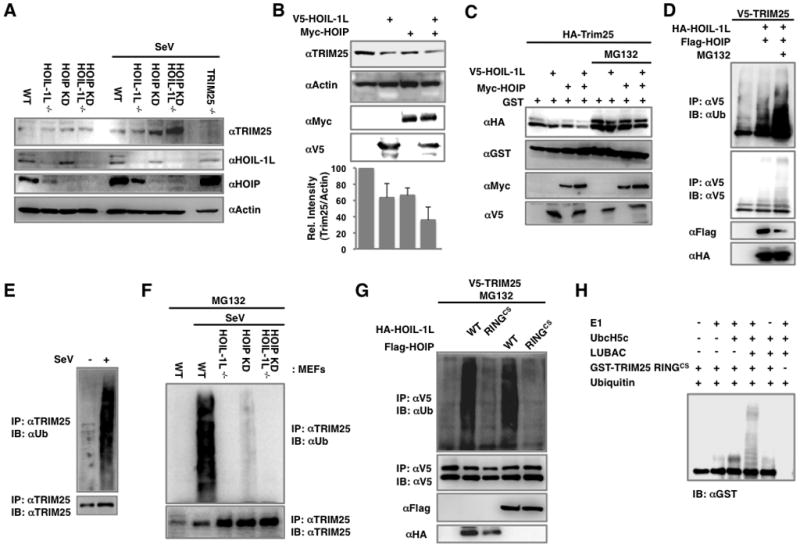
(A) Increment of TRIM25 levels in HOIL-1L/HOIP depleted MEFs. WT, HOIL-1L−/−, HOIP KD, HOIL-1L−/−-HOIP KD MEFs were mock-infected or infected with SeV for 12 h. Cell lysates were subjected to IB. (B) Decreased endogenous TRIM25 levels upon HOIL-1L/HOIP overexpression. Endogenous TRIM25 levels were analyzed by IB and densitometry. IBs show representative data, and graph shows the averages of triplicate. (C) Reduction of TRIM25 by HOIL-1L/HOIP expression. At 36 h after the transfection with V5-TRIM25 and GST together with HOIL-1L and HOIP as indicated, 293T cells were mock-treated or treated with MG132 for 6 h and were subjected to IB. (D) TRIM25 ubiquitination induced by HOIL-1L/HOIP. 293T cells were transfected with V5-TRIM25, HOIL-1L and HOIP and MG132 treatment for IP. (E) Endogenous TRIM25 ubiquitination upon SeV infection. MEFs were mock-infected or infected with SeV for 10 h and cell lysates were subjected to IP using anti-TRIM25 antibody. (F) TRIM25 ubiquitination levels in HOIL-1L/HOIP depleted MEFs. MEFs were infected with SeV and treated with MG132, followed by IP with anti-TRIM25 and IB with anti-Ub or anti-TRIM25. (G) TRIM25 ubiquitination by HOIL-1L/HOIP RINGCS mutants. 293T cells were transfected with V5-TRIM25 and WT or RINGCS mutants of HOIL-1L or HOIP, followed by IP with anti-V5 and IB with anti-Ub or anti-V5. (H) In vitro ubiquitination of TRIM25 by LUBAC. Purified GST-TRIM25 RINGCS was subjected to an in vitro ubiquitination with baculovirus-purified HOIL-1L/HOIP LUBAC, followed by IB with anti-GST antibody. See also Figure S3.
An in vitro ubiquitination assay showed that the HOIL-1L/HOIP LUBAC purified from a baculovirus expression system specifically ubiquitinated bacterially purified GST-TRIM25 RINGCS, but not GST-RIG-2CARD (Figures 5H and S3C). The TRIM25 ΔSPRY mutant, lacking the C-terminal SPRY domain and thereby no longer interacting with HOIL-1L and HOIP, did not undergo ubiquitination upon viral infection (Figure S3D). Conversely, the TRIM25 SPRY domain alone, which is sufficient to interact with HOIL-1L and HOIP, underwent robust ubiquitination that was decreased upon shRNA-mediated knockdown of HOIL-1L or HOIP gene expression (Figures S3E, F, G and H). The Lys48 (K48)-linked ubiquitin chain was the original type identified, however, more recent works have uncovered a wide variety of linkages including K63-linked and linear ubiquitin chains (Ikeda and Dikic, 2008). Since LUBAC is known to generate linear ubiquitination, we tested whether LUBAC can conjugate a linear ubiquitin chain onto TRIM25. An in vitro assay with a ubiquitin KO mutant containing no lysine residues showed that HOIL-1L/HOIP LUBAC was able to efficiently incorporate linear ubiquitin chains onto TRIM25 (Figure S3I). In addition, the linear ubiquitination of TRIM25 was detectably increased upon SeV infection or ectopic expression of HOIL-1L/HOIP LUBAC (Figure S3J). Furthermore, TRIM25 also underwent K48-linked ubiquitination upon SeV infection, which was significantly attenuated by shRNA-mediated knockdown of HOIP expression (Figure S3K). Conversely, HOIL-1L/HOIP LUBAC expression induced the K48-linked ubiquitination of TRIM25, which was further augmented by treatment with MG132 (Figure S3L). In contrast, K63-linked ubiquitination of TRIM25 was not detected in vivo (Figure S3K and data not shown).
