

Abstract
Background & Aims
Liver inflammation and injury are mediated by the innate immune response, which is regulated by Toll-like receptors (TLR). Activation of TLR9 induces Type I interferons (IFNs) via the interferon regulatory factor (IRF)-7. We investigated the roles of Type I IFNs in TLR9-associated liver injury in mice.
Methods
Liver injury was induced in wild-type (WT), IRF7-deficient, and IFN-α/s receptor-1 (IFNAR1)-deficient mice by administration of ligands for TLR9 or TLR2. Findings from mice were verified in cultured hepatocytes and liver mononuclear cells, and in vivo experiments using recombinant Type-I IFN and interleukin-1 receptor antagonist (IL-1ra).
Results
Type I IFNs were upregulated during TLR9-associated liver injury in WT mice. IRF7- and IFNAR1-deficient mice, which have disruptions in Type I IFN production or signaling, respectively, had greater amounts of liver damage and inflammation, decreased recruitment of dendritic cells, and increased production of TNF-α by liver mononuclear cells (LMNC). These findings indicate that Type I IFNs have anti-inflammatory activities in liver. The IL-1ra, which is produced by LMNC and hepatocytes, is an IFN-regulated antagonist of the pro-inflammatory cytokine IL-1β; IRF7- and IFNAR1-deficient mice had decreased levels of IL-1ra, compared with WT mice. IL-1ra protected cultured hepatocytes from IL-1β-mediated sensitization to cytotoxicity from TNF-α. In vivo exposure to Type I IFN, which induced IL-1ra, or administration of IL-1ra reduced TLR9-associated liver injury; the protective effect of Type-I IFNs therefore appears to be mediated by IFN-dependent induction of IL-1ra.
Conclusions
Type I IFNs have anti-inflammatory effects mediated by endogenous IL-1ra which regulates the extent of TLR9-induced liver damage. Type I interferon signaling is therefore required for protection from immune-mediated liver injury.
Keywords: liver disease, immunology, innate immunity, bacterial DNA
Liver-related etiologies are among the top ten leading causes of death in the United States.1 The common hallmark of most liver diseases is inflammation and injury, which may result in acute liver failure, or, if persist, promote the development of fibrosis and eventually cirrhosis.2 Presently, liver inflammation and injury have no pathogenesis-specific treatment, but they share a common pathway of induction of innate immune responses, triggered by Toll-like receptors (TLRs).3
For example, TLR9 is activated by unmethylated DNA rich in cytidine-phosphate-guanosine (CpG). This motif is present in bacterial DNA and also in DNA from apoptotic mammalian cells.4 Emerging data provide evidence for the role of CpG DNA and TLR9-mediated inflammation in acute and chronic liver injury of diverse origin, including alcoholic liver disease,5 primary biliary cirrhosis,6 primary sclerosing cholangitis7 and acetaminophen-induced liver injury.8 TLR9-initiated signals are also involved in general processes such as liver fibrosis,9, 10 liver cirrhosis,11 ischemia-reperfusion injury,12 and liver graft rejection13. TLR9 acts synergistically with the TLR2 ligand lipoteichoic acid (LTA),14 and further sensitizes the liver to injury induced by the TLR4 ligand lipopolysaccharide (LPS).15, 16
Activation of TLR9 by CpG results in increased production of inflammatory mediators via the nuclear factor κB (NFκB), and in a strong induction of Type I interferons (IFN-α and IFN-β) via the interferon regulatory factor 7 (IRF-7),17 which together with IRF3 also plays a role in TLR4-dependent induction of Type I IFNs. 18 While NFκB-dependent induction of inflammatory cytokines by CpG is considered the key event in the TLR9-mediated liver injury,5, 6, 8, 13 little is known about the role of IRF7-dependent Type-I IFN induction and signaling after TLR9 stimulation.
Here we demonstrate that induction of Type I IFNs and Type I IFN-mediated signaling has a protective role in TLR9-induced liver inflammation and injury. We report that impaired induction of interferon-stimulated genes, including the anti-inflammatory cytokine, interleukin 1 receptor antagonist (IL-1ra), increases liver inflammation and injury. Our results also demonstrate a protective effect of the IL-1ra in vivo in TLR9-associated liver injury.
Experimental procedures
Animals and experimental protocol
The B6.129F2 and C57Bl/6 wild-type (WT) mice were purchased from Jackson Laboratory. IRF7-deficient (IRF7-/-) mice on B6.129F2 background were provided by Tadagatsu Tanaguichi (Tokyo) and type I interferon α/β receptor 1-deficient (IFNAR1−/−) mice on the C57Bl/6 background were the kind gift of Jonathan Sprent (Scripps Research Institute, La Jolla, CA). All animals (3–6/experimental group) were 6–8 weeks old and received proper care in agreement with animal protocols approved by the Institutional Animal Use and Care Committee of the University of Massachusetts Medical School.
We employed a previously described model of TLR9-associated liver injury induced by administration of TLR9 and TLR2 ligands.15, 16 After acclimatization, WT, IRF7−/− and IFNAR1−/− mice were injected intraperitoneally (i.p.) with saline or the combination of 2.5 mg/kg unmethylated DNA rich in cytidine-phosphate-guanosine (CpG, ODN1826 murine TLR9 ligand; InvivoGen, San Diego, CA), and 5 mg/kg lipoteichoic acid (LTA, from Staphylococcus aureus; Sigma, Saint Louis, MO). Three days after the above priming stimulus, the mice were injected i.p. with either saline or 0.5 mg/kg lipopolysaccharide (LPS, from Escherichia coli 0111:B4, Sigma, St. Louis, MO) and sacrificed as indicated. Some C57Bl/6 WT mice received a single i.p. injection of 100,000 IU human pegylated interferon alpha-2b (pegIFNα2, Pegintron, Schering, Kenilworth, NJ) two hours prior to LPS. Others were pretreated with recombinant human interleukin-1 receptor antagonist (IL-1ra) 25 mg/kg i.p. every six hours (Anakinra, Amgen, Thousand Oaks, CA) for 24 hours before CpG+LTA, and the treatment with IL-1ra was ongoing until sacrifice. Serum was separated by centrifugation. Livers were snap frozen, stored in RNAlater (Qiagen GmbH, Hilden, Germany) or fixed in 10% neutral-buffered formalin. ALT was quantified by biochemical assay (D-Tek Analytical Laboratories Inc, San Diego, CA).
