

Abstract
DNA copy-number variations (CNVs) underlie many neuropsychiatric conditions, but they have been less studied in cancer. We report the association of a 17p13.1 CNV, childhood-onset developmental delay (DD), and cancer. Through a screen of over 4000 patients with diverse diagnoses, we identified eight probands harboring microdeletions at TP53 (17p13.1). We used a purpose-built high-resolution array with 93.75% breakpoint accuracy to fine map these microdeletions. Four patients were found to have a common phenotype including DD, hypotonia, and hand and foot abnormalities, constituting a unique syndrome. Notably, these patients were not affected with cancer. Moreover, none of the TP53-deletion patients affected with cancer (n = 4) had neurocognitive impairments. DD patients have larger deletions, which encompass but do not disrupt TP53, whereas cancer-affected patients harbor CNVs with at least one breakpoint within TP53. Most 17p13.1 deletions arise by Alu-mediated nonallelic homologous recombination. Furthermore, we identify a critical genomic region associated with DD and containing six underexpressed genes. We conclude that, although they overlap, 17p13.1 CNVs are associated with distinct phenotypes depending on the position of the breakpoint with respect to TP53. Further, detailed characterization of breakpoints revealed a common formation signature. Future studies should consider whether other loci in the genome also give rise to phenotypically distinct disorders by means of a common mechanism, resulting in a similar formation signature.
Introduction
As the range of diseases associated with copy-number variation (CNV) expands, it has become apparent that specific CNV loci can be associated with a spectrum of unrelated conditions. For example, CNVs at 1q21.1 predispose one to schizophrenia (MIM 181500),1,2 tetralogy of Fallot (MIM 187500),3 cancer (including neuroblastoma [MIM 256700]4), and a range of pediatric conditions.5 It is unclear whether individuals harboring structural changes at these and other hotspots are predisposed to one or many diseases. High-resolution copy-number platforms, such as tiling oligonucleotide arrays, offer increased accuracy6 and can therefore be applied to disentangle overlapping CNV-based diseases. Furthermore, characterization of CNVs at the single-base-pair level can unearth common sequence elements, which represent signatures of the various DNA repair processes that led to their formation.
Most pediatric cancers arise sporadically; however, at least 5%–10% harbor an underlying germline defect,7 and there is an emerging link between CNVs and cancer susceptibility.4,8,9 We have previously shown that germline TP53 (MIM 191170) missense mutations predispose one to the autosomal-dominant cancer-susceptibility condition known as Li-Fraumeni syndrome (LFS [MIM 151623]),10 in which an excess of CNVs across the genome are observed.8 Here, we investigate whether 17p13.1 CNVs, which include TP53, are sufficient to cause LFS.
To improve our understanding of 17p13.1 CNVs, we constructed an oligonucleotide comparative genomic hybridization (CGH) array to interrogate this genomic region at ultrahigh resolution; overlapping probes covering all exons of every gene in the region, were designed to achieve 93.75% breakpoint accuracy. Using this platform, we set out to determine whether patients with 17p13.1 CNVs contain shared breakpoint sequences, critically deleted genes, or common clinical features.
Material and Methods
Sample Recruitment
Samples were collected from The Hospital for Sick Children, Stanford University Hospital, The Children's Hospital of Philadelphia, The University Health Network (Toronto), Hospital do Câncer A.C. Camargo, and Emory University. Research was approved by each center's institutional review board.
TP53 Sequencing
All exons and at least 50 bp into exon-intron boundaries of TP53 were sequenced in all available samples (7/8). No base pair sequence mutations were found.
CGH Microarray Design and Hybridization
Six of eight patients' DNA was hybridized to custom arrays. Two samples (from cancer patients) had insufficient DNA for custom array analysis, and their breakpoints were not fine mapped. We instead used multiplex ligation-dependent probe amplification (MLPA) to determine the extent of TP53 exon deletions. Because their exact breakpoints are not known, we excluded them from statistics regarding formation signature (see below). Array CGH was performed with the use of a customized 4×44K microarray platform (Agilent Technologies, Santa Clara, CA, USA), with genomic DNA extracted from peripheral blood via standard methods. A total of 40,577 oligonucleotide probes were placed on the short arm of chromosome 17, in which an 8 Mb target region around TP53 was covered in ultrahigh resolution. Of the 38,061 in the target region, 15,762 (41%) were designed on exons. Exonic probes were overlapping and tiled across all exons, of all alternative splice variants, for every gene. A minimum of one probe per 350 bp was placed in intronic and nongenic regions. An additional set of probes was designed to also extend our coverage to the telomere and centromere of chromosome 17p but at a reduced density (Figure S3, available online). With this array design, we hoped to capture all copy-number changes anywhere within 8 Mb of TP53, from small, single-exon-sized alterations (45–350 bp) to large macroscopic events, and to quickly obtain breakpoint information of copy-number changes, especially those within protein-coding regions. The lab performing array experiments was blinded to all previous results (sequencing, MLPA, quantitative PCR [qPCR], etc.). Patient and male reference DNA were labeled with Cy3-dCTP and Cy5-dCTP (PerkinElmer, Waltham, MA, USA), respectively, with the use of the BioPrime genomic labeling module (Invitrogen, Carlsbad, CA, USA) and were hybridized to the array platform, as recommended by the manufacturer's protocol (Agilent Technologies). The arrays were washed and scanned with the Agilent G2505B microarray scanner. Data analysis was performed with DNA Analytics version 4.0 (Agilent Technologies). Although this approach is capable of ascertaining both forms of copy-number change (namely deletions and duplications), only deletions were found in this cohort.
Gene-Expression Arrays and Analysis
RNA was extracted from blood via standard methods, assessed by Bioanalyzer (Agilent Technologies), and hybridized to Affymetrix Exon 1.0 microarrays (Affymetrix, Santa Clara, CA, USA). High-quality RNA was available for one individual with a large 17p13.1 deletion (encompassing TP53), two individuals with small 17p13.1 deletions (disrupting TP53), and two individuals harboring germline TP53 missense mutations. RNA samples from three noncarrier siblings were used as controls. Gene-expression analysis was performed with the use of the Partek Genomics Suite (Partek, St. Louis, MO, USA).
Breakpoint Simulation
Custom software was developed for the simulation of CNV deletions across all autosomal chromosomes in the human genome. For each size range (from 10 Kb to 2 Mb), 10,000 simulated CNVs were assessed for intersection with Alu elements at both breakpoints.
Quantitative PCR
To obtain better size information on these deletions, we developed a high-throughput quantitative assay by using an automated liquid handling system in a 384-well plate format with 176 qPCR probes (Table S2) to target and detect the copy number of a large region of chromosome 17 (1.3 Mb). qPCR assays were performed on a Roche LightCycler by relative quantification. qPCR plates were set up with the use of a custom script and an automated liquid handling system. Primers were designed with the use of Primer3 and the human genome reference assembly (UCSC Genome Browser, version hg18). Deletion sizes were found to be larger than that reported by array CGH or MLPA and, on average, were improved by 31% with the use of this assay. The developmental delay (DD)-associated deletions were nearly 100 times larger than those involved in early-onset cancer.