TRIM25 undergoes self-mono-ubiquitination
It was noted that TRIM25 WT, but not the TRIM25CS E3 ligase dead mutant, was present as a doublet (70 and 78 kDa), and that the upper band of TRIM25 often disappeared upon HOIL-1L and HOIP expression (Figure 1A, B and S3M), suggesting that TRIM25 undergoes auto-mono-ubiquitination and that the mono-ubiquitinated form of TRIM25 may be highly susceptible to HOIL-1L and HOIP. Mass spectrometry showed that the upper band of TRIM25 is mono-ubiquitinated at the K117, K264, K320 and K416 residues. To corroborate the mono-ubiquitination of TRIM25, we individually replaced these four lysine residues with arginine (K → R) and then tested these mutants for their ubiquitination levels. The K117R mutation of TRIM25 caused the near complete loss of mono-ubiquitination, whereas the K264R, K320R and K416R mutations led to little or no reduction of mono-ubiquitination (Figures S3N). Intriguingly, the TRIM25 K117R mutant was functionally similar to WT TRIM25 with respect to its ability to ubiquitinate RIG-I-2CARD and induce RIG-I mediated IFN-β promoter activation (Figures S3O and P). Furthermore, the TRIM25 K117R mutant was still sensitive to the ubiquitination and signaling inhibition induced by the HOIL-1L/HOIP LUBAC (Figures S3Q and R). These data indicate that the mono-ubiquitination of TRIM25 is not essential for its RIG-I activation function and that LUBAC targets both non- and mono-ubiquitinated forms of TRIM25.
HOIL-1L/HOIP LUBAC complex interrupts the interaction between TRIM25 and RIG-I
To determine whether the E3 ligase activities of HOIL-1L and HOIP are sufficient to negatively regulate RIG-I mediated IFN-β signaling, the HOIL-1L and HOIP RINGCS E3-ligase defective mutants were tested for their abilities to downregulate RIG-I ubiquitination and IFN-β signaling. Surprisingly, the RINGCS mutants of both HOIL-1L and HOIP still suppressed SeV- or RIG-I-induced IFN-β promoter activation and TRIM25-mediated RIG-I ubiquitination (Figures S4A, S4B and 6A), suggesting that HOIP/HOIL-1L LUBAC might have additional mechanisms to downregulate RIG-I ubiquitination and signaling activity other than TRIM25 degradation. We hypothesized that the diverse interacting activities of HOIL-1L and HOIP could ultimately influence the interaction between TRIM25 and RIG-I. Indeed, the interaction between RIG-I-2CARD and TRIM25 was noticeably reduced by HOIL-1L or HOIP expression where the reduction was further decreased by HOIL-1L and HOIP coexpression (Figure 6B, left). Moreover, increasing levels of HOIL-1L and HOIP coexpression inhibited the interaction between RIG-I-2CARD and TRIM25 in a dose dependent manner (Figure 6B, right). Conversely, the interaction between endogenous RIG-I and TRIM25 was elevated in HOIL-1L−/− MEFs and HOIL-1L−/−-HOIP KD MEFs. (Figure 6C). The interaction between endogenous RIG-I and TRIM25 reached a peak at 8 hours post SeV infection and declined afterward, coinciding with the significant increase in HOIL-1L and HOIP protein levels after 12 hrs of SeV infection (Figure 6D). These results were further confirmed in in vitro binding assays using bacterially purified GST fusions of RIG-2CARD, TRIM25, HOIL-1L ΔRBR and HOIP ΔRBR. Increasing amounts of LUBAC ΔRBR (HOIL-1L ΔRBR/HOIP ΔRBR) proteins led to the decreased interaction between RIG-I-2CARD and TRIM25 in a dose-dependent manner, while the interaction between RIG-I and LUBAC ΔRBR increased under the same conditions (Figure 6E).
Figure 6. HOIL-1L/HOIP LUBAC inhibits the interaction between RIG-I and TRIM25.