Histopathology analysis
Sections of formalin-fixed, paraffin-embedded livers were stained with hematoxylin and eosin (H&E), and assessed for inflammatory infiltrate; area of inflammatory infiltrates was calculated with Microsuite (Olympus Soft Imaging Solution GmbH, Munster, Germany) image analysis software in 20 high power fields.
Isolation of hepatocytes and liver mononuclear cells
Animals received anesthesia with ketamine (100 mg/kg) and xylazine (10 mg/kg); the livers were perfused with saline solution followed by in vivo digestion, as we previously described.19 The hepatocytes and liver mononuclear cells (LMNCs) were purified by centrifugation at slow speed (500g) and in Percoll gradient, respectively.
Phenotype analysis by flow cytometry
Cells were washed in PBS and incubated with anti-CD68 (FITC), anti-CD11c (FITC) or anti-PDCA1 (Alexa Fluor 647) antibodies for 30 minutes on ice. After incubation, cells were washed with PBS, fixed in paraformaldehyde and analyzed by flow cytometry. All antibodies were from eBioscience (eBioscience, Inc., San Diego, CA).
In vitro cell culture
Primary hepatocytes were cultured in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS), 1% insulin, transferrin, and selenium supplement on collagen-coated plates (Becton Dickinson Labware, Bedford, MA). Primary LMNCs were cultured in DMEM with 10% FBS. Hepatocytes were treated with recombinant murine IFN-α2b (500 IU/mL, eBioscience, San Diego, CA), murine IL-1ra (100 pg/mL, R&D Systems, Minneapolis, MN), murine IL-1β (100 IU/mL, Peprotech Inc., Rocky Hill, NJ) or murine TNF-α (0-100 ng/mL, Peprotech Inc., Rocky Hill, NJ). LMNCs were treated with mouse IFN-α2b or LPS (100 ng/mL, Sigma, St. Louis, MO).
Hepatocyte cytotoxicity assay
Lactate dehydrogenase (LDH) release from the hepatocyte into the culture supernatants was measured using the LDH-cytotoxicity assay kit (Abcam, Cambridge, MA), and normalized to total LDH (determined after treatment of cells with detergent-based lysis solution).
Cytokine and chemokine measurement
Serum concentrations of the secreted forms of interleukin-1β (IL-1β) and IL-1 receptor antagonist (IL-1ra) were analyzed with ELISA (R&D Systems, Minneapolis, MN). Monocyte chemotactic protein 1 (MCP-1) in the liver lysate was measured with ELISA from R&D, and TNF-α in cell culture supernatants was analysed using ELISA from BD Biosciences (BD Biosciences, San Jose, CA).
RNA analysis
RNA was extracted from liver tissue using the RNeasy kit (Qiagen Sciences, Maryland, USA) and on-column DNase digestion was performed using the DNase Set (Qiagen GmbH, Hilden, Germany); cDNA was synthesized with Reverse Transcription System (Promega Corp., Madison, WI). Real-time quantitative polymerase chain reaction (qPCR) was performed using the iCycler iQ Cycler (Bio-Rad Laboratories, Inc., Hercules, CA) and specific primers (Supplementary Table 1).
Statistical Analysis
The data are presented as mean ± SEM. Comparison of the means was performed using Student’s T-test or Kruskall-Wallis test, when appropriate. P values less than 0.05 were considered significant.
Results
Type I IFNs are induced in TLR9-associated liver injury
TLR9 signaling induces Type I IFNs, which may play a role in tissue injury and regeneration. 20, 21 We have previously demonstrated that MyD88, a common adaptor to TLR9, TLR2 and TLR4 is critical in sensitization to liver injury.16 Here we employed a model of TLR9-associated liver injury owed to sensitization by the synergistic effect of the TLR9 ligand CpG and the TLR2 ligand LTA, followed by a second hit with LPS,15, 16 and asked if the integrity of the Type I IFN pathway was involved in liver damage. We found that combined administration of TLR9 and TLR2 ligands induced and sensitized the liver to injury by the TLR4 ligand LPS, as indicated by increased serum ALT levels in WT mice (Fig. 1A). More importantly, TLR9+TLR2 ligands induced expression of IFNα (Fig. 1B) and IFNβ (Fig. 1C) mRNA in the liver and expression of both IFNα and IFNβ was further upregulated upon TLR4 stimulation in WT mice. We also found that expression of the Type I IFN-inducible gene, interferon regulated gene ISG15, was induced by TLR9+TLR2 ligand treatment and the expression was further increased upon subsequent LPS stimulation (Fig. 1D). Collectively, these data suggested that Type I IFNs and IFN-triggered signaling pathways were upregulated in TLR9-associated liver injury.
Fig. 1. Type I IFNs are induced in TLR9-associated liver injury.