Fluorescence In Situ Hybridization
Fluorescence in situ hybridization (FISH) was performed with the use of standard protocols.11
Parent-of-Origin Analysis
SNP genotyping was performed with Affymetrix GeneChip 250 Sty arrays or by direct sequencing. Microsatellite-marker genotyping was performed by The Center for Applied Genomics at The Hospital for Sick Children.
Breakpoint Mapping
After custom-array processing and analysis, breakpoints were mapped by first designing primers flanking the predicted breakpoints and then amplifying junction-specific fragments with the use of long-range PCR (Roche Expand Long Template PCR System). Junction fragments were subjected to sequencing. Putative breakpoints were analyzed by BLAST, BLAT, and manual inspection.
Results
Rare CNVs at TP53 Are Associated with Cancer Predisposition or Developmental Delay
Our six diagnostic labs screened 4524 patients with diverse clinical phenotypes for DNA dosage changes via array CGH or MLPA (Table S1). Eight probands with a microdeletion at TP53 (17p13.1), a tumor-suppressor gene that predisposes one to early-onset cancer when mutated in LFS, were identified.10 We performed interphase and metaphase FISH, using TP53 and 17ptel probes (Figure 1A). The interstitial deletion could be seen in all cells and was therefore not due to mosaicism.
Figure 1.
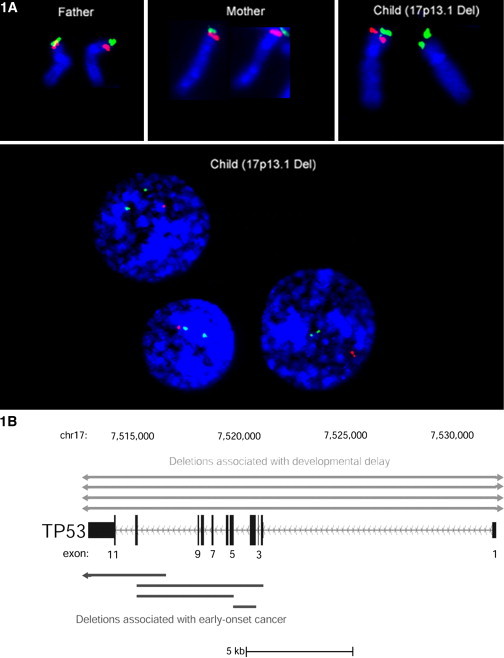
Discovery of a 17p13.1 CNV Leading to Two Distinct Phenotypes
(A) FISH experiments using TP53 (red) and 17ptel (green) probes. The fluorescent signals in this representative family trio confirm a de novo hemizygous TP53 deletion in the child's metaphase and interphase nuclei. Two hundred nuclei were scored, and no evidence of mosaicism for the CNV was observed. TP53 microdeletions were not observed by conventional Giemsa banded karyotyping.
(B) Results of MLPA, qPCR, and clinical array revealed two isoforms of the 17p13.1 CNV. Among DD patients, the CNV includes and extends past TP53 in the telomeric and centromeric directions (n = 4; top); Among the cancer-affected patients, the 17p13.1 CNV deletes some—but not all—of TP53's exons. TP53 is transcribed off of the minus DNA strand; therefore, its introns and exons are drawn from last to first.
Individuals with microdeletions at TP53 had cancer (n = 4) or a noncancer phenotype (n = 4) comprising a spectrum of congenital anomalies (Table 1 and Figure S2) that included pervasive DD and mental retardation, speech difficulties, hypotonia, hand and foot abnormalities, and facial dysmorphisms.
Table 1.
Phenotypic Features of Four Patients with 17p13.1 CNVs and Developmental Delay
| Patient ID | 3026 | 2723 | 3148 | 3354 |
|---|---|---|---|---|
| Sex | F | F | F | M |
| Age (yrs) | 33.67 | 7.58 | 5.75 | 3.50 |
| Inheritance | – | de novo | de novo | de novo |
| Parental origin | paternal | paternal | maternal | – |
| Cognitive | GDD; nonverbal; severe MR | GDD; speech apraxia | GDD; limited speech development | GDD |
| Growth (percentile) | height, < 3rd; weight, < 3rd; HC, 50th | height, 25th; weight, 70th; HC, 25th | height, 10th–25th; weight, 75th–90th; HC, 97th | height, 50th–75th; weight, 50th–75th; HC, 10th–25th |
| Facial features | prominent nasal bridge; high-arched palate; thin lips; high forehead; bilateral low-set ears; earlobe pits; downslanting palpebral fissures; short neck with webbing; highly arched eyebrows that extend laterally; low hairline; left-sided mild ptosis; recessed chin; bitemporal narrowing | upturned nasal tip; high-arched palate; thin, puckered lips; brachycephaly | wide nasal bridge; high-arched palate; high forehead; downslanting palpebral fissures; broad, flat epicanthal folds; small, recessed chin; downturned corners of mouth; telecanthus; depressed nasal tip; bifid uvula; posterior hair whorl | broad, upturned nose with small nares; upswept ear lobules with earlobe pits; short neck with no webbing; unusually arched eyebrows; epicanthal folds; small, recessed chin; downturned corners of mouth; mildly upslanting palpebral fissures; short columella with prominent ala nasi |
| MSK features | ligamentous laxity; bilateral elbow contractures | extra flexion creases (calves, arms); mild pectus deformity | ligamentous laxity; contractures of the elbow and knees; dimpling at ankles elbows, and knees; sacral crease; bilat vagus; deformity of ankles | asymmetric crease with deep sacral dimple; 13 pairs of ribs; mild spine curvature; partial sacralization of lower lumbar spine |
| Cardiovascular | VSD | PDA (self-resolved) | normal | no echocardiogram |
| Ocular | strabismus | strabismus; legally blind; right eye hamartoma (CHRPE-like lesion); iris hypoplasia; astigmatism; decreased lacrimation | bilateral alternating exotropia; myopia | lateral vision difficulty and difficulty tracking (11 mo of age) |
| Bone marrow | hemolytic anemia of infancy (self-resolved); pure red cell aplasia (onset age 15 yrs) | N/A | N/A | N/A |
| Neurological | hypotonia; brisk DTRs; ankle clonus; hydrocephalus; broad-based gait; brain MRI: arrested | hypotonia; brisk DTRs; ankle clonus; external hydrocephalus; choreoathetoid movements; brain MRI: thinned CC, delayed myelination, tethered cord; upgoing plantar responses | hypotonia; brain MRI: normal | hypotonia; DTRs difficult to elicit |
| Audiology | normal | normal | decreased hearing | normal |
| Psychiatric | PDD; bipolar disorder | PDD | N/A | N/A |
| Behavioral | self-injurious; aggressive | intermittent hand-wringing and hand clapping | N/A | N/A |
| Hands | thumbs proximally placed; short hands; bilateral deep palmar creases; bilateral first-finger clinodactyly; bilateral 5th finger IP joint contracture | left thumb proximally placed; short hands (3rd–25th percentile); broad thumbs | left transverse palmar crease; right “hockey stick”-shaped crease | normal |
| Feet | short feet; big toe abnormally long and narrow bilaterally; bilateral deep plantar creases; flat feet; bilateral shortened fourth toe | small feet (< 5th percentile); big toe large and broad bilaterally; pollicization of big toes | normal | shortened feet with broad big toes |
| GU | normal | neurogenic bladder; ovarian cysts; resolved renal cysts | normal | shawl scrotum |
| Skin | normal | dermoid cyst above left eye; compound melanocytic nevus; epithelioid cell type of scalp | normal | sacral Mongolian spot |
| Nipples | normal | bilateral inverted supernumerary nipple | N/A | bilateral inverted nipples |
| Other | failure to thrive and feeding difficulties; sleep disturbances; hypothyroidism and iron overload secondary to blood transfusions every 3–4 weeks | feeding difficulties; sleep disturbances; GERD; chronic constipation; hypogammaglobinemia | feeding difficulties; GERD; benign paroxysmal torticollis | N/A |
Bolded and italicized text indicates features shared by more than one patient. Abbreviations are as follows: HC, head circumference; CC, corpus callosum; CHRPE, congenital hypertrophy of the retinal pigment epithelium; DTR, deep tendon reflexes; GDD, global developmental delay; GERD, gastro-esophageal reflux disease; IP, interphalangeal; MR, mental retardation; PDD, pervasive developmental disorder.