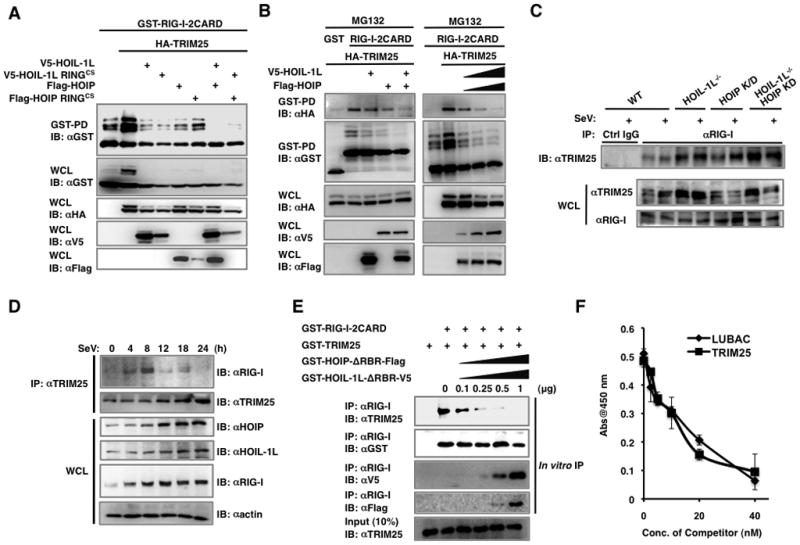
(A) Inhibition of RIG-I ubiquitination by HOIL-1L/HOIP RINGCS mutants. 293T cells were transfected with GST-RIG-I-2CARD and TRIM25 together with HOIL-1L, HOIP, or RINGCS mutants and treated with MG132, followed by GST-PD. (B) Inhibition of RIG-I-2CARD and TRIM25 interaction by HOIL-1L/HOIP. 293T cells were transfected with GST-RIG-I-2CARD and TRIM25 together with HOIL-1L and/or HOIP as indicated, followed by GST-PD. (C) Increased interaction between RIG-I and TRIM25 in HOIL-1L/HOIP depleted MEFs. MEFs were mock-infected or infected with SeV (50 HAU/ml) for 12 h and subjected to co-IP using control IgG or anti-RIG-I. (D) Time-course analysis of endogenous RIG-I and TRIM25 interaction. At different time points after SeV infection, the same amounts of proteins from WT MEFs were subjected to co-IP using anti-TRIM25. (E) In vitro competition assay. Bacterially purified GST-RIG-I-2CARD and GST-TRIM25 were incubated with increasing amounts of GST-HOIL-1L ΔRBR-V5 or GST-HOIP ΔRBR-Flag, followed by IP with anti-RIG-I. (F) Inhibition of RIG-I and TRIM25 interaction by LUBAC. Biotin-labeled GST-TRIM25 were added to GST-RIG-I coated wells with increasing amounts of GST-HOIL-1L ΔRBR-V5/GST-HOIP ΔRBR-Flag (LUBAC) or unlabeled GST-TRIM25. Bound biotin-labeled GST-TRIM25 was detected using streptavidin-HRP. Experiments were performed in triplicate and graph shows the mean ± SD. See also Figure S4.
To further analyze this binding competition, biotin-labeled GST-TRIM25 was added to immobilized GST-RIG-I-2CARD, followed by the addition of increasing amounts of unlabeled TRIM25 or LUBAC ΔRBR (HOIL-1L ΔRBR/HOIP ΔRBR) as binding competitors. Biotin-labeled TRIM25 interaction with RIG-I-2CARD was inhibited by unlabeled TRIM25 or LUBAC ΔRBR to similar extent, suggesting that TRIM25 and LUBAC may have similar binding affinities to RIG-I (Figure 6F). On the other hand, the HOIL-1L ΔNZF mutant, which does not interact with RIG-I, failed to compete with TRIM25 for RIG-I-2CARD binding. (Figure S5A). Consequently, the HOIL-1L ΔNZF mutant downregulated RIG-I ubiquitination and IFN-β promoter activation at much lesser extent than HOIL-1L WT (Figures S5B and C). Interestingly, the HOIL-1L ΔUBL mutant that lost the LUBAC formation with HOIP only weakly inhibited RIG-I ubiquitination and signaling activity compared to its wild-type, suggesting that the LUBAC formation between HOIL-1L and HOIP is important for its suppressive activity (Figures S5B and C).
To further corroborate the importance of HOIL-1L NZF domain for RIG-I inhibition, we generated the NZFCS mutant that lost its zinc finger structure due to the mutations of the conserved cysteines to serines. As seen with its ΔNZF mutant, HOIL-1L NZFCS mutant did not bind to RIG-I-2CARD (Figure 7A). Consequently, HOIL-1L NZFCS mutant was incapable of suppressing the interaction between RIG-I and TRIM25 (Figure 7B) and thus showed reduced ability to downregulate RIG-I ubiquitination and IFN-β promoter activation (Figure 7C and D). These results suggest that HOIL-1L suppresses the RIG-I-TRIM25 interaction by binding RIG-I in a competitive fashion, resulting in an inhibitory effect on RIG-I signaling activity.
Figure 7. Roles of the NZF and RBR domains of HOIL-1L in RIG-I mediated IFN-β signaling.