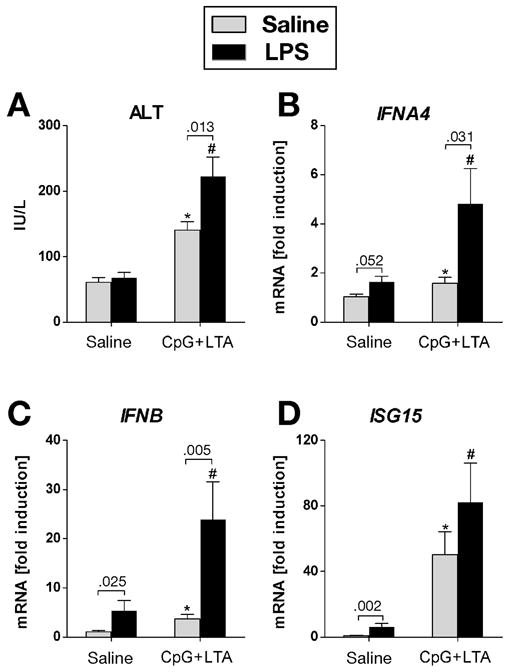
Wild-type mice were injected i.p. with 2.5 mg/kg CpG DNA and 5 mg/kg LTA. Three days later mice were injected with saline or 0.5 mg/kg LPS i.p. and sacrificed after 2 hours. Serum ALT levels (A) were measured and messenger RNA levels of (B) liver interferon α-4 (IFNA4), (C) interferon β (IFNB) and (D) interferon-stimulated gene 15 (ISG15) were analyzed by real-time PCR and normalized to 18s. Values are shown as mean ± SEM fold increase over saline-primed group (3–6 mice per group). Numbers in graphs denote p values. *) p < 0.05 vs. saline-stimulated control mice; #) p < 0.05 vs. LPS-stimulated control mice
Deficiency in Type I IFN induction exacerbates liver injury
Our data indicated activation of Type I IFN signaling in TLR9-associated liver injury. While both TLR9 and TLR2 ligands activate production of proinflammatory cytokines, TLR9 stimulation also triggers Type I IFNs.17 Type I IFN production induced by TLR9 is largely dependent on activation of intracellular pathways involving interferon regulatory factor-7 (IRF7).22 To define the importance of Type I IFN in TLR9-associated liver injury, we tested IRF7-deficient mice.
In sharp contrast to their wild-type littermates, IRF7-deficient mice showed exacerbation of TLR9-associated liver injury, as indicated by serum ALT elevations (Fig. 2A). Histopathology analysis (Fig. 2B) of IRF7-deficient livers revealed an about 5-fold increase in the number of inflammatory infiltrates (10.33 ± 3.71 in IRF7-KO vs. 1.75 ± 0.48 in WT, P = 0.026) and an about 2.5-fold increase in total area of inflammatory infiltrates (0.67 ± 0.05 vs. 0.28 ± 0.05 mm2, P = 0.017), compared to wild-type controls.
Fig. 2. Deficiency of Type I IFN induction exacerbates liver injury.

B6.129F2 wild-type mice and IRF7-deficient mice were treated with CpG DNA + LTA ± LPS as in figure 1. Serum ALT levels were measured (A). Assessment of liver inflammatory infiltrate (B) was performed in histology samples stained with H&E. Arrows point at inflammatory infiltrates, magnification 200x. mRNA levels of liver (C) interferon α-4 (IFNA4), (D) interferon β (IFNB), (E) interferon-stimulated gene 15 (ISG15) and (F) IP-10 were analyzed by real-time PCR. Values are shown as mean ± SEM fold increase over saline-primed control group (3–6 mice per group). *) p < 0.05 vs. saline-primed wild-type mice; #) p < 0.05 vs. saline-primed IRF7-/- mice
The increased TLR9-associated liver injury in IRF7-deficient mice was accompanied by deficient induction of IFNα (Fig. 2C) and IFNβ (Fig. 2D), compared to controls. LPS stimulation could partially overcome the deficit in IFNα and IFNβ expression in TLR9+TLR2 primed mice, presumably by the direct effect of TLR4 signaling on Type I IFN induction.23 However, there was decreased induction of the IFN-inducible genes, ISG15 (Fig. 2E) and IP-10 (Fig. 2F), in IRF7-deficient mice and further expression of these molecules upon stimulation with LPS was limited. These data supported our hypothesis that Type I IFN induction plays a role in the pathogenesis of TLR9-induced liver damage and suggested a protective role for Type I IFNs in liver injury.
Deficiency in Type I IFN signaling exacerbates liver injury
The effects of Type I IFNs are mediated by a cognate receptor composed of two chains, IFNAR1 and IFNAR2, both of which are essential to initiate production of the interferon-stimulated genes (ISGs).24 To differentiate between the direct and indirect protective effects of Type-I IFNs in TLR9-associated liver injury, we tested mice deficient in type I interferon receptor expression (IFNAR1-deficient mice).
Similar to findings in IRF7-deficient mice, mice deficient in IFNAR1 expression showed significantly increased serum ALT (Fig. 3A), widespread liver inflammation, about 5-fold increase in the number of inflammatory foci (4.80 ± 0.49 in IFNAR1-KO vs. 1.0 ± 0.58 in WT, P = 0.018) and about 7-fold increase in total area of inflammatory infiltrate (0.40 ± 0.11 vs. 0.06 ± 0.04 mm2, P = 0.036) after TLR9+TLR2 stimulation compared to wild-type controls (Fig. 3B).
Fig. 3. Deficiency of Type I IFN signaling exacerbates liver injury.

C57Bl6 wild-type mice and IFNAR1-deficient mice were treated with CpG DNA + LTA ± LPS as in figure 1. Serum ALT levels were measured (A), and H&E-stained samples assessed for liver inflammatory infiltrate (B) was performed in histology samples stained with H+E. Arrows point at inflammatory infiltrates, magnification 200x. mRNA levels of liver (C) interferon α-4 (IFNA4), (D) interferon β (IFNB), (E) interferon-stimulated gene 15 (ISG15) and (F) IP-10 were analyzed by real-time PCR. Values are shown as mean ± SEM (3–6 mice per group). *) p < 0.05 vs. saline-primed wild-type mice; #) p < 0.05 vs. saline-primed IFNAR1-/- mice
Type I IFNs per se are IFN-sensitive and are strictly regulated by a self-initiated amplification loop involving IRF7 and both IFN receptor chains.24 Consistent with this mechanism, IFNAR1-deficient mice showed substantially lower induction of liver IFNα (Fig. 3C), IFNβ (Fig. 3D) and minimal expression of ISG15 (Fig. 3E) and IP-10 (Fig. 3F), compared to controls. This difference was not overcome by LPS stimulation. Collectively, these data suggested that both Type I IFN induction and IFN-induced signaling may play a protective role in the liver by limiting inflammatory infiltrate and liver injury.