Different 17p13.1 Breakpoints Are Related to Two Distinct Phenotypes
Several congenital syndromes are known to also occur in association with cancer predisposition. Such dual phenotypes are frequently caused by gene dosage mutations, either through numerical chromosomal abnormalities or through specific structural changes (e.g., trisomy 2112). In contrast, LFS patients do not show increased rates of neurocognitive disability or any phenotype besides cancer. Consistent with this, none of the TP53-deletion patients affected with cancer had DD or congenital anomalies. Similarly, none of the patients with DD exhibited any neoplastic growth that might suggest an underlying susceptibility, nor did they have family histories of cancer consistent with LFS. Therefore, although they share genomic alterations at TP53, their distinct clinical presentations suggest that these patients fall into two nonoverlapping groups.
Next, we determined the genetic basis of this dichotomy. Our initial patient discovery was performed on two platforms, with complementary depth and breadth. Clinical array CGH provides low resolution at TP53 but provides more information on the extent of 17p13.1 CNVs beyond TP53, whereas MLPA provides high resolution across TP53's 11 exons but provides little information for the surrounding regions. To determine whether CNVs defined by MLPA extend beyond TP53, we used qPCR to determine the copy number of the genes immediately flanking TP53 (Figure S1). Both ATP1B2 (telomeric; MIM 182331) and WRAP53 (centromeric; MIM 612661) were disomic in all cancer patients (mean copy numbers = 2.03 and 2.11, respectively). However, all patients with DD were hemizygously deleted for both flanking genes (mean copy numbers = 0.87 [ATP1B2] and 1.13 [WRAP53]), a significant reduction in comparison to the cancer patients (p = 2.90 × 10-4 [ATP1B2] and 2.42 × 10-8 [WRAP53]). We also carried out MLPA experiments on all array-CGH-ascertained samples, and we found that in every DD case all 11 exons of TP53 were contiguously deleted. In contrast, no cancer case harbored a CNV that included all 11 exons. These results demonstrate that our cohort of DD and cancer patients have overlapping but genotypically distinct CNVs at TP53; whereas DD-associated CNVs include all exons of TP53 as well as flanking genes, cancer-associated CNVs are within TP53, causing a change in copy number to some— but not all—of its exons (Figure 1B).
17p13.1 Genomic Deletions Can Be Inherited or Arise De Novo
A review of the eight probands' pedigrees showed that families of DD patients did not have neurocognitive impairment and that the pedigrees of the four cancer patients were consistent with LFS (Figure S2). All DD patients with available parental samples (n = 3) had a de novo deletion, as shown by CGH, MLPA, and FISH analysis (200 nuclei tested, with no evidence of low-level mosaicism in parents). Among the cancer patients with deletions, familial samples were available in two cases. Of these, one family's samples were sufficiently informative to establish inheritance of the deletion. No apparent parent-of-origin bias was observed for deletions in either group of our cohort.
Design of a Custom Ultrahigh-Resolution Tiling Array
Obtaining sequence-level resolution is the most definitive method of validating rearrangements,13 because it leads to precise definitions of the CNVs' breakpoints and gene content, provides clues as to the mechanism underlying their formation,14,15 and reveals their potential architectural complexity.16
We designed an ultrahigh-resolution array covering 8 Mb of chromosome 17 to get close to sequence-level resolution and to use it as a clinical diagnostic platform for identifying all possible rearrangements in future patients. The array comprises ∼45,000 oligonucleotide probes spanning 4 Mb upstream and 4 Mb downstream of the TP53 locus (7,512,444 to 7,531,588). All exons within this region are tiled, representing the entire coding sequence of 182 genes and all possible alternative transcripts (2,130 exons; Figures S3A and S3B). The precise array design and probe placement are described in Material and Methods and Figure S3.
We tested our custom array on patients whose breakpoints we had already successfully sequenced. These experiments yielded highly precise size and breakpoint information. For example, a patient with DD was found, by the 17p13.1 array, to have a contiguous genomic deletion of 923,492 bp, a difference in size of only 2183 bp (0.2%) from that established by sequencing. The 5′ and 3′ breakpoints of the deletion were 2341 bp and 153 bp away from the true breakpoints, respectively.
Alu Short Interspersed Nuclear Repeats Are Associated with Breakpoints
Using this array, we determined the size and breakpoints of the remaining samples. Using long-range PCR, we amplified junction fragments spanning putative breakpoints. Then, in cases for which high-molecular-weight DNA was available, we sequenced junction fragments and determined the breakpoint and size of the deletions. The average difference between actual CNV sizes and the arrays' predicted sizes was 6.25% (i.e., 93.75% accuracy).
By array, one patient was revealed to harbor another deletion in 17p13.1. The secondary deletion, which is also heterozygous, is 24 Kb in length and is located downstream of the primary deletion's distal breakpoint (Figure S4). In another instance, an identical deletion was found in the proband and the proband's sibling, indicating the inheritance of the same pathogenic CNV (Figure 3, patient 3332). Although asymptomatic, the sibling is now undergoing routine biochemical and radiographic surveillance for cancer.