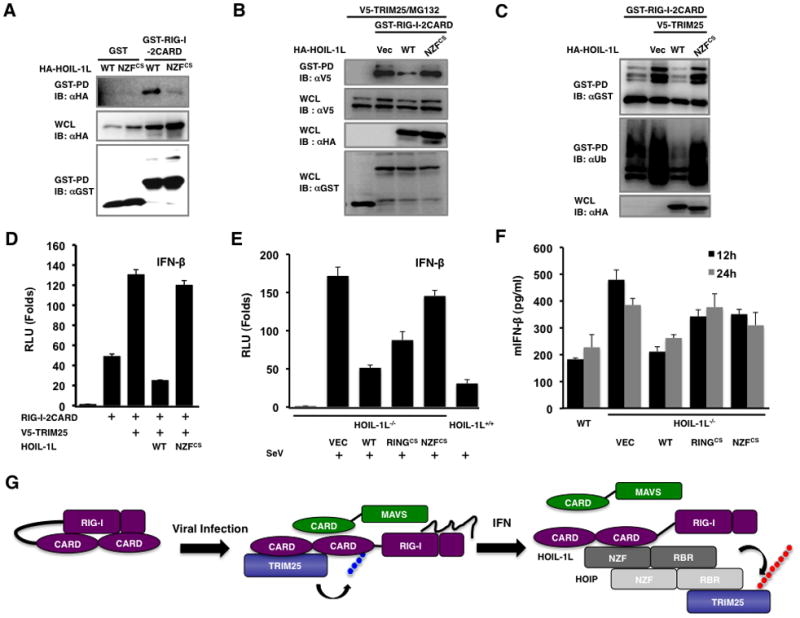
(A) Inability of HOIL-1L NZFCS mutant to bind RIG-I. 293T cells were transfected with GST-RIG-I-2CARD together with HA-HOIL-1L WT or NZFCS mutant, followed by GST-PD and IB with indicated antibodies. (B) Inability of HOIL-1L NZFCS mutant to interfere the RIG-I-TRIM25 interaction. 293T cells were transfected with GST-RIG-I-2CARD and TRIM25 together with HA-HOIL-1L WT or NZFCS mutants and treated with MG132, followed by GST-PD and IB. (C) Inability of HOIL-1L NZFCS mutant to inhibit RIG-I ubiquitination. 293T cells were transfected with GST-RIG-I-2CARD and TRIM25 together with HOIL-1L WT or NZFCS mutant as indicated, followed by GST-PD and IB. (D) Effect of HOIL-1L WT or NZFCS mutant on RIG-I-mediated IFN-β promoter activity. IFN-β promoter activities were determined from 293T cells transfected with RIG-I-2CARD, and TRIM25 together with HOIL-1L WT or NZFCS mutant. Experiments were performed in triplicate and graph shows the mean ± SD. (E-F) Complementation of HOIL-1L−/− MEFs. HOIL-1L−/− MEFs were infected with recombinant retrovirus containing HOIL-1L WT, RINGCS, or NZFCS mutant, followed by the puromycin antibiotic selection. (E) Complemented MEFs were transfected with IFN-β reporter, followed by mock-infection or SeV infection and luciferase assay. Experiments were performed in triplicate and graph shows the mean ± SD. (F) Complemented MEFs were mock-infected or infected with SeV (50 HAU/ml) and supernatants were subjected to IFN-β ELISA. Experiments were performed in triplicate and graph shows the mean ± SD. (G) Hypothetical model for LUBAC-mediated inhibition of RIG-I and TRIM25 pathway. See also Figure S5.
Finally, HOIL-1L−/− MEFs were complemented with wild-type HOIL-1L or its mutants (NZFCS or RINGCS) and subsequently infected with SeV to measure IFN-β promoter activity and production. A high basal level of SeV infection-induced IFN-β production of HOIL-1L−/− MEFs was robustly dampened by complementation with the HOIL-1L WT (Figure 7E). In contrast, the complementation with either the HOIL-1L NZFCS or the HOIL-1L RINGCS mutant resulted in significantly weaker suppression of anti-viral response than that with the HOIL-1L WT as assayed by IFN-β promoter activity and IFN-β production (Figure 7E and F). These data collectively demonstrate that LUBAC comprehensively suppresses RIG-I anti-viral activity by inducing the C-terminal RBR-E3 ligase-mediated degradation of TRIM25 and the central NZF-mediated inhibition of TRIM25-RIG-I interaction (Figure 7G).
Discussion
Tight regulation of immune signaling pathways is essential for a successful immune response against viral infections; otherwise, excessive production of IFNs or pro-inflammatory cytokines would be destructive rather than protective. In this report, we unveil the potential feedback inhibitory role of the HOIL-1L/HOIP LUBAC, which specifically suppresses RIG-I ubiquitination and activation by inducing TRIM25 degradation and inhibiting TRIM25 and RIG-I interaction.
Similar to the HOIL-1L/HOIP LUBAC, numerous intracellular proteins have been demonstrated to contribute to the negative regulation or feedback inhibition of RIG-I signal transduction. First, the RIG-I-like RNA helicase, Lgp2, which lacks the CARD domains, inhibits virus-induced RIG-I downstream signaling (Komuro and Horvath, 2006; Rothenfusser et al., 2005) and RNF125 E3 ligase catalyzes RIG-I proteasomal degradation (Arimoto et al., 2007), whereas CYLD deubiquitinase eliminates the K63-linked polyubiquitin chain from RIG-I to downregulate its signaling activity (Friedman et al., 2008). In addition, A20, NLRX1, PIN1, DUBA and Triad3A deregulate RIG-I mediated signaling by inhibiting MAVS, IRF3 and TRAF3 (Kayagaki et al., 2007; Lin et al., 2006; Moore et al., 2008; Nakhaei et al., 2009b; Saitoh et al., 2006). Furthermore, the alternative splicing or ISGylation of RIG-I also negatively regulates its function on virus-triggered IFN production (Gack et al., 2008; Kim et al., 2008). Finally, the influenza A virus non-structural protein 1 (NS1) specifically inhibits TRIM25-mediated RIG-I ubiquitination, thereby suppressing RIG-I signal transduction (Gack et al., 2009). This indicates that the host dedicates a number of its intracellular components to tightly control RIG-I signaling for the successful elimination of viral infections.