Deficiency of Type I IFN signaling results in decreased dendritic cell recruitment to the liver in TLR9-associated injury
The exaggerated liver inflammatory infiltrate in IRF7- and IFNAR1-deficient mice suggested a role for Type I IFNs in recruitment of inflammatory cells in TLR9-associated liver injury. We thus aimed to define the cell composition of these inflammatory infiltrates. Flow cytometric analysis of liver mononuclear cells (LMNCs) showed equal enrichment of CD68+ monocytes/macrophages in the livers of both WT and IFNAR1-deficient mice after TLR9+TLR2 priming (Fig. 4A). Similarly, liver expression of chemokines MCP-1, MCP-2, MIP-1α and MIP-1β (Suppl. Fig. 1A-E) and chemokine receptors CCR1, CCR2 and CCR5 (Suppl. Fig. 1F-H), which are involved in macrophage recruitment, was upregulated in TLR9-associated liver injury to a comparable extent in both WT and IFNAR1-deficient mice.
Fig. 4. Deficiency of Type I IFN signaling results in decreased dendritic cell recruitment to the liver in TLR9-associated liver injury.
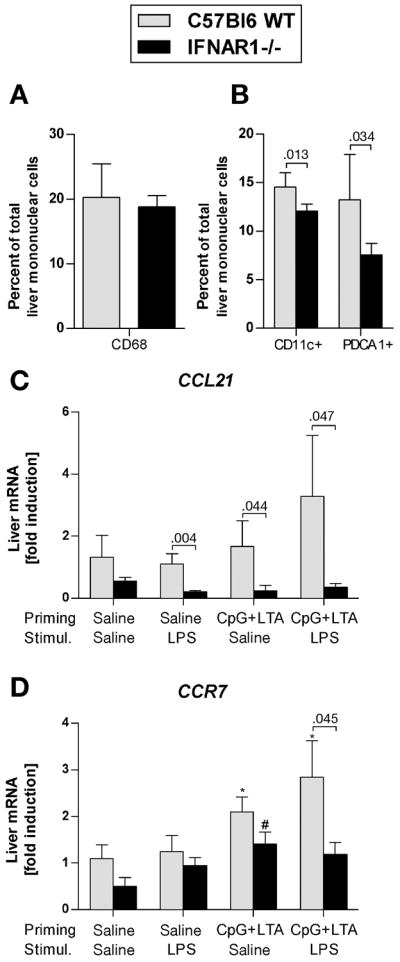
(A,B) Wild-type and IFNAR1-deficient mice were injected i.p. with CpG DNA + LTA ± LPS as indicated. Liver mononuclear cells were isolated, stained with anti-CD68 (A), anti-CD11c and anti-PDCA1 (B) monoclonal antibodies and analyzed using flow cytometry (N= 5 mice per group). mRNA levels of (C) liver chemokine ligand 21 (CCL-21) and (D) chemokine receptor 7 (CCR7) were analyzed by real-time PCR. Values are shown as mean ± SEM. *) p < 0.05 vs. saline-primed wild-type mice; #) p < 0.05 vs. saline-primed IFNAR1-deficient mice
In contrast to WT mice, LMNCs isolated from mice deficient in IFNAR1 showed a significantly lower proportion of CD11c+ (myeloid) and PDCA1+ (plasmacytoid) dendritic cells (DC) (Fig. 4B). This finding was associated with a significantly decreased liver expression of the chemokine ligand 21 (CCL-21) (Fig. 4C), a key molecule involved in recruitment of DC, 25 and decreased expression of the CCL-21 receptor CCR7 (Fig. 4D). These data support our hypothesis that in TLR9-associated liver injury, Type I IFNs are required for dendritic cell recruitment to the liver.
Deficient Type I interferon signaling results in an imbalance in IL-1β / IL-1 receptor antagonist induction in TLR9-associated liver injury
The protective effect of Type I IFN induction and signaling against TLR9-associated liver injury implied the involvement of Type I IFN-dependent anti-inflammatory factors. We thus analyzed the expression of another IFN-dependent gene, IL-1 receptor antagonist (IL-1ra), which is a natural endogenous antagonist of the proinflammatory interleukin-1β (IL-1β) at the receptor level.26 Pro-IL-1β gene in the liver (Fig. 5A) and IL-1β protein (Fig. 5B) in the serum were significantly induced by TLR9+TLR2 ligands and further upregulated by LPS to a comparable extent in WT, IRF7- and IFNAR-deficient mice (Fig. 5A,B). Induction of IL-1ra mRNA in the liver and of the secreted IL-1ra protein in serum were decreased both in IRF7- and IFNAR1-deficient mice treated with CpG+LTA, compared to controls (Fig. 5C,D). More important, sensitization via TLR9+TLR2 ligands resulted in less IL-1ra protein production upon stimulation with LPS in both IRF7- and IFNAR1-deficient mice compared to their wild-type controls (Fig. 5D). This finding suggested that aggravated TLR9-associated liver injury in Type I IFN deficient animals is associated with a significant imbalance in IL-1β/IL-1ra signaling.
Fig. 5. Deficient Type I interferon signaling results in an imbalance in IL-1β / IL1 receptor antagonist induction in TLR9-associated liver injury.

Mice were treated with CpG DNA + LTA ± LPS as in figure 1. mRNA levels of liver pro-interleukin-1β (IL-1β) and interleukin 1-receptor antagonist (IL-1ra) were analyzed by real-time PCR (A,C). Serum IL-1ra levels were measured by ELISA (B,D). Values are shown as mean ± SEM fold increase over saline-primed group (3-6 mice per group). *) p < 0.05 vs. saline-primed wild-type B6.129F2 or C57Bl6 mice; #) p < 0.05 vs. saline-primed IRF7-/- or IFNAR1-/- mice
IL-1ra protects hepatocytes from IL-1β-dependent sensitization to cell death induced by TNF-α
Our data suggested a protective effect of IL-1ra in TLR9-associated liver injury. To gain a mechanistic insight, we first investigated the source of IL-1ra in the liver. We observed that WT primary hepatocytes and LMNCs stimulated with IFN-α in vitro produced significantly more IL-1ra, compared to non-stimulated cells (Fig. 6A). Next, we asked whether deficiency of Type I IFNs results in differential production of inflammatory cytokines by LMNCs. In vivo priming of mice with TLR9+TLR2 ligands sensitized LMNCs to ex vivo stimulation with LPS and resulted in significantly greater induction of inflammatory cytokines TNF-α (Fig. 6B) and IL-1β (Fig. 6C), compared with LPS-stimulated LMNCs of non-sensitized mice. In contrast to WT mice, LMNCs isolated from IRF7- and IFNAR-deficient mice showed significantly increased induction of TNF-α upon ex vivo stimulation with LPS (Fig. 6B); induction of IL-1β did not significantly differ between genotypes (Fig. 6C).