Figure 3.
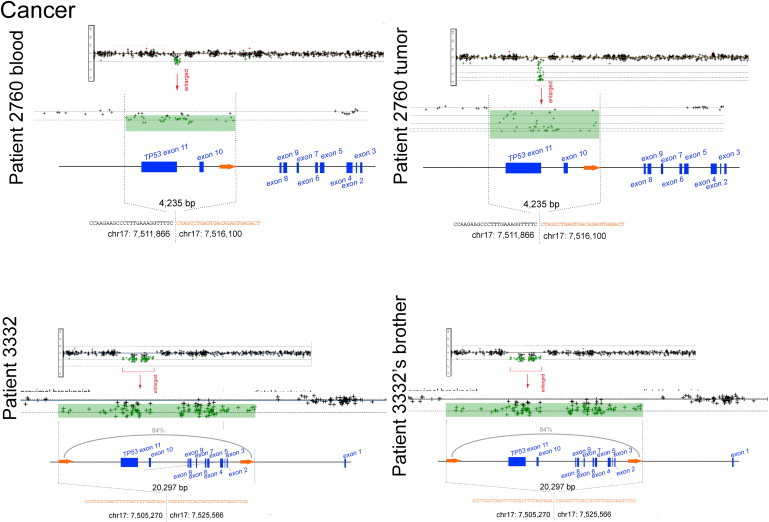
Breakpoint Maps, Sequence Resolution, and Inferred Mechanism of Cancer-Associated 17p13.1 CNVs
An ultrahigh-resolution CGH array (see Figure S3) was used to obtain breakpoint-level information on 17p13.1 CNVs in two cancer-affected patients. Log2 ratios from the array are shown, each dot representing one probe and deletions indicated in green. The precise breakpoint positions, their sizes, and the nucleotide sequence of the disrupted regions are shown. The presence of two Alu elements (orange arrows and orange-colored nucleotides) at the junctions is consistent with the formation of the CNV by Alu-Alu-mediated NAHR (patient 3332 and brother). The percentage of homology between directly oriented Alus is indicated for NAHR CNVs. The proximal and distal breakpoints were always either intronic in TP53 or intragenic, never disrupting other genes besides TP53 or leading to gene fusions. Using high-quality DNA from one patient's frozen tumor, we observed a second deletion on the opposite allele, conforming to the classical two-hit hypothesis of tumorigenesis42 (patient 2760). This custom array was used to test for the presence of the CNV in two asymptomatic siblings of an index case affected with cancer (patient 3332). One sibling (shown) was found to harbor the identical deletion.
We looked for repeat elements coinciding with CNV breakpoints. Of the 12 sequenced breakpoints, ten directly intersect with an Alu short interspersed nuclear repeat element (one from the oldest AluJ family, seven from the intermediate AluS family, and two from the young AluY family).
Most 17p13.1 CNVs Arise by Alu-Mediated Nonallelic Homologous Recombination
Analysis of sequenced CNVs revealed the mechanisms by which they arose. Four of six deletions involved nonallelic homologous recombination (NAHR) between Alu elements present at both the proximal and the distal ends (Figure 2, patients 3026, 3148, 3354; Figure 3, patient 3332). The Alu elements flanking these deletions were in the same orientation and shared a moderate degree of homology (81%–84% similarity by BLAST). The remaining two patients did not exhibit extensive homologies spanning their breakpoints. Of these, one breakpoint showed a 4 bp microinsertion (CAAG), an “information scar” that is a hallmark of nonhomologous DNA end joining17 (NHEJ; Figure 2, patient 2723).
Figure 2.
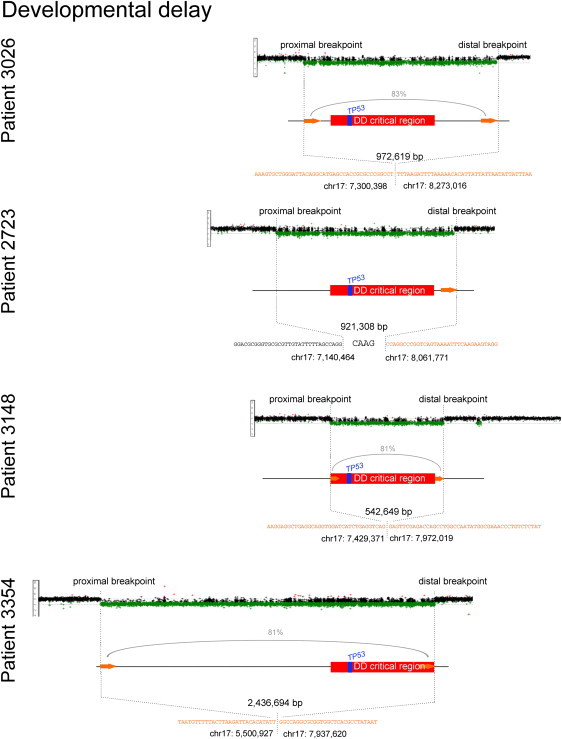
Breakpoint Maps, Sequence Resolution, and Inferred Mechanism of DD-Associated 17p13.1 CNVs
We developed an ultrahigh-resolution CGH array (see Figure S3) to obtain breakpoint-level information on 17p13.1 CNVs. Shown are the array results for all four DD patients. Log2 ratios from the array are shown, each dot representing one probe and deletions indicated in green. The proximal and distal breakpoints were determined for all samples, revealing that all DD patients shared a critical region including TP53 and 23 other genes (red). The precise breakpoint positions, their sizes, and the nucleotide sequence of the disrupted regions are shown. The presence of two Alu elements (orange arrows and orange-colored nucleotides) at the junctions is consistent with the formation of the CNV by Alu-Alu-mediated NAHR (patients 3026, 3148, and 3354). The percentage of homology between directly oriented Alus is indicated for NAHR CNVs. In one instance an NHEJ signature could be seen at the at the breakpoint sequence: Four additional base pairs incorporated at the junction (patient 2723).
To evaluate the significance of the observed number of Alus at 17p13.1 CNV breakpoints, we performed 10,000 permutation experiments using randomly distributed CNVs of different sizes (10 Kb to 2 Mb). In these simulations, less than 1% of breakpoints coincided with an Alu pair in the same orientation. In contrast, the majority of 17p13.1 CNVs coincide with directly oriented Alus (67%; Figure S5).
A Common Region Implicates Candidate Genes in Developmental Delay
In our study cohort, all CNVs associated with occurrence of childhood cancer were limited to the TP53 locus, deleting between one and ten of 11 exons. Such deletions are predicted to cause protein truncation, thus interfering with the gene's tumor-suppressive activity. Indeed, in a paired tumor specimen we observed an additional copy-number alteration of the same size, thus inactivating the wild-type allele (Figure 3, patient 2760).