Mutational analysis showed that the C-terminal RBR domains of HOIL-1L and HOIP are essential for TRIM25 binding, whereas the central NZF domain of HOIL-1L is necessary for RIG-I binding, indicating that HOIL-1L and HOIP interact with TRIM25 and RIG-I in different manners. While the detailed resolution of the HOIL-1L/HOIP LUBAC interactions with TRIM25 or RIG-I needs to be further characterized, these differential interactions of HOIL-1L and HOIP with TRIM25 and RIG-I eventually lead to the competition between the LUBAC and TRIM25 for RIG-I binding, resulting in the reduction of the K63-ubiquitination of RIG-I. Furthermore, LUBAC expression induces TRIM25 ubiquitination and proteasomal degradation, thereby suppressing RIG-I mediated IFN signaling. Conversely, HOIL-1L knockout and HOIP knockdown cells showed markedly higher IFN production than WT cells, suggesting that LUBAC serves as an important negative factor to tightly control RIG-I signaling activity.
The HOIL-1L/HOIP LUBAC has been shown to negatively regulate various signaling pathways by degrading its interacting partners such as IRP2, TAB1/2, and Bach1 through ubiquitin-dependent proteasomal pathway (Ishikawa et al., 2005; Tian et al., 2007; Zenke-Kawasaki et al., 2007). In addition, a recent study showed that linear ubiquitination of a protein can lead to its proteasomal degradation (Zhao and Ulrich, 2010). Our study has added TRIM25 into the increasing substrate list of HOIL-1L and HOIP LUBAC. However, both the RINGCS and ΔRBR mutants of HOIL-1L and HOIP that lose ubiquitin-incorporating activity were still capable of downregulating RIG-I signaling, suggesting that LUBAC might have additional mechanism(s) to suppress RIG-I ubiquitination and signaling activity. Indeed, we found that the interaction between HOIL-1L and RIG-I competitively suppresses the interaction between TRIM25 and RIG-I. Particularly, this action is dependent on the central NZF domain of HOIL-1L: the HOIL-1L ΔNZF or NZFCS mutant, which lost RIG-I binding ability, was incapable of suppressing the interaction between RIG-I and TRIM25 and thus showed the reduced activity to downregulate the RIG-I ubiquitination and IFN-β promoter activation. The NZF domain is frequently found in proteins involved in ubiquitin-dependent pathways, such as TAB2, TAB3 (Kanayama et al., 2004), Npl4 (Meyer et al., 2002), and yeast Vps36 (Alam et al., 2004). The ubiquitin-binding NZF domain comprises of a highly conserved TF/Φ motif, where Φ represents a hydrophobic residue and 10 amino-acid residues are inserted between the -Thr-Phe (TF)- sequence and Φ (Alam et al., 2004). Certain NZF domains can discriminate against different types of ubiquitin chains, as exemplified by the NZF domain of TAB2, which displays a high affinity for the K63-linked ubiquitin moiety (Kanayama et al., 2004). It is particularly intriguing that since free unanchored K63-linked ubiquitin chain is able to activate RIG-I signaling (Zeng et al., 2010), the NZF domain may potentially function as a K63-linked ubiquitin chain-anchoring motif for RIG-I signaling. However, while the central NZF domain of HOIL-1L plays an important role in RIG-I CARD binding, this binding is not dependent on the K63-linked ubiquitination of RIG-I CARD. Further structural studies are necessary to delineate the interaction between the N-terminal RIG-I CARD and the central HOIL-1L NZF domain.
During our research, it has been reported that RBCK1, an alternative name of HOIL-1L, downregulates RIG-I signaling through IRF3 degradation in transient overexpression conditions (Zhang et al., 2008). However, we were unable to observe the detectable reduction of endogenous IRF3 levels upon HOIL-1L and/or HOIP transient expression (Data not shown) or in HOIL-1L−/− MEFs, HOIP KD MEFs or HOIL-1L−/−-HOIP KD MEFs (data not shown), suggesting that IRF3 may not be a direct substrate for HOIL-1L/HOIP LUBAC. In summary, our data collectively demonstrate that HOIL-1L/HOIP LUBAC comprehensively suppresses RIG-I antiviral activity by inducing the C-terminal RBR E3 ligase-mediated degradation of TRIM25 and/or the central NZF-mediated inhibition of TRIM25 and RIG-I interaction (Figure 7G). Thus, the HOIL-1L/HOIP complex represents a negative regulator of RIG-I and TRIM25-mediated innate immunity that may serve to prevent an excessive and prolonged IFN production.
Experimental Procedures
Plasmids
GST-RIG-2CARD, GST-RIG-I 1st CARD, GST-RIG-I 2nd CARD, pBOS-Flag-RIG-I, MAVS-CARD-PRD, pIRES-V5-TRIM25 and its deletion mutants were previously described (Gack et al., 2007). HOIL-1L-His6-HA wild-type, Flag-, Myc-HOIP wild type, their deletion mutants, and their RINGCS and NZFCS mutants were described previously (Kirisako et al., 2006). pBabe-HOIL-1L and mutants for complementation assay were obtained by standard PCR and cloning method.