Fig. 6. IL-1ra protects hepatocytes from IL-1β dependent sensitization to cell death induced by TNF-α.

(A) Hepatocytes and liver mononuclear cells (LMNCs), isolated from WT mice, were stimulated with murine IFN-α2b, and IL-1ra in supernatants was measured with ELISA (N=5 mice per group). (B,C) WT, IRF7- and IFNAR1-deficient mice were treated with i.p. with CpG DNA + LTA. Three days later, LMNCs were isolated, ex vivo stimulated with LPS, and TNF-α and IL-1β in supernatant were measured after 6 hours. Representative values from total N= 4 mice per group are shown. *,#,§,†) p < 0.05 vs WT cells from the respective treatment group. (D) Primary WT hepatocytes were ex vivo pretreated with murine IL-1ra and IL-1β. After four hours, murine TNF-α was added. LDH release into cell culture supernatant was measured at 24 hours and normalized to total LDH. Representative values from total N= 4 mice are shown. *) p < 0.05 vs. cells not treated with IL-1β; #) p < 0.05 vs. cells not treated with IL-1ra. (E) Primary hepatocytes from WT, IRF7- and IFNAR1-deficient mice were isolated and treated ex vivo as indicated for 4 hours, followed by murine TNF-α. LDH release into cell culture supernatant was measured at 24 hours and normalized to total LDH. Representative values from total N= 4 mice per group are shown. *,#) p < 0.05 vs. WT hepatocytes of the respective treatment group.
Consequently, we asked whether IL-1ra could play a protective role in hepatocyte death induced by synergistic activities of TNF-α and IL-1β. Treatment of primary WT hepatocytes with TNF-α did not induce hepatocyte death (Fig. 6D), consistent with previous reports that healthy WT hepatocytes are resistant to stimulation with TNF-α alone.27 However, pretreatment with IL-1β sensitized hepatocytes to TNF-α induced death (Fig. 6D). More important, the sensitizing effect of IL-1β was significantly reduced by IL-1ra (Fig. 6D). Stimulation with IL-1β + TNF-α lead to a significantly greater extent of hepatocyte death in IRF7- and IFNAR1-deficient hepatocytes, compared to WT; significant protection from hepatocyte death was observed after co-treatment with IL-1ra in all genotypes (Fig. 6E).
Taken together, these data suggested that Type I IFNs induce IL-1ra production both in hepatocytes and LMNCs, and that IL-1ra protects hepatocytes from IL-1β-dependent sensitization to TNF-α-induced cell death.
Type I interferon induces endogenous IL-1ra in vivo and ameliorates TLR9-associated liver injury
To evaluate the protective role of Type I IFN in TLR9-associated liver injury, we first administered pegylated IFNα2a (pegIFNα2) to WT mice and observed a significant induction of serum IL-1ra (Suppl. Fig. 2). Next, we showed that pegIFNα2 administered to TLR9+TLR2-primed mice two hours prior to LPS significantly prolonged survival (Fig. 7A) and ameliorated TLR9-associated liver injury (Fig. 7B), compared to control mice. Furthermore, pegIFNα2 prevented aggravation of LPS-induced liver injury after TLR9+TLR2-priming (Fig. 7B). Importantly, serum IL-1ra positively correlated with the length of survival (Suppl. Fig. 3A), and negatively correlated with the extent of liver injury (Suppl. Fig. 3B). Accordingly, histopathology analysis revealed reduction in liver inflammatory infiltrate and the extent of necrosis in mice pretreated with pegIFNα2 compared to controls (Fig. 7C). Taken together, these data suggest that type I IFNs induce IL-1ra which has hepatoprotective and anti-inflammatory effects in TLR9-induced liver injury.
Fig. 7. Type I interferon induces endogenous IL-1ra and ameliorates TLR9-associated liver injury.

(A-C) Wild-type mice were injected with CpG DNA + LTA i.p., followed 3 days later by saline or 100,000 IU pegIFNα2, and followed by LPS i.p. for the last 2 hours. Survival (A) and serum ALT were analysed at indicated time points (B). Livers were stained with H&E, magnification 200x (C). Values are shown as mean ± SEM (11 mice per group). *) p < 0.05 vs. control mice, #) p < 0.05 vs. control mice treated with pegIFNα2. (D-F) Wild-type mice were treated with saline or with recombinant IL-1ra 25 mg/kg i.p. every six hours. Twenty-four hours after IL-1ra initiation, mice were injected with CpG DNA + LTA, followed by LPS 3 days later. Survival (D) and serum ALT (E) were analysed at indicated time points. Livers were stained with H&E, magnification 200x (F). Values are shown as mean ± SEM (15 mice per group). *) p < 0.05 vs. control mice, #) p < 0.05 vs. control mice treated with IL-1ra
IL-1ra ameliorates TLR9-associated liver injury
To demonstrate the direct protective effect of IL-1ra, we initiated treatment with recombinant IL-1ra in WT mice 24 hours prior to administration of TLR9+TLR2 ligands and continued with IL-1ra throughout the experiment. Mice pretreated with IL-1ra showed significantly prolonged survival after injection of LPS (Fig. 7D), which was associated with lower ALT levels (Fig. 7E) and reduction in liver inflammatory infiltrate and extent of necrosis compared to controls (Fig. 7F). Collectively, these data suggested that Type I IFNs are needed to elicit a strong anti-inflammatory response by induction of IL-1ra, and that Type I IFN-regulated IL-1β/IL-1ra system is key to the protective role of Type I IFNs in liver injury.