In contrast, in the four patients with DD we found, by fine mapping, a common deleted region (Table 2) that includes 24 genes (critical region shown in Figure 2). There are a number of candidate genes for the observed phenotypes. The four patients with DD harbored between 27 and 86 fully deleted genes. Additionally, fine mapping revealed that two DD patients carried partial deletions of genes, disrupting some but not all of their exons (Table 2).
Table 2.
Deleted and Disrupted Genes in 17p13.1-Deletion Patients
| Patient | Chr. | Start | End | No. of Deleted Genes | Disrupted Gene |
|---|---|---|---|---|---|
| Developmental Delay | |||||
| 1 | 17 | 7,300,398 | 8,273,016 | 55 | CHRNB1 |
| 2 | 17 | 7,140,464 | 8,061,771 | 58 | – |
| 3 | 17 | 7,429,371 | 7,972,019 | 28 | MPDU1 |
| 4 | 17 | 5,500,927 | 7,937,620 | 86 | – |
| Patients 1–4: Critical region | 17 | 7,429,371 | 7,937,620 | 24 | MPDU1 |
| Cancer | |||||
| 1 | 17 | 7,511,866 | 7,516,100 | 1 | TP53 |
| 2 | 17 | 7,505,270 | 7,525,566 | 1 | TP53 |
| 3 | 17 | 7,512,445 | 7,519,262 | 1 | TP53 |
| 4 | 17 | 7,520,037 | 7,520,315 | 1 | TP53 |
The four patients with DD harbored between 27 and 86 fully deleted genes and two partially deleted genes. The minimally deleted region includes 24 genes.
We evaluated the effect of this 17p13.1 CNV on mRNA levels by using expression arrays (see Material and Methods). Among the genes in the minimally deleted region, the expression of TP53 was significantly underexpressed in the patient affected with cancer but not in the patient with DD (p = 6.82 × 10−3; fold change = −1.85797; Figures 4A and 4B).
Figure 4.
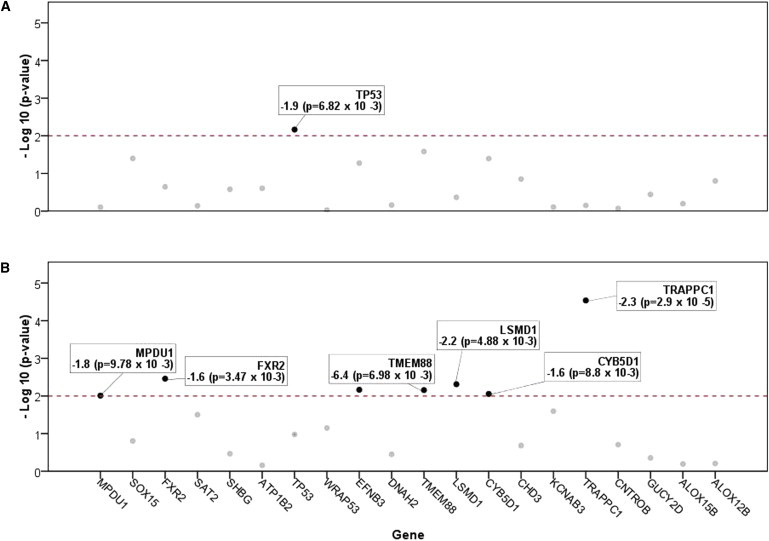
Gene-Expression Differences Distinguish between Cancer-Affected an DD Patients with 17p13.1 Deletions
We used Affymetrix exon arrays to look for gene-expression differences in available blood-derived RNA. We first evaluated which of the 24 genes in our critical region (commonly deleted in patients with DD) is significantly under- or overexpressed. Twenty of these 24 genes could be assayed with the array and are shown. On the y axis is depicted the significance of each gene's expression change relative to controls (plotted in reverse order). The red dotted line represents the p value threshold of 0.01, above which all significant changes in gene expression are highlighted (black dots).
(A) Among patients with small 17p13.1 CNV only TP53's expression is significantly changed (p = 6.82 × 10−3; fold change = −1.9).
(B) Notably, a similar analysis of RNA from a DD patient did not show TP53 underexpression, despite the gene being fully deleted in a large 17p13.1 CNV. There are, however, six significantly changed genes (all underexpressed). As shown, these are TRAPPC1 (p = 2.90 × 10−5, fold change = −2.3), FXR2 (p = 3.47 × 10−3, fold change = −1.6), LSMD1 (p = 4.88 × 10−3, fold change = −2.2), KDM6B (p = 6.98 × 10−3, fold change = −6.4), CYB5D1 (p = 8.80 × 10−3, fold change = −1.6), and MPDU1 (p = 9.78 × 10−3, fold change = −1.8). In a separate analysis (Figure S6), TRAPPC1 was found to be the most significantly underexpressed gene in the transcriptome.
The expression of six other genes (of the 24 candidates in the region) were significantly changed in the patient with DD but not in the cancer-affected patient (Figure 3B; p < 0.01; fold change < −1.5 or > 1.5). Of these DD-specific genes, which are both hemizygously deleted and underexpressed in all patients, the trafficking protein particle complex 1 gene (TRAPPC1 [MIM 610969]) is particularly intriguing. TRAPPC1, involved in vesicular transport from the endoplasmic reticulum to the Golgi apparatus as part of the TRAPP complex, is the most significantly changed at 17p13.1 and, of note, is also the most significantly changed gene genome-wide (Figure S6; p = 2.90 × 10−5; fold change = −2.34146).
Three additional DD-specific genes are noteworthy: MPDU1 (MIM 604041), mutations of which result in congenital disorder of glycosylation type If involving severe mental and psychomotor retardation;18 FXR2 (MIM 605339), a homolog of the fragile X mental retardation gene, FMRP (MIM 309550), which itself may play a role in that disease;19 and EFNB3 (MIM 602297), known to be important in the development of normal locomotor behavior.20 To determine whether the deletion unmasks a recessive mutation, we sequenced these four genes, but we did not find additional mutations.
Having found reduced TP53 expression in the cancer-affected individual, we examined whether other TP53 signaling-pathway members were altered. We first measured the mRNA levels of a proband from an LFS family carrying an established deleterious base pair mutation (Arg273Cys) and conducted pathway analysis. Using the well-annotated Ingenuity Pathway Analysis “Core Pathways,” we noted a subtle but significant difference of genes in the TP53 pathway in individuals with either an established mutation or an internal deletion of TP53, but not in those with complete deletions and DD (p = 4.74 × 10−2 and 3.01 × 10−2, respectively). This shows that TP53 is aberrantly expressed in individuals affected with cancer but not in those affected with DD. Furthermore, we find that disregulated genes common to the missense- and internally-deleted-mutation carriers are associated with known molecular mechanisms of cancer (p = 2.59 × 10−3). Together, these data highlight gene-expression differences between individuals having large or small CNVs at 17p13.1.