Antibodies and other reagents
Mouse monoclonal antibody 2E2 against HOIL-1L (Kirisako et al., 2006), and anti-linear ubiquitin antibody (Tokunaga et al., 2009) were described previously. Antibodies against TRIM25 (BD Biosciences), RIG-I (mouse monoclonal: Alexis; rabbit: IBL), HOIP (goat: IMGENEX; rabbit: Abcam), MAVS (Cell Signaling), Ubiquitin (SantaCruz, P4D1), Lys63-polyubiquitin chain (HWA4C4: Biomol), Lys48-polyubiquitin chain (Apu2: Millipore) and actin (Abcam) were purchased from the indicated manufacturer. VSV-eGFP was described previously (Gack et al., 2007).
Yeast two-hybrid screen
A human B-lymphocyte cDNA library was screened using TRIM25 ΔRING as bait. Matchmaker yeast two-hybrid system was obtained from Clontech.
Cell lines
HOIL-1L−/− MEF cell line was described previously (Tokunaga et al., 2009). HOIP-K/D and HOIL-1L−/−-HOIP KD MEFs were established by stable knock-down of HOIP using shRNA (Openbiosystems; TRCN0000037277; CAGAGAAACAACGCCAAGATACTCGAGTATCTTGGCGTTGTTTCTCTG). Cells transduced with lentivirus were selected with puromycin (2 μg/ml) 2 days after infection. Control cell lines were generated using scrambled control shRNA vector. HOIL-1L stable-expressing HEK293T cell line was generated by transfecting pcDNA3-HOIL-1L, followed by selection with G418 (400 μg/ml). For the complementation, HOIL-1L−/− MEFs were infected with pBabe-HOIL-1L WT or mutants retrovirus, followed by 4-day selection with puromycin.
IFN-β production and virus replication assay
Cell culture supernatants were collected from cells infected with SeV as indicated in the figure legends and subjected to IFN-β ELISA (PBL Biomedical Laboratories). To assay the viral replication, culture supernatants were collected from VSV-eGFP infected cells, and viral titer was determined in Vero cells by standard plaque assay.
Reporter Assays
All reporter assays were performed using Dual-luciferase Assay kit (Promega). Cells were transfected with the indicated reporters together with pRL-TK reporter. Twenty-four hrs after transfection, cells were treated as indicated in the figure legends.
Immunoprecipitation and immunoblot analysis
For co-immunoprecipitations, cells were lysed with NP40 lysis buffer (25 mM Tris-pH-7.5, 150 mM NaCl, 1 mM EDTA, 0.6% Nonidet P-40). After clarification and pre-clearing, protein amounts were determined using BCA assay. Cell extracts were incubated for 12–16 hours with the indicated antibodies, followed by further incubation with protein A/G bead for 2–4 hours. The immune complexes were washed with lysis buffer with various concentrations of NaCl and subjected to western blot analysis. For endogenous co-IP, antibodies were conjugated to resin using a co-IP kit (Pierce). For ubiquitination assay, cells were initially lysed with RIPA buffer containing 1% SDS and the cell extracts were diluted with RIPA buffer until 0.1% SDS concentration before IP, and immunoblottings were performed as described previously (Tokunaga et al., 2009).
Confocal Microscopy
Cells grown on chamber slides were mock-infected or infected with SeV for 10 hours, followed by fixation with 2% paraformaldehyde solution at room temperature for 20 minutes. For co-localization of TRIM25 and HOIL-1L/HOIP, mouse anti-TRIM25 (anti-EFP; BD Biosciences), rabbit anti-EFP (Santa Cruz), goat anti-HOIL-1L (anti-RBCK1; Santa Cruz), and mouse anti-HOIP (mN1) were used.
Protein purification and in vitro ubiquitination assay
TRIM25 WT was cloned into pGEX-4T-1 vector. TRIM25 RINGCS-V5, HOIL-1LΔRBR-V5 and HOIPΔRBR-flag were cloned into pGEX-6P-1 vector. BL21 E.coli were transformed with each plasmid. Proteins were purified using Glutathione Sepharose4B resin according to the manufacturer’s instruction. LUBAC complex, E1, and E2 proteins were purified, and in vitro ubiquitination assays were carried out as described previously (Kirisako et al., 2006). In vitro translation was performed using TnT Quick Coupled Transcription/Translation system (Promega) according to the manufacturer’s instruction.
In vitro binding assays
Purified GST-TRIM25 (250 ng) and GST-RIG-I-2CARD (250 ng) were incubated together with increasing amount of purified GST-HOIL-1L ΔRBR-V5/GST-HOIPΔRBR-Flag for 2 hrs at 4°C. R37 anti-RIG-I antibody was added and further incubated for 2 hrs, followed by incubation with protein A/G bead for 2 hrs. Samples were eluted by SDS-sample buffer and subjected to IB analysis. For TRIM25 in vitro binding to immobilized RIG-I, GST-RIG-I-2CARD were diluted in bicarbonate buffer and coated onto Maxisorb ELISA plates (200 ng/well). Biotin-labeled GST-TRIM25 was added to wells together with increasing amount of unlabeled GST-TRIM25 or GST-HOIL-1L ΔRBR-V5/GST-HOIPΔRBR-Flag. After washing, bound TRIM25 was detected using Streptavidin-HRP.