Discussion
TLR9-dependent liver damage is involved in the pathogenesis of several liver diseases, such as alcoholic liver disease,5 primary biliary cirrhosis,6 primary sclerosing cholangitis7 and acetaminophen-induced liver injury,8 as well as in pathological processes such as liver fibrosis,10 liver cirrhosis,11 ischemia-reperfusion injury,12 and liver graft rejection.13 Thus, TLR9 holds the common link between different processes which lead to liver diseases; therefore, unraveling the pathogenesis of TLR9-induced liver injury may aid in identification of novel, efficient pathogenesis-based management or cure. Here we report the novel finding that Type I IFNs have an important regulatory role in TLR9-induced liver injury by limiting inflammation and liver injury. Further, we demonstrate that Type I IFN-induced IL-1ra could serve as a potential therapeutic target in liver injury.
In the current liver injury model, three TLR agonists are being used. Our recent and previous 15, 16 data show that liver inflammation and injury induced in this model are primarily dependent on TLR9; liver damage elicited by CpG is aggravated by co-stimulation with TLR2 and a secondary stimulation with TLR4. Of these TLRs, TLR2, 4 and 9 activate the MyD88-dependent pathway, while IRF3 activation is exclusive to TLR4. Type I IFN and IRF7 induction can occur via both TLR9 and TLR4. 18
We first demonstrated that Type I IFNs, IFNα and IFNs, were induced at the mRNA level in the TLR9-associated liver injury. This Type I IFN was biologically active as suggested by the increased expression of the IFN-inducible genes, ISG15 and IP-10 in the liver. Our novel observation of the increased liver inflammatory infiltrate and exacerbated liver injury in IRF7 and IFNAR1-deficient mice suggested a protective role for the Type I IFN induction in the liver. Indeed, previous studies suggested that Type-I IFNs can mediate anti-inflammatory effects (summarized in 28).
Compared to IFNAR1-deficient mice, where the protective Type I IFN signaling was fully inhibited, IRF7-deficient mice could still produce TLR4-induced Type I IFNs (preferentially IFNβ) via the TLR4-IRF3 pathway. In our study, TLR9-associated liver injury was associated with induction of IFNα, IFNβ and interferon-stimulated genes in wild-type mice. Notably, IFNAR1-deficiency abrogated the induction of Type I IFN response to much greater extent than IRF7 deficiency. Also, IFNAR1-deficient mice showed the most severe TLR9-associated histological liver damage, which exceeded the extent present in IRF7-deficient mice. The different observation between IRF7- and IFNAR1-deficient mice could be explained by differential composition of inflammatory liver infiltrate, or by the fact that Type I IFNs are potentially inducible by multiple pathways, including IRF1, IRF3 and IRF7.23, 29 Therefore, protective Type I IFNs could still be induced in IRF7-deficient mice by TLR9 in an IRF7- independent, IRF3-dependent manner, while the downstream protective effect of Type I IFNs is completely abolished in IFNAR1-deficient mice which cannot respond to type I IFNs.
We found that deficient Type I IFN signaling was associated with decreased liver recruitment of dendritic cells and with decreased expression of CCL-21and CCR7, a key chemokine-receptor pair involved in dendritic cell trafficking.30 Our novel findings indicating a role of Type I IFNs in recruitment of dendritic cells and in expression of CCL-21, together with data showing that TLR9-activated dendritic cells are key producers of Type I IFNs,31 suggest a novel self-sustaining mechanism for dendritic cell recruitment into the liver. It has been reported that dendritic cells are essential for Th1 response in the liver induced by TLR9 + TLR2 ligands 32 and contribute to liver fibrosis,9 Consistent with earlier observations15 TLR9-induced inflammatory infiltrates, that require dendritic cell recruitment to the liver,32 amplified pro-inflammatory cytokine induction by LPS. Our novel data suggest a dual role for dendritic cells in the liver. First, dendritic cells are necessary for TLR9-induced inflammatory cell infiltrates and sensitization to TLR4 ligands. Second, dendritic cells provide anti-inflammatory signals mediated by Type I IFNs.
We observed a synergistic effect of TLR9 + TLR2 and TLR4 ligands in upregulation of IL-1ra in wild-type animals; both IRF7-deficient and IFNAR1-deficient mice showed decreased induction of IL-1ra, compared to WT. One can speculate that this synergistic effect of TLR ligands on IL-1ra induction is mediated by Type I IFNs and IRF7. This notion is consistent with our data showing a synergistic effect of TLR9+2 and TLR4 ligands on Type I IFN expression in wild-type mice. We observed that Type I IFN induces IL-1ra in hepatocytes and liver mononuclear cells, and that IL-1ra protects hepatocytes against the sensitizing effect of IL-1β towards cytotoxicity induced by TNF-α. Our findings support previous reports that in order to become susceptible to the cytotoxic effect of TNF-α, hepatocytes require priming with ligands that interfere with their proliferation.27 Indeed, IL-1β has been shown to inhibit DNA synthesis in rat hepatocytes.33
We further showed that administration of recombinant pegIFNα2 induced IL-1ra in vivo. This finding was in agreement with our in vitro data from hepatocytes and liver mononuclear cells, with in vitro data that IL-1RA promoter region contains Type I IFN-inducible elements,34 and with data demonstrating increased in vivo production of IL-1ra upon treatment with recombinant IFN in humans.35 In addition, we observed that in vivo administration of pegIFNα2 significantly ameliorated TLR9-associated survival, liver injury and inflammation.
Our novel data demonstrated that pretreatment with recombinant IL-1ra significantly ameliorated liver injury and inflammation in WT mice. These findings confirm the anti-inflammatory effect of IL-1ra in liver injury and are complementary to study of Iizasa et al.36 who showed that deficiency of IL-1ra resulted in exacerbated liver injury and inflammation induced by Propionibacterium Acnes, which activates TLR9 and TLR2 receptors.15, 16 Our results show for the first time that Type I IFNs potentially protect from liver injury by inducing anti-inflammatory IL-1ra.