Discussion
LFS is a highly penetrant susceptibility to cancer that disproportionately affects the young. Children with germline TP53 mutations are at a 20% risk of developing cancer by 15 years of age and, over a lifetime, have a 73% to 100% risk.21 However, the four DD patients in this report, ranging in age from 3.5 to 33.67 yrs, are not affected with cancer despite harboring complete deletions of TP53. Other case reports highlight an additional six patients with 17p13.1 deletions,22–25 of whom none are affected with cancer. Although these reports support our contention that DD-associated deletions involve reduced cancer risk, it is premature to discount the possibility that these patients may have a high risk of developing cancers due to somatic TP53 mutation, which may become manifest only at later ages.
The molecular basis for this apparent absence or reduction of cancer risk remains to be elucidated. Studies of mouse models of LFS as well as the somatic mutation spectra of TP53 in human cancers provide evidence that tumorigenesis is accelerated when TP53 is altered by point mutations or short insertions or deletions, rather than completely lost. In contrast, a number of nonsense mutations that predict total absence of TP53 expression are strongly associated with cancer. It should be noted that in the cancer-prone patients described here the deletions do not include exon 1 and the long intron 1. It is possible that sequences in the latter region may contribute to regulate TP53 suppressor function. In particular, the proximal region of intron 1 contains sequences encoding a natural antisense transcript of TP53, WRAP53, which regulates endogenous TP53 mRNA by targeting the 5′ untranslated region of TP53 mRNA.26 The exact role of this sequence in predisposition to cancer deserves further study. Notwithstanding this caveat, we show here that mRNA expression levels of TP53 and TP53-dependent genes are altered in patients with partial, but not complete, deletions—consistent with mutant TP53-initiated tumorigenesis in the former group but not in the latter. In contrast, the neurocognitive-delay phenotype is characterized by the dysregulation of a different set of six genes at 17p13.1 and, in particular, TRAPPC1, which is also the most significantly underexpressed gene in the transcriptome.
Our data support a model in which partial deletions lead to the expression of a truncated protein, rather than the complete absence of it due to nonsense-mediated decay. Truncated and wild-type protein (from the opposite allele) would oligomerize to form a defective TP53 tetramer, leading to a dominant-negative or gain-of-function effect similar to that observed with certain missense mutations, resulting in inhibition of wild-type TP53 function.27,28 We and others have previously shown that TP53 alterations, whether somatic or inherited, are more commonly missense than nonsense or truncating mutations.29 Tumors will frequently accumulate dysfunctional TP53 that is structurally intact. As such, tumors derive a greater advantage from retaining dysfunctional TP53 instead of eliminating it entirely. Likewise, in this study we demonstrate that partial deletions lead to a stronger cancer-predisposition phenotype than full-length deletions of TP53. Like their somatic equivalents, these “first hits” to TP53 involve the expression of dysfunctional TP53, which as a transcription factor leads to the aberrant expression of a number of targets. We go on to show that more TP53 targets are dysfunctional in this group than in persons harboring large and complete TP53 deletions. In light of the existing literature, we view these results as aberrant transcription of TP53 targets resulting from qualitative difference in the expressed TP53 protein, not necessarily because of a quantitative difference in its abundance. Among its many targets, TP53 also regulates its own mRNA. This feedback loop is made possible by the direct binding of TP53 and its mRNA, which forms a stable-stem loop structure.30 Additionally, TP53 induces genes that regulate its mRNA, such as Wig-1, which stabilizes TP53 mRNA.31 Wrap53 also stabilizes TP53 mRNA, but instead of being induced by TP53, it lies immediately proximal to it on chromosome 17p13.1.26 Mutant TP53 disrupts many of these autoregulatory loops through aberrant binding or transactivaion. In this study, we show that this disruption, which leads to reduced TP53 mRNA (Figure 4A), is associated with partial but not large TP53-deletion mutants.
We designed a tiling array to determine accurate breakpoints of CNVs at 17p13.1, the locus that we also show to be responsible for a unique congenital syndrome. By achieving base pair resolution, we gained insight into the genomic basis of this dysmorphology syndrome, including the precise determination of deletion length and gene content, the definition of a critical region, and the recognition of a shared mechanism of CNV formation in multiple probands. Alu retrotransposons are nearly ubiquitous at 17p13.1 breakpoints, which is highly suggestive of Alu-mediated NAHR.32 Alus make up the largest family of mobile elements in the human genome and have been implicated in a number of diseases, such as neurofibromatosis and breast cancer.33,34 A large proportion of MLH1 (chromosome 3p22.2) and MSH2 (chromosome 2p21) deletions, which predispose one to Lynch syndrome, are mediated by Alu elements present at both breakpoints (24% and 85%, respectively, in one analysis),35 whereas other non-Alu-mediated events are associated with the remaining breakpoints. Somatically acquired rearrangements are common in cancer, and it has been shown that regions with high levels of Alus are more susceptible to recombination in tumors.32,36 Disruptions of TP53, by somatic mutation or loss of heterozygosity, are a virtual prerequisite for transformation of incipient cancer cells. Although the breakpoint resolution achieved in our study has yet to be examined in many cancer samples, at least one report has demonstrated that Alus can indeed mediate somatic rearrangements at TP53.37 Future studies of cancer38 will determine whether Alu-mediated recombination at 17p13.1 is as widespread in tumors as we show them to be in the germline.
This report adds 17p13.1 deletions—which result in two seemingly distinct phenotypes—to the list of disease loci associated with Alus. As more CNV-associated disorders are discovered, it will be intriguing to consider whether other loci in the genome also give rise to phenotypically distinct disorders by means of a common mechanism.
All cancer-associated TP53 deletions reported to date are, to our knowledge, small (< 50 Kb)39–41 and, except for one particularly complex Alu-mediated 45 Kb rearrangement,40 involve only partial deletion of the gene. Whereas the cancer-specific susceptibility of LFS is well recognized, we show that 17p13.1 deletions are associated with a contiguous deletion syndrome involving a recognizable phenotype with DD, hypotonia, and hand and foot abnormalities. Furthermore, we demonstrate that a high-resolution array platform improves detection of previously unrecognized microdeletions, suggesting that it could provide a valuable tool in the molecular diagnosis of TP53 wild-type LFS and for patients with cognitive-delay phenotypes.