Supplementary Material
Acknowledgments
This work was partly supported by CA082057, AI083025, AI083355, KRIBB, GRL Program (K20815000001) from National Research Foundation of Korea, Hastings Foundation, and Fletcher Jones Foundation (JUJ), AI087846-01A1 and RR00168 (to M.U.G.). We thank Stacy Lee for manuscript preparation and all of JJ’s lab members for their discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arimoto K, Takahashi H, Hishiki T, Konishi H, Fujita T, Shimotohno K. Negative regulation of the RIG-I signaling by the ubiquitin ligase RNF125. Proc Natl Acad Sci U S A. 2007;104:7500–7505. doi: 10.1073/pnas.0611551104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhoj VG, Chen ZJ. Ubiquitylation in innate and adaptive immunity. Nature. 2009;458:430–437. doi: 10.1038/nature07959. [DOI] [PubMed] [Google Scholar]
- Friedman CS, O’Donnell MA, Legarda-Addison D, Ng A, Cardenas WB, Yount JS, Moran TM, Basler CF, Komuro A, Horvath CM, et al. The tumour suppressor CYLD is a negative regulator of RIG-I-mediated antiviral response. EMBO Rep. 2008;9:930–936. doi: 10.1038/embor.2008.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gack MU, Albrecht RA, Urano T, Inn KS, Huang IC, Carnero E, Farzan M, Inoue S, Jung JU, Garcia-Sastre A. Influenza A Virus NS1 Targets the Ubiquitin Ligase TRIM25 to Evade Recognition by the Host Viral RNA Sensor RIG-I. Cell Host Microbe. 2009;5:439–449. doi: 10.1016/j.chom.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gack MU, Kirchhofer A, Shin YC, Inn KS, Liang C, Cui S, Myong S, Ha T, Hopfner KP, Jung JU. Roles of RIG-I N-terminal tandem CARD and splice variant in TRIM25-mediated antiviral signal transduction. Proc Natl Acad Sci U S A. 2008;105:16743–16748. doi: 10.1073/pnas.0804947105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gack MU, Shin YC, Joo CH, Urano T, Liang C, Sun L, Takeuchi O, Akira S, Chen Z, Inoue S, Jung JU. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature. 2007;446:916–920. doi: 10.1038/nature05732. [DOI] [PubMed] [Google Scholar]
- Ikeda F, Dikic I. Atypical ubiquitin chains: new molecular signals. ‘Protein Modifications: Beyond the Usual Suspects’ review series. EMBO Rep. 2008;9:536–542. doi: 10.1038/embor.2008.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H, Kato M, Hori H, Ishimori K, Kirisako T, Tokunaga F, Iwai K. Involvement of heme regulatory motif in heme-mediated ubiquitination and degradation of IRP2. Mol Cell. 2005;19:171–181. doi: 10.1016/j.molcel.2005.05.027. [DOI] [PubMed] [Google Scholar]
- Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441:101–105. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- Kawai T, Akira S. Toll-like receptor and RIG-I-like receptor signaling. Ann N Y Acad Sci. 2008;1143:1–20. doi: 10.1196/annals.1443.020. [DOI] [PubMed] [Google Scholar]
- Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, Ishii KJ, Takeuchi O, Akira S. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol. 2005;6:981–988. doi: 10.1038/ni1243. [DOI] [PubMed] [Google Scholar]
- Kayagaki N, Phung Q, Chan S, Chaudhari R, Quan C, O’Rourke KM, Eby M, Pietras E, Cheng G, Bazan JF, et al. DUBA: a deubiquitinase that regulates type I interferon production. Science. 2007;318:1628–1632. doi: 10.1126/science.1145918. [DOI] [PubMed] [Google Scholar]
- Kim MJ, Hwang SY, Imaizumi T, Yoo JY. Negative feedback regulation of RIG-I-mediated antiviral signaling by interferon-induced ISG15 conjugation. J Virol. 2008;82:1474–1483. doi: 10.1128/JVI.01650-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirisako T, Kamei K, Murata S, Kato M, Fukumoto H, Kanie M, Sano S, Tokunaga F, Tanaka K, Iwai K. A ubiquitin ligase complex assembles linear polyubiquitin chains. EMBO J. 2006;25:4877–4887. doi: 10.1038/sj.emboj.7601360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komuro A, Horvath CM. RNA- and virus-independent inhibition of antiviral signaling by RNA helicase LGP2. J Virol. 2006;80:12332–12342. doi: 10.1128/JVI.01325-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin R, Yang L, Nakhaei P, Sun Q, Sharif-Askari E, Julkunen I, Hiscott J. Negative regulation of the retinoic acid-inducible gene I-induced antiviral state by the ubiquitin-editing protein A20. J Biol Chem. 2006;281:2095–2103. doi: 10.1074/jbc.M510326200. [DOI] [PubMed] [Google Scholar]
- Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R, Tschopp J. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature. 2005;437:1167–1172. doi: 10.1038/nature04193. [DOI] [PubMed] [Google Scholar]