IL-1ra acts as a natural antagonist of IL-1β,26 and we show that in the liver IL-1ra is produced by both hepatocytes and liver mononuclear cells. Our data also demonstrates the protective effect of IL-1ra in TLR9-associated liver injury and survival. In previous studies, Imaeda et al.8 showed that in acetaminophen hepatotoxicity, inflammatory response is triggered by apoptotic mammalian DNA that increases transcription of IL-1β, and that inflammatory response is ameliorated in mice deficient for TLR9 and in mice treated with anti-IL-1β antibody. Importantly, IL-1ra has been reported for the treatment of hepatic failure in rats using a bioartificial liver device.37 These studies support our finding that the balance between IL-1β/IL-1ra is of crucial importance in TLR9-induced liver damage (Suppl. Fig. 4).
In conclusion, our findings suggest that the endogenous anti-inflammatory signaling induced by Type I IFNs and mediated by IL-1ra regulates the extent of TLR9-induced liver damage, and support the indispensable role of Type I interferon signaling in immune mediated liver injury. Finally, we suggest the potential role of IL-1ra in therapy of TLR9-associated liver diseases.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
Financial support: This work was supported by PHS grant # RO1DK075653 from NIDDK (to G.S.). Core resources supported by the Diabetes Endocrinology Research Center grant DK32520 were also used.
Abbreviations
- CpG
cytidine-phosphate-guanosine-rich DNA
- IFN
interferon
- IFNAR1
type I interferon receptor
- IL-1ra
interleukin 1 receptor antagonist
- IL-1β
interleukin 1 beta
- IRF
interferon regulatory factor
- ISG
interferon stimulated gene
- LPS
lipopolysaccharide
- LTA
lipoteichoic acid
- NFκB
nuclear factor κB
- TLR
toll-like receptor
Footnotes
Jan Petrasek - Acquisition of data, analysis and interpretation of data, statistical analysis and manuscript writing
Angela Dolganiuc - Acquisition of data, analysis and interpretation of data, and manuscript writing
Evelyn A. Kurt-Jones - Intellectual content, critical revision of the manuscript
Timea Csak - Acquisition of data, analysis and interpretation of data
Gyongyi Szabo - Study concept and design, critical revision of the manuscript, and obtained funding
Disclosures: nothing to disclose
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Minino AM, Heron MP, Murphy SL, et al. Deaths: final data for 2004. Natl Vital Stat Rep. 2007;55:1–119. [PubMed] [Google Scholar]
- 2.Polson J, Lee WM. AASLD position paper: the management of acute liver failure. Hepatology. 2005;41:1179–97. doi: 10.1002/hep.20703. [DOI] [PubMed] [Google Scholar]
- 3.Seki E, Brenner DA. Toll-like receptors and adaptor molecules in liver disease: update. Hepatology. 2008;48:322–35. doi: 10.1002/hep.22306. [DOI] [PubMed] [Google Scholar]
- 4.Huck S, Deveaud E, Namane A, et al. Abnormal DNA methylation and deoxycytosine-deoxyguanine content in nucleosomes from lymphocytes undergoing apoptosis. Faseb J. 1999;13:1415–22. doi: 10.1096/fasebj.13.11.1415. [DOI] [PubMed] [Google Scholar]
- 5.Gustot T, Lemmers A, Moreno C, et al. Differential liver sensitization to toll-like receptor pathways in mice with alcoholic fatty liver. Hepatology. 2006;43:989–1000. doi: 10.1002/hep.21138. [DOI] [PubMed] [Google Scholar]
- 6.Moritoki Y, Lian ZX, Wulff H, et al. AMA production in primary biliary cirrhosis is promoted by the TLR9 ligand CpG and suppressed by potassium channel blockers. Hepatology. 2007;45:314–22. doi: 10.1002/hep.21522. [DOI] [PubMed] [Google Scholar]
- 7.Karrar A, Broome U, Sodergren T, et al. Biliary epithelial cell antibodies link adaptive and innate immune responses in primary sclerosing cholangitis. Gastroenterology. 2007;132:1504–14. doi: 10.1053/j.gastro.2007.01.039. [DOI] [PubMed] [Google Scholar]
- 8.Imaeda AB, Watanabe A, Sohail MA, et al. Acetaminophen-induced hepatotoxicity in mice is dependent on Tlr9 and the Nalp3 inflammasome. J Clin Invest. 2009;119:305–14. doi: 10.1172/JCI35958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Connolly MK, Bedrosian AS, Mallen-St Clair J, et al. In liver fibrosis, dendritic cells govern hepatic inflammation in mice via TNF-alpha. J Clin Invest. 2009;119:3213–25. doi: 10.1172/JCI37581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Watanabe A, Hashmi A, Gomes DA, et al. Apoptotic hepatocyte DNA inhibits hepatic stellate cell chemotaxis via toll-like receptor 9. Hepatology. 2007;46:1509–18. doi: 10.1002/hep.21867. [DOI] [PubMed] [Google Scholar]
- 11.Mencin A, Kluwe J, Schwabe RF. Toll-like receptors as targets in chronic liver diseases. Gut. 2009;58:704–20. doi: 10.1136/gut.2008.156307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tsuchihashi S, Zhai Y, Bo Q, et al. Heme oxygenase-1 mediated cytoprotection against liver ischemia and reperfusion injury: inhibition of type-1 interferon signaling. Transplantation. 2007;83:1628–34. doi: 10.1097/01.tp.0000266917.39958.47. [DOI] [PubMed] [Google Scholar]
- 13.Ma LL, Gao X, Liu L, et al. CpG oligodeoxynucleotide triggers the liver inflammatory reaction and abrogates spontaneous tolerance. Liver Transpl. 2009;15:915–23. doi: 10.1002/lt.21771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee KS, Scanga CA, Bachelder EM, et al. TLR2 synergizes with both TLR4 and TLR9 for induction of the MyD88-dependent splenic cytokine and chemokine response to Streptococcus pneumoniae. Cell Immunol. 2007;245:103–10. doi: 10.1016/j.cellimm.2007.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Romics L, Jr, Dolganiuc A, Velayudham A, et al. Toll-like receptor 2 mediates inflammatory cytokine induction but not sensitization for liver injury by Propioni- bacterium acnes. J Leukoc Biol. 2005;78:1255–64. doi: 10.1189/jlb.0804448. [DOI] [PubMed] [Google Scholar]