Acknowledgments
Thanks to members of the Malkin Lab for assistance and proofreading and to Stephen Meyn, Stephen Scherer, Christian Marshall, and Dalila Pinto for critical review of the manuscript and discussion. Thanks also to Megan Kowalski for help with sample coordination and pedigree ascertainment and to the Cytogenomics Laboratory at the Children's Hospital of Philadelphia. The Centre for Applied Genomics (Toronto, Canada) is acknowledged for technical support. The research was supported by grants from the Canadian Cancer Society Research Institute (with funds from the Canadian Cancer Society), the Canadian Institutes for Health Research (D.M.), the SickKids Foundation, Canada Foundation for Innovation (D.M), and the Grundy Vision for Life fund (K.E.N.). A.S. is supported by a Canadian Institutes of Health Research Frederick Banting & Charles H. Best Doctoral Studentship Award.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
The International Agency for Research on Cancer (IARC) TP53 Mutation Database, http://www-p53.iarc.fr/
References
- 1.Stefansson H., Rujescu D., Cichon S., Pietiläinen O.P., Ingason A., Steinberg S., Fossdal R., Sigurdsson E., Sigmundsson T., Buizer-Voskamp J.E., GROUP Large recurrent microdeletions associated with schizophrenia. Nature. 2008;455:232–236. doi: 10.1038/nature07229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.International Schizophrenia Consortium Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature. 2008;455:237–241. doi: 10.1038/nature07239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Greenway S.C., Pereira A.C., Lin J.C., DePalma S.R., Israel S.J., Mesquita S.M., Ergul E., Conta J.H., Korn J.M., McCarroll S.A. De novo copy number variants identify new genes and loci in isolated sporadic tetralogy of Fallot. Nat. Genet. 2009;41:931–935. doi: 10.1038/ng.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Diskin S.J., Hou C., Glessner J.T., Attiyeh E.F., Laudenslager M., Bosse K., Cole K., Mossé Y.P., Wood A., Lynch J.E. Copy number variation at 1q21.1 associated with neuroblastoma. Nature. 2009;459:987–991. doi: 10.1038/nature08035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mefford H.C., Sharp A.J., Baker C., Itsara A., Jiang Z., Buysse K., Huang S., Maloney V.K., Crolla J.A., Baralle D. Recurrent rearrangements of chromosome 1q21.1 and variable pediatric phenotypes. N. Engl. J. Med. 2008;359:1685–1699. doi: 10.1056/NEJMoa0805384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Conrad D.F., Pinto D., Redon R., Feuk L., Gokcumen O., Zhang Y., Aerts J., Andrews T.D., Barnes C., Campbell P. Origins and functional impact of copy number variation in the human genome. Nature. 2009;464:704–712. doi: 10.1038/nature08516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nagy R., Sweet K., Eng C. Highly penetrant hereditary cancer syndromes. Oncogene. 2004;23:6445–6470. doi: 10.1038/sj.onc.1207714. [DOI] [PubMed] [Google Scholar]
- 8.Shlien A., Tabori U., Marshall C.R., Pienkowska M., Feuk L., Novokmet A., Nanda S., Druker H., Scherer S.W., Malkin D. Excessive genomic DNA copy number variation in the Li-Fraumeni cancer predisposition syndrome. Proc. Natl. Acad. Sci. USA. 2008;105:11264–11269. doi: 10.1073/pnas.0802970105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shlien A., Malkin D. Copy number variations and cancer. Genome Med. 2009;1:62. doi: 10.1186/gm62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Malkin D., Li F.P., Strong L.C., Fraumeni J.F., Jr., Nelson C.E., Kim D.H., Kassel J., Gryka M.A., Bischoff F.Z., Tainsky M.A. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science. 1990;250:1233–1238. doi: 10.1126/science.1978757. [DOI] [PubMed] [Google Scholar]
- 11.Bayani J., Squire J.A. Fluorescence in situ Hybridization (FISH) Curr Protoc. Cell. Biol. 2004 doi: 10.1002/0471143030.cb2204s23. Chapter 22, Unit 22.24. [DOI] [PubMed] [Google Scholar]
- 12.Malinge S., Izraeli S., Crispino J.D. Insights into the manifestations, outcomes, and mechanisms of leukemogenesis in Down syndrome. Blood. 2009;113:2619–2628. doi: 10.1182/blood-2008-11-163501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scherer S.W., Lee C., Birney E., Altshuler D.M., Eichler E.E., Carter N.P., Hurles M.E., Feuk L. Challenges and standards in integrating surveys of structural variation. Nat. Genet. 2007;39(7, Suppl):S7–S15. doi: 10.1038/ng2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kidd J.M., Cooper G.M., Donahue W.F., Hayden H.S., Sampas N., Graves T., Hansen N., Teague B., Alkan C., Antonacci F. Mapping and sequencing of structural variation from eight human genomes. Nature. 2008;453:56–64. doi: 10.1038/nature06862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Conrad D.F., Bird C., Blackburne B., Lindsay S., Mamanova L., Lee C., Turner D.J., Hurles M.E. Mutation spectrum revealed by breakpoint sequencing of human germline CNVs. Nat. Genet. 2010;42:385–391. doi: 10.1038/ng.564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Perry G.H., Ben-Dor A., Tsalenko A., Sampas N., Rodriguez-Revenga L., Tran C.W., Scheffer A., Steinfeld I., Tsang P., Yamada N.A. The fine-scale and complex architecture of human copy-number variation. Am. J. Hum. Genet. 2008;82:685–695. doi: 10.1016/j.ajhg.2007.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lieber M.R., Ma Y., Pannicke U., Schwarz K. Mechanism and regulation of human non-homologous DNA end-joining. Nat. Rev. Mol. Cell Biol. 2003;4:712–720. doi: 10.1038/nrm1202. [DOI] [PubMed] [Google Scholar]
- 18.Kranz C., Denecke J., Lehrman M.A., Ray S., Kienz P., Kreissel G., Sagi D., Peter-Katalinic J., Freeze H.H., Schmid T. A mutation in the human MPDU1 gene causes congenital disorder of glycosylation type If (CDG-If) J. Clin. Invest. 2001;108:1613–1619. doi: 10.1172/JCI13635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Darnell J.C., Fraser C.E., Mostovetsky O., Darnell R.B. Discrimination of common and unique RNA-binding activities among Fragile X mental retardation protein paralogs. Hum. Mol. Genet. 2009;18:3164–3177. doi: 10.1093/hmg/ddp255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kullander K., Butt S.J., Lebret J.M., Lundfald L., Restrepo C.E., Rydström A., Klein R., Kiehn O. Role of EphA4 and EphrinB3 in local neuronal circuits that control walking. Science. 2003;299:1889–1892. doi: 10.1126/science.1079641. [DOI] [PubMed] [Google Scholar]