- Moore CB, Bergstralh DT, Duncan JA, Lei Y, Morrison TE, Zimmermann AG, Accavitti-Loper MA, Madden VJ, Sun L, Ye Z, et al. NLRX1 is a regulator of mitochondrial antiviral immunity. Nature. 2008;451:573–577. doi: 10.1038/nature06501. [DOI] [PubMed] [Google Scholar]
- Nakhaei P, Genin P, Civas A, Hiscott J. RIG-I-like receptors: sensing and responding to RNA virus infection. Semin Immunol. 2009a;21:215–222. doi: 10.1016/j.smim.2009.05.001. [DOI] [PubMed] [Google Scholar]
- Nakhaei P, Mesplede T, Solis M, Sun Q, Zhao T, Yang L, Chuang TH, Ware CF, Lin R, Hiscott J. The E3 ubiquitin ligase Triad3A negatively regulates the RIG-I/MAVS signaling pathway by targeting TRAF3 for degradation. PLoS Pathog. 2009b;5:e1000650. doi: 10.1371/journal.ppat.1000650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickart CM, Fushman D. Polyubiquitin chains: polymeric protein signals. Curr Opin Chem Biol. 2004;8:610–616. doi: 10.1016/j.cbpa.2004.09.009. [DOI] [PubMed] [Google Scholar]
- Rahighi S, Ikeda F, Kawasaki M, Akutsu M, Suzuki N, Kato R, Kensche T, Uejima T, Bloor S, Komander D, et al. Specific recognition of linear ubiquitin chains by NEMO is important for NF-kappaB activation. Cell. 2009;136:1098–1109. doi: 10.1016/j.cell.2009.03.007. [DOI] [PubMed] [Google Scholar]
- Rothenfusser S, Goutagny N, DiPerna G, Gong M, Monks BG, Schoenemeyer A, Yamamoto M, Akira S, Fitzgerald KA. The RNA helicase Lgp2 inhibits TLR-independent sensing of viral replication by retinoic acid-inducible gene-I. J Immunol. 2005;175:5260–5268. doi: 10.4049/jimmunol.175.8.5260. [DOI] [PubMed] [Google Scholar]
- Saitoh T, Tun-Kyi A, Ryo A, Yamamoto M, Finn G, Fujita T, Akira S, Yamamoto N, Lu KP, Yamaoka S. Negative regulation of interferon-regulatory factor 3-dependent innate antiviral response by the prolyl isomerase Pin1. Nat Immunol. 2006;7:598–605. doi: 10.1038/ni1347. [DOI] [PubMed] [Google Scholar]
- Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell. 2005;122:669–682. doi: 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- Tian Y, Zhang Y, Zhong B, Wang YY, Diao FC, Wang RP, Zhang M, Chen DY, Zhai ZH, Shu HB. RBCK1 negatively regulates tumor necrosis factor- and interleukin-1-triggered NF-kappaB activation by targeting TAB2/3 for degradation. J Biol Chem. 2007;282:16776–16782. doi: 10.1074/jbc.M701913200. [DOI] [PubMed] [Google Scholar]
- Tokunaga F, Sakata S, Saeki Y, Satomi Y, Kirisako T, Kamei K, Nakagawa T, Kato M, Murata S, Yamaoka S, et al. Involvement of linear polyubiquitylation of NEMO in NF-kappaB activation. Nat Cell Biol. 2009;11:123–132. doi: 10.1038/ncb1821. [DOI] [PubMed] [Google Scholar]
- Wang H, Matsuzawa A, Brown SA, Zhou J, Guy CS, Tseng PH, Forbes K, Nicholson TP, Sheppard PW, Hacker H, et al. Analysis of nondegradative protein ubiquitylation with a monoclonal antibody specific for lysine-63-linked polyubiquitin. Proc Natl Acad Sci U S A. 2008;105:20197–20202. doi: 10.1073/pnas.0810461105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu LG, Wang YY, Han KJ, Li LY, Zhai Z, Shu HB. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol Cell. 2005;19:727–740. doi: 10.1016/j.molcel.2005.08.014. [DOI] [PubMed] [Google Scholar]
- Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S, Fujita T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol. 2004;5:730–737. doi: 10.1038/ni1087. [DOI] [PubMed] [Google Scholar]
- Zeng W, Sun L, Jiang X, Chen X, Hou F, Adhikari A, Xu M, Chen ZJ. Reconstitution of the RIG-I pathway reveals a signaling role of unanchored polyubiquitin chains in innate immunity. Cell. 2010;141:315–330. doi: 10.1016/j.cell.2010.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenke-Kawasaki Y, Dohi Y, Katoh Y, Ikura T, Ikura M, Asahara T, Tokunaga F, Iwai K, Igarashi K. Heme induces ubiquitination and degradation of the transcription factor Bach1. Mol Cell Biol. 2007;27:6962–6971. doi: 10.1128/MCB.02415-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Tian Y, Wang RP, Gao D, Zhang Y, Diao FC, Chen DY, Zhai ZH, Shu HB. Negative feedback regulation of cellular antiviral signaling by RBCK1-mediated degradation of IRF3. Cell Res. 2008;18:1096–1104. doi: 10.1038/cr.2008.277. [DOI] [PubMed] [Google Scholar]
- Zhao S, Ulrich HD. Distinct consequences of posttranslational modification by linear versus K63-linked polyubiquitin chains. Proc Natl Acad Sci U S A. 2010;107:7704–7709. doi: 10.1073/pnas.0908764107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.