- 16.Velayudham A, Hritz I, Dolganiuc A, et al. Critical role of toll-like receptors and the common TLR adaptor, MyD88, in induction of granulomas and liver injury. J Hepatol. 2006;45:813–24. doi: 10.1016/j.jhep.2006.06.017. [DOI] [PubMed] [Google Scholar]
- 17.Kawai T, Akira S. TLR signaling. Semin Immunol. 2007;19:24–32. doi: 10.1016/j.smim.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 18.Moynagh PN. TLR signalling and activation of IRFs: revisiting old friends from the NF-kappaB pathway. Trends Immunol. 2005;26:469–76. doi: 10.1016/j.it.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 19.Hritz I, Mandrekar P, Velayudham A, et al. The critical role of toll-like receptor (TLR) 4 in alcoholic liver disease is independent of the common TLR adapter MyD88. Hepatology. 2008;48:1224–31. doi: 10.1002/hep.22470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhai Y, Qiao B, Gao F, et al. Type I, but not type II, interferon is critical in liver injury induced after ischemia and reperfusion. Hepatology. 2008;47:199–206. doi: 10.1002/hep.21970. [DOI] [PubMed] [Google Scholar]
- 21.Lim R, Knight B, Patel K, et al. Antiproliferative effects of interferon alpha on hepatic progenitor cells in vitro and in vivo. Hepatology. 2006;43:1074–83. doi: 10.1002/hep.21170. [DOI] [PubMed] [Google Scholar]
- 22.Honda K, Yanai H, Negishi H, et al. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature. 2005;434:772–7. doi: 10.1038/nature03464. [DOI] [PubMed] [Google Scholar]
- 23.Schafer SL, Lin R, Moore PA, et al. Regulation of type I interferon gene expression by interferon regulatory factor-3. J Biol Chem. 1998;273:2714–20. doi: 10.1074/jbc.273.5.2714. [DOI] [PubMed] [Google Scholar]
- 24.Sato M, Hata N, Asagiri M, et al. Positive feedback regulation of type I IFN genes by the IFN-inducible transcription factor IRF-7. FEBS Lett. 1998;441:106–10. doi: 10.1016/s0014-5793(98)01514-2. [DOI] [PubMed] [Google Scholar]
- 25.Itakura M, Tokuda A, Kimura H, et al. Blockade of secondary lymphoid tissue chemokine exacerbates Propionibacterium acnes-induced acute lung inflammation. J Immunol. 2001;166:2071–9. doi: 10.4049/jimmunol.166.3.2071. [DOI] [PubMed] [Google Scholar]
- 26.Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol. 2009;27:519–50. doi: 10.1146/annurev.immunol.021908.132612. [DOI] [PubMed] [Google Scholar]
- 27.Cosgrove BD, Cheng C, Pritchard JR, et al. An inducible autocrine cascade regulates rat hepatocyte proliferation and apoptosis responses to tumor necrosis factor-alpha. Hepatology. 2008;48:276–88. doi: 10.1002/hep.22335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Benveniste EN, Qin H. Type I interferons as anti-inflammatory mediators. Sci STKE 2007. 2007:pe70. doi: 10.1126/stke.4162007pe70. [DOI] [PubMed] [Google Scholar]
- 29.Honda K, Ohba Y, Yanai H, et al. Spatiotemporal regulation of MyD88-IRF-7 signalling for robust type-I interferon induction. Nature. 2005;434:1035–40. doi: 10.1038/nature03547. [DOI] [PubMed] [Google Scholar]
- 30.Yoneyama H, Matsuno K, Zhang Y, et al. Regulation by chemokines of circulating dendritic cell precursors, and the formation of portal tract-associated lymphoid tissue, in a granulomatous liver disease. J Exp Med. 2001;193:35–49. doi: 10.1084/jem.193.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu YJ. IPC: professional type 1 interferon-producing cells and plasmacytoid dendritic cell precursors. Annu Rev Immunol. 2005;23:275–306. doi: 10.1146/annurev.immunol.23.021704.115633. [DOI] [PubMed] [Google Scholar]
- 32.Yoneyama H, Narumi S, Zhang Y, et al. Pivotal role of dendritic cell-derived CXCL10 in the retention of T helper cell 1 lymphocytes in secondary lymph nodes. J Exp Med. 2002;195:1257–66. doi: 10.1084/jem.20011983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang Z, Wang M, Carr BI. The inhibitory effect of interleukin 1beta on rat hepatocyte DNA synthesis is mediated by nitric oxide. Hepatology. 1998;28:430–5. doi: 10.1002/hep.510280221. [DOI] [PubMed] [Google Scholar]
- 34.Wan L, Lin CW, Lin YJ, et al. Type I IFN induced IL1-Ra expression in hepatocytes is mediated by activating STAT6 through the formation of STAT2: STAT6 heterodimer. J Cell Mol Med. 2008;12:876–88. doi: 10.1111/j.1582-4934.2008.00143.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Naveau S, Emilie D, Borotto E, et al. Interleukin-1 receptor antagonist plasma concentration is specifically increased by alpha-2A-interferon treatment. J Hepatol. 1997;27:272–5. doi: 10.1016/s0168-8278(97)80171-7. [DOI] [PubMed] [Google Scholar]
- 36.Iizasa H, Yoneyama H, Mukaida N, et al. Exacerbation of granuloma formation in IL-1 receptor antagonist-deficient mice with impaired dendritic cell maturation associated with Th2 cytokine production. J Immunol. 2005;174:3273–80. doi: 10.4049/jimmunol.174.6.3273. [DOI] [PubMed] [Google Scholar]
- 37.Shinoda M, Tilles AW, Kobayashi N, et al. A bioartificial liver device secreting interleukin-1 receptor antagonist for the treatment of hepatic failure in rats. J Surg Res. 2007;137:130–40. doi: 10.1016/j.jss.2006.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.