- 21.Chompret A., Brugières L., Ronsin M., Gardes M., Dessarps-Freichey F., Abel A., Hua D., Ligot L., Dondon M.G., Bressac-de Paillerets B. P53 germline mutations in childhood cancers and cancer risk for carrier individuals. Br. J. Cancer. 2000;82:1932–1937. doi: 10.1054/bjoc.2000.1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schluth-Bolard C., Sanlaville D., Labalme A., Till M., Morin I., Touraine R., Edery P. 17p13.1 microdeletion involving the TP53 gene in a boy presenting with mental retardation but no tumor. Am. J. Med. Genet. A. 2010;152A:1278–1282. doi: 10.1002/ajmg.a.33316. [DOI] [PubMed] [Google Scholar]
- 23.Adam M.P., Justice A.N., Schelley S., Kwan A., Hudgins L., Martin C.L. Clinical utility of array comparative genomic hybridization: uncovering tumor susceptibility in individuals with developmental delay. J. Pediatr. 2009;154:143–146. doi: 10.1016/j.jpeds.2008.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krepischi-Santos A.C., Rajan D., Temple I.K., Shrubb V., Crolla J.A., Huang S., Beal S., Otto P.A., Carter N.P., Vianna-Morgante A.M., Rosenberg C. Constitutional haploinsufficiency of tumor suppressor genes in mentally retarded patients with microdeletions in 17p13.1. Cytogenet. Genome Res. 2009;125:1–7. doi: 10.1159/000218743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schwarzbraun T., Obenauf A.C., Langmann A., Gruber-Sedlmayr U., Wagner K., Speicher M.R., Kroisel P.M. Predictive diagnosis of the cancer prone Li-Fraumeni syndrome by accident: new challenges through whole genome array testing. J. Med. Genet. 2009;46:341–344. doi: 10.1136/jmg.2008.064972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mahmoudi S., Henriksson S., Corcoran M., Méndez-Vidal C., Wiman K.G., Farnebo M. Wrap53, a natural p53 antisense transcript required for p53 induction upon DNA damage. Mol. Cell. 2009;33:462–471. doi: 10.1016/j.molcel.2009.01.028. [DOI] [PubMed] [Google Scholar]
- 27.Brosh R., Rotter V. When mutants gain new powers: news from the mutant p53 field. Nat. Rev. Cancer. 2009;9:701–713. doi: 10.1038/nrc2693. [DOI] [PubMed] [Google Scholar]
- 28.de Vries A., Flores E.R., Miranda B., Hsieh H.M., van Oostrom C.T., Sage J., Jacks T. Targeted point mutations of p53 lead to dominant-negative inhibition of wild-type p53 function. Proc. Natl. Acad. Sci. USA. 2002;99:2948–2953. doi: 10.1073/pnas.052713099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hainaut P., Wiman K.G. 30 years and a long way into p53 research. Lancet Oncol. 2009;10:913–919. doi: 10.1016/S1470-2045(09)70198-6. [DOI] [PubMed] [Google Scholar]
- 30.Mosner J., Mummenbrauer T., Bauer C., Sczakiel G., Grosse F., Deppert W. Negative feedback regulation of wild-type p53 biosynthesis. EMBO J. 1995;14:4442–4449. doi: 10.1002/j.1460-2075.1995.tb00123.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vilborg A., Wilhelm M.T., Wiman K.G. Regulation of tumor suppressor p53 at the RNA level. J. Mol. Med. 2010;88:645–652. doi: 10.1007/s00109-010-0609-2. [DOI] [PubMed] [Google Scholar]
- 32.Sen S.K., Han K., Wang J., Lee J., Wang H., Callinan P.A., Dyer M., Cordaux R., Liang P., Batzer M.A. Human genomic deletions mediated by recombination between Alu elements. Am. J. Hum. Genet. 2006;79:41–53. doi: 10.1086/504600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Batzer M.A., Deininger P.L. Alu repeats and human genomic diversity. Nat. Rev. Genet. 2002;3:370–379. doi: 10.1038/nrg798. [DOI] [PubMed] [Google Scholar]
- 34.Deininger P.L., Batzer M.A. Alu repeats and human disease. Mol. Genet. Metab. 1999;67:183–193. doi: 10.1006/mgme.1999.2864. [DOI] [PubMed] [Google Scholar]
- 35.Li L., McVety S., Younan R., Liang P., Du Sart D., Gordon P.H., Hutter P., Hogervorst F.B., Chong G., Foulkes W.D. Distinct patterns of germ-line deletions in MLH1 and MSH2: the implication of Alu repetitive element in the genetic etiology of Lynch syndrome (HNPCC) Hum. Mutat. 2006;27:388. doi: 10.1002/humu.9417. [DOI] [PubMed] [Google Scholar]
- 36.Smith T.M., Lee M.K., Szabo C.I., Jerome N., McEuen M., Taylor M., Hood L., King M.C. Complete genomic sequence and analysis of 117 kb of human DNA containing the gene BRCA1. Genome Res. 1996;6:1029–1049. doi: 10.1101/gr.6.11.1029. [DOI] [PubMed] [Google Scholar]
- 37.Slebos R.J., Resnick M.A., Taylor J.A. Inactivation of the p53 tumor suppressor gene via a novel Alu rearrangement. Cancer Res. 1998;58:5333–5336. [PubMed] [Google Scholar]
- 38.Hudson T.J., Anderson W., Artez A., Barker A.D., Bell C., Bernabé R.R., Bhan M.K., Calvo F., Eerola I., Gerhard D.S., International Cancer Genome Consortium International network of cancer genome projects. Nature. 2010;464:993–998. doi: 10.1038/nature08987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bougeard G., Sesboüé R., Baert-Desurmont S., Vasseur S., Martin C., Tinat J., Brugières L., Chompret A., de Paillerets B.B., Stoppa-Lyonnet D., French LFS working group Molecular basis of the Li-Fraumeni syndrome: an update from the French LFS families. J. Med. Genet. 2008;45:535–538. doi: 10.1136/jmg.2008.057570. [DOI] [PubMed] [Google Scholar]
- 40.Bougeard G., Brugières L., Chompret A., Gesta P., Charbonnier F., Valent A., Martin C., Raux G., Feunteun J., Bressac-de Paillerets B., Frébourg T. Screening for TP53 rearrangements in families with the Li-Fraumeni syndrome reveals a complete deletion of the TP53 gene. Oncogene. 2003;22:840–846. doi: 10.1038/sj.onc.1206155. [DOI] [PubMed] [Google Scholar]
- 41.Plummer S.J., Santibáñez-Koref M., Kurosaki T., Liao S., Noble B., Fain P.R., Anton-Culver H., Casey G. A germline 2.35 kb deletion of p53 genomic DNA creating a specific loss of the oligomerization domain inherited in a Li-Fraumeni syndrome family. Oncogene. 1994;9:3273–3280. [PubMed] [Google Scholar]
- 42.Knudson A.G., Jr. Mutation and cancer: statistical study of retinoblastoma. Proc. Natl. Acad. Sci. USA. 1971;68:820–823. doi: 10.1073/pnas.68.4.